用于筛选文库的方法
发明领域
1.本发明涉及鉴定内切核酸酶的dna靶序列的方法。用于该方法的底物文库和工程化改造内切核酸酶以具有改善的对特定底物的切割效率的方法形成本发明的其他方面。
2.发明背景
3.能够切割基因组内单个位点的内切核酸酶通过刺激非同源末端连接或同源重组而具有巨大的基因组编辑的潜力,并且多种工程化改造的内切核酸酶已经进入临床试验,包括ccr5-2246(靶向人ccr5)和vr24684(靶向人vegf-a启动子)。尽管有能力工程化改造某些核酸酶(例如rna引导的核酸酶,诸如cas9/向导rna、talen或锌指核酸酶)以对特定的独特靶序列具有特异性,但很明显这些核酸酶也可能与其他脱靶序列相互作用并在其他脱靶序列处切割。事实上,一些工程化改造的核酸酶与细胞毒性和致瘤性有关。在2016年11月15日的科学委员会会议上,fda指出批准治疗性内切核酸酶时,需要考虑切割的特异性。
4.鉴于对脱靶切割的担忧,已经进行了各种尝试来表征与内切核酸酶经工程化改造以切割的靶位点密切相关的靶位点上的内切核酸酶的切割。wo2018/119010描述了使用寡核苷酸文库的方法,该方法易于大规模生产,并且该方法与自动化相容。然而,该方法具有低信噪比,并且需要高切割率以检测信号。因此,该方法使用非生理酶:dna化学计量学进行,其本身可能导致人为现象(artefact)。
5.需要能够克服与现有技术相关的缺点的方法。
技术实现要素:
6.在第一方面,本发明提供了底物文库,其包含多种dna底物,其中该文库内的每个底物含有推定的靶序列,该推定的靶序列是能够独特地鉴定所述推定的靶序列的标识符dna序列的5’,该标识符dna序列是与反向pcr引物相同的序列的5’,并且其中文库内的双链dna底物彼此之间的不同仅在于推定的靶序列和标识符dna序列。
7.在一个实施方案中,底物文库包含多个双链dna底物。
8.在另一个方面,本发明提供了用于制备包含如本文所定义的多个双链dna底物的底物文库的方法。该方法包括用文库正向引物和文库反向引物对多个推定的靶序列进行pcr扩增的步骤,该推定的靶序列侧接有a)与所述文库正向引物互补的序列和b)与所述文库反向引物序列的部分相同的序列,其中文库反向引物是含有不同标识符序列的dna序列的异质混合物,该不同标识符序列位于与反向引物互补的所有序列共有的序列的5’,并且其中不同标识符序列的数目以摩尔超过推定的靶序列的数目。
9.另一方面,本发明提供了用于鉴定内切核酸酶的dna靶序列的方法,其包括以下步骤:
10.a)在合适的条件下使如本文所述的底物文库与内切核酸酶接触以允许切割;
11.b)将经内切核酸酶处理的文库与包括与“切割”pcr引物互补的序列的dna序列连接;
12.c)用切割和反向pcr引物对经切割的底物进行pcr扩增;和
13.d)测序扩增的pcr产物;
14.其中通过标识符序列的序列鉴定切割产物中的dna靶序列。
15.在又一方面,本发明提供了用于工程化改造内切核酸酶的方法,其包括:
16.a)使用相同的底物文库,用第一内切核酸酶和至少两种其他内切核酸酶进行如本文所述的鉴定内切核酸酶的dna靶序列的方法,该其他内切核酸酶与第一内切核酸酶的不同在于内切核酸酶氨基酸序列内不同位置处的单个氨基酸变化;
17.b)比较步骤a)中测试的每种内切核酸酶在特定底物处的切割效率;
18.c)鉴定提高切割效率的不同位置处的至少两个氨基酸变化;
19.d)产生含有步骤c)中鉴定的至少两个氨基酸变化的变体内切核酸酶。
20.在又一方面,本发明提供了通过本文所述方法获得的变体内切核酸酶。
21.附图简述
22.图1显示用于鉴定内切核酸酶的dna靶序列的方法的步骤a)至c)的示意图。图1a显示双链底物文库,包括推定的靶序列、标识符序列和与反向引物互补的序列。图1b显示经切割的底物文库。在此表示中,推定的靶序列2是仅有的经切割的推定靶序列。图1c显示与切割引物连接后的文库。图1d显示使用切割和反向引物进行pcr扩增所得的扩增pcr产物。
23.图2显示实施例3中表征的底物文库中单个dna底物的频率。y轴显示单个寡核苷酸计数频率。
24.图3a显示使用野生型cas9-rnp和r691a突变体cas9-rnp两者的未切割pcr反应的4%琼脂糖凝胶。图3b显示使用野生型cas9-rnp和r691a突变体cas9-rnp两者的切割pcr反应的4%琼脂糖凝胶。
25.图4显示所得探针文库中各种错配的数目的相对丰度以及dna的pam或grna区域中具有n=4、5或6个错配的未采样序列的相对比例。
26.图5和图6显示beesem衍生的结合谱(a和b)以及在1:1和1:5dna:rnp比率下hifi cas9相对于wt cas9的比较(c)。
27.图7显示证明在相同条件下两次测定运行的高重现性的重现性试验,以及池中每个寡核苷酸之间的相对高相关性的比较(相互比较)。
28.图8显示实施例6中表征的底物文库中单个dna底物的频率。y轴显示单个寡核苷酸计数频率。
29.图9显示i-scei探针文库中各种错配的数目的相对丰度(a)和文库靶区域中具有n=4、5或6个错配的未采样序列的相对比例(b)。
30.图10显示在50单位/ug的dna文库(a)、5单位/ug的dna文库(b)和0.5单位/ug的dna文库(c)下i-scei的beesem衍生的结合谱。
31.发明详述
32.如上所述,本发明提供了底物文库,其包含多种dna底物,其中文库中的每种底物含有推定的靶序列,该推定的靶序列是能够独特地鉴定所述推定的靶序列的标识符序列的5’,该标识符序列是与反向pcr引物相同的序列的5’,并且其中文库内的双链dna底物彼此之间的不同仅在于推定的靶序列和标识符dna序列。
33.在本发明的上下文中,推定的靶序列是可以潜在地由内切核酸酶进行切割的dna序列。在dna底物是双链的情况下,推定的靶序列是可以潜在地由内切核酸酶进行双链切割
id no:2)。在一个实施方案中,可以制备基于该已知靶序列的底物文库。在一个实施方案中,本发明提供了底物文库,其中在文库中存在的推定的靶序列作为整体包括seq id no:2和seq id no:x1的所有可能的单个变体。在另一个实施方案中,本发明提供了底物文库,其中在文库中存在的推定的靶序列作为整体包括seq id no:2和seq id no:2的所有可能的单个变体和双变体。在另一个实施方案中,本发明提供了底物文库,其中在文库中存在的推定的靶序列作为整体包括seq id no:2和seq id no:2的所有可能的单个变体、双变体和三变体。基于该靶序列的底物文库的制备描述于实施例6。
39.在底物文库包含双链dna底物的一个实施方案中,底物文库针对锌指核酸酶进行定制。虽然这些核酸酶不是天然存在的,但已经生成了许多。事实上,有许多公开可用的生成锌指核酸酶的系统,包括寡聚化池工程(open)、上下文相关组装(coda)和基于细菌单杂交(b1h)选择的系统。open策略已经用于生成识别内源性人和烟草基因中的特定序列的锌指核酸酶(maeder et al.,molecular cell,2008,31(2):294-301)。在该研究中,生成了许多锌指核酸酶,包括能够识别序列ctaccccgaccacatgaagcagcac(seq id no:3)的锌指核酸酶。在一个实施方案中,可以制备基于锌指核酸酶的靶序列的底物文库。在一个实施方案中,本发明提供了底物文库,其中在文库中存在的推定的靶序列作为整体包括seq id no:3和seq id no:3的所有可能的单个变体。在另一个实施方案中,本发明提供了底物文库,其中在文库中存在的推定的靶序列作为整体包括seq id no:3和seq id no:3的所有可能的单个变体和双变体。在另一个实施方案中,本发明提供底物文库,其中在文库中存在的推定的靶序列作为整体包括seq id no:3和seq id no:3的所有可能的单个变体、双变体和三变体。
40.在底物文库包含双链dna底物的一个实施方案中,底物文库针对talen进行定制。同样,这些不是天然存在的,但已经生成了许多,并且存在设计和合成具有特定特异性的talen的软件和平台(例如,talen targetter、e-talen、flash、golden gate)。reyon和同事使用flash系统来产生靶向许多人基因的talen(reyon et al.,nature biotechnology,2012,30:460-465)。靶向ercc2的talen识别序列tccggccggcgccatgaagtgagaagggggctgggggtcgcgctcgcta(seq id no:4)。在一个实施方案中,可以制备基于该已知靶序列的底物文库。在一个实施方案中,本发明提供了底物文库,其中在文库中存在的推定的靶序列作为整体包括seq id no:4和seq id no:4的所有可能的单个变体。在另一个实施方案中,本发明提供了底物文库,其中在文库中存在的推定的靶序列作为整体包括seq id no:4和seq id no:4的所有可能的单个变体和双变体。在另一个实施方案中,本发明提供了底物文库,其中在文库中存在的推定的靶序列作为整体包括seq id no:4和seq id no:4的所有可能的单个变体、双变体和三变体。
41.在一个实施方案中,底物文库针对rna引导的核酸酶进行定制。rna引导的核酸酶是指与向导rna(grna)相互作用并且与grna缔合来切割可以是双链或单链的靶区域的核酸酶。grna可以是单分子的(包括单个rna分子)或模块化的,包括crispr rna(crrna)和反式激活crrna(tracrrna)两者。无论是单分子的还是模块化的grna,都包含与期望被切割的dna序列互补的向导序列。在一个实施方案中,rna引导的核酸酶包括但不限于天然存在的2类crispr核酸酶,诸如cas9或cpf1,以及切割双链dna的这些的变体。在一个实施方案中,ii类crispr核酸酶是天然存在的。此类核酸酶的靶序列将以众所周知的方式取决于核酸酶类
别的性质和grna。例如,通常cas9核酸酶识别其中pam(前间隔区相邻基序)序列是与向导序列互补的前间隔区序列的3’的序列。实施例1举例说明了含有作为推定的靶序列的22bp tcrα靶区域和该22bp序列的所有可能的单个变体、双变体和三变体的底物文库。实施例1中制备的底物文库用于实施例5以表征来自酿脓链球菌(s.pyogenes)的野生型crispr cas9酶或其展现出显示脱靶活性降低的点突变r691a的变体,与tcrαcrrna和经修饰以增加核酸酶抗性的市售67bp tracrrna组合的切割。在另一个实施方案中,rna引导的核酸酶包括但不限于天然存在的ii型c亚类的cas9酶或其变体,或切割单链dna的cas3酶或其变体。
42.在另一个实施方案中,底物文库针对已知与特定疾病适应症相关的已知snp进行定制。在该实施方案中,推定的靶序列将包括长度为9-50个核苷酸的序列,包括snp和周围的野生型序列,其中在不同的推定的靶序列中,序列内snp的位置移动了单个核苷酸位置。除此之外,推定的靶序列将包括对应于所有天然存在的同种型的上述所有序列的野生型序列。
43.在一个实施方案中,底物文库将含有超过1000个dna底物。在更特定的实施方案中,底物文库将含有超过10000个dna底物。在更特定的实施方案中,底物文库将含有超过60000个dna底物。在更特定的实施方案中,底物文库将含有超过100000个dna底物。在本文所述的底物文库的一个实施方案中,dna底物将是双链的。在本文所述的底物文库的另一个实施方案中,dna底物将是单链的。发明人已经生成了用于本发明的方法的约290000个底物的文库。
44.在本发明的特定实施方案中,底物文库中的每种底物以大致相同的拷贝数存在,换言之,底物的丰度大致相等。可以通过实施例3中描述的方法评估底物的丰度。在一个实施方案中,文库中至少99%底物的丰度变化小于5倍,并且在更特定的实施方案中,小于2倍。
45.除了含有推定的靶序列外,文库中的每个底物还含有标识符序列。这是结合特定的靶序列而独特存在的dna序列,因此它可以充当条形码。标识符序列的确切序列并不重要,并且可以使用任何序列,只要它可以链接回特定的推定靶序列。然而,有一个重要的限制,即推定的靶序列的序列不应该与标识符序列的序列相同。在一个实施方案中,每个推定的靶序列的序列与相关标识符序列的序列不同。在另一个实施方案中,标识符序列的序列与文库中存在的任何推定的靶序列的序列不同。这确保了当推定的靶序列经内切核酸酶切割时,标识符序列将保持完整。
46.在一个实施方案中,存在于底物文库中的每个独特标识符序列的长度相同。在一个实施方案中,每个独特标识符与每个其他独特标识符序列在至少1个核苷酸上不同。在更特定的实施方案中,每个独特标识符与每个其他独特标识符序列在至少2或至少3个核苷酸上不同。具有一个以上不同的核苷酸最小化测序错误生成文库中另一个标识符序列的序列的可能性。
47.在一个实施方案中,独特标识符不包括内部互补或与其自身、引物或底物的其他部分具有同源性的序列。
48.在一个实施方案中,底物文库中的推定的靶序列中的每一个与底物文库中的其他推定的靶序列长度相同,并且底物文库中的标识符dna序列中的每一个与底物文库中的其他标识符dna序列的长度相同。
49.文库中的每个底物还含有与反向pcr引物互补的序列。该序列在文库的每个成员中相同。如果反向pcr引物能够在适当的条件下扩增底物,该确切序列不重要。
50.在dna底物是双链的某些实施方案中,文库中的每个底物还含有与正向引物互补的序列,其中该序列位于推定的靶序列的5’。该序列在文库的每个成员中相同。如与反向引物互补的序列,如果正向pcr引物能够在适当条件下扩增底物,与正向引物互补的区域的确切序列不重要。
51.在某些实施方案中,在dna底物是双链或单链的情况下,dna底物在其5’末端具有亲和标签。在dna底物为单链的某些实施方案中,所选择的5’亲和标签防止5’末端与双链dna连接,例如,通过其5’羟基附接至dna底物。
52.每个底物内的序列元件的相对定位也很重要。重要的是,与正向和反向引物互补的序列分别在5’和3’末端,使得它们在推定的靶标和标识符靶序列的侧翼。因此,使用正向和反向引物的pcr扩增将扩增推定的靶序列和标识符序列两者。
53.此外,重要的是推定的靶序列是标识符序列的5’(或上游)。这对于在鉴定靶序列的方法中使用文库很重要,这将在后面解释。在dna底物是双链的情况下,技术人员将理解序列元件的顺序将仅出现在两条链之一中。为避免疑义,双链dna底物是本发明的dna底物,其中序列元件在两条链之一中的顺序正确。
54.各种序列元件(推定的靶序列、标识符靶序列、与反向引物互补的序列)可以通过dna间隔物彼此分开。通常,这些长度在1-20个核苷酸之间。任何间隔物的精确序列均不重要,尽管应当理解它们不应该具有与推定的靶序列或标识符序列相同的序列。在一个实施方案中,间隔物序列不含有内切核酸酶的已知靶序列。在一个实施方案中,标识符靶序列和推定的靶序列之间的间隔物在1-20个核苷酸之间。在另一个实施方案中,标识符靶序列和推定的靶序列之间的间隔物在1-10个核苷酸之间。在进一步的实施方案中,标识符靶序列和推定的靶序列之间的间隔物在1-5个核苷酸之间。在进一步的实施方案中,标识符靶序列和推定的靶序列之间的间隔物不超过2个核苷酸。
55.底物文库可以通过常规方法,例如诱变方法合成。
56.生成简并寡核苷酸的常用方法是在寡核苷酸中的期望位置处使用混合亚磷酰胺(又名亚酰胺,寡核苷酸合成的构件),例如使用“n”以掺入da、dc、dg和dt核苷酸,或“y”用于嘧啶,“r”用于嘌呤。在寡核苷酸的自动化学合成过程中,合成器以预设的比率(例如,每种25%)连续添加dt、da、dc或dg(在“n”的情况下)。此过程并不总是导致每种亚酰胺的预期使用,因为不同的亚酰胺具有不同的偶联效率,并且添加顺序也可能偏向于稍后添加的亚酰胺。
57.使用如上所述的混合碱基可能导致有限的控制以实现特定氨基酸的密码子的比率。通过使用可以用于在每个合成循环中添加3个核苷酸的三聚体亚酰胺,可以创建以预定百分比编码选定氨基酸的寡核苷酸。然而,这个规程很难执行,因为三聚体亚酰胺庞大且难以与延长的寡核苷酸偶联;合成期间存在的任何水分将具有比使用常规亚酰胺更严重的不利影响。
58.另一种制备文库寡核苷酸的方法是“分裂和合并(split-and-pool)”方法,该方法特别适用于将多种氨基酸嵌入其他常见序列如抗体可变区内的cdr中。
59.此外,dna池可以通过易错pcr生成,或更具体地,使用简并引物通过重叠pcr生成。
60.此处所公开类型的文库也可以从商业供应商诸如twist bioscience处订购。
61.在底物文库包含双链dna底物的一个实施方案中,底物文库在两步过程中获得。第一步包括制备推定的靶序列。推定的靶序列可以通过诱变方法从已知的靶序列或从商业供应商诸如twist bioscience衍生。重要的是,在推定的靶序列侧翼,该序列必须包含与引物序列(其用于本文所述的鉴定dna靶序列的方法)互补的序列以及文库中存在的另外的序列元件。通过使用5’和3’引物对推定的靶序列进行pcr扩增生成底物文库。重要的是,这意味着3’引物不是单个序列,而是含有不同标识符序列以及与反向引物互补的序列的多个序列。在其中底物文库含有与正向引物互补的序列的实施方案中,该序列由所用的5’引物编码。
62.因此,在一个实施方案中,本发明提供了用于制备包含本文所定义的双链dna底物的底物文库的方法,该方法包括用文库正向引物和文库反向引物对多个推定的靶序列进行pcr扩增的步骤,该推定的靶序列侧接有a)所述文库正向引物和b)与所述文库反向引物序列的部分相同的序列,其中文库反向引物是含有不同标识符序列的dna序列的异质混合物,该不同标识符序列位于与反向引物序列互补的所有序列共有的序列的5’,并且其中不同标识符序列的数目以摩尔超过推定的靶序列的数目。
63.在一个实施方案中,多个推定的靶序列是所有相同长度的序列。
64.本文所述的底物文库可以用于鉴定内切核酸酶的dna靶序列的方法。因此,在第二方面,本发明提供了用于鉴定内切核酸酶的dna靶序列的方法,其包括以下步骤:
65.a)在合适的条件下使本文所定义的底物文库与内切核酸酶接触以允许切割;
66.b)将经内切核酸酶处理的文库与包括与“切割”pcr引物互补的序列的dna序列连接;
67.c)用切割和反向pcr引物对经切割的底物进行pcr扩增;和
68.d)对扩增的pcr产物进行测序;
69.其中通过标识符序列的序列鉴定切割产物中的dna靶序列。
70.虽然不是必需的,但可以匹配内切核酸酶和文库的选择(即,使得推定的靶序列包括相关内切核酸酶的任何已知靶序列及其变体)。例如,在测定中使用的核酸酶是i-crei的情况下,合适的底物文库将是包括序列tgttctcaggtacctcagccag(seq id no:1)及其变体的文库。
71.方法步骤a)至d)能够鉴定经内切核酸酶切割的推定的靶序列。一经鉴定,推定的靶序列称为dna靶序列。
72.该测定能够如下鉴定那些被切割的序列和因此所包含的dna靶序列。首先,文库与内切核酸酶接触。在底物的子集含有由内切核酸酶识别的dna靶序列的情况下,这些底物将被切割。然后将该文库连接到与切割pcr引物互补的dna序列。这种连接使得能够使用切割和反向引物选择性地扩增经切割的底物。然后可以对扩增的dna进行测序以鉴定dna靶序列。鉴于该序列已被切割,这不能直接进行,但可以通过dna靶序列独有的标识符序列间接鉴定dna靶序列。
73.步骤a)涉及用内切核酸酶处理底物文库。内切核酸酶可以是工程化改造的核酸酶,诸如talen或锌指核酸酶。在一个实施方案中,核酸酶是天然存在的核酸酶,诸如大范围核酸酶(归巢内切核酸酶)或天然存在的rna引导的核酸酶。在另一个实施方案中,内切核酸
酶可以是有机化合物核酸酶、烯二炔、抗生素核酸酶、达内霉素(dynemicin)、新制癌菌素(neocarzinostatin)、卡奇霉素(calicheamicin)、埃斯培拉霉素(esperamicin)或博来霉素(bleomycin)。在一个实施方案中,核酸酶是工程化改造的大范围核酸酶或rna引导的核酸酶,其与天然存在的大范围核酸酶在一个或多个残基上不同。
74.重要的是要注意文库中的不同底物不需要经物理分离才能使该方法有效。步骤(a)、(b)和(c)可以在整个底物文库上进行。将步骤(c)的pcr产物稀释至乳剂中约60-70%的随机形成的液滴将精确含有1个dna片段的水平,从而允许对单个片段进行测序。
75.因此,步骤(a)可以作为一锅反应进行,其中将整个底物文库与内切核酸酶接触。技术人员将充分理解合适的条件并且可以对每种内切核酸酶进行优化,但总而言之,步骤(a)在合适的温度下的溶液中,在合适的缓冲溶液中进行合适的时间。在方法的步骤(a)和(b)使用相同的缓冲液的情况下,重要的是所选择的缓冲液适合允许这两个步骤中的反应发生(例如与dna切除和连接广泛相容)。
76.在底物文库包含双链dna底物的一些实施方案中,内切核酸酶切割导致平切割末端。在其他实施方案中,双链dna底物的内切核酸酶切割导致突出端或粘性末端。在某些实施方案中,其中内切核酸酶切割导致粘性末端,该方法可以包括将粘性末端转化为平末端的步骤。这可以通过任何适当的方法来实现。钝化5’突出端的方法是本领域已知的。例如,可以通过使用5’至3’dna聚合酶诸如t4聚合酶或dna聚合酶i或其功能片段(例如dna聚合酶i的大klenow片段)填充来钝化5’突出端。例如,在实施例7中,通过用klenow聚合酶填充来钝化5’突出端。或者,可以使用5’至3’外切核酸酶诸如绿豆核酸酶或其功能片段来钝化5’突出端。通过填充和/或3’至5’外切核酸酶消化来钝化3’突出端的方法也是众所周知的。
77.在一个实施方案中,在步骤(a)之后、在(任选的)生成平末端的步骤之后或在这两者之后淬灭反应以使酶失活。可以使用任何合适的使酶失活的方法,但应注意避免引入可能干扰该方法后续步骤的物质。在一个实施方案中,通过加热到适合使酶失活但不使dna底物变性的温度,例如在65-70℃之间,来淬灭反应。在另一个实施方案中,例如使用螯合剂诸如edta除去酶的必需辅助因子。在另一个实施方案中,例如使用捕捉树脂诸如ni-nta-琼脂糖或链霉亲和素-琼脂糖来物理除去酶。在另一个实施方案中,通过使用混杂蛋白酶诸如蛋白酶k破坏酶。
78.步骤(b)涉及在包括与切割pcr引物互补的序列的dna序列存在下进行连接。包括与切割pcr引物互补的序列的dna序列应该以摩尔过量存在。在一个实施方案中,包括与切割pcr引物互补的序列的dna序列相对于文库dna以至少3:1的摩尔比存在。在一个实施方案中,包括与切割pcr引物互补的序列的dna序列另外含有切割事件标识符序列(在实施例中称为孔条形码寡核苷酸)。
79.在步骤(b)中可以使用任何合适的dna连接酶(或其功能片段)。许多dna连接酶(例如t4、t3、t7)在本领域中是已知的并且许多是市售的。为步骤(b)选择的连接酶的类型将取决于步骤(a)之后存在的切割末端的性质以及包括与切割pcr引物互补的序列的dna序列的末端的性质。在内切核酸酶切割生成平末端的情况下(或在步骤b之前钝化突出端的情况下)并且在包括与切割pcr引物互补的序列的dna序列也具有平3’末端的情况下,可以选择适用于连接平末端的连接酶。在内切核酸酶切割生成粘性末端的情况下,可以选择适用于粘性末端的连接酶。在底物文库是单链的实施方案中,连接步骤可以采用适用于连接单链
和双链dna的连接酶,例如环化酶(circligase)。在底物文库是单链的另一个实施方案中,包括与切割pcr引物互补的序列的dna序列可以包括能够与切割的单链dna底物杂交的简并粘性末端。在使用此类dna序列的情况下,可以使用能够连接粘性末端的连接酶。
80.如上所述,在一个实施方案中,包括与切割pcr引物互补的序列的dna序列是平末端的。在另一个实施方案中,包括与切割pcr引物互补的序列的dna序列具有3’突出端。在更特定的实施方案中,突出端的长度在1-10个核苷酸之间,更特定地,长度在3-6个核苷酸之间,更特定地长度为4个核苷酸。在dna序列包括突出端的情况下,与切割序列的连接减少,但噪音(由与未切割序列连接所导致)程度显著降低,从而提高了信噪比。
81.在一个实施方案中,将连接酶和包括与切割pcr引物互补的序列的dna序列直接添加到步骤(a)的经核酸酶处理的文库中。在替代的实施方案中,连接酶和包括与切割pcr引物互补的dna序列在钝化5’或3’突出端的步骤之后直接添加到经核酸酶处理的文库中。在缓冲液中不存在反应所需的辅助因子的情况下,也可以在此阶段添加这些辅助因子。
82.在另一个实施方案中,将连接酶和包括与切割pcr引物互补的序列的dna序列添加到步骤(a)的经核酸酶处理的文库或步骤(a)的反应淬灭(和/或在适当的情况下,钝化步骤的淬灭)之后的钝化文库中。在这些实施方案中,步骤(b)的反应在合适的温度下进行合适的时间。在缓冲液中不存在反应所需的辅助因子的情况下,也可以在此阶段添加这些辅助因子。还必须确保先前使用的淬灭方法不干扰步骤(b)的反应。
83.在一个实施方案中,底物文库中的dna底物包括亲和标签,该标签能够用于将底物在其5’末端附接至固相。在一个实施方案中,亲和标签是生物素、链霉亲和素或组氨酸标签。也可以采用共价捕捉标签,诸如巯基、二硫化物、环氧化物或醛底物。在连接到经核酸酶处理的文库上的dna序列附接到能够用于将底物附接到固相的亲和标签上的情况下,这可以用于将切割的序列和连接的底物与文库的其余部分分开。通过解链或分离双链体dna的单个链,只有那些已被活性内切核酸酶切割的链将能够从固相中解离,导致切割的dna级分的选择性富集。在一个实施方案中,该解链过程由将溶液的ph提高到9以上,导致dna双链体的解离组成。在其他实施方案中,该过程可以通过加热或用离液剂诸如氯化胍、高氯酸锂或尿素处理来实现。虽然在柱或板上捕捉将是可能的,但在珠上捕捉与规程的后续步骤最相容。技术人员将理解捕捉将在经包被的珠(其中标签对包被具有亲和力)上实现。虽然不是必需的,但该步骤增加了该方法的信噪比。
84.步骤c)涉及用切割和反向pcr引物对经切割的底物进行pcr扩增。在一个实施方案中,步骤(b)的产物直接用于步骤(c),简单地添加pcr所需要的组分(聚合酶、核苷酸、引物、任何必需的辅助因子)。在其中将未切割的序列捕捉在珠上的实施方案中,dna可以从珠上洗脱下来并用所需的pcr组分(包括合适的缓冲液)重悬浮,或者可以将珠本身重悬浮于所需的pcr组分(包括合适的缓冲液)中。本领域技术人员将充分理解pcr的要求。
85.在一个实施方案中,反向引物包括衔接物以促进特定测序平台的后续下一代测序,诸如,例如life technologies s5 sequencer上的ion torrent ngs、roche 454a或454b测序平台、illumina solexa测序平台、applied biosystems solid测序平台、pacific biosciences'mrt测序平台、pollonator polony测序平台、a helicos测序平台、complete genomics测序平台、intelligent biosystems测序平台或任何其他测序平台。在一个特定实施方案中,反向引物包括反向illumina衔接物(例如i7)。
86.在一个实施方案中,将反向引物附接到能够用于将基底附接到固相的亲和标签上。在一个实施方案中,亲和标签是生物素、链霉亲和素或组氨酸标签。在另一个实施方案中,亲和标签是共价捕捉系统并且标签是巯基、二硫化物、环氧化物或醛底物。
87.步骤(d)涉及对扩增的pcr产物进行测序。这里可以使用下一代测序技术。通常,这些要求产物包括在固相上。在需要的情况下,反向引物上的亲和标签用于以合适的形式(例如在板或珠上)捕捉pcr产物以进行测序。对于熟练的阅读者来说显而易见的是,捕捉是通过用标签对其具有亲和性的物质包被板或珠来完成的。对于熟练的阅读者来说也将显而易见的是,在捕捉前适当的稀释允许对单个dna片段进行测序。
88.虽然有时与切割引物互补的序列将连接到未切割的底物上,但这种情况极为罕见,绝大多数仅经切割的底物并通过pcr扩增。pcr产物的序列在步骤(d)中鉴定。虽然dna靶序列不完全存在于该序列中,但通过标识符序列的序列可以明确鉴定。在那些未切割的底物被扩增的罕见事件中,dna测序将显示这包括完整的推定的靶序列。因此,可以将此类“假阳性”排除在考虑之外(即,可以理解,所含有的推定的靶序列不是dna靶序列)。
89.在一个实施方案中,在底物文库包含双链dna底物并另外含有与位于推定的靶序列的5’的正向引物互补的序列的情况下,步骤c)进一步包括用正向和反向pcr引物对未切割的底物进行pcr扩增。未切割的底物是唯一用正向引物扩增的底物。此外,未切割的底物可以通过测序步骤(d)与经切割的序列进一步区分。未切割的序列将不含有切割引物序列并且将含有完整的推定的靶序列和标识符序列,而经切割的序列将含有切割引物序列和完整的标识符序列。
90.在一个实施方案中,在底物文库包含单链dna底物(其包含防止双链dna连接的5’亲和标签)的情况下,步骤c)进一步包括从5’亲和标签上释放未切割的dna底物,然后是将未切割的dna底物连接到与切割引物序列不同的双链dna序列(在5’到3’方向上含有与未切割的正向引物序列互补的序列),以及使用未切割的正向引物和反向引物进行pcr扩增的步骤。在双链dna序列(在5’至3’方向上含有与未切割的正向引物序列互补的序列)具有平末端的情况下,连接步骤可以采用适用于连接单链和双链dna的连接酶,例如环化酶。在另一个实施方案中,双链dna序列(在5’至3’方向含有与未切割的正向引物序列互补的序列)具有与未切割单链dna底物5’末端处的已知序列杂交的粘性末端。在其发生的情况下,可以使用能够连接粘性末端的连接酶。
91.关于上述步骤(b)和(c)讨论的条件也适用于此处使用的连接和pcr步骤。
92.从亲和标签相继除去经切割和未切割的底物允许分别扩增未切割和经切割的底物。此外,可以通过测序步骤(d)进一步区分未切割的底物与经切割的序列。未切割的序列将不含有切割引物序列并且将含有完整的推定的靶序列和标识符序列,而经切割的序列将含有切割引物序列和完整的标识符序列。
93.该方法的这个实施方案提供了关于哪些序列被切割以及哪些没有被切割的信息。在文库含有每个底物的多个拷贝的情况下,可能是推定的靶序列的一些拷贝被切割而另一些没有被切割的情况。关于被切割的具有特定标识符的序列的比例的信息给予关于哪些dna靶序列被核酸酶优先靶向的信息。
94.在一个实施方案中,包括与切割pcr引物互补的序列的dna序列另外包含在与切割pcr引物互补的序列的5’的独特标识符序列。很明显,使用切割pcr引物和反向引物进行的
pcr扩增将产生含有两个独特标识符序列的产物,一个鉴定推定的靶序列并且另一个鉴定连接事件。这控制了扩增偏倚,并因此允许更准确地鉴定切割事件的数目。
95.在一个实施方案中,未切割的正向引物另外含有独特标识符序列。很明显,使用未切割的正向引物和反向引物进行的pcr扩增将产生含有两个独特标识符序列的产物,一个鉴定推定的靶序列并且另一个鉴定连接事件。这控制了扩增偏倚。
96.类似地,重复该方法但改变步骤(a)的条件以降低切割效率也将给予关于哪些序列被优先切割的信息。
97.应当理解,测序步骤不仅提供关于推定的靶序列是否是dna靶序列的信息,而且还提供关于dna靶序列内的精确切割位点的信息。因此,在一个实施方案中,本发明还提供了用于鉴定dna靶序列中的内切核酸酶切割位点的方法,其包括以下步骤:
98.a)在合适的条件下使本文所定义的底物文库与内切核酸酶接触以允许切割;
99.b)将经内切核酸酶处理的文库与包括与“切割”pcr引物互补的序列的dna序列连接;
100.c)用切割和反向pcr引物对经切割的底物进行pcr扩增;和
101.d)对扩增的pcr产物进行测序;
102.其中通过标识符序列的序列鉴定经切割的产物中的dna靶序列,并且其中通过对扩增的pcr产物进行测序鉴定dna靶序列中的内切核酸酶切割的位点。
103.重要的是,这种方法可以鉴定切割的确切位点是否不变,或者这是否会因任何特定的内切核酸酶而不同。
104.熟练的阅读者将理解,除了鉴定在单个位置优先切割的序列之外,该方法还生成关于可能与脱靶结合和切割相关的相关序列的切割的信息。显而易见的是,由该测定生成的信息将能够鉴定对特定序列具有活性并且另外对基因组中另外存在的任何相关序列没有活性的酶。这是旨在用于期望在单个位点进行切割的基因疗法应用的酶中的特别期望的特征。
105.实施例7证实本发明的方法适合鉴定脱靶序列。本实施例中鉴定的前25个dna靶序列包括先前其他研究人员使用替代方法强调的那些所鉴定的那些。值得注意的是,这使得使用严格的体外测定能够直接鉴定真正的体内倾向(liabilities),显著简化了分类具有显著倾向性的酶并追踪人细胞中这些倾向的过程。
106.在另一方面,使用相同的底物文库和变体内切核酸酶进行多次该方法。例如,在内切核酸酶是大范围核酸酶的情况下,使用野生型大范围核酸酶和其中一个或多个残基变化的大范围核酸酶的工程化改造形式进行该方法。
107.核对来自相同底物上的变体内切核酸酶的信息可以确定内切核酸酶的哪些变化改变靶序列特异性,并且可以用于引导内切核酸酶的进一步修饰以提高对特定dna靶序列的特异性和/或降低对相关序列的特异性。
108.例如,该方法经常使用已知的内切核酸酶和与该核酸酶相差一个氨基酸残基的一组变体内切核酸酶(单个变体)进行。在一个实施方案中,内切核酸酶组包括所有可能的单个氨基酸变体(即,其中每个位置的氨基酸突变为该位置的每个其他可能的残基)。这允许比较变体内切核酸酶在特定底物处的切割效率。在这种情况下,切割效率是指在测序步骤中鉴定的经切割序列的百分比。在方法的步骤d)中获得的具有相关标识符的序列的100%
被切割的情况下,认为这是100%的效率。在具有相关标识符的序列的50%被切割,而50%未切割的情况下,则效率为50%。类似地,在具有相关标识符的序列的30%被切割,而70%未切割的情况下,则效率为30%。
109.因此,在另一个方面,本发明提供了用于工程化改造内切核酸酶的方法,其包括:a)使用相同底物文库,用第一内切核酸酶和至少两种其他内切核酸酶进行用于鉴定本文所定义的dna靶序列的方法,该其他内切核酸酶与第一内切核酸酶的不同在于内切核酸酶氨基酸序列内不同位置处的单个氨基酸变化;b)比较步骤a)中测试的每种内切核酸酶在特定底物上的切割效率;c)鉴定提高切割效率的不同位置处的至少两个氨基酸变化;和d)产生含有步骤c)中鉴定的至少两个氨基酸变化的变体内切核酸酶。
110.熟练的阅读者会理解,该方法可以扩展以鉴定可以提高底物的切割效率的多个氨基酸取代,允许鉴定和产生含有3个或更多个氨基酸取代的变体内切核酸酶。
111.可以以相同的方式比较多个底物。在一种底物是期望靶序列且其他序列是基因组中存在的相关序列的情况下,可以鉴定可能提高期望靶序列的切割效率同时使相关基因组序列的切割效率最小化的变体序列。因此,在另一个方面,本发明提供了用于工程化改造内切核酸酶的方法,其包括:a)使用相同底物文库,用第一内切核酸酶和至少两种其他内切核酸酶进行用于鉴定本文定义的内切核酸酶的dna靶序列的方法,该其他内切核酸酶与第一内切核酸酶的不同在于内切核酸酶氨基酸序列内不同位置的单个氨基酸变化;b)比较步骤a)中测试的每种内切核酸酶在两个单独的底物上的切割效率,其中一个是期望的靶序列,另一个是基因组中存在的相关序列;c)鉴定不同位置的至少两个氨基酸变化,其提高期望靶序列的切割效率,或降低基因组中存在的相关序列的切割效率;和d)产生含有步骤c)中鉴定的至少两个氨基酸变化的变体内切核酸酶。
112.熟练的阅读者将理解,该方法可以扩展以鉴定多个氨基酸取代,这些取代可以提高期望靶底物的切割效率和/或降低基因组中存在的相关序列的切割效率,从而允许鉴定和产生含有3个或更多氨基酸取代的变体内切核酸酶。
113.根据上述方法产生的变体内切核酸酶也构成本发明的一个方面。变体内切核酸酶可能在基因编辑领域具有实用性。因此,在一个实施方案中,本发明提供了用于基因编辑的变体内切核酸酶。在一个实施方案中,本发明提供了基因编辑方法,该方法包括在体外将编码变体内切核酸酶的dna转染到细胞中的步骤。在另一个实施方案中,本发明提供了用于基因疗法的编码变体内切核酸酶的dna。在另一个实施方案中,本发明提供了用于基因疗法的方法,该方法包括将包含编码变体内切核酸酶的dna的载体施用至有此需要的患者的步骤。在进一步的实施方案中,本发明提供了包含编码变体内切核酸酶的dna的载体在制备用于基因疗法的药物中的用途。
实施例
114.实施例1:tcrα底物文库制备
115.从twist biosciences订购了基于tcrα基因中存在的序列的推定的靶序列的文库。特别地,从包括3’pam序列ggn的起始22bp的靶序列,计算机诱变用于生成其中的所有单、双和三突变体,以及连续5个或更多突变的所有串,总计143,452个靶序列。突变包括3’pam序列,以及作为对照碱基添加到池的末端的4bp aaaa片段。然后进行以下反应以将推定
0.1x清洗缓冲液清洗一次,然后用50μl 150mm naoh清洗两次。将珠重悬浮于50μl 10mm tris ph 7.5中。然后如下使用珠来制备50μl pcr反应物:
144.1. 5μl珠
145.2. 10μl 5x phusion hf缓冲液(new england biosciences b0518s)
146.3. 0.25μl含有板条形码的引物
147.4. 0.25μl引物
148.5. 1μl 10mm dntp
149.6. 0.5μl phusion hs2
150.7. 33μl水
151.根据以下程序进行pcr扩增:
152.1. 98℃ 30秒
153.2. 98℃ 10秒
154.3. 60℃ 5秒
155.4. 72℃ 5秒
156.重复2-4 9x
157.5. 12℃保持
158.根据制造商的说明,使用3%盒使用pippin ht净化来分离产物。然后进行定量pcr,并使用整个540芯片将50pm产物加载到ion chef上。然后根据制造商的说明,使用life technologies ion s5测序仪(a27212)对540芯片进行测序。
159.图2显示tcrα底物文库中存在的dna序列的相对丰度。
160.实施例4:tcrαcas9-核糖核蛋白制备
161.将具有序列gagaaucaaaaucggugaau(seq id no:5)的crrna和67bp通用tracrrna(美国专利号9840702中的seq id no:134;可商购自idt,目录号1072532)各自在水中重建至100mm。通过混合等摩尔量的crrna和tracrrna制备双链体grna,加热至95℃达3分钟,并允许冷却至室温。将等摩尔量的双链体grna与来自酿脓链球菌的野生型crispr cas9酶或r691a突变体酶混合以形成活性cas9-核糖核蛋白。
162.实施例5:鉴定tcrαcas9-核糖核苷酸的dna靶序列
163.如下在多孔板中制备10μl切割反应物:
164.1.实施例1中制备的30ng/μl底物文库
165.2. 1mm mgcl2166.3. 1mg/ml牛血清白蛋白
167.4. 10mm tris ph 7.5
168.5.tcrαcas9-核糖核蛋白(不同浓度
–
8μm、4μm、2μm和0.4μm)
169.将切割反应物在37℃下温育1小时,然后在65℃下温育20分钟。然后将以下添加到每个孔中:
170.1. 5μl孔条形码寡核苷酸
171.2. 5μl连接预混物(2.5μl 10x t4连接酶缓冲液、0.5μl t4连接酶、2μl去离子水)
172.将反应物在30.5℃下温育1.5小时,然后在65℃下温育20分钟。合并使用相同条件的反应物(包括孔条形码寡核苷酸)以确保至少50μl的总体积。
173.上述步骤涉及以上描述的用于鉴定dna靶序列的方法的步骤(a)和(b)。
174.通过在链霉亲和素珠(如实施例2中所述制备)上捕捉来纯化文库(切割的和未切割的序列两者)。
175.将50μl珠与50μl反应物相结合。混合物用100μl 1x清洗缓冲液清洗4次,然后用50μl 0.1x清洗缓冲液清洗一次。
176.然后通过将珠与50μl 150mm naoh温育1分钟来洗脱切割的dna,然后将上清液置于含有12μl 1.25m乙酸和6μl 1m tris ph 7.5的受体孔中,然后再与另外的50μl 150mm naoh第二次温育1分钟,然后与第一洗脱物合并。然后将珠(含有未切割的dna)重悬浮并贮存在50μl 10mm tris ph 7.5中。
177.使用切割和未切割的dna样品两者均以如下制备pcr反应物:
178.用于切割样品的pcr反应
179.1. 5μl含有切割产物的珠纯化的上清液,
180.2. 10μl 5x phusion hf缓冲液(new england biosciences b0518s)
181.3. 5μl引物混合物(5μm与孔条形码互补并含有板条形码的引物 5μm与寡核苷酸文库的3’末端互补的引物)
182.4. 1μl 10mm dntp
183.5. 0.5μl phusion hs2
184.6. 28.5μl水
185.根据以下程序进行pcr扩增:
186.1. 98℃ 3分钟
187.2. 98℃ 10秒
188.3. 60℃ 30秒
189.4. 72℃ 30秒
190.重复2-4 11x
191.5. 4℃保持
192.用于未切割样品的pcr反应
193.1. 5μl珠
194.2. 10μl 5x phusion hf缓冲液(new england biosciences b0518s)
195.3. 0.25μl含有板条形码的引物
196.4. 0.25μl引物
197.5. 1μl 10mm dntp
198.6. 0.5μl phusion hs2
199.7. 33μl水
200.根据以下程序进行pcr扩增:
201.6. 98℃ 30秒
202.7. 98℃ 10秒
203.8. 60℃ 5秒
204.9. 72℃ 5秒
205.重复2-4 9x
206.10. 12℃保持
207.在4%琼脂糖凝胶上运行未切割/切割pcr反应物的样品。这些显示于图3。凝胶证实cas-9 rnp切割,并显示野生型cas9-rnp似乎比r691a突变体cas9-rnp具有更多的非特异性切割。
208.根据制造商的说明,使用3%盒使用pippin ht净化来分离切割/未切割的dna。然后进行定量pcr,并使用整个540芯片将50pm产物加载到ion chef上。然后根据制造商的说明,使用life technologies ion s5测序仪(a27212)对540芯片进行测序。
209.根据它们的孔条形码对池进行去卷积,并分析整体切割频率。在每个实验中,将每个寡核苷酸的原始丰度与真正的grna靶定寡核苷酸的丰度进行比较,并评估潜在的脱靶。这些结果列于表1。
210.表1
211.[cas9]/[dna]#》靶标,cas9#》靶标,hifi2016,528(30)16,556(34)1023,599(27)18,603(29)527,821(27)18,227(22)117,092(30)7,559(27)0.114,268(40)9,889(26)0.0110,642(84)10,208(30)0.00110,498(48)10,642(67)0.00019,713(32)10,516(58)
[0212]
与预期一致,我们观察到脱靶的数目随着酶:dna化学计量的减少而减少。令人惊讶的是,我们在每种情况下均观察到“基线特异性”,其出现在大约0.01-0.1rnp:dna比率处,其中特异性随着酶载荷的减少而停止改善。这表明系统的酶/grna依赖性的不可还原背景。
[0213]
此外,使用zhao和stormo报道的beesem方法,使用切割丰度以生成每个碱基对结合能(nature biotechnology 29,pages480
–
483(2011))。样品作为生物一式三份运行,然后平均以生成整体脱靶结合惩罚。1:1rnp:dna化学计量的结果显示于图5,并且5:1rnp:dna化学计量的结果显示于图6。该分析显示了hifi和野生型spcas9两者的每个碱基对结合亲和力中的明显对应,但我们观察到整体化学势项在统计上显著增加,表明活性普遍丧失。这与先前报道的通过对hifi spcas9突变体的生物物理分析衍生的hifi spcas9的作用模式一致(nature medicine 24,pages 1216
–
1224(2018))。
[0214]
为了测试单个寡核苷酸切割率的再现性,使用beesem估计值对这些进行计算并将其显示于图7,表明该方法具有高度的再现性。
[0215]
实施例6:i-scei底物文库制备和表征
[0216]
i-scei具有18个碱基对的识别序列tagggataacagggtaat(seq id no:2)。列举了含有seq id no:2和来自集[a,c,t,g]的seq id no:2的所有单、双和三突变体的文库,即[aagg
…
taat、tagg
…
tgga]。为此,包括了所有可能的n大小的突变体的大小n=4至6的运行窗口的所有列举,即[cccc
…
taat,tagg
…
cccccc]。所得的文库包含59,914个成员,每个成员代表推定的靶序列。该文库是从twist biosciences订购的。
[0217]
基本上使用实施例1中描述的方法从获得自twist biosciences的推定的靶序列的文库生成i-scei底物文库(微小差异在于pcr反应中使用的推定的靶序列的浓度为约9ng/μl)。基本上,对于池的每个成员,将具有序列cacgagcgtagcagagtatgtc(seq id no:6)的参考寡核苷酸预先添加到推定的靶序列的5’末端,将“cg”间隔物置于推定的靶序列和独特标识符dna序列之间,并最后将具有序列gagcatgctctatcgtctgatg(seq id no:7)的第二参考寡核苷酸附加到3’末端。示例池成员将具有以下构建形式:seq id no:6-推定的靶序列-cg-标识符dna序列-seq id no:7。
[0218]
根据实施例3中概述的方法表征i-scei底物文库。图8显示了存在于i-scei底物文库中的dna序列的相对丰度。
[0219]
实施例7:鉴定i-scei的dna靶序列
[0220]
如下所列连续稀释市售的i-scei:
[0221]
1.纯的(neat)
[0222]
2. 1:10-4ul的纯的至36ul h2o
[0223]
3. 1:100-4ul的1:10至36ul h2o
[0224]
4. 1:1,000-4ul的1:100至36ul h2o
[0225]
5. 1:10,000-4ul的1:1000至36ul h2o
[0226]
6. 1:100,000-4ul的1:10000至36ul h2o
[0227]
7. 1:1,000,000-4ul的1:100000至36ul h2o
[0228]
8. 1:10,000,000-4ul的1:1000000至36ul h2o
[0229]
将如实施例6中所列制备的i-scei底物文库稀释至约1000ng/ul并用于制备10μl切割反应,如下所列:
[0230]
1. 30ng/μl i-scei底物文库
[0231]
2. 1mm mgcl2[0232]
3. 1mg/ml bsa
[0233]
4. 10mm tris ph7.5
[0234]
5. 3μl i-scei(可变浓度-以上制备的稀释物)
[0235]
将板在37℃下温育1小时,然后在65℃下温育20分钟。该切割反应涉及上述用于鉴定dna靶序列的方法的步骤(a)。
[0236]
i-scei切割导致突出的单链。这些使用klenow聚合酶“填充”,其通过向每个切割反应中添加5ul klenow混合物(31.9μl 10mm dntp、42.5μl klenow dna聚合酶、456.9μl去离子水),密封并在室温下温育约30min,然后在65℃下加热杀死酶20min。
[0237]
然后将以下添加到每个孔中:
[0238]
1. 5μl孔条形码寡核苷酸
[0239]
2. 5μl连接预混物(2.5μl 10x t4连接酶缓冲液、0.5μl t4连接酶、2μl去离子水)
[0240]
将反应物在30.5℃下温育1.5小时,然后在65℃下温育20分钟,然后在4℃下贮存。合并使用相同条件的反应物(包括孔条形码寡核苷酸),以确保至少50μl的总体积。这涉及上述用于鉴定dna靶序列的方法的步骤(b)。
[0241]
通过在链霉亲和素珠(如实施例2中所述制备)上捕捉来纯化文库(切割的和未切割的序列两者)。
[0242]
将50μl珠与50μl反应物相结合。混合物用100μl 1x清洗缓冲液清洗4次,然后用50μl 0.1x清洗缓冲液清洗一次。
[0243]
然后通过将珠与50μl 150mm naoh温育1分钟来洗脱切割的dna,然后将上清液置于含有12μl 1.25m乙酸和6μl 1m tris ph7.5的受体孔中,然后再与另外的50μl 150mm naoh第二次温育1分钟,然后将其与第一洗脱物合并。然后将珠(含有未切割的dna)重悬浮并贮存在50μl 10mm tris ph7.5中。
[0244]
使用切割和未切割的dna样品两者以如下制备pcr反应:
[0245]
用于切割样品的pcr反应
[0246]
1. 5μl含有切割产物的珠纯化的上清液
[0247]
2. 10μl 5x phusion hf缓冲液(new england biosciences b0518s)
[0248]
3. 5μl引物混合物(5μm与孔条形码互补并含有板条形码的引物 5μm与寡核苷酸文库的3’末端互补的引物)
[0249]
4. 1μl 10mm dntp
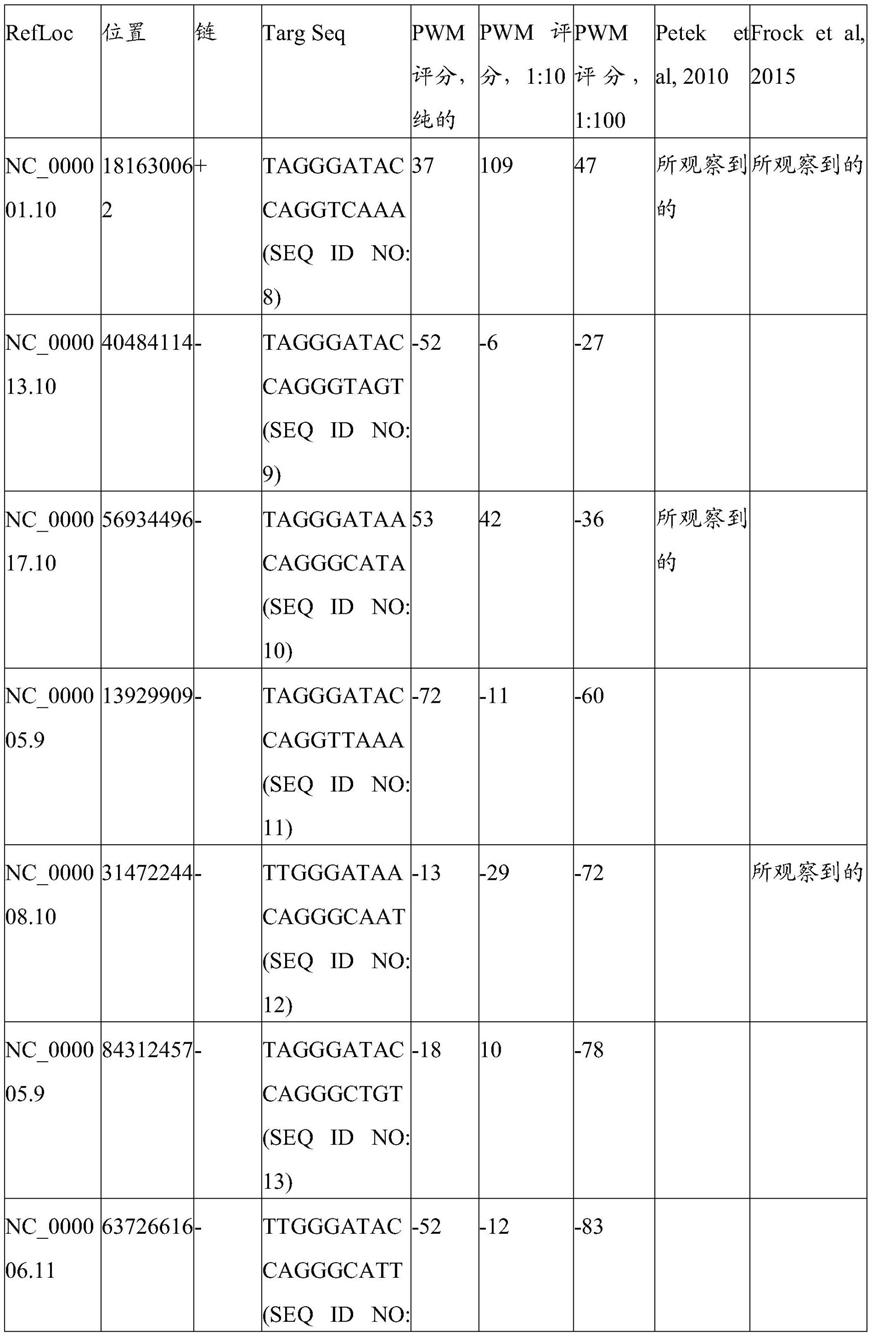
[0250]
5. 0.5μl phusion hs2
[0251]
6. 28.5μl水
[0252]
根据以下程序进行pcr扩增:
[0253]
1. 98℃ 3分钟
[0254]
2. 98℃ 10秒
[0255]
3. 60℃ 30秒
[0256]
4. 72℃ 30秒
[0257]
重复2-4 15x
[0258]
5. 4℃保持
[0259]
用于未切割样品的pcr反应
[0260]
1. 5μl珠
[0261]
2. 10μl 5x phusion hf缓冲液(new england biosciences b0518s)
[0262]
3. 0.25μl含有板条形码的引物
[0263]
4. 0.25μl引物
[0264]
5. 1μl 10mm dntp
[0265]
6. 0.5μl phusion hs2
[0266]
7. 33μl水
[0267]
根据以下程序进行pcr扩增:
[0268]
1. 98℃ 30秒
[0269]
2. 98℃ 10秒
[0270]
3. 60℃ 5秒
[0271]
4. 72℃ 5秒
[0272]
重复2-4 9x
[0273]
5. 12℃保持
[0274]
在4%琼脂糖凝胶上运行未切割/切割pcr反应物的样品。在切割反应物中不存在180bp片段的情况下,进行另外两个循环的pcr。
[0275]
根据制造商的说明,使用3%盒使用pippin ht净化来分离切割/未切割的dna。然后进行定量pcr,并使用整个540芯片将50pm产物加载到ion chef上。然后根据制造商的说明,使用life technologies ion s5测序仪(a27212)对540芯片进行测序。
[0276]
根据它们的孔条形码对池进行去卷积,并分析整体切割频率。在每个实验中,将每个寡核苷酸的原始丰度与真正的grna靶定寡核苷酸的丰度进行比较,并评估潜在的脱靶。使用来自这些数据的衍生评分矩阵,评估人参考基因组的潜在脱靶dna序列,并对各种酶稀释物中的每一种鉴定前25个推定的脱靶。值得注意的是,该方法能够正确检测出在前25个推定的脱靶中使用体内方法检测到的除一个之外的所有脱靶,大大简化了鉴定和评估潜在基因组脱靶序列的工作流程。
[0277]
表2鉴定了使用该方法鉴定的人基因组中存在的i-scei的前25个dna靶序列。值得注意的是,该方法鉴定了先前工作中所观察到的所有5个位点(petek,lisa m et al,"frequent endonuclease cleavage at off-target locations in vivo."molecular therapy 18.5(2010):983-986.)以及在二次研究中所观察到的9个位点中的8个(frock,richard l.,et al."genome-wide detection of dna double-stranded breaks induced by engineered nucleases."nature biotechnology 33.2(2015):179-186.)。
[0278]
表2
[0279]
[0280]
[0281]
[0282]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。