1.本发明属于固相萃取技术领域,具体涉及一种固相萃取填料及其制备方法。
背景技术:
2.固相萃取技术是指利用固体吸附剂对液体样品中的目标物进行吸附并富集,或对液体样品中的杂质进行吸附,从而达到将目标物与杂质进行分离的技术。固相萃取技术作为样品的前处理技术,在残留检测中得到越来越广泛的应用。
3.目前的固相萃取技术普遍通过范德华力或静电力等物理吸附完成,这些物理吸附的相互作用力很小,使得被吸附的目标物易被较弱的溶剂洗脱,因此,在使用时需十分注意淋洗液的洗脱能力甚至需要控制淋洗液的用量。在残留检测过程中,由于目标物残留量极低,基质复杂,杂质多种并且含量高,许多杂质在固相萃取柱中与目标物分离效果差,其纯化效果极为有限;纯化后的上机液杂质含量高,影响色谱柱效率,也可能降低检测器对目标物的响应值。
4.为了尽量除去杂质,本领域技术人员开发了多种固相净化的方法。
5.cn112730672a公开了一种土壤中有机膦农药残留量的快速测定方法。使用了c18固相萃取小柱与hlb固相萃取小柱串联的方法,对提取液进行净化,之后再分别进行洗脱、浓缩,气相色谱-电子捕获检测器进行测定。该发明使用了两支固相萃取柱串联,成本较高,由于两支固相萃取柱的性能完全不同,需要使用两种洗脱液洗脱,步骤繁琐,两种洗脱液在不同固相萃取柱中的洗脱能力不同,部分目标物可能无法完全洗脱,造成检测值偏低。cn105032381b公开了一种复合分子印迹固相萃取柱及其制备方法,但是分子印迹技术选择性太强,其应用范围窄,难以在通用检测中大量推广。
技术实现要素:
6.为了克服上述现有技术的缺陷,本发明所要解决的技术问题是:如何提供一种与操作简单且分离效果好的固相萃取填料及其制备方法。
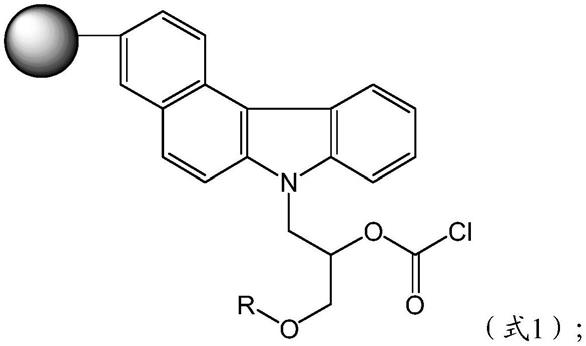
7.为了解决上述技术问题,本发明采用的技术方案为:一种固相萃取填料,所述固相萃取填料为苯并咔唑基甲氧羰酰氯改性超顺磁聚合物微球,所述固相萃取填料的结构通式如式1所示:
[0008][0009]
其中,r为甲基、乙基、异丙基、叔丁基和正丁基中的任意一种。
[0010]
本发明采用的另一种技术方案为:所述的固相萃取填料的制备方法,包括以下步骤:
[0011]
s1、按重量份计,将100份单分散超顺磁交联聚苯乙烯微球和5-9份1,4-丁内酯混合冷却至0-10℃;加入9-14份无水三氯化铝和60-80份硝基苯,升温至170-180℃,反应后降温至0-10℃;调节ph至体系ph=1-2,调节完毕后搅拌,得到悬浊液,将悬浊液转入砂芯玻璃层析柱中淋洗,得浅红色微球;
[0012]
s2、按重量份计,将15-23份浓盐酸和100份n,n-二甲基甲酰胺混合,加热至70-80℃,加入8-13份苯肼,升温至100℃回流;加入所述浅红色微球,保持温度继续反应,反应后降温至20-30℃,得冷却液;将冷却液中的固体洗涤后,抽干溶剂,得到微球;向微球中加入1倍体积的1,3,5-三甲苯和15-25份1,4-四氯苯醌,升温至135-140℃反应,反应后再降温至20-30℃,调节ph至体系ph=8-9,得到悬浊液;将悬浊液全部转入砂芯玻璃层析柱中淋洗,淋洗后加入3倍体积的乙醇,常温真空干燥过夜,得黄色微球;
[0013]
s3、按重量份计,将100份所述黄色微球与1倍体积的乙醇混合,冷却至0-5℃,加入3-12.5份40%naoh溶液,再滴加15-30份烷基缩水甘油醚,升温至55-60℃,反应3-5小时,再冷却至20-25℃,得到悬浊液;将悬浊液中的固体洗涤后,加入1倍体积的无水二氯甲烷,降温至0-5℃;再加入4-6份三光气、0.05份n-甲基咪唑和0.1份n,n-二甲基-4-吡啶胺,在0-5℃下避光搅拌4小时后升温至32-35℃,继续反应,反应后以1倍体积的无水二氯甲烷洗涤5次后过滤,常温真空干燥过夜,得固相萃取填料。
[0014]
其中,所述单分散超顺磁交联聚苯乙烯微球的直径为50μm,外观及官能团结构如式2所示:
[0015][0016]
所述微球的超顺磁性由内部含有的超顺磁四氧化三铁纳米粒子提供,超顺磁四氧化三铁纳米粒子在微球内部分布均匀,超顺磁四氧化三铁纳米颗粒被交联聚苯乙烯紧密包裹而形成直径50μm的单分散交联聚苯乙烯微球。
[0017]
本发明的有益效果在于:本发明提供的固相萃取填料,可在常温下迅速与伯胺和仲胺进行反应,将胺基化合物与基质有效分离,在对含伯胺和仲胺的农药残留检测中实际
应用效果良好,检测周期短,灵敏度高,检测限低,回收率高,不会对设备造成污染。
具体实施方式
[0018]
为详细说明本发明的技术内容、所实现目的及效果,以下结合实施方式予以说明。
[0019]
本发明提供一种固相萃取填料,固相萃取填料为苯并咔唑基甲氧羰酰氯改性超顺磁聚合物微球,固相萃取填料的结构通式如式1所示:
[0020][0021]
其中r为甲基、乙基、异丙基、叔丁基和正丁基中的任意一种。
[0022]
从上述描述可知,本发明的有益效果在于:固相萃取填料中的苯并咔唑基甲氧羰酰氯为胺基保护基,可迅速与含胺基的化合物结合形成牢固的共价键;随后利用固相萃取填料的磁性,使用磁场将已结合了胺基化合物的固相萃取填料和基质分离开来,分离后以10%哌啶乙腈溶液将胺基化合物从固相萃取填料中释放出来,即可完成分离;分离过程中,结合了胺基化合物的固相萃取填料耐各类中性溶剂洗涤,能将杂质去除干净,最终使得分离后的进样液的杂质含量低,检测效果好。
[0023]
上述固相萃取填料的制备方法,包括以下步骤:
[0024]
s1、按重量份计,将100份单分散超顺磁交联聚苯乙烯微球和5-9份1,4-丁内酯混合冷却至0-10℃;加入9-14份无水三氯化铝和60-80份硝基苯,升温至170-180℃,反应后降温至0-10℃;调节ph至体系ph=1-2,调节完毕后搅拌,得到悬浊液,将悬浊液转入砂芯玻璃层析柱中淋洗,得浅红色微球;
[0025]
s2、按重量份计,将15-23份浓盐酸和100份n,n-二甲基甲酰胺混合,加热至70-80℃,加入8-13份苯肼,升温至100℃回流;加入所述浅红色微球,保持温度继续反应,反应后降温至20-30℃,得冷却液;将冷却液中的固体洗涤后,抽干溶剂,得到微球;向微球中加入1倍体积的1,3,5-三甲苯和15-25份1,4-四氯苯醌,升温至135-140℃反应,反应后再降温至20-30℃,调节ph至体系ph=8-9,得到悬浊液;将悬浊液全部转入砂芯玻璃层析柱中淋洗,淋洗后加入3倍体积的乙醇,常温真空干燥过夜,得黄色微球;
[0026]
s3、按重量份计,将100份所述黄色微球与1倍体积的乙醇混合,冷却至0-5℃,加入3-12.5份40%naoh溶液,再滴加15-30份烷基缩水甘油醚,升温至55-60℃,反应3-5小时,再冷却至20-25℃,得到悬浊液;将悬浊液中的固体洗涤后,加入1倍体积的无水二氯甲烷,降温至0-5℃;再加入4-6份三光气、0.05份n-甲基咪唑和0.1份n,n-二甲基-4-吡啶胺,在0-5℃下避光搅拌4小时后升温至32-35℃,继续反应,反应后以1倍体积的无水二氯甲烷洗涤5
次后过滤,常温真空干燥过夜,得固相萃取填料。
[0027]
从上述描述可知,本发明的有益效果在于:本发明提供的固相萃取填料的制备方法,操作精确,能有效制备出本发明的固相萃取填料,制备得到的固相萃取填料灵敏度高,检测周期短,实际应用效果良好。
[0028]
进一步地,单分散超顺磁交联聚苯乙烯微球的直径为50μm,外观及官能团结构如式2所示:
[0029][0030]
从上述描述可知,使用单分散超顺磁交联聚苯乙烯微球作为改性基球,是因为该改性基球在磁场中可带磁性,一旦撤掉磁场改性基球即不带磁性,利用这一特点,可方便地使用磁铁等永磁体将固相萃取填料吸附于容器壁,待测物基质则由于重力作用沉淀在容器底部,达到物理分离的目的。此操作不需要专门的设备,高效简便。
[0031]
进一步地,步骤(1)中滴加1mol/l盐酸溶液时,需要将1mol/l盐酸溶液预先冷却至0℃,并且滴加过程中保证温度不超过10℃。
[0032]
从上述描述可知,通过控制盐酸溶液的温度,可以精确控制混合物的反应温度,使得反应顺利进行。
[0033]
进一步地,步骤(1)中淋洗的方式为从上至下依次以3倍体积的40%乙醇溶液,3倍体积的乙醇,3倍体积的n,n-二甲基甲酰胺淋洗。
[0034]
进一步地,步骤(2)中淋洗的方式为从上至下依次以10倍体积的40%乙醇溶液、3倍体积的n,n-二甲基甲酰胺淋洗。
[0035]
进一步地,步骤(2)中洗涤的方式为以1倍体积无水乙醇洗涤3次后,再以1倍体积1,3,5-三甲苯洗涤3次;
[0036]
进一步地,步骤(3)中洗涤的方式为以去离子水洗涤微球至ph=7-8,以1倍体积的无水乙醇洗涤3次,再以1倍体积的无水二氯甲烷洗涤3次。
[0037]
从上述描述可知,在不同制备步骤中选用不同的溶剂进行洗涤或淋洗,有利于杂质与目标物物分离,提高目标物的纯度。
[0038]
进一步地,所述的固相萃取填料的制备方法,包括以下步骤:
[0039]
s1、按重量份计,将100份粒径单分散超顺磁交联聚苯乙烯微球和5-9份1,4-丁内酯混合,冷却至0-10℃;加入9-14份无水三氯化铝和60-80份硝基苯,升温至170-180℃,反应6-12小时后降温至0-10℃;滴加预先冷却至0℃的1mol/l盐酸溶液至体系ph=1-2,保持温度不超过10℃,滴加完毕后搅拌1小时,得到悬浊液,将悬浊液全部转入砂芯玻璃层析柱中,从上至下依次以3倍体积40%乙醇溶液,3倍体积乙醇,3倍体积n,n-二甲基甲酰胺淋洗,得浅红色微球;
[0040]
s2、按重量份计,将15-23份浓盐酸和100份n,n-二甲基甲酰胺混合,加热至70-80℃,加入8-13份苯肼,升温至100℃回流;加入所述浅红色微球,保持温度继续反应4-6小时,再降温至20-30℃,得冷却液;将冷却液以1倍体积无水乙醇洗涤3次后,再以1倍体积1,3,5-三甲苯洗涤3次,抽干溶剂,得到微球;向微球中加入1倍体积的1,3,5-三甲苯和15-25份1,4-四氯苯醌,升温至135-140℃,反应5-8小时,再降温至20-30℃,调节ph至体系ph=8-9,得
到悬浊液;将悬浊液全部转入砂芯玻璃层析柱中,从上至下依次以10倍体积40%乙醇溶液,3倍体积n,n-二甲基甲酰胺淋洗,淋洗后加入3倍体积的乙醇,常温真空干燥过夜,得黄色微球;
[0041]
s3、按重量份计,将100份所述黄色微球与乙醇混合,冷却至0-5℃,加入3-12.5份40%naoh溶液,再滴加15-30份烷基缩水甘油醚,升温至55-60℃,反应3-5小时,再冷却至20-25℃,得到悬浊液;将悬浊液中的固体以去离子水洗涤微球至ph=7-8,以1倍体积无水乙醇洗涤3次,再以1倍体积无水二氯甲烷洗涤3次后,加入1倍体积的无水二氯甲烷,降温至0-5℃;再加入4-6份三光气、0.05份n-甲基咪唑和0.1份n,n-二甲基-4-吡啶胺,在0-5℃下避光搅拌4小时后升温至32-35℃,继续反应5-7小时;反应完成后以1倍体积的无水二氯甲烷洗涤5次后过滤,常温真空干燥过夜,得固相萃取填料。
[0042]
本发明的实施例一为:
[0043]
一种固相萃取填料的制备方法,包含以下步骤:
[0044]
s1、在四口瓶中加入100g粒径为50μm的单分散超顺磁交联聚苯乙烯微球和5g 1,4-丁内酯,在搅拌条件下冷却至10℃,分批次加入9g无水三氯化铝和60g硝基苯,升温至170℃,反应12小时后降温至0℃;缓慢滴加预先冷却至0℃的1mol/l盐酸溶液至体系ph=2,保持温度不超过10℃,滴加完毕后搅拌1小时;将悬浊液全部转入砂芯玻璃层析柱中,从上至下依次以3倍体积40%乙醇溶液,3倍体积乙醇,3倍体积n,n-二甲基甲酰胺淋洗,得浅红色微球;
[0045]
s2、在四口瓶中加入15g浓盐酸和100g n,n-二甲基甲酰胺,加热至80℃,缓慢加入8g苯肼,升温至100℃回流2小时后,加入浅红色微球,保持温度继续反应4小时;降温至20℃,以1倍体积无水乙醇洗涤3次后,再以1倍体积1,3,5-三甲苯洗涤3次;抽干溶剂,向固体中加入1倍体积1,3,5-三甲苯和15g 1,4-四氯苯醌,升温至135℃,反应8小时;降温至30℃,加入1mol/l naoh至体系ph=9,将悬浊液全部转入砂芯玻璃层析柱中,从上至下依次以10倍体积40%乙醇溶液,3倍体积n,n-二甲基甲酰胺淋洗,3倍体积乙醇,置于真空干燥箱中,常温真空干燥过夜,得黄色微球;
[0046]
s3、将100g黄色微球,1倍体积乙醇搅拌后冷却至0℃,加入7.5g 40%naoh溶液,缓慢滴加15g乙基缩水甘油醚,升温至55℃,反应5小时;冷却至25℃,以去离子水洗涤微球至ph=7,以1倍体积无水乙醇洗涤3次,再以1倍体积无水二氯甲烷洗涤3次后,加入1倍体积无水二氯甲烷,降温至0℃,迅速加入4g三光气,加入0.05g n-甲基咪唑和0.1g n,n-二甲基-4-吡啶胺,在0℃下避光搅拌4小时后缓慢升温至32℃,继续反应7小时,以1倍体积无水二氯甲烷洗涤5次后过滤,置于真空干燥箱中,常温真空干燥过夜,得固相萃取填料a。
[0047]
本发明的实施例二为:
[0048]
一种固相萃取填料的制备方法,包含以下步骤:
[0049]
s1、在四口瓶中加入100g粒径为50μm的单分散超顺磁交联聚苯乙烯微球和9g 1,4-丁内酯,在搅拌条件下冷却至0℃,分批次加入14g无水三氯化铝和80g硝基苯,升温至180℃,反应6小时后降温至10℃;缓慢滴加预先冷却至0℃的1mol/l盐酸溶液至体系ph=1,保持温度不超过10℃,滴加完毕后搅拌1小时;将悬浊液全部转入砂芯玻璃层析柱中,从上至下依次以3倍体积40%乙醇溶液,3倍体积乙醇,3倍体积n,n-二甲基甲酰胺淋洗,得浅红色微球;
[0050]
s2、在四口瓶中加入23g浓盐酸和100g n,n-二甲基甲酰胺,加热至70℃,缓慢加入13g苯肼,升温至100℃回流1小时后,加入浅红色微球,保持温度继续反应6小时;降温至20℃,以1倍体积无水乙醇洗涤3次后,再以1倍体积1,3,5-三甲苯洗涤3次;抽干溶剂,向固体中加入1倍体积1,3,5-三甲苯和25g 1,4-四氯苯醌,升温至140℃,反应5小时;降温至20℃,加入1mol/l naoh至体系ph=8.5,将悬浊液全部转入砂芯玻璃层析柱中,从上至下依次以10倍体积40%乙醇溶液,3倍体积n,n-二甲基甲酰胺淋洗,3倍体积乙醇,置于真空干燥箱中,常温真空干燥过夜,得黄色微球;
[0051]
s3、将100g黄色微球,1倍体积乙醇搅拌后冷却至5℃,加入12.5g 40%naoh溶液,缓慢滴加30g异丙基缩水甘油醚,升温至60℃,反应3小时;冷却至20℃,以去离子水洗涤微球至ph=8,以1倍体积无水乙醇洗涤3次,再以1倍体积无水二氯甲烷洗涤3次后,加入1倍体积无水二氯甲烷,降温至5℃,迅速加入6g三光气,加入0.05g n-甲基咪唑和0.1g n,n-二甲基-4-吡啶胺,在5℃下避光搅拌4小时后缓慢升温至35℃,继续反应5小时,以1倍体积无水二氯甲烷洗涤5次后过滤,置于真空干燥箱中,常温真空干燥过夜,得固相萃取填料b。
[0052]
本发明的实施例三为:
[0053]
一种固相萃取填料的制备方法,包含以下步骤:
[0054]
s1、在四口瓶中加入100g粒径为50μm的单分散超顺磁交联聚苯乙烯微球和7g 1,4-丁内酯,在搅拌条件下冷却至5℃,分批次加入12g无水三氯化铝和70g硝基苯,升温至175℃,反应9小时后降温至6℃;缓慢滴加预先冷却至0℃的1mol/l盐酸溶液至体系ph=2,保持温度不超过10℃,滴加完毕后搅拌1小时;将悬浊液全部转入砂芯玻璃层析柱中,从上至下依次以3倍体积40%乙醇溶液,3倍体积乙醇,3倍体积n,n-二甲基甲酰胺淋洗,得浅红色微球;
[0055]
s2、在四口瓶中加入20g浓盐酸和100g n,n-二甲基甲酰胺,加热至77℃,缓慢加入11g苯肼,升温至100℃回流1小时后,加入浅红色微球,保持温度继续反应5小时;降温至25℃,以1倍体积无水乙醇洗涤3次后,再以1倍体积1,3,5-三甲苯洗涤3次;抽干溶剂,向固体中加入1倍体积1,3,5-三甲苯和21g 1,4-四氯苯醌,升温至140℃,反应6小时;降温至25℃,加入1mol/l naoh至体系ph=8,将悬浊液全部转入砂芯玻璃层析柱中,从上至下依次以10倍体积40%乙醇溶液,3倍体积n,n-二甲基甲酰胺淋洗,3倍体积乙醇,置于真空干燥箱中,常温真空干燥过夜,得黄色微球;
[0056]
s3、将100g黄色微球,1倍体积乙醇搅拌后冷却至3℃,加入10g 40%naoh溶液,缓慢滴加25g丁基缩水甘油醚,升温至55℃,反应5小时;冷却至20℃,以去离子水洗涤微球至ph=8,以1倍体积无水乙醇洗涤3次,再以1倍体积无水二氯甲烷洗涤3次后,加入1倍体积无水二氯甲烷,降温至0℃,迅速加入5g三光气,加入0.05g n-甲基咪唑和0.1g n,n-二甲基-4-吡啶胺,在0℃下避光搅拌4小时后缓慢升温至34℃,继续反应7小时,以1倍体积无水二氯甲烷洗涤5次后过滤,置于真空干燥箱中,常温真空干燥过夜,得固相萃取填料c。
[0057]
对实施例1-3的甲氧羰酰氯含量进行测定:
[0058]
测定方式为:在50ml离心管中加入1.000g实施例1-3中的固相萃取填料、15ml乙醇和15ml 0.1mol/l的二正丁胺水溶液,混合振荡1小时后过滤;用250ml锥形瓶收集滤液,用20ml 50%乙醇溶液洗涤离心管和固相萃取填料,合并洗涤液和滤液;以0.5ml 5%铬酸钾水溶液为指示剂,使用0.02mol/l硝酸银溶液滴定洗涤液和滤液至桔红色,30秒内不消失确
定为终点。测定结果如表1所示。
[0059]
表1
[0060]
名称甲氧羰酰氯含量(mmol/g)固相萃取填料a0.24固相萃取填料b0.47固相萃取填料c0.36
[0061]
从上述数据可知,本发明的固相萃取填料的制备方法,能够有效地使用苯并咔唑基甲氧羰酰氯改性单分散超顺磁交联聚苯乙烯微球,得到一种苯并咔唑基甲氧羰酰氯改性超顺磁聚合物微球固相萃取填料。
[0062]
将实施例1得到的固相萃取填料a应用于大豆中嗪草酮残留的测定,包含以下步骤:
[0063]
s1、前处理:在12支50ml离心管中分别称取5.0g大豆粉末样品,以两个样品为平行,分别加入0ng、5ng、10ng、25ng、100ng和250ng嗪草酮标准溶液,旋紧离心管,涡旋振荡10分钟后静置24小时。再分别依次加入15ml乙腈,2ml 5%硼酸盐缓冲溶液(ph=9.0)和50mg固相萃取填料a,旋紧离心管,振荡30分钟;将一块磁铁贴着离心管壁,轻微振摇离心管,固相萃取填料a将被磁铁吸引在离心管壁聚集;弃去残渣和溶液,分别以15ml去离子水,15ml无水乙醇,15ml正己烷,15ml二氯甲烷,15ml乙腈洗涤,转移至6ml空管柱中,以烧结pp垫片压实后,5.0ml 10%哌啶乙腈溶液洗脱,洗脱液以0.22μm滤膜过滤。
[0064]
s2、滤液在高效液相色谱-串联质谱中进行检测:
[0065]
液相色谱条件:色谱柱:acquity uplc beh c18柱,50mm*2.1mm,1.7μm;流动相:乙腈-0.1%甲酸水,梯度条件如表2所示;流速:0.2ml/min;进样量:5μl;柱温:30℃。
[0066]
质谱条件:质谱型号:waters quattro ultima pt质谱仪;监测模式:多反应监测(mrm);质谱设备条件见表3;嗪草酮的多反应监测参数见表4。
[0067]
表2
[0068]
洗脱时间(min)乙腈(%)0.1%甲酸水(%)0208038020480206208072080
[0069]
表3
[0070]
电离方式esi 毛细管电压(kv)3.0离子源温度(℃)120去溶剂温度(℃)350锥孔气流氮气,100l/h去溶剂气流氮气,600l/h碰撞电压氩气,2.4*10-6pa
[0071]
表4
[0072][0073]
测得大豆样品中嗪草酮残留的平均回收率见表5。
[0074]
表5
[0075][0076]
从上述数据可知,本发明所得的固相萃取填料,实际应用效果良好,检测周期短,使用方便,灵敏度高,检测限低,回收率高,不会对设备造成污染。
[0077]
综上所述,本发明所述的固相萃取填料通过使用苯并咔唑基甲氧羰酰氯改性单分散超顺磁交联聚苯乙烯微球上得到一种苯并咔唑基甲氧羰酰氯改性超顺磁聚合物微球固相萃取填料。其中苯并咔唑基甲氧羰酰氯为胺基保护基,可在常温下、碱性缓冲液中迅速与含胺基的化合物结合形成牢固的共价键,将胺基化合物从复杂的基质中提取出来;随后利用固相萃取填料的超顺磁交联聚苯乙烯微球基团的磁性,使用磁场将已结合了胺基化合物的固相萃取填料和基质分离开来;再将固相萃取填料用水去除水溶性盐和水溶性有机物、非极性溶剂去除非极性有机物、极性溶剂去除极性有机物,最大限度除去各种杂质;最后以10%哌啶乙腈溶液将胺基化合物从固相萃取填料中释放出来,经定容过滤后即可上机测试。
[0078]
本发明所述的固相萃取填料,克服了常规固相萃取填料由于物理吸附无法使用强溶剂淋洗的缺陷,以共价键与基质中的胺基化合物结合,耐各类中性溶剂洗涤,其杂质去除彻底,最终进样液的杂质含量低,可高效防止色谱设备及质谱设备被杂质污染,延长设备的维护周期,降低设备的维护成本,提高设备的使用年限。
[0079]
本发明所述固相萃取填料在与胺基化合物结合后,利用苯并咔唑甲氧羰基不耐哌啶的原理,在常温下迅速彻底地将胺基化合物释放,所释放的胺基化合物与原胺基化合物结构完全相同,所使用的溶液不含无机盐和其他不挥发物质等,不损伤质谱设备,可直接进入质谱进行定量分析。
[0080]
本发明所述的固相萃取填料,使用超顺磁聚合物微球作为改性基球,超顺磁聚合物微球的特点是在磁场中可带磁性,一旦撤掉磁场,超顺磁聚合物微球即不带磁性,利用这一特点,可方便地使用磁铁等永磁体将固相萃取填料吸附于容器壁,待测物基质则由于重
力作用沉淀在容器底部,达到物理分离的目的。此操作不需要专门的设备,高效简便。
[0081]
本发明所述的固相萃取填料,可在常温下迅速与伯胺和仲胺进行反应,在对含伯胺和仲胺的农药残留检测中实际应用效果良好,检测周期短,使用方便,灵敏度高,检测限低,回收率高,不会对设备造成污染。
[0082]
本发明所述的固相萃取填料,在使用完毕后以各种溶剂洗涤除去污染物和体系中的水分后以氯化试剂如亚硫酰氯等进行氯化再生,性能可恢复,实际应用中可重复使用20次以上性能无明显变化,大大降低使用成本。
[0083]
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书内容所作的等同变换,或直接或间接运用在相关的技术领域,均同理包括在本发明的专利保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。