1.本发明属于有机硅技术领域,涉及一种有机硅组合物及其应用。
背景技术:
2.陶瓷材料因具有硬度高、耐高温、物理化学性质稳定等优点,在航空航天、工业、化工等诸多领域获得应用。传统的陶瓷粉末成型方法涉及微粉制备、成型(包括压延、挤塑、干压、等静压、浇注、注射等方式)、烧结(包括热压烧结、反应烧结、常压烧结、气氛压烧结、热等静压烧结、放电等离子体烧结等方式)到加工等过程。但这一传统方法仍存在难以超越的局限性,包括:难以获得均匀的化学成分、可精加工性差、不易制造复杂构件、难以解决陶瓷材料本征脆性问题等,已影响到陶瓷材料应用领域的拓展。
3.有机硅先驱体转化法制备陶瓷是以经高温热解可转化为陶瓷的有机硅聚合物为原料,经成型后再通过高温热解转化为陶瓷。该方法突破了传统陶瓷粉末冶金制备技术的一些不足,且还具有诸多优点:如分子的可设计性,进而实现对陶瓷组成、结构与性能的调控;可移植高分子材料成熟的加工工艺和设备用于陶瓷材料的成型,从而可制备传统粉末工艺难以获取的如陶瓷纤维、陶瓷涂层、复杂陶瓷构件等;可较低温下实现陶瓷化,有利于降低生产能耗和成本;可通过浸渍-裂解工艺制备复杂、大型、近净成型的高强度纤维增强陶瓷基复合材料,已成为高性能陶瓷成型和制造的一项变革性技术。已报道的可转化为陶瓷的有机硅聚合物包括聚硅氧烷、聚硅氮烷、聚硅碳烷、聚硼硅烷、聚硼硅氮烷、聚硅基碳氮亚酰胺等。
4.以有机硅先驱体转化法制备陶瓷,其关键之处在于能否制备出合适的有机硅聚合物。理想的转化为陶瓷的有机硅聚合物应具备以下性质:易存储且稳定性好、杂质元素含量少、陶瓷产率高、易加工成型、价格低、无毒或低毒等。
5.聚碳硅烷是一类有机硅高分子化合物,其主链由硅和碳原子交替组成,硅和碳原子上连接有氢或有机基团,分子链为线形或支化结构,经高温处理可转化为碳化硅陶瓷。结构简式为[(sih2ch2)
x
(sihrch2)y]n(r为不饱和基团;x、y和n为大于0的数值)或其支化结构的聚碳硅烷,其不仅陶瓷产率高、流动性好且易成型,可用于碳化硅陶瓷基复合材料、多孔碳化硅、3d打印碳化硅等材料的制备。但是该类陶瓷先驱体稳定性差,室温易交联固化。为了延长该类有机硅聚合物的存储期,现有技术普遍采用低温冷藏和改变有机硅聚合物结构。如starfire相关的液体聚碳硅烷产品(商品名为smp-10,是含烯丙基的聚碳硅烷)要求真空存储、惰性气氛存储或者冷藏。使用不方便,且在使用过程中难免与空气接触。中国专利cn104177621认为,该类聚碳硅烷中含有丰富的sih3端基,在存储过程中易自缩聚或与空气中的水分反应而产生氢气,导致粘度增加,不利于安全稳定地储存。为延长存储期,采用的方式是引入环体结构以控制sih3基团含量。但该方案对有机硅聚合物结构要求高,适用范围有限。
技术实现要素:
[0006]
本发明针对现有技术的现状,提供一种易存储的有机硅组合物,其具有室温空气环境中存储稳定性好、有机硅聚合物含量高、对有机硅聚合物结构适用性广、且能转化为碳化硅陶瓷的特征。
[0007]
本发明的一个方面提供了一种有机硅组合物,所述有机硅组合物包括有机硅聚合物和稳定剂;所述有机硅聚合物为含有不饱和基团的聚碳硅烷;所述稳定剂为能消耗自由基或者降低自由基活性的化合物。
[0008]
作为优选,所述有机硅聚合物具有如下式(1)、式(2)、式(3)和式(4)所示的结构单元:
[0009][0010]
其中,r1、r2、r3、r4、r5、r6、r7、r8、r9、r
10
彼此独自地选自c1-c6烷基、不饱和基团中的一种,但r1、r2、r3、r4、r5、r6、r7、r8、r9、r
10
中的至少一个选自不饱和基团并且其所在的结构单元存在于所述的有机硅聚合物结构中。
[0011]
上述结构式,式(1)为结构-csih3的结构式表达;式(2)为结构-c2sih2的结构式表达;式(3)为结构-c3sih的结构式表达。
[0012]
c1-c6烷基可以为c1-c6的直链烷烃,也可以为c3-c6的支链烷烃。
[0013]
作为优选,所述不饱和基团为含c=c和/或c≡c的不饱和基团。
[0014]
作为优选,所述含c=c和/或c≡c的不饱和基团为乙烯基、乙炔基、炔丙基或烯丙基。
[0015]
作为优选,有机硅聚合物中不包括c1-c6烷基。
[0016]
通过实验研究发现,对于室温空气环境下易交联固化的含不饱和基团与si-h基团的有机硅聚合物,中性条件下水分对其交联固化影响有限,惰性气氛下稳定性也很好,推测氧气的存在是导致其室温空气环境中易交联固化的决定性因素。进一步研究发现,室温空气环境中易交联的主因并不是si-h氧化后自缩合,而是推测为自由基的形成并引发结构中的不饱和基团交联。基于上述实验设计与分析,发现通过添加能消耗自由基或者降低自由基活性的稳定剂,可提高所述有机硅聚合物室温空气环境下的存储稳定性。
[0017]
当有机硅聚合物中不含c1-c6烷基基团而仅含有h和含c=c和/或c≡c的不饱和基团时,在该有机硅聚合物中添加所述稳定剂,仍可以有效提高有机硅聚合物在室温空气环境下的存储稳定性。
[0018]
当有机硅聚合物中-csih3结构中si-h基团的含量占所有si-h基团总量的10%以上时,在该有机硅聚合物中添加所述稳定剂,可以有效提高有机硅聚合物在室温空气环境下的存储稳定性。
[0019]
当有机硅聚合物中-csih3结构中si-h基团的含量占所有si-h基团总量的30%以上时,在该有机硅聚合物中添加所述稳定剂,仍然可以有效提高有机硅聚合物在室温空气环境下的存储稳定性。
[0020]
当有机硅聚合物中-csih3结构中si-h基团的含量占所有si-h基团总量的50%以上时,在该有机硅聚合物中添加所述稳定剂,仍然可以有效提高有机硅聚合物在室温空气环境下的存储稳定性。
[0021]
作为优选,所述稳定剂选自芳胺类化合物、酚类化合物、醌类化合物、硝基化合物、亚硝基化合物中的一种或多种。
[0022]
作为优选,所述稳定剂为酚类化合物。酚类化合物易被氧化,从而消耗有机硅聚合物吸收的氧气,且酚类化合物的氧化产物醌类具有与自由基结合的能力,从而显著提高所述有机硅聚合物的存储稳定性。
[0023]
作为优选,所述酚类化合物为含两个或两个以上酚羟基的多元酚,进一步优选,所述酚类化合物为含有烷基取代基的多元酚。含有烷基取代基的多元酚可以列举为5-叔丁基顺苯三酚、4-丙基邻苯二酚、4-叔丁基邻苯二酚、2,5-二叔丁基对苯二酚、2,5-二叔戊基对苯二酚等中的一种或多种。酚类化合物中烷基取代基的存在,有助于提高多元酚在有机硅聚合物中的溶解性。
[0024]
作为优选,所述稳定剂为酚类化合物和醌类化合物以质量比(1~3):1混合的混合物。在酚类化合物中添加醌类化合物,可更早捕捉形成的自由基,进一步提高有机硅聚合物的存储稳定性。
[0025]
作为优选,所述醌类化合物为含有烷基取代基的醌类化合物。含有烷基取代基的醌类化合物可以列举为2,5-二叔丁基-1,4-苯醌、2,5-二叔丁基-邻苯二醌、2,5-二叔戊基-1,4-苯醌等中的一种或多种。醌类化合物中烷基取代基的存在,有助于提高醌类化合物在有机硅聚合物中的溶解性。
[0026]
作为优选,稳定剂的质量与有机硅聚合物的质量关系为:0%<稳定剂的质量/有机硅聚合物的质量≤1%。
[0027]
作为优选,稳定剂的质量与有机硅聚合物的质量关系为:0%<稳定剂的质量/有机硅聚合物的质量≤0.2%。少量稳定剂的添加就能显著提高有机硅聚合物的存储稳定性。
[0028]
作为优选,所述有机硅组合物还包括陶瓷填料或金属化合物填料。陶瓷填料或金属化合物填料可以列举为碳化硅、氮化硅、二氧化硅、氧化铝、氧化钇、氮化硼、碳化硼、氧化锆的颗粒或晶须。
[0029]
本发明的另一个方面提供了一种有机硅组合物在制备碳化硅陶瓷的应用,有机硅组合物在高温热解下转化为碳化硅陶瓷材料,碳化硅陶瓷材料包括但不限于碳化硅陶瓷基复合材料、多孔碳化硅、碳化硅连接层材料、碳化硅涂层。
[0030]
与现有技术相比,本发明具有如下有益效果:
[0031]
(1)本发明通过在含不饱和基团与si-h基团的有机硅聚合物中加入能消耗自由基或者降低自由基活性的化合物作为稳定剂,可提高所述有机硅聚合物室温空气环境下的存储稳定性;
[0032]
(2)本发明在含不饱和基团与si-h基团的有机硅聚合物中加入≤1%的稳定剂,在少量添加稳定剂的情况下稳定效果显著;
[0033]
(3)本发明在含不饱和基团与si-h基团的有机硅聚合物中加入酚类化合物和醌类化合物以质量比(1~3):1形成的混合物作为稳定剂,相对于加入单成分的稳定剂具有更好的存储稳定性;
[0034]
(4)本发明采用的添加稳定剂的技术方案,有效避免有机硅聚合物低温存储、隔绝空气环境下存储或使用等苛刻条件,减少对相关设备的依赖;
[0035]
(5)有机硅聚合物存储稳定性的提高,对提高其使用寿命、减少因存储时粘度增加或者固化导致的加工困难与经济损失;
[0036]
(6)本发明对所述有机硅聚合物结构适用性广、稳定剂添加量少、效果显著、不影响有机硅聚合物高温热解转化为碳化硅陶瓷的应用。
附图说明
[0037]
图1为本发明对比实施例1中所述的含烯丙基聚碳硅烷的核磁氢谱;
[0038]
图2为本发明对比实施例1中所述的含烯丙基聚碳硅烷于35℃空气氛围中放置24h前后的红外谱图;
[0039]
图3为本发明对比实施例2中所述的含烯丙基聚碳硅烷于40℃空气氛围中保温时质量随时间变化;
[0040]
图4为本发明对比实施例3中所述的含烯丙基聚碳硅烷于50℃空气氛围中保温时质量随时间变化;
[0041]
图5为本发明对比实施例4中所述的含烯丙基聚碳硅烷核磁氢谱;
[0042]
图6为本发明对比实施例5中所述的含乙烯基聚碳硅烷核磁氢谱;
[0043]
图7为本发明对比实施例6中所述的含乙炔基和烯丙基聚碳硅烷核磁氢谱;
[0044]
图8为本发明实施例1-4中所述有机硅组合物的复数粘度随剪切速率变化曲线;
[0045]
图9为本发明实施例4所述有机硅组合物1600℃裂解产物的x射线衍射图;
[0046]
图10为本发明实施例23所得碳化硅纤维增强碳化硅复合材料管;
[0047]
图11为本发明实施例24所得多孔碳化硅块体及内部形貌;
[0048]
图12为本发明实施例24所得多孔碳化硅块体的压缩应力-应变曲线。
具体实施方式
[0049]
下面通过具体实施例及附图,对本发明的技术方案作进一步描述说明,应当理解的是,此处所描述的具体实施例仅用于帮助理解本发明,不用于本发明的具体限制,且本文中所使用的附图,仅仅是为了更好地说明本发明所公开内容,对保护范围并不具有限制作用。
[0050]
对比实施例1
[0051]
以cl
1.25
(ch3o)
1.75
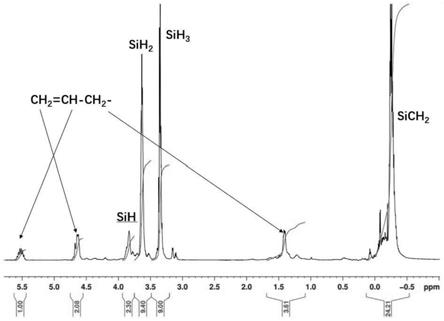
sich2cl和氯丙烯等为原料,参照us7714092b2制备方法,经格氏反应和还原反应合成含烯丙基液态聚碳硅烷,其数均分子量为922,重均分子量为2490,其核磁氢谱如图1所示。依据核磁氢谱,其结构中,包含如说明书中所示的三种结构单元(结构式(1)、结构式(2)与结构式(3))以及不饱和基团烯丙基;此外,核磁硅谱显示其结构中包含如说明书所示的结构单元(4)。其中,结构式(1)、结构式(2)与结构式(3)三种结构单元中si-h基团的积分比为3.91:4.09:1,结构式(1)中的si-h基团的含量占所有si-h基团含量的43.4%。烯丙基与si-h基团摩尔比为1:20.7。
[0052]
将该含烯丙基液态聚碳硅烷,在氮气气氛中,80℃加热24小时,粘度、核磁及红外光谱未发现可见变化。在氮气气氛中,加入5wt%水并于35℃搅拌24小时,分离后,该含烯丙基液态聚碳硅烷的粘度、核磁及红外光谱也未发现可见变化。将该含烯丙基液态聚碳硅烷置于35℃鼓风烘箱中,24h后凝胶化,凝胶化后,并未出现气泡。其放置前后的红外谱图如图2所示,发现与不饱和双键相关联的红外吸收峰显著降低。
[0053]
对比实施例2
[0054]
将对比实施例1中所示结构的含烯丙基液态聚碳硅烷,置于40℃鼓风烘箱中,10h后凝胶化并变脆,凝胶化后,未出现气泡。采用热失重监测其保温过程中质量变化,见图3,当于40℃保温时,随保温时间延长,质量逐渐减少,但减少幅度并不大,10h后质量保留率为98.68%。并采用元素分析检测其氧含量变化,元素分析表明,40℃保温前,样品中氧含量为1.60wt%,10h后,氧含量为3.81wt%。在保温过程,并未出现含烯丙基液态聚碳硅烷结构中si-h基团较多氧化致使氧含量提高及出现增重的现象。但红外谱图显示与不饱和双键相关联的红外吸收峰显著降低。
[0055]
对比实施例3
[0056]
将对比实施例1中所示结构的含烯丙基液态聚碳硅烷,置于50℃鼓风烘箱中,6h后凝胶化并变脆,凝胶化后,未出现气泡。采用tga监测其保温过程中质量变化,见图4,当于50℃保温时,随保温时间延长,质量逐渐减少,但减少幅度并不大,6h后质量保留率为98.65%。并采用元素分析检测其氧含量变化,元素分析表明,50℃保温6h后,氧含量为3.61wt%,而保温前氧含量为1.60wt%。在50℃保温过程,也未出现结构中si-h基团较多氧化致使氧含量提高及出现增重的现象。但红外谱图也显示与不饱和双键相关联的红外吸收峰显著降低。
[0057]
对比实施例4
[0058]
以cl
0.5
(ch3o)
2.5
sich2cl和氯丙烯等为原料,参照us7714092b2制备方法,经格氏反应和还原反应合成含烯丙基液态聚碳硅烷,其数均分子量为1462,重均分子量为9642,其核磁氢谱如图5所示。结构式(1)、结构式(2)与结构式(3)三种结构单元中si-h基团的积分比为3.82:1.91:1,结构式(1)中的si-h基团的含量占所有si-h基团含量的56.8%,烯丙基与si-h基团摩尔比为1:29.87。将该结构的含烯丙基液态聚碳硅烷,置于35℃鼓风烘箱中,15h后凝胶化。元素分析表明,35℃保温15h后,氧含量为3.35wt%,而保温前氧含量为1.72wt%。红外谱图显示与不饱和双键相关联的红外吸收峰显著降低。
[0059]
对比实施例5
[0060]
以cl(ch3o)2sich2cl和乙烯基溴化镁等为原料,参照us7714092b2制备方法,经格氏反应和还原反应合成含乙烯基液态聚碳硅烷,其数均分子量为1321,重均分子量为4371。
核磁谱图如图6所述,依据核磁,结构式(1)、结构式(2)与结构式(3)三种结构单元中si-h基团的积分比为2.56:3.39:1,结构式(1)中的si-h基团的含量占所有si-h基团含量的36.8%,乙烯基与si-h基团摩尔比为1:17.1。将该结构的液态聚碳硅烷,置于40℃鼓风烘箱中,37h后凝胶化。元素分析表明,40℃保温37h后,氧含量为2.98wt%,而保温前氧含量为1.47wt%。红外谱图显示与不饱和双键相关联的红外吸收峰显著降低。
[0061]
对比实施例6
[0062]
以cl
1.25
(ch3o)
1.75
sich2cl、氯丙烯和乙炔基氯化镁等为原料,参照us7714092b2制备方法,经格氏反应和还原反应合成含乙炔基与烯丙基液态聚碳硅烷,其数均分子量为1031,重均分子量为3867。核磁谱图如图7所述,依据核磁,结构式(1)、结构式(2)与结构式(3)三种结构单元中si-h基团的积分比为2.82:2.76:1,结构式(1)中的si-h基团的含量占所有si-h基团含量的42.9%。烯丙基、乙炔基与si-h基团摩尔比为1:0.38:41.72。将该结构的液态聚碳硅烷,置于35℃鼓风烘箱中,31h后凝胶化。元素分析表明,35℃保温31h后,氧含量为3.79wt%,而保温前氧含量为1.52wt%。红外谱图显示与不饱和基团相关联的红外吸收峰显著降低。
[0063]
通过对比实施例1-6可知,对于含不饱和基团和si-h基团的有机硅聚合物液态聚碳硅烷,在氮气氛围下稳定性好,中性水的存在对其稳定性的影响也不显著。室温空气环境下放置易交联,且随着温度的升高凝胶化时间缩短。但是室温凝胶化前后氧含量增加的幅度并不大且质量还略微有降低,比较显著的特征是不饱和基团含量的降低。据此推测,导致其室温存储不稳定的关键要素是不饱和基团的存在并参与了交联反应。对于所述有机硅聚合物中的不饱和基团,其参与交联反应的机理包括si-h加成不饱和基团、自身自由基连锁聚合。在无催化剂存在的情况下,si-h加成双键在80℃加热24小时也难以进行,证明了所述有机硅聚合物结构中si-h加成双键在低温下不易进行。因此,推测最可能的交联机理是不饱和基团自身自由基连锁聚合。
[0064]
实施例1
[0065]
向对比实施例1中所述含烯丙基液态聚碳硅烷中加入0.20wt%含量的4-叔丁基邻苯二酚(即4-叔丁基邻苯二酚的质量为含烯丙基液态聚碳硅烷的质量的0.2%),混合后,置于35℃鼓风烘箱中,保温10天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量以及采用流变仪表征其复数粘度。其数均分子量从922增加到1026,重均分子量从2490增加到2546。复数粘度随剪切速率变化的结果在图8中。在线性粘弹性区,实施例1的粘度从保温前的0.017pa
·
s仅增加到0.022pa
·
s。
[0066]
实施例2
[0067]
向对比实施例1中所述含烯丙基液态聚碳硅烷中加入0.20wt%含量的4-叔丁基邻苯二酚,混合后,置于35℃鼓风烘箱中,保温20天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量以及采用流变仪表征其复数粘度。其数均分子量从922增加到1091,重均分子量从2490增加到2742。复数粘度随剪切速率变化的结果在图8中。在线性粘弹性区,实施例2的粘度从0.017pa
·
s增加到0.024pa
·
s。
[0068]
实施例3
[0069]
向对比实施例1中所述含烯丙基液态聚碳硅烷中加入0.20wt%含量的4-叔丁基邻苯二酚,混合后,置于35℃鼓风烘箱中,保温30天,未发生凝胶化,流动性好。采用凝胶渗透
色谱仪表征其分子量以及采用流变仪表征其复数粘度。其数均分子量从922增加到1138,重均分子量从2490增加到2778。复数粘度随剪切速率变化的结果在图8中。在线性粘弹性区,实施例3的粘度从0.017pa
·
s增加到0.025pa
·
s。
[0070]
实施例4
[0071]
向对比实施例1中所述含烯丙基液态聚碳硅烷中加入0.20wt%含量的4-叔丁基邻苯二酚,混合后,置于35℃鼓风烘箱中,保温60天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量以及采用流变仪表征其复数粘度。其数均分子量从922增加到1138,重均分子量从2490增加到2999。复数粘度随剪切速率变化的结果在图8中。在线性粘弹性区,实施例4的粘度从0.017pa
·
s增加到0.028pa
·
s。将其氩气氛围下裂解至1600℃,其x射线衍射峰如图9所示,表明所述有机硅组合物经高温处理可转化为结晶性碳化硅陶瓷。
[0072]
实施例5
[0073]
向对比实施例1中所述含烯丙基液态聚碳硅烷中加入0.5wt%含量的4-叔丁基邻苯二酚,置于35℃鼓风烘箱中,保温60天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量,其数均分子量从922增加到1104,重均分子量从2490增加到2658。
[0074]
实施例6
[0075]
向对比实施例1中所述含烯丙基液态聚碳硅烷中加入1.0wt%含量的4-叔丁基邻苯二酚,置于40℃鼓风烘箱中,保温60天,未发生凝胶化,可流动。采用凝胶渗透色谱仪表征其分子量,其数均分子量从922增加到1352,重均分子量从2490增加到5729。
[0076]
实施例7
[0077]
向对比实施例4中所述含烯丙基液态聚碳硅烷加入0.25wt%含量的4-叔丁基邻苯二酚,置于35℃鼓风烘箱中,保温60天,未发生凝胶化,可流动。采用凝胶渗透色谱仪表征其分子量,其数均分子量从1462增加到1526,重均分子量从9642增加到13824。
[0078]
实施例8
[0079]
向对比实施例5所述含乙烯基液态聚碳硅烷加入0.5wt%含量的4-丙基邻苯二酚,置于40℃鼓风烘箱中,保温30天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量,其数均分子量从1321增加到1374,重均分子量从4371增加到4865。
[0080]
实施例9
[0081]
向对比实施例6所述含乙炔基与烯丙基液态聚碳硅烷加入0.25wt%含量的1,3,5-三硝基苯,置于35℃鼓风烘箱中,保温60天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量。其数均分子量从1031增加到1182,重均分子量从3867增加到4739。
[0082]
实施例10
[0083]
向对比实施例1中所述含烯丙基液态聚碳硅烷加入0.2wt%含量的2,5-二叔戊基-1,4-苯醌,置于35℃鼓风烘箱中,保温60天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量。其数均分子量从922增加到1249,重均分子量从2490增加到3318。
[0084]
实施例11
[0085]
向对比实施例1中所述含烯丙基液态聚碳硅烷加入0.06wt%含量的4-叔丁基邻苯二酚和0.14wt%含量的2,5-二叔戊基-1,4-苯醌,置于35℃鼓风烘箱中,保温60天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量。其数均分子量从922增加到1101,重均分子量从2490增加到3006。
[0086]
实施例12
[0087]
向对比实施例1中所述含烯丙基液态聚碳硅烷加入0.1wt%含量的4-叔丁基邻苯二酚和0.1wt%含量的2,5-二叔戊基-1,4-苯醌,置于35℃鼓风烘箱中,保温60天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量。其数均分子量从922增加到1036,重均分子量从2490增加到2647。
[0088]
实施例13
[0089]
向对比实施例1中所述含烯丙基液态聚碳硅烷加入0.13wt%含量的4-叔丁基邻苯二酚和0.07wt%含量的2,5-二叔戊基-1,4-苯醌,置于35℃鼓风烘箱中,保温60天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量。其数均分子量从922增加到1023,重均分子量从2490增加到2590。
[0090]
实施例14
[0091]
向对比实施例1中所述含烯丙基液态聚碳硅烷加入0.15wt%含量的4-叔丁基邻苯二酚和0.05wt%含量的2,5-二叔戊基-1,4-苯醌,置于35℃鼓风烘箱中,保温60天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量。其数均分子量从922增加到1065,重均分子量从2490增加到2751。
[0092]
实施例15
[0093]
向对比实施例1中所述含烯丙基液态聚碳硅烷加入0.17wt%含量的4-叔丁基邻苯二酚和0.03wt%含量的2,5-二叔戊基-1,4-苯醌,置于35℃鼓风烘箱中,保温60天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量。其数均分子量从922增加到1136,重均分子量从2490增加到2993。
[0094]
通过实施例4和实施例10-15可证明,当采用的稳定剂为酚类化合物和醌类化合物以质量比(1~3):1形成的混合物时,有机硅组合物具有更优异的稳定性。
[0095]
实施例16
[0096]
向对比实施例1中所述含烯丙基液态聚碳硅烷加入0.2wt%含量的4,4
’‑
二氨基二苯醚,置于35℃鼓风烘箱中,保温30天,未发生凝胶化,流动性好。采用凝胶渗透色谱仪表征其分子量。其数均分子量从922增加到1374,重均分子量从2490增加到3862。将其氩气氛围下裂解至1600℃,x射线衍射结果显示可转化为结晶性碳化硅。
[0097]
实施例17
[0098]
将碳化硅纤维编织管件放入量筒中,加入实施例1所述有机硅组合物,然后抽真空浸渍。取出后,擦去表面附着的有机硅组合物。放入热解炉中,在氮气气氛中,按如下程序升温:以10℃/min升至1200℃,期间于200℃和1200℃各保温1h。重复真空浸渍和裂解过程9次,实现碳化硅纤维增强碳化硅复合材料管件致密化。计算所得复合材料中碳化硅纤维含量为55.3wt%。所得管件外形与内部形貌如图10所示。
[0099]
实施例18
[0100]
将2g碳化硅晶须加入20ml实施例11所述有机硅组合物的正庚烷溶液中(所述有机硅组合物含量为20wt%)。高速机械搅拌1小时,以分散sic晶须。搅拌均匀后倒入模具中并抽滤得到柱形块体。之后放入热解炉中,在氩气气氛中,以10℃/min升至1500℃,并保温1小时,期间于200℃保温1h。得到密度为0.36g/cm3的多孔碳化硅。其外观及扫描电镜照片如图11所示,碳化硅晶须通过裂解后的液态聚碳硅烷相互粘结,并形成块体。压缩应力-应变曲
线如图12所示,显示出较优异的机械强度。
[0101]
通过实施例17和实施例18可证明,所述有机硅组合物可应用于碳化硅陶瓷基复合材料、多孔碳化硅等碳化硅材料的制备中。
[0102]
以上对比实施例和实施例中的性能检测采用以下仪器进行:
[0103]
红外光谱:采用美国thermo nicolet 6700傅立叶变换红外光谱分析仪测试;
[0104]
核磁:采用德国bruker公司bruker avanceⅲ核磁共振仪测试;
[0105]
分子量:采用日本tosoh公司hlc-8320gpc凝胶渗透色谱仪测试;
[0106]
氧含量:采用日本horiba公司emga-620型氧氮分析仪测试;
[0107]
热失重:采用日本精工生产tg-dta 6300热重分析仪测试;
[0108]
流变行为:采用奥地利physica mcr 301流变仪测试;
[0109]
x-衍射:采用德国布鲁克公司bruker d8 advance多晶x射线衍射仪测试;
[0110]
扫描电镜:采用日本hitachi公司s-4800型场发射扫描电子显微镜测试;
[0111]
压缩应力-应变曲线:采用美国instron公司instron model5567万能材料试验机测试。
[0112]
最后应说明的是,本文中所描述的具体实施例仅仅是对本发明精神作举例说明,而并非对本发明的实施方式的限定。本发明所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,这里无需也无法对所有的实施方式予以全例。而这些属于本发明的实质精神所引申出的显而易见的变化或变动仍属于本发明的保护范围,把它们解释成任何一种附加的限制都是与本发明精神相违背的。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。