n,3-二取代-1-异吲哚啉酮类化合物的高产率合成方法
技术领域
1.本发明涉及有机合成技术领域,具体涉及n,3-二取代-1-异吲哚啉酮类化合物的高产率合成方法。
背景技术:
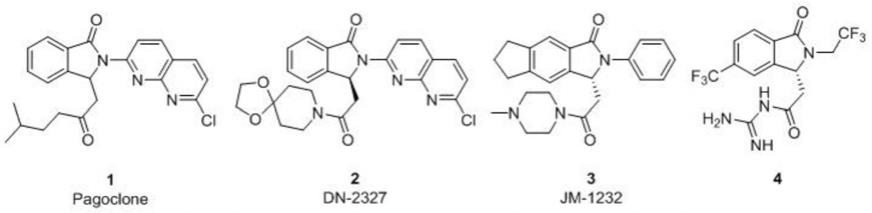
2.异吲哚啉酮骨架是一种重要的结构,这种结构存在于多种天然以及合成的具有生物和药学活性的化合物中。n,3-二取代-1-异吲哚啉酮类化合物有着许多重要的生物和药物活性,如pagoclone、dn-2327、jm-1232都可作为苯二氮卓受体激动剂治疗焦虑症;化合物4作为nhe1抑制剂已得到充分研究。除此之外,n,3-二取代-1-异吲哚啉酮衍生物也是合成许多非常有用的有机化合物和天然产物的关键中间体,因此合成n,3-二取代-1-异吲哚啉酮衍生物有着重大的意义。
[0003][0004]
目前,合成n,3-二取代-1-异吲哚啉酮类化合物多从邻苯二甲酰亚胺或者已有的3-羟基异吲哚啉酮出发,利用催化剂催化分子内或者分子间成环的反应制备。beller等人通过[ru(acac)3]催化体系,在h2和过量的醇或仲胺中,催化邻苯二甲酰亚胺进行还原烷氧基化或胺基化反应,以中高收率得到n,3-二取代异吲哚啉酮,需用到高压氢气。miura等人使用[ru(p-cymene)cl2]2和一水合醋酸铜作为氧化剂,使得n-ph苯甲酰胺和丙烯酸丁酯以80%的产率偶联合成n,3-二取代-1-异吲哚啉酮,虽然避免了高压氢气带来的危险,但仍需使用贵金属催化剂。李阳等人利用cu(otf)2催化邻氰基苯甲醛、末端炔烃和二苯基碘鎓三氟甲磺酸盐合成n,3-二取代异吲哚啉酮,但反应需较高温度,容易造成浪费,而且产率不高。
[0005]
上述合成n,3-二取代-1-异吲哚啉酮类化合物的方法仍存在着缺陷,如需要贵金属催化剂、高压,或者反应复杂、产率不高。
技术实现要素:
[0006]
为弥补现有技术的不足,本发明提供了一种高性价比、反应条件温和的n,3-二取代-1-异吲哚啉酮类化合物的高产率合成方法,以有效解决背景技术中所提及的技术问题,通过以下技术方案实现:
[0007]
本发明在铜盐-膦配体形成的催化剂作用下,使用硅烷作为还原剂,对2-((烃基亚氨基)甲基)苯甲酸甲酯(ⅰ)和α,β-不饱和酸酯(ⅱ)进行共轭还原/mannich反应/内酰胺化串联反应,直接得到n,3-二取代-1-异吲哚啉酮类化合物(ⅲ)。该方法使用催化量的cu-膦
催化剂,不需分离反应中间体,避免减少了分步反应中的繁琐的分离操作过程,并且具有反应条件温和,具有反应迅速、产率高等优点。
[0008]
n,3-二取代-1-异吲哚啉酮类化合物的高产率合成方法,合成路线是:
[0009][0010]
进一步的,所述式ⅰ化合物与式ⅲ化合物中的r1为c
1-c
10
的烷基、芳基、取代芳基中的一种;式ⅱ化合物与式ⅲ化合物中的r2为c
1-c
12
的直链烷基、c
3-c
12
的支链烷基、c
3-c6的环烷基中的一种,r3和r4为h或c
1-c6的直链烷基、c
3-c6的支链烷基、c
3-c6的环烷基中的一种。
[0011]
合成方法具体为:在休良克瓶1中加入铜盐、膦配体,休良克瓶2中加入式ⅰ化合物,抽真空,n2置换三次;保持氮气氛围往休良克瓶2中加入式ⅱ化合物,之后向休良克瓶1和2中分别加入反应溶剂并搅拌10min;再加入硅烷至休良克瓶1中搅拌20min;将休良克瓶2中溶液经双头针加入至休良克瓶1中,即可合成式ⅲ的化合物n,3-二取代-1-异吲哚啉酮类化合物。
[0012]
进一步的,所述的铜盐为cuf(pph3)3·
2meoh、cu(r5co2)2、cu(r5co2)2·
mh2o、cuor5中的一种,或cux、cux2、cui中的一种与mor5的混合物,其中x为f、cl、br中的一种;m=1或2或3;r5为c
1-c
12
的烷基;m为na或k。
[0013]
进一步的,所述的膦配体为有如下结构的膦化合物l1-l4的一种;
[0014][0015]
l1-l4中r6为ph、2-mec6h4、4-mec6h4、
t
bu、cy、3,5-me
2-c6h3、3,5-me
2-4-meo-c6h2、3,5-(
t
bu)
2-c6h3、4-meo-3,5-(
t
bu)
2-c6h2、3,4,5-(meo)
3-c6h2的一种;l3中n=1-5。
[0016]
进一步的,所述的硅烷包括(ipr)3sih、(
t
bu)3sih、(ipro)3sih、(
t
bu)2sih2、
t
bume2sih、phsih3、ph2sih2、ph3sih、ph2mesih、(sihme2)2o、et3sih、(meo)3sih、(sihme2)2nh、((ch3)2osi)n、r8(osihme)
p
or8中的一种,p=1,2,3,
…
,100的整数;r8为h或si(ch3)3或si(ch3)2bu
t
。
[0017]
进一步的,所述的反应溶剂为醚类或芳烃类或卤代烷类,包括但并不限于乙醚、丁醚、四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、苯、甲苯、乙苯、二甲苯、二氯甲烷、三氯甲烷、1,2-二氯乙烷中的一种或一种以上的混合物。
[0018]
进一步的,反应中溶剂/式i化合物的质量比为(1-1000)ml/g。
[0019]
进一步的,所述的铜盐、膦配体、硅烷、式i化合物、式ⅱ化合物的摩尔比为(0.02~
0.08):(0.02~0.16):(2~5):1:(2-3)。
[0020]
进一步的,在休良克瓶1中分别加入铜盐、膦配体时,休良克瓶内的温度为-10-45℃。
[0021]
本发明与现有技术相比的有益效果是:
[0022]
本发明可以高产率地合成n,3-二取代-1-异吲哚啉酮类化合物,反应条件温和、性价比高、后处理简单,该方法只使用了催化量的cu-膦催化剂,不需要贵金属催化剂、反应操作步骤少,成本低。
附图说明
[0023]
图1为实施例1的hplc色谱图;
[0024]
图2为实施例2的hplc色谱图;
[0025]
图3为实施例4的hplc色谱图。
具体实施方式
[0026]
下面通过具体实施例详述本发明,但不限制本发明的保护范围。如无特殊说明,本发明所采用的实验方法均为常规方法,所用实验器材、材料、试剂等均可从商业途径获得。
[0027]
当r
1-r4为相同类型的烷基、芳基和取代芳基时,其制备方法相同,区别仅在于碳的个数不同,不再赘述。
[0028]
实施例1
[0029]
在干燥的休良克瓶1中加入0.0075g cu(pph3)3f
·
2meoh和0.0069gl1(r6=ph),休良克瓶2中加入0.0470g 2-((苯亚氨基)甲基)苯甲酸甲酯,抽真空,n2置换三次;保持氮气氛围往休良克瓶2中加入0.03620g丙烯酸甲酯,之后往休良克瓶1和2中分别加入0.5ml无水甲苯后在20℃搅拌10min;再加入3equiv的pmhs至休良克瓶1中并搅拌20min;将休良克瓶2中溶液经双头针加入至休良克瓶1中。反应6h后加入3ml饱和氯化铵水溶液并搅拌1h,过滤、分液,水相用3ml二氯甲烷萃取2次。合并的有机相先旋干再加入5ml二氯甲烷溶解,后经2ml饱和氯化钠溶液洗涤2次,无水硫酸钠干燥、旋蒸、柱层析纯化,得到产物n-苯基-3-(1-甲氧基酰基乙基)-1-异吲哚啉酮类化合物
ⅲ‑
1和
ⅲ‑
2。
[0030][0031]
实施例2
[0032]
在干燥的休良克瓶1中依次加入0.0075g cu(pph3)3f
·
2meoh和0.0085g l1(r6=ph),休良克瓶2中加入0.0575g 2-((对氯苯亚氨基)甲基)苯甲酸甲酯,抽真空,n2置换三次;保持氮气氛围往休良克瓶2中加入0.0380g丙烯酸甲酯,之后往休良克瓶1和2中分别加入0.8ml无水甲苯保持25℃搅拌10min;再加入3equiv的pmhs至休良克瓶1中并搅拌20min;将休良克瓶2中溶液经双头针加入至休良克瓶1中。反应6h后加入3ml饱和氯化铵水溶液并搅拌1h,过滤、分液,水相用3ml二氯甲烷萃取2次。合并的有机相先旋干再加入5ml二氯甲烷溶解,经2ml饱和氯化钠溶液洗涤2次,无水硫酸钠干燥、旋蒸、柱层析纯化,得到产物n-对氯苯基-3-(1-甲氧基酰基乙基)-1-异吲哚啉酮类化合物
ⅲ‑
3和
ⅲ‑
4。
[0033][0034]
实施例3
[0035]
在干燥的休良克瓶1中依次加入0.0060g cu(pph3)3f
·
2meoh和0.0056g l3(r6=ph,n=3),休良克瓶2中加入0.0561g 2-((3,4-二甲苯亚氨基)甲基)苯甲酸甲酯,抽真空,n2置换三次;保持氮气氛围往休良克瓶2中加入0.0450g甲基丙烯酸甲酯,之后往休良克瓶1和2中分别加入0.8ml无水四氢呋喃降温至0℃并搅拌10min;再加入3equiv的pmhs至休良克瓶1中并搅拌20min;将休良克瓶2中溶液经双头针加入至休良克瓶1中。反应6h后加入3ml饱和氯化铵水溶液并搅拌1h,过滤、分液,水相用3ml乙酸乙酯萃取2次。合并的有机相先旋干再加入5ml二氯甲烷溶解,经2ml饱和氯化钠溶液洗涤2次,无水硫酸钠干燥、旋蒸、柱层析纯化,得到产物n-(3,4-二甲基苯基)-3-(1-甲基-1-甲氧基酰基乙基)-1-异吲哚啉酮类化合物
ⅲ‑
5。
[0036][0037]
实施例4
[0038]
在干燥的休良克瓶1中依次加入0.0060g cu(pph3)3f
·
2meoh和0.0052g l1(r6=ph),休良克瓶2中加入0.0640g 2-(([1,1'-联苯基]-4-亚氨基)甲基)苯甲酸甲酯,抽真空,n2置换三次;保持氮气氛围往休良克瓶2中加入0.0360g丙烯酸甲酯,之后往休良克瓶1和2中分别加入0.8ml无水四氢呋喃降温至-10℃并搅拌10min;再加入3equiv的ph2sih2至休良克瓶1中并搅拌20min;将休良克瓶2中溶液经双头针加入至休良克瓶1中。反应6h后加入3ml饱和氯化铵水溶液并搅拌1h,过滤、分液,水相用3ml二氯甲烷萃取2次。合并的有机相先旋干再加入5ml二氯甲烷溶解,经2ml饱和氯化钠溶液洗涤2次,无水硫酸钠干燥、旋蒸、柱层析纯化,得到产物n-联苯基-3-(1-甲氧基酰基乙基)-1-异吲哚啉酮类化合物
ⅲ‑
6和
ⅲ‑
7。
[0039][0040]
实施例5
[0041]
在干燥的休良克瓶1中依次加入0.0020g cubr、0.0062g l2(r6=ph),和0.0480g kobu
t
休良克瓶2中加入0.0510g 2-((对甲苯亚氨基)甲基)苯甲酸甲酯,抽真空,n2置换三次;保持氮气氛围往休良克瓶2中加入0.0420g甲基丙烯酸甲酯,之后往休良克瓶1和2中分别加入0.8ml无水甲苯并搅拌10min;再加入3equiv的(me2sih)2o至休良克瓶1中并搅拌
20min;将休良克瓶2中溶液经双头针加入至休良克瓶1中。反应6h后加入3ml饱和氯化铵水溶液并搅拌1h,过滤、分液,水相用3ml乙酸乙酯萃取2次。合并的有机相先旋干再加入5ml二氯甲烷溶解,经2ml饱和氯化钠溶液洗涤2次,无水硫酸钠干燥、旋蒸、柱层析纯化,得到产物n-对甲苯基-3-(1-甲基-1-甲氧基酰基乙基)-1-异吲哚啉酮类化合物
ⅲ‑
8。
[0042][0043]
实施例6
[0044]
在干燥的休良克瓶1中依次加入0.0056g cu(pph3)3f
·
2meoh和0.0058g l4(r6=ph),休良克瓶2中加入0.0490g 2-((苯亚氨基)甲基)苯甲酸甲酯,抽真空,n2置换三次;保持氮气氛围往休良克瓶2中加入0.0570g丙烯酸叔丁酯,之后往休良克瓶1和2中分别加入0.8ml无水四氢呋喃降温至0℃并搅拌10min;再加入3equiv的pmhs至休良克瓶1中并搅拌20min;将休良克瓶2中溶液经双头针加入至休良克瓶1中。反应6h后加入3ml饱和氯化铵水溶液并搅拌1h,过滤、分液,水相用3ml二氯甲烷萃取2次。合并的有机相先旋干再加入5ml二氯甲烷溶解,经2ml饱和氯化钠溶液洗涤2次,无水硫酸钠干燥、旋蒸、柱层析纯化,得到产物n-苯基-3-(1-叔丁氧基酰基乙基)-1-异吲哚啉酮类化合物
ⅲ‑
9和
ⅲ‑
10。
[0045][0046]
以上所述,仅为本发明创造较佳的具体实施方式,但本发明创造的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明创造披露的技术范围内,根据本发明创造的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明创造的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
