羟基琥珀酰亚胺,搅 拌后获得混合溶液c;将混合溶液c和cs-da溶液混合,得到混合溶液d进行反应,将反应后的溶液透析 后冻干,获得cdl;
11.步骤3,将癸二酸和聚乙二醇反应后获得混合物e;在混合物e中加入甘油,反应后生成混合物f,将 混合物f纯化并干燥后获得pegs共聚物;
12.将pegs共聚物、对羧基苯甲醛和4-羧基苯基硼酸频哪醇酯在无水dmf中混合,形成混合物g;将1-(3
‑ꢀ
二甲基丙基)-3-乙基碳二亚胺盐酸盐和4-二甲氨基吡啶加入至混合物g中,反应后,将反应产物沉淀,将沉 淀产物纯化,将纯化后的产物透析、冷冻干燥后,获得ppb;
13.步骤4,将go和多巴胺盐酸盐溶解在tris-hcl缓冲液中,超声处理后搅拌,将搅拌获得混合物过滤纯 化和分离后,获得反应产物,将反应产物洗涤后获得rgo@pda溶液,将rgo@pda溶液干燥后获得 rgo@pda共聚物;
14.步骤5,将ppb溶于水中,获得ppb溶液;将rgo@pda共聚物和二甲双胍加入至ppb溶液中,获得 rgo@pda修饰且负载有二甲双胍的ppb溶液;
15.步骤6,将cdl溶于水中,获得cdl溶液;将rgo@pda修饰且负载有二甲双胍的ppb溶液和cdl 溶液混合均匀后,获得具有ph/葡萄糖双响应性的水凝胶。
16.本发明的进一步改进在于:
17.优选的,步骤1中,混合溶液a中,壳聚糖溶液中壳聚糖和二氢咖啡酸的质量比为0.523:(0.3-1.2); 壳聚糖溶液的ph值为5;
18.所述1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和壳聚糖溶液中壳聚糖的质量比为(0.62-2.48):0.523。
19.优选的,步骤1中,所述ph值为5.0~6.0;
20.步骤2中,l-精氨酸溶液的ph值为5.0~6.0。
21.优选的,步骤2中,混合溶液c中l-精氨酸、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和n-羟基琥 珀酰亚胺的混合质量比为(0.28-1.13):(0.62-2.48):(0.37-1.5)。
22.优选的,步骤3中,癸二酸和聚乙二醇的混合质量比为3.63:9;二者的反应过程为:癸二酸和聚乙二醇 混合后,在125℃下反应12h,然后在5kpa下反应24h,获得混合物e;
23.步骤3中,加入的甘油和癸二酸的质量比为3.3:3.63;加入甘油后,反应过程为:在125℃下反应12h, 然后在5kpa下反应48h,获得混合物f;
24.混合物f纯化的过程为:将混合物f溶解在氯仿中,离心后去除上清液,将残留的溶液在乙醚中沉淀, 获得甘油封端的pegs。
25.优选的,步骤3中,pegs共聚物、对羧基苯甲醛和4-羧基苯基硼酸频哪醇酯的混合质量比为1:(0.1-0.4): (0.1-0.4);加入的1-(3-二甲基丙基)-3-乙基碳二亚胺盐酸盐、4-二甲氨基吡啶和pegs共聚物的混合质量比 为(0.67-2.68):(0.13-0.53):1;
26.混合物g的反应温度为室温,反应时间为48-96h,反应产物在乙醚中沉淀;
27.沉淀产物纯化的过程为:将沉淀产物在四氢呋喃中离心,取上清液,将上清液的ph值调至2,获得纯 化后的产物。
28.优选的,步骤4中,go和多巴胺盐酸盐溶液的混合质量比为1:1;反应产物通过水和乙醇洗涤数次获 得rgo@pda溶液,rgo@pda溶液的干燥温度为60℃,干燥时间为48h。
29.优选的,步骤5中,ppb溶液的浓度为30wt%;
30.步骤6中cdl溶液的浓度为1wt%~3wt%;rgo@pda修饰和负载有二甲双胍的ppb溶液和cdl溶液 的混合体积比为2:1。
31.一种通过上述任意一项制备方法制得的具有ph/葡萄糖双响应性释放二甲双胍的水凝胶敷料。
32.一种上述的具有ph/葡萄糖双响应性释放二甲双胍的水凝胶的应用,作为水凝胶敷料,用于促进糖尿病 伤口愈合。
33.与现有技术相比,本发明具有以下有益效果:
34.本发明涉及一种具有ph/葡萄糖双响应性二甲双胍释放的水凝胶的制备方法。该制备方法将聚多巴胺包 被的还原氧化石墨烯(rgo@pda)和二甲双胍(met)同时添加到苯甲醛和苯硼酸双修饰的聚乙二醇共聚(甘 油癸二酸酯)(ppb)溶液,随后将其与二氢咖啡酸和l-精氨酸共接枝的壳聚糖(cdl)溶液混合,通过席 夫碱和苯基硼酸酯交联形成具有ph/葡萄糖双响应性pc/go/met水凝胶。该方法原料来源广泛、制备工艺 简单、制备成本低廉,通过两条大分子链交联而成,其中一条大分子链是在壳聚糖主链上接枝二氢咖啡酸 和l-精氨酸(cdl),通过化学接枝二氢咖啡酸到主链上,增加体系的溶解度、粘附性、抗氧化性和机械强度; 将l-精氨酸接枝到壳聚糖上赋予材料以修复血管和促进血液流通的能力;另一条大分子链苯硼酸和苯甲醛 双功能化修饰的聚乙二醇共聚(甘油癸二酸酯)(ppb)和主链cdl交联时能够形成席夫碱和苯基硼酸酯的双动 态键,使体系具有ph和葡萄糖双响应性释放二甲双胍的能力;将氧化石墨烯(go)用聚多巴胺包被形成聚多 巴胺包被的还原性氧化石墨烯(rgo@pda)加入到水凝胶体系中,为体系提供了良好的导电性,促进伤口的 愈合。
35.本发明还公开了一种具有ph/葡萄糖双响应性二甲双胍释放的水凝胶,该水凝胶敷料具有ph/葡萄糖双 响应性二甲双胍释放的特性,可以促进糖尿病伤口愈合,,还具有良好的粘附性、抗菌、抗氧化、凝血和体 内止血能力。
36.进一步的,经本发明方法制得的水凝胶有以下优点:
37.(1)将二氢咖啡酸接枝到主链上,增加体系的溶解度、粘附性、抗氧化性和机械强度;
38.(2)将l-精氨酸接枝到壳聚糖上赋予材料以修复血管和促进血液流通的能力;
39.(3)两条大分子链苯硼酸和苯甲醛双功能化修饰聚乙二醇共聚(甘油癸二酸酯)(ppb)和接枝了二氢咖啡 酸和l-精氨酸的壳聚糖(cdl)交联时,能够形成席夫碱和苯基硼酸酯的双动态键,使体系具有ph和葡萄糖 双响应性释放二甲双胍的能力;
40.(4)将氧化石墨烯(go)引入水凝胶体系,增加了材料的导电性,有助于皮肤表面形成电池促进细胞迁 移,加速伤口愈合。
41.本发明还公开了一种具有ph/葡萄糖双响应性二甲双胍释放的水凝胶的应用,该水凝胶敷料具有双响应 性,可以释放有助于ii型糖尿病伤口愈合的药物,促进糖尿病伤口愈合。
附图说明
42.图1(a)是水凝胶的溶胀曲线图;图1(b)是水凝胶的降解曲线图;图1(c)是水凝胶的流变特性图。
43.图2(a)是以pc2为基础不同含量rgo@pda水凝胶的导电率;图2(b)是不同ph条件下
水凝胶对二甲双 胍的释放率;图2(c)是同ph不同葡萄糖浓度条件下水凝胶对二甲双胍的释放率;图2(d)是pc1、pc2、pc3 和pc2/gp2水凝胶的黏附能力;图2(e)是水凝胶的止血能力;图2(f)是pc1、pc2、pc3和pc2/gp2水凝胶 的dpph清除率。
44.图3是pc1、pc2、pc3和pc2/go2水凝胶对金黄色葡萄球菌和大肠杆菌的体外抗菌活性。
45.图4(a)是细胞相容性的评估,*p<0.05;图4(b)是溶血率的定量数据。
46.图5(a)是tegaderm
tm
薄膜和pc2,pc2/go2,pc2/met,pc2/go2/met水凝胶组分别在3天,7天,14 天和21天的伤口愈合情况统计图,*p<0.05;图5(b)第7天和第14天表皮再生率;图5(c)是第7天肉芽组 织厚度统计;图5(d)是第7天血管再生统计;图5(e)是第14天和第21天毛囊的相对数量统计;图5(f)是第 14天新生组织中羟脯氨酸含量,*p<0.05。
具体实施方式
47.下面结合附图和具体实施例对本发明做进一步详细描述:
48.本发明公开了一种具有ph/葡萄糖双响应性二甲双胍释放的水凝胶的制备方法,具体包括以下步骤:
49.步骤1,在1-(3-二甲基丙基)-3-乙基碳二亚胺盐酸盐(edc)的作用下,将二氢咖啡酸接枝到壳聚糖主链 上生成壳聚糖接枝二氢咖啡酸(cs-da);
50.壳聚糖接枝二氢咖啡酸的具体制备步骤包括:
51.(1a)将0.523g壳聚糖溶于45ml去离子水中,室温条件下搅拌使其充分溶解;
52.(1b)向(1a)所得的溶液中加入1m盐酸调节溶液ph至5.0左右,形成壳聚糖溶液;
53.(1c)将0.30-1.2g二氢咖啡酸加入上述(1b)所得的壳聚糖溶液中,形成混合溶液a;在混合溶液a中 快速加入0.62-2.48g的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc),充分搅拌使溶液混匀;
54.(1d)向(1c)所得溶液中在加入1m盐酸调节溶液ph值在5.0~6.0之间,搅拌8小时,得到混合溶液b;
55.(1e)用1m盐酸调节透析水至其ph=5,将(1d)所得溶液至于其中透析72小时,透析后得到壳聚糖接枝 二氢咖啡酸(cs-da)溶液。
56.(2)向步骤(1)所得的cs-da上接枝l-精氨酸,生成壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl);
57.合成cdl的具体制备步骤包括:
58.(2a)将0.28-1.13g l-精氨酸溶于20ml去离子水中,形成l-精氨酸溶液,依次加入0.62-2.48g edc和 0.37-1.5g n-羟基琥珀酰亚胺(nhs),将混合溶液搅拌30分钟以激活l-精氨酸上的羧基,获得混合溶液 c;
59.(2b)将混合溶液c加入到步骤(1)获得的cs-da溶液中,得到混合溶液d,将混合溶液d在室温下搅拌 12小时并透析3天;
60.(2c)最后将(2b)所得溶液通过冻干获得所需的壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl)。
61.(3)将对甲酰基苯甲酸和苯硼酸同时接枝到聚乙二醇共聚(甘油癸二酸酯)上生成苯甲醛苯硼酸双修饰聚 乙二醇共聚(甘油癸二酸酯);具体的,以edc作为脱水剂,4-二甲氨
基吡啶(dmap)作为催化剂,通过4
‑ꢀ
羧基苯基硼酸频哪醇酯(pba)和4-羧基苯甲醛(ba)合成苯硼酸和苯甲醛双修饰的聚乙二醇共聚(甘油癸二酸 酯)(ppb);
62.合成ppb的具体制备步骤包括:
63.(3a)将3.63g癸二酸和9g聚乙二醇(peg)添加到50ml圆底烧瓶中,在氮气气氛下,125℃反应12小 时后,将圆底烧瓶压力降至5kpa,再反应24小时,获得混合物e;
64.(3b)将3.3g甘油引入混合物e中,使混合物在氮气环境下,125℃反应12小时,然后在5kpa压力下 再反应48小时,获得混合物f;
65.(3c)将混合物f进行2次重复纯化,过程如下:溶解在氯仿中并在4500rpm下离心5分钟,之后,去 除上清液(未反应的甘油)并将残留溶液在过量预冷的乙醚中沉淀以获得聚乙二醇共聚(甘油癸二酸酯)共聚 物(pegs);
66.(3d)将(3c)所得的纯化的pegs在真空烘箱中室温干燥48小时得到干燥的pegs共聚物;
67.(3e)将1g pegs共聚物、0.1-0.4g对羧基苯甲醛(fa)和0.1-0.4g pba溶解在10ml无水dmf中形成 混合物g,然后将0.67-2.68g edc和0.13-0.53g dmap溶解在上述混合物g中;
68.(3f)将(3e)最终所得混合物置于室温下在氮气气氛下反应48-96小时,之后将混合物在过量的预冷乙醚 中沉淀;
69.(3g)将(3f)所得沉淀物进行如下纯化:将沉淀物溶解在thf(四氢呋喃)中并以4500rpm离心10分钟后, 取上清液,用1m盐酸调节上清液的ph调至2;
70.(3h)通过透析和冷冻干燥得到所需的ppb。
71.(4)利用多巴胺(da)在碱性条件下的自聚合在氧化石墨烯(go)表面制备聚多巴胺涂层,生成还原 性氧化石墨烯(rgo@pda);
72.合成rgo@pda的具体制备步骤如下:
73.(4a)将go(20mg)和多巴胺盐酸盐(20mg)溶解在40ml tris-hcl缓冲液(ph=8.5)中并超声处理30分 钟;
74.(4b)将(4a)所得混合物在室温下剧烈搅拌24小时;
75.(4c)将(4b)所得混合物通过过滤纯化和分离,并进一步用水和乙醇洗涤数次,得到rgo@pda溶液;
76.(4d)将(4c)所得的rgo@pda溶液在真空烘箱中60℃干燥48小时,得到rgo@pda共聚物。
77.(5)将ppb溶解在蒸馏水中,配制成质量浓度为30wt%的ppb溶液;
78.(6)将一定量的rgo@pda和二甲双胍依次加入到步骤(4a)配制好的ppb溶液中,使得rgo@pda和二 甲双胍的浓度均为6mg/ml,得到rgo@pda修饰和met负载的ppb溶液;
79.(7)将cdl溶解在蒸馏水,配制成质量浓度为1wt%~3wt%的cdl溶液;
80.(8)将(6)和(7)所得溶液以体积比为2:1混匀,通过席夫碱和苯基硼酸酯交联,得到有助于糖尿病伤口 愈合且具有ph/葡萄糖双响应性释放二甲双胍的水凝胶。
81.本发明所制得的具有ph/葡萄糖双响应性释放二甲双胍的水凝胶能够应用在糖尿病伤口愈合中,如伤口 敷料方面。
82.实施例1
83.(1)在1-(3-二甲基丙基)-3-乙基碳二亚胺盐酸盐(edc)的作用下将二氢咖啡酸接枝到壳聚糖主链上生成 壳聚糖接枝二氢咖啡酸(cs-da):
84.(1a)将0.523g壳聚糖溶于45ml去离子水中,室温条件下搅拌使其充分溶解;
85.(1b)向(1a)所得的溶液中加入1m盐酸调节溶液ph至5.0左右;
86.(1c)将0.591g二氢咖啡酸加入上述(1b)所得溶液中,并快速加入1.239g edc,充分搅拌使溶液混匀;
87.(1d)向(1c)所得溶液中在加入1m盐酸调节溶液ph值在5.0~6.0之间,搅拌8小时;
88.(1e)用1m盐酸调节透析水的ph=5,将(1d)所得溶液至于其中透析72小时。
89.(2a)将0.565g l-精氨酸溶于20ml去离子水中,依次加入1.239g edc和0.746g nhs,将混合溶液搅 拌30分钟以激活l-精氨酸上的羧基;
90.(2b)将(2a)所得溶液加入到步骤(1)最终所得的溶液中,将合并的溶液在室温下搅拌12小时并透析3天;
91.(2c)最后将(2b)所得溶液通过冻干获得所需的壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl)。
92.(3)以edc作为脱水剂,4-二甲氨基吡啶(dmap)作为催化剂,用4-羧基苯基硼酸频哪醇酯(pba)和4
‑ꢀ
羧基苯甲醛(ba)合成苯硼酸和苯甲醛双修饰的聚乙二醇共聚(甘油癸二酸酯)(ppb);
93.合成ppb的具体制备步骤包括:
94.(3a)将3.63g癸二酸和9g聚乙二醇(peg)添加到50ml圆底烧瓶中,在氮气气氛下,125℃反应12小 时后,将圆底烧瓶压力降至5kpa,再反应24小时;
95.(3b)将3.3g甘油引入(3a)所得混合物中,使混合物在氮气环境下,125℃反应12小时,然后在5kpa 压力下再反应48小时;
96.(3c)将(3b)反应最终所得混合物进行2次重复纯化,过程如下:溶解在氯仿中并在4500rpm下离心5 分钟,之后,去除上清液(未反应的甘油)并将残留溶液在过量预冷的乙醚中沉淀以获得聚乙二醇共聚(甘 油癸二酸酯)共聚物(pegs);
97.(3d)将(3c)所得的纯化的pegs在真空烘箱中室温干燥48小时得到干燥的pegs共聚物;
98.(3e)将1g pegs共聚物、0.206g对羧基苯甲醛(fa)和0.194g pba溶解在10ml无水dmf中形成混合 物,然后将1.34g edc和0.265g dmap溶解在上述混合物中;
99.(3f)将(3e)最终所得混合物置于室温下在氮气气氛下反应72小时,之后将混合物在过量的预冷乙醚中 沉淀;
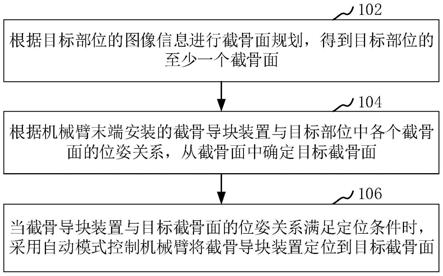
100.(3g)将(3f)所得沉淀物进行如下纯化:将沉淀物溶解在thf(四氢呋喃)中并以4500rpm离心10分钟后, 取上清液,用1m盐酸调节上清液的ph调至2;
101.(3h)通过透析和冷冻干燥得到所需的ppb。
102.(4)将ppb溶解在蒸馏水中,配制成质量浓度为30wt%的ppb溶液;
103.(5)将cdl溶解在蒸馏水,配制成质量浓度为1.5wt%的cdl溶液;
104.(6)将(4)和(5)所得溶液以体积比为2:1混匀,得到1.0wt%cdl-ppb水凝胶,将其命名为pc1。
105.实施例2
106.与实施例1不同的是,将步骤(5)中1.5wt%的cdl溶液替换成2.25wt%,制得的水凝胶命名为pc2。
107.实施例3
108.与实施例1不同的是,将步骤(5)中1.5wt%的cdl溶液替换成3wt%,制得的水凝胶命名为pc3。
109.实施例4
110.(1)在edc的作用下将二氢咖啡酸接枝到壳聚糖主链上生成壳聚糖接枝二氢咖啡酸(cs-da);
111.壳聚糖接枝二氢咖啡酸的具体制备步骤包括:
112.(1a)将0.523g壳聚糖溶于45ml去离子水中,室温条件下搅拌使其充分溶解;
113.(1b)向(1a)所得的溶液中加入1m盐酸调节溶液ph至5.0左右;
114.(1c)将0.591g二氢咖啡酸加入上述(1b)所得溶液中,并快速加入1.239g edc,充分搅拌使溶液混匀;
115.(1d)向(1c)所得溶液中在加入1m盐酸调节溶液ph值在5.0~6.0之间,搅拌8小时;
116.(1e)用1m盐酸调节透析水的ph=5,将(1d)所得溶液至于其中透析72小时。
117.(2)向步骤(1)所得的cs-da上接枝l-精氨酸,生成壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl);
118.合成cdl的具体制备步骤包括:
119.(2a)将0.565g l-精氨酸溶于20ml去离子水中,依次加入1.239g edc和0.746g nhs,将混合溶液搅 拌30分钟以激活l-精氨酸上的羧基;
120.(2b)将(2a)所得溶液加入到步骤(1)最终所得的溶液中,将合并的溶液在室温下搅拌12小时并透析3天;
121.(2c)最后将(2b)所得溶液通过冻干获得所需的壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl)。
122.(3)以edc作为脱水剂,4-二甲氨基吡啶(dmap)作为催化剂,用4-羧基苯基硼酸频哪醇酯(pba)和4
‑ꢀ
羧基苯甲醛(ba)合成苯硼酸和苯甲醛双修饰的聚乙二醇共聚(甘油癸二酸酯)(ppb);
123.合成ppb的具体制备步骤包括:
124.(3a)将3.63g癸二酸和9g聚乙二醇(peg)添加到50ml圆底烧瓶中,在氮气气氛下,125℃反应12小 时后,将圆底烧瓶压力降至5kpa,再反应24小时;
125.(3b)将3.3g甘油引入(3a)所得混合物中,使混合物在氮气环境下,125℃反应12小时,然后在5kpa 压力下再反应48小时;
126.(3c)将(3b)反应最终所得混合物进行2次重复纯化,过程如下:溶解在氯仿中并在4500rpm下离心5 分钟,之后,去除上清液(未反应的甘油)并将残留溶液在过量预冷的乙醚中沉淀以获得聚乙二醇共聚(甘 油癸二酸酯)共聚物(pegs);
127.(3d)将(3c)所得的纯化的pegs在真空烘箱中室温干燥48小时得到干燥的pegs共聚物;
128.(3e)将1g pegs共聚物、0.206g对羧基苯甲醛(fa)和0.194g pba溶解在10ml无水dmf中形成混合 物,然后将1.34g edc和0.265g dmap溶解在上述混合物中;
129.(3f)将(3e)最终所得混合物置于室温下在氮气气氛下反应72小时,之后将混合物在过量的预冷乙醚中 沉淀;
130.(3g)将(3f)所得沉淀物进行如下纯化:将沉淀物溶解在thf(四氢呋喃)中并以4500rpm离心10分钟后, 取上清液,用1m盐酸调节上清液的ph调至2;
131.(3h)通过透析和冷冻干燥得到所需的ppb。
132.(4)用氧化石墨烯(go)和多巴胺(da)生成还原性氧化石墨烯(rdo@pda);
133.合成rdo@pda的具体制备步骤如下:
134.(4a)将go(20mg)和多巴胺盐酸盐(20mg)溶解在40ml tris-hcl缓冲液(ph=8.5)中并超声处理30分 钟;
135.(4b)将(4a)所得混合物在室温下剧烈搅拌24小时;
136.(4c)将(4b)所得混合物通过过滤纯化和分离,并进一步用水和乙醇洗涤数次,得到rgo@pda溶液;
137.(4d)将(4c)所得的rgo@pda溶液在真空烘箱中60℃干燥48小时,得到rgo@pda共聚物。
138.(5)将ppb溶解在蒸馏水中,配制成质量浓度为30wt%的ppb溶液;
139.(6)将一定量的rgo@pda加入到步骤(4a)配制好的ppb溶液中,使得rgo@pda浓度为6mg/ml,得 到rgo@pda修饰的ppb溶液;
140.(7)将cdl溶解在蒸馏水,配制成质量浓度为2.25wt%的cdl溶液;
141.(8)将(6)和(7)所得溶液以体积比为2:1混匀,得到1.5wt%cdl-ppb-rgo@pda水凝胶,将其命名为 pc2/go2。
142.实施例5
143.与实施例4不同的是,将步骤(6)中多余加一定量的met,使得met的浓度为6mg/ml,制得的水凝胶 命名为pc2/go2/met。
144.本发明中拥有黏附、止血、导电、抗氧化、ph和葡萄糖双响应的有助于糖尿病伤口修复的水凝胶敷料, 性能稳定,其抗菌性能在体外抗菌测试中表现良好,机械性能和导电性在测试中均表现优异,空白凝胶和 负载二甲双胍的凝胶均对糖尿病大鼠足部创口有优于商用敷料tegaderm^tm的促进愈合作用,而且该方法 制备的水凝胶能够显著促进人脐静脉内皮细胞(huvec)的增殖以及对小鼠成纤维细胞(l929)具有良好的生 物相容性,下面结合附图及实验数据详细分析:
145.附图部分实验选取ppb浓度30wt%,cdl浓度为1.5wt%,2.25wt%,3wt%,rgo/pda和met浓度 为0.6wt%。不添加rgo/pda和met,根据水凝胶中cdl浓度为1.5wt%,2.25wt%,3wt%,依次将水 凝胶命名为pc1,pc2,pc3;使用cdl浓度为2.25wt%,ppb浓度恒定,只添加rgo/pda,生成水凝 胶pc2/go2;使用cdl浓度为2.25wt%,ppb浓度恒定,添加rgo/pda和met,成水凝胶pc2/go2/met。
146.对比实施例1、实施例2、实施例3和实施例4图1(a)的水凝胶溶胀行为测试结果表明在pc水凝胶中 随着水凝胶中cdl含量的增加,水凝胶的溶胀率逐渐降低;加入rgo@pda后的pc2/go2比pc2溶胀率 低,但比pc3溶胀率高。
147.图1(b)的水凝胶降解行为测试结果表明本方法制得的所有水凝胶都有良好的降解性能,各组分间降解差 异不明显。
148.图1(c)的水凝胶流变学测试获得的储存模量(g’)和消耗模量(g”)交叉点的时间被认为是成胶时间。当 pc水凝胶中cdl的质量百分比从1.5wt%,2.25wt%增加到3wt%,成胶时间逐渐减小,表明cdl在水凝 胶网络中浓度越高越有利于凝胶化效率。当加入组分rgo@pda后,凝胶时间进一步减小,且水凝胶的最 终强度(存储模量)也最高。
149.图2(a)为本发明制得的水凝胶的电导率的测试结果,电导率随着rgo@pda含量的增加而增加。
150.图2(b)为本发明所制得的水凝胶敷料在不同ph条件下水凝胶对二甲双胍的释放曲线,随着ph的降低 水凝胶对二甲双胍的释放含量明显增加且具有缓释行为。ph为5.5时释药率比ph 7.4时高30.4%,ph为 6.8时释药率比ph 7.4时高12.7%。ph为5.5时对二甲双胍的释放过程持续近200小时,最终释药率将近 100%。
151.图2(c)为本发明所制得的水凝胶敷料在不同葡萄糖浓度条件下水凝胶对二甲双胍的释放率,葡萄糖越高 的组分水凝胶释放的药物越多,二甲双胍的最终累积释放量比无糖组高23.3%。
152.图2(d)为本发明所制得的水凝胶敷料的黏附测试实验结果,通过实验估算了这些水凝胶对皮肤的黏附性 能。在cdl含量分别为1.5wt%,2.25wt%和3wt%的pc水凝胶中,pc2的黏附性能最强;并且加入rgo@pda 后所获得的pc2/go2进一步提高了黏附性能。
153.图2(e)为本发明所制得的水凝胶敷料的止血性能测试结果,实验显示和不采取任何措施的对照组相比, 水凝胶具有显著的降低血液损失的效果。
154.图2(f)为本发明所制得的水凝胶敷料的自由基清除实验结果,即使浓度低至0.5mg/ml的pc1水凝胶也 具有32.1%的dpph清除率,2mg/ml浓度下,所有水凝胶的dpph清除率均大于90%,而4mg/ml浓度 下,pc2和pc3的dpph清除率基本接近100%。证明了pc/go水凝胶优异的抗氧化活性。
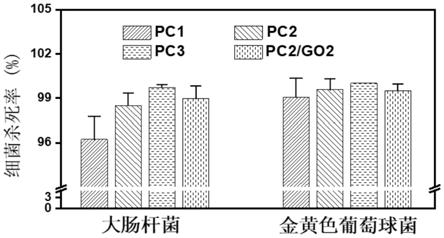
155.图3为本发明所制得的水凝胶敷料在体外对金黄色葡萄球菌和大肠杆菌的抗菌效果测试,结果表明随 着水凝胶中cdl浓度的增加,pc1、pc2和pc3对大肠杆菌的抑菌效果依次增加,pc2、pc3和pc2/go2 水凝胶组的琼脂糖培养板上几乎看不到细菌菌落。对于金黄色葡萄球菌pc水凝胶似乎有更好的抗菌效果。 除pc1水凝胶外,pc2、pc3和pc2/go2水凝胶组中几乎没有明显的菌落。证明了水凝胶良好的体外抗菌 性能。
156.图4(a)为本发明所制得的水凝胶敷料在体外对细胞相容性的结果,共培养1天、2天和3天,第1天各 组细胞无明显差异,表明所制得的水凝胶都无毒;第2天pc1、pc2、pc2/go2和pc3组细胞依次增多; 第3天与第2天结果相似,但组间结果差异更为显著,pc2/go2和pc3组结果相近都具有较高的细胞增值 促进效果。证明了水凝胶良好的细胞相容性。
157.图4(b)为本发明所制得的水凝胶敷料在体外溶血实验的结果,将阳性组(0.1%triton)的溶血率设为100%, 所有水凝胶组的溶血率均小于4%,显示了良好的血液相容性。
158.图5(a)为本发明所制得的水凝胶敷料在糖尿病创伤修复实验中的伤口闭合率统计结果经过3天的治疗 后,与tegaderm
tm
薄膜组相比,pc2,pc2/go2,pc2/met和pc2/go2/met水凝胶的伤口闭合度明显更高 (p<0.05),且在伤口修复第21天时,pc2/go2/met组愈合效果最好,几乎没有伤口痕迹,还长出皮肤上的 毛发。与tegaderm^tm薄膜对比,显示了pc2/go2/met水凝胶有更好的伤口愈合效果;各水凝胶组之间相 比,显示了pc2/go2/met组中
rgo@pda和met都对糖尿病伤口愈合有积极的促进作用。
159.图5(b)为本发明所制得的水凝胶敷料在糖尿病创伤修复实验中的表皮再生率的统计,在伤口修复的第7 天,与tegaderm
tm
薄膜组相比,pc2,pc2/go2,pc2/met和pc2/go2/met水凝胶的表皮再生率逐渐增高, 在伤口修复的第14天,除tegaderm
tm
组以外,各水凝胶组对于表皮的再生率都接近100%。显示了本发明 所制的水凝胶对于糖尿病伤口愈合中表皮的再生有良好的促进效果。
160.图5(c)为本发明所制得的水凝胶敷料在糖尿病创伤修复实验中的肉芽组织厚度统计结果,肉芽组织的厚 度可以反应创面修复的质量。与tegaderm
tm
薄膜组相比,4种水凝胶组肉芽组织厚度依次增厚(p<0.5);各 水凝胶组之间相比,pc2/go2/met组的肉芽组织最厚。显示了本发明所制的水凝胶对于糖尿病创伤修复实 验中的肉芽组织良好的促进形成效果。
161.图5(d)为本发明所制得的水凝胶敷料在糖尿病创伤修复实验中的血管再生的统计,在伤口修复第7天, 与tegaderm^tm薄膜组相比,pc2,pc2/go2,pc2/met和pc2/go2/met水凝胶的血管再生率依次增加。 显示了本发明所制的水凝胶对于糖尿病创伤修复实验中的具有优良的改善血管形成速度的效果(p<0.5)。
162.图5(e)为本发明所制得的水凝胶敷料在糖尿病创伤修复实验中的毛囊相对数量的统计,在伤口修复第7 天和21天,tegaderm^tm薄膜,pc2,pc2/go2,pc2/met和pc2/go2/met组的毛囊数量都是依次增加的, 且在第21天时,pc2/go2/met组的毛囊数量增加的比较明显。显示了本发明所制的水凝胶对于糖尿病创伤 修复实验中的具有更好的修复毛囊的效果。
163.图5(f)为本发明所制得的水凝胶敷料在糖尿病创伤修复实验中胶原蛋白的沉积情况统计。在伤口修复的 第14天,tegaderm^tm薄膜,pc2,pc2/go2,pc2/met和pc2/go2/met组的胶原沉积水平依次增加。结 果表明,本发明所制的水凝胶对于糖尿病创伤修复实验中的具有显著的促进胶原代谢的作用(p<0.5)。
164.本发明公开了一种具有ph/葡萄糖双响应性二甲双胍释放的水凝胶的制备方法及其在糖尿病伤口敷料 方面的应用。
165.实验结果表明:可以通过改变水凝胶中cdl的含量来调节本发明所制备的水凝胶流变性、溶胀率和粘 附性等。实验结果验证了双动态键交联赋予水凝胶ph和葡萄糖双响应性释放二甲双胍的特性,二氢咖啡酸 赋予水凝胶的组织黏附、止血和抗氧化性能,l-精氨酸赋予水凝胶广谱的抗菌活性,以及促进修复血管的 能力,聚多巴胺包被的还原性氧化石墨烯赋予水凝胶良好的导电能力。此外,溶血实验和l929细胞共培养 实验验证其具有良好的体外生物相容性。组织学结果:胶原代谢、肉芽组织厚度、表皮再生、毛囊数量和 血管再生以及il-6、cd31和α-sma免疫荧光染色结果证实了水凝胶良好的促进糖尿病伤口愈合的作用。 因此,该系统多功能性水凝胶在促进糖尿病伤口愈合领域有着良好的应用前景。
166.实施例6
167.(1)在edc的作用下将二氢咖啡酸接枝到壳聚糖主链上生成壳聚糖接枝二氢咖啡酸(cs-da);
168.壳聚糖接枝二氢咖啡酸的具体制备步骤包括:
169.(1a)将0.523g壳聚糖溶于45ml去离子水中,室温条件下搅拌使其充分溶解;
170.(1b)向(1a)所得的溶液中加入1m盐酸调节溶液ph至5.0左右;
171.(1c)将0.591g二氢咖啡酸加入上述(1b)所得溶液中,并快速加入1.239g edc,充分搅拌使溶液混匀;
172.(1d)向(1c)所得溶液中在加入1m盐酸调节溶液ph值在5.0~6.0之间,搅拌8小时;
173.(1e)用1m盐酸调节透析水的ph=5,将(1d)所得溶液至于其中透析72小时。
174.(2)向步骤(1)所得的cs-da上接枝l-精氨酸,生成壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl);
175.合成cdl的具体制备步骤包括:
176.(2a)将1g l-精氨酸溶于20ml去离子水中,依次加入1.239g edc和0.746g nhs,将混合溶液搅拌 30分钟以激活l-精氨酸上的羧基;
177.(2b)将(2a)所得溶液加入到步骤(1)最终所得的溶液中,将合并的溶液在室温下搅拌12小时并透析3天;
178.(2c)最后将(2b)所得溶液通过冻干获得所需的壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl)。
179.(3)以edc作为脱水剂,4-二甲氨基吡啶(dmap)作为催化剂,用4-羧基苯基硼酸频哪醇酯(pba)和4
‑ꢀ
羧基苯甲醛(ba)合成苯硼酸和苯甲醛双修饰的聚乙二醇共聚(甘油癸二酸酯)(ppb);
180.合成ppb的具体制备步骤包括:
181.(3a)将3.63g癸二酸和9g聚乙二醇(peg)添加到50ml圆底烧瓶中,在氮气气氛下,125℃反应12小 时后,将圆底烧瓶压力降至5kpa,再反应24小时;
182.(3b)将3.3g甘油引入(3a)所得混合物中,使混合物在氮气环境下,125℃反应12小时,然后在5kpa 压力下再反应48小时;
183.(3c)将(3b)反应最终所得混合物进行2次重复纯化,过程如下:溶解在氯仿中并在4500rpm下离心5 分钟,之后,去除上清液(未反应的甘油)并将残留溶液在过量预冷的乙醚中沉淀以获得聚乙二醇共聚(甘 油癸二酸酯)共聚物(pegs);
184.(3d)将(3c)所得的纯化的pegs在真空烘箱中室温干燥48小时得到干燥的pegs共聚物;
185.(3e)将1g pegs共聚物、0.2g对羧基苯甲醛(fa)和0.2g pba溶解在10ml无水dmf中形成混合物, 然后将1g edc和0.2g dmap溶解在上述混合物中;
186.(3f)将(3e)最终所得混合物置于室温下在氮气气氛下反应90小时,之后将混合物在过量的预冷乙醚中 沉淀;
187.(3g)将(3f)所得沉淀物进行如下纯化:将沉淀物溶解在thf(四氢呋喃)中并以4500rpm离心10分钟后, 取上清液,用1m盐酸调节上清液的ph调至2;
188.(3h)通过透析和冷冻干燥得到所需的ppb。
189.(4)用氧化石墨烯(go)和多巴胺(da)生成还原性氧化石墨烯(rdo@pda);
190.合成rdo@pda的具体制备步骤如下:
191.(4a)将go(20mg)和多巴胺盐酸盐(20mg)溶解在40ml tris-hcl缓冲液(ph=8.5)中并超声处理30分 钟;
192.(4b)将(4a)所得混合物在室温下剧烈搅拌24小时;
193.(4c)将(4b)所得混合物通过过滤纯化和分离,并进一步用水和乙醇洗涤数次,得到rgo@pda溶液;
194.(4d)将(4c)所得的rgo@pda溶液在真空烘箱中60℃干燥48小时,得到rgo@pda共聚物。
195.(5)将ppb溶解在蒸馏水中,配制成质量浓度为30wt%的ppb溶液;
196.(6)将一定量的rgo@pda加入到步骤(4a)配制好的ppb溶液中,使得rgo@pda浓度为6mg/ml,得 到rgo@pda修饰的ppb溶液;
197.(7)将cdl溶解在蒸馏水,配制成质量浓度为1wt%的cdl溶液;
198.(8)将(6)和(7)所得溶液以体积比为2:1混匀,得到0.66wt%cdl-ppb-rgo@pda水凝胶,将其命名为 p1c11’/go2。
199.(9)将步骤(6)中多余加一定量的met,使得met的浓度为6mg/ml,制得的水凝胶命名为 p1c11’/go2/met。
200.实施例7
201.(1)在edc的作用下将二氢咖啡酸接枝到壳聚糖主链上生成壳聚糖接枝二氢咖啡酸(cs-da);
202.壳聚糖接枝二氢咖啡酸的具体制备步骤包括:
203.(1a)将0.523g壳聚糖溶于45ml去离子水中,室温条件下搅拌使其充分溶解;
204.(1b)向(1a)所得的溶液中加入1m盐酸调节溶液ph至5.0左右;
205.(1c)将0.591g二氢咖啡酸加入上述(1b)所得溶液中,并快速加入1.239g edc,充分搅拌使溶液混匀;
206.(1d)向(1c)所得溶液中在加入1m盐酸调节溶液ph值在5.0~6.0之间,搅拌8小时;
207.(1e)用1m盐酸调节透析水的ph=5,将(1d)所得溶液至于其中透析72小时。
208.(2)向步骤(1)所得的cs-da上接枝l-精氨酸,生成壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl);
209.合成cdl的具体制备步骤包括:
210.(2a)将1.13g l-精氨酸溶于20ml去离子水中,依次加入1.239g edc和0.746g nhs,将混合溶液搅 拌30分钟以激活l-精氨酸上的羧基;
211.(2b)将(2a)所得溶液加入到步骤(1)最终所得的溶液中,将合并的溶液在室温下搅拌12小时并透析3天;
212.(2c)最后将(2b)所得溶液通过冻干获得所需的壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl)。
213.(3)以edc作为脱水剂,4-二甲氨基吡啶(dmap)作为催化剂,用4-羧基苯基硼酸频哪醇酯(pba)和4
‑ꢀ
羧基苯甲醛(ba)合成苯硼酸和苯甲醛双修饰的聚乙二醇共聚(甘油癸二酸酯)(ppb);
214.合成ppb的具体制备步骤包括:
215.(3a)将3.63g癸二酸和9g聚乙二醇(peg)添加到50ml圆底烧瓶中,在氮气气氛下,125℃反应12小 时后,将圆底烧瓶压力降至5kpa,再反应24小时;
216.(3b)将3.3g甘油引入(3a)所得混合物中,使混合物在氮气环境下,125℃反应12小时,然后在5kpa 压力下再反应48小时;
217.(3c)将(3b)反应最终所得混合物进行2次重复纯化,过程如下:溶解在氯仿中并在4500rpm下离心5 分钟,之后,去除上清液(未反应的甘油)并将残留溶液在过量预冷的乙醚中沉淀以获得聚乙二醇共聚(甘 油癸二酸酯)共聚物(pegs);
218.(3d)将(3c)所得的纯化的pegs在真空烘箱中室温干燥48小时得到干燥的pegs共聚物;
219.(3e)将1g pegs共聚物、0.4g对羧基苯甲醛(fa)和0.4g pba溶解在10ml无水dmf中形成混合物, 然后将2.68g edc和0.53g dmap溶解在上述混合物中;
220.(3f)将(3e)最终所得混合物置于室温下在氮气气氛下反应96小时,之后将混合物在过量的预冷乙醚中 沉淀;
221.(3g)将(3f)所得沉淀物进行如下纯化:将沉淀物溶解在thf(四氢呋喃)中并以4500rpm离心10分钟后, 取上清液,用1m盐酸调节上清液的ph调至2;
222.(3h)通过透析和冷冻干燥得到所需的ppb。
223.(4)用氧化石墨烯(go)和多巴胺(da)生成还原性氧化石墨烯(rdo@pda);
224.合成rdo@pda的具体制备步骤如下:
225.(4a)将go(20mg)和多巴胺盐酸盐(20mg)溶解在40ml tris-hcl缓冲液(ph=8.5)中并超声处理30分 钟;
226.(4b)将(4a)所得混合物在室温下剧烈搅拌24小时;
227.(4c)将(4b)所得混合物通过过滤纯化和分离,并进一步用水和乙醇洗涤数次,得到rgo@pda溶液;
228.(4d)将(4c)所得的rgo@pda溶液在真空烘箱中60℃干燥48小时,得到rgo@pda共聚物。
229.(5)将ppb溶解在蒸馏水中,配制成质量浓度为30wt%的ppb溶液;
230.(6)将一定量的rgo@pda加入到步骤(4a)配制好的ppb溶液中,使得rgo@pda浓度为6mg/ml,得 到rgo@pda修饰的ppb溶液;
231.(7)将cdl溶解在蒸馏水,配制成质量浓度为3wt%的cdl溶液;
232.(8)将(6)和(7)所得溶液以体积比为2:1混匀,得到2wt%cdl-ppb-rgo@pda水凝胶,将其命名为 p2c22/go2。
233.(9)将步骤(6)中多余加一定量的met,使得met的浓度为6mg/ml,制得的水凝胶命名为 p2c22/go2/met。
234.实施例8
235.(1)在edc的作用下将二氢咖啡酸接枝到壳聚糖主链上生成壳聚糖接枝二氢咖啡酸(cs-da);
236.壳聚糖接枝二氢咖啡酸的具体制备步骤包括:
237.(1a)将0.523g壳聚糖溶于45ml去离子水中,室温条件下搅拌使其充分溶解;
238.(1b)向(1a)所得的溶液中加入1m盐酸调节溶液ph至5.0左右;
239.(1c)将0.591g二氢咖啡酸加入上述(1b)所得溶液中,并快速加入1.239g edc,充分搅拌使溶液混匀;
240.(1d)向(1c)所得溶液中在加入1m盐酸调节溶液ph值在5.0~6.0之间,搅拌8小时;
241.(1e)用1m盐酸调节透析水的ph=5,将(1d)所得溶液至于其中透析72小时。
242.(2)向步骤(1)所得的cs-da上接枝l-精氨酸,生成壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl);
243.合成cdl的具体制备步骤包括:
244.(2a)将0.28g l-精氨酸溶于20ml去离子水中,依次加入1.239g edc和0.746g nhs,将混合溶液搅 拌30分钟以激活l-精氨酸上的羧基;
245.(2b)将(2a)所得溶液加入到步骤(1)最终所得的溶液中,将合并的溶液在室温下搅拌12小时并透析3天;
246.(2c)最后将(2b)所得溶液通过冻干获得所需的壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl)。
247.(3)以edc作为脱水剂,4-二甲氨基吡啶(dmap)作为催化剂,用4-羧基苯基硼酸频哪醇酯(pba)和4
‑ꢀ
羧基苯甲醛(ba)合成苯硼酸和苯甲醛双修饰的聚乙二醇共聚(甘油癸二酸酯)(ppb);
248.合成ppb的具体制备步骤包括:
249.(3a)将3.63g癸二酸和9g聚乙二醇(peg)添加到50ml圆底烧瓶中,在氮气气氛下,125℃反应12小 时后,将圆底烧瓶压力降至5kpa,再反应24小时;
250.(3b)将3.3g甘油引入(3a)所得混合物中,使混合物在氮气环境下,125℃反应12小时,然后在5kpa 压力下再反应48小时;
251.(3c)将(3b)反应最终所得混合物进行2次重复纯化,过程如下:溶解在氯仿中并在4500rpm下离心5 分钟,之后,去除上清液(未反应的甘油)并将残留溶液在过量预冷的乙醚中沉淀以获得聚乙二醇共聚(甘 油癸二酸酯)共聚物(pegs);
252.(3d)将(3c)所得的纯化的pegs在真空烘箱中室温干燥48小时得到干燥的pegs共聚物;
253.(3e)将1g pegs共聚物、0.1g对羧基苯甲醛(fa)和0.1g pba溶解在10ml无水dmf中形成混合物, 然后将0.67g edc和0.13g dmap溶解在上述混合物中;
254.(3f)将(3e)最终所得混合物置于室温下在氮气气氛下反应48小时,之后将混合物在过量的预冷乙醚中 沉淀;
255.(3g)将(3f)所得沉淀物进行如下纯化:将沉淀物溶解在thf(四氢呋喃)中并以4500rpm离心10分钟后, 取上清液,用1m盐酸调节上清液的ph调至2;
256.(3h)通过透析和冷冻干燥得到所需的ppb。
257.(4)用氧化石墨烯(go)和多巴胺(da)生成还原性氧化石墨烯(rdo@pda);
258.合成rdo@pda的具体制备步骤如下:
259.(4a)将go(20mg)和多巴胺盐酸盐(20mg)溶解在40ml tris-hcl缓冲液(ph=8.5)中并超声处理30分 钟;
260.(4b)将(4a)所得混合物在室温下剧烈搅拌24小时;
261.(4c)将(4b)所得混合物通过过滤纯化和分离,并进一步用水和乙醇洗涤数次,得到rgo@pda溶液;
262.(4d)将(4c)所得的rgo@pda溶液在真空烘箱中60℃干燥48小时,得到rgo@pda共聚物。
263.(5)将ppb溶解在蒸馏水中,配制成质量浓度为30wt%的ppb溶液;
264.(6)将一定量的rgo@pda加入到步骤(4a)配制好的ppb溶液中,使得rgo@pda浓度为6mg/ml,得 到rgo@pda修饰的ppb溶液;
265.(7)将cdl溶解在蒸馏水,配制成质量浓度为1.5wt%的cdl溶液;
266.(8)将(6)和(7)所得溶液以体积比为2:1混匀,得到1.00wt%cdl-ppb-rgo@pda水凝胶,将其命名为 p3c31/go2。
267.(9)将步骤(6)中多余加一定量的met,使得met的浓度为6mg/ml,制得的水凝胶命名为 p3c31/go2/met。
268.实施例9
269.(1)在edc的作用下将二氢咖啡酸接枝到壳聚糖主链上生成壳聚糖接枝二氢咖啡酸(cs-da);
270.壳聚糖接枝二氢咖啡酸的具体制备步骤包括:
271.(1a)将0.523g壳聚糖溶于45ml去离子水中,室温条件下搅拌使其充分溶解;
272.(1b)向(1a)所得的溶液中加入1m盐酸调节溶液ph至5.0左右;
273.(1c)将0.591g二氢咖啡酸加入上述(1b)所得溶液中,并快速加入1.239g edc,充分搅拌使溶液混匀;
274.(1d)向(1c)所得溶液中在加入1m盐酸调节溶液ph值在5.0~6.0之间,搅拌8小时;
275.(1e)用1m盐酸调节透析水的ph=5,将(1d)所得溶液至于其中透析72小时。
276.(2)向步骤(1)所得的cs-da上接枝l-精氨酸,生成壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl);
277.合成cdl的具体制备步骤包括:
278.(2a)将0.8g l-精氨酸溶于20ml去离子水中,依次加入1.239g edc和0.746g nhs,将混合溶液搅拌 30分钟以激活l-精氨酸上的羧基;
279.(2b)将(2a)所得溶液加入到步骤(1)最终所得的溶液中,将合并的溶液在室温下搅拌12小时并透析3天;
280.(2c)最后将(2b)所得溶液通过冻干获得所需的壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl)。
281.(3)以edc作为脱水剂,4-二甲氨基吡啶(dmap)作为催化剂,用4-羧基苯基硼酸频哪醇酯(pba)和4
‑ꢀ
羧基苯甲醛(ba)合成苯硼酸和苯甲醛双修饰的聚乙二醇共聚(甘油癸二酸酯)(ppb);
282.合成ppb的具体制备步骤包括:
283.(3a)将3.63g癸二酸和9g聚乙二醇(peg)添加到50ml圆底烧瓶中,在氮气气氛下,125℃反应12小 时后,将圆底烧瓶压力降至5kpa,再反应24小时;
284.(3b)将3.3g甘油引入(3a)所得混合物中,使混合物在氮气环境下,125℃反应12小时,然后在5kpa 压力下再反应48小时;
285.(3c)将(3b)反应最终所得混合物进行2次重复纯化,过程如下:溶解在氯仿中并在4500rpm下离心5 分钟,之后,去除上清液(未反应的甘油)并将残留溶液在过量预冷的乙醚中沉淀以获得聚乙二醇共聚(甘 油癸二酸酯)共聚物(pegs);
286.(3d)将(3c)所得的纯化的pegs在真空烘箱中室温干燥48小时得到干燥的pegs共聚物;
287.(3e)将1g pegs共聚物、0.3g对羧基苯甲醛(fa)和0.3g pba溶解在10ml无水dmf中形成混合物, 然后将2g edc和0.4g dmap溶解在上述混合物中;
288.(3f)将(3e)最终所得混合物置于室温下在氮气气氛下反应60小时,之后将混合物在过量的预冷乙醚中 沉淀;
289.(3g)将(3f)所得沉淀物进行如下纯化:将沉淀物溶解在thf(四氢呋喃)中并以4500rpm离心10分钟后, 取上清液,用1m盐酸调节上清液的ph调至2;
290.(3h)通过透析和冷冻干燥得到所需的ppb。
291.(4)用氧化石墨烯(go)和多巴胺(da)生成还原性氧化石墨烯(rdo@pda);
292.合成rdo@pda的具体制备步骤如下:
293.(4a)将go(20mg)和多巴胺盐酸盐(20mg)溶解在40ml tris-hcl缓冲液(ph=8.5)中并超声处理30分 钟;
294.(4b)将(4a)所得混合物在室温下剧烈搅拌24小时;
295.(4c)将(4b)所得混合物通过过滤纯化和分离,并进一步用水和乙醇洗涤数次,得到rgo@pda溶液;
296.(4d)将(4c)所得的rgo@pda溶液在真空烘箱中60℃干燥48小时,得到rgo@pda共聚物。
297.(5)将ppb溶解在蒸馏水中,配制成质量浓度为30wt%的ppb溶液;
298.(6)将一定量的rgo@pda加入到步骤(4a)配制好的ppb溶液中,使得rgo@pda浓度为6mg/ml,得 到rgo@pda修饰的ppb溶液;
299.(7)将cdl溶解在蒸馏水,配制成质量浓度为2.50wt%的cdl溶液;
300.(8)将(6)和(7)所得溶液以体积比为2:1混匀,得到1.66wt%cdl-ppb-rgo@pda水凝胶,将其命名为 p4c
42’
/go2。
301.(9)将步骤(6)中多余加一定量的met,使得met的浓度为6mg/ml,制得的水凝胶命名为 p4c
42’
/go2/met。
302.实施例10
303.(1)在edc的作用下将二氢咖啡酸接枝到壳聚糖主链上生成壳聚糖接枝二氢咖啡酸(cs-da);
304.壳聚糖接枝二氢咖啡酸的具体制备步骤包括:
305.(1a)将0.523g壳聚糖溶于45ml去离子水中,室温条件下搅拌使其充分溶解;
306.(1b)向(1a)所得的溶液中加入1m盐酸调节溶液ph至5.0左右;
307.(1c)将0.591g二氢咖啡酸加入上述(1b)所得溶液中,并快速加入1.239g edc,充分搅拌使溶液混匀;
308.(1d)向(1c)所得溶液中在加入1m盐酸调节溶液ph值在5.0~6.0之间,搅拌8小时;
309.(1e)用1m盐酸调节透析水的ph=5,将(1d)所得溶液至于其中透析72小时。
310.(2)向步骤(1)所得的cs-da上接枝l-精氨酸,生成壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl);
311.合成cdl的具体制备步骤包括:
312.(2a)将0.5g l-精氨酸溶于20ml去离子水中,依次加入1.239g edc和0.746g nhs,将混合溶液搅拌 30分钟以激活l-精氨酸上的羧基;
313.(2b)将(2a)所得溶液加入到步骤(1)最终所得的溶液中,将合并的溶液在室温下搅拌12小时并透析3天;
314.(2c)最后将(2b)所得溶液通过冻干获得所需的壳聚糖接枝二氢咖啡酸和l-精氨酸(cdl)。
315.(3)以edc作为脱水剂,4-二甲氨基吡啶(dmap)作为催化剂,用4-羧基苯基硼酸频哪醇酯(pba)和4
‑ꢀ
羧基苯甲醛(ba)合成苯硼酸和苯甲醛双修饰的聚乙二醇共聚(甘油癸二酸酯)(ppb);
316.合成ppb的具体制备步骤包括:
317.(3a)将3.63g癸二酸和9g聚乙二醇(peg)添加到50ml圆底烧瓶中,在氮气气氛下,125℃反应12小 时后,将圆底烧瓶压力降至5kpa,再反应24小时;
318.(3b)将3.3g甘油引入(3a)所得混合物中,使混合物在氮气环境下,125℃反应12小时,然后在5kpa 压力下再反应48小时;
319.(3c)将(3b)反应最终所得混合物进行2次重复纯化,过程如下:溶解在氯仿中并在4500rpm下离心5 分钟,之后,去除上清液(未反应的甘油)并将残留溶液在过量预冷的乙醚中沉淀以获得聚乙二醇共聚(甘 油癸二酸酯)共聚物(pegs);
320.(3d)将(3c)所得的纯化的pegs在真空烘箱中室温干燥48小时得到干燥的pegs共聚物;
321.(3e)将1g pegs共聚物、0.25g对羧基苯甲醛(fa)和0.25g pba溶解在10ml无水dmf中形成混合物, 然后将1.5g edc和0.3g dmap溶解在上述混合物中;
322.(3f)将(3e)最终所得混合物置于室温下在氮气气氛下反应50小时,之后将混合物在过量的预冷乙醚中 沉淀;
323.(3g)将(3f)所得沉淀物进行如下纯化:将沉淀物溶解在thf(四氢呋喃)中并以4500rpm离心10分钟后, 取上清液,用1m盐酸调节上清液的ph调至2;
324.(3h)通过透析和冷冻干燥得到所需的ppb。
325.(4)用氧化石墨烯(go)和多巴胺(da)生成还原性氧化石墨烯(rdo@pda);
326.合成rdo@pda的具体制备步骤如下:
327.(4a)将go(20mg)和多巴胺盐酸盐(20mg)溶解在40ml tris-hcl缓冲液(ph=8.5)中并超声处理30分 钟;
328.(4b)将(4a)所得混合物在室温下剧烈搅拌24小时;
329.(4c)将(4b)所得混合物通过过滤纯化和分离,并进一步用水和乙醇洗涤数次,得到rgo@pda溶液;
330.(4d)将(4c)所得的rgo@pda溶液在真空烘箱中60℃干燥48小时,得到rgo@pda共聚物。
331.(5)将ppb溶解在蒸馏水中,配制成质量浓度为30wt%的ppb溶液;
332.(6)将一定量的rgo@pda加入到步骤(4a)配制好的ppb溶液中,使得rgo@pda浓度为6mg/ml,得 到rgo@pda修饰的ppb溶液;
333.(7)将cdl溶解在蒸馏水,配制成质量浓度为2wt%的cdl溶液;
334.(8)将(6)和(7)所得溶液以体积比为2:1混匀,得到1.33wt%cdl-ppb-rgo@pda水凝胶,将其命名为 p5c52”/go2。
335.(9)将步骤(6)中多余加一定量的met,使得met的浓度为6mg/ml,制得的水凝胶命名为 p5c52”/go2/met。
336.以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作 的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。