本发明涉及作为c-abl之抑制剂的式(i)化合物,并且特别是式(ii)化合物。本发明还涉及包含这些化合物的药物组合物,以及其在治疗或预防其中c-abl的抑制是有益的医学病症中的用途。这样的医学病症包括神经退行性疾病和癌症。
背景技术:
abl1(艾贝尔森鼠白血病病毒癌基因同源物1(abelson murine leukaemia viral oncogene homolog 1))是显示酪氨酸激酶酶活性并与多种细胞功能相关的蛋白质。在人中,该蛋白质由位于染色体9上的abl1基因编码。在哺乳动物基因组内发现的abl1基因的形式表示为c-abl。费城染色体(philadelphia chromosome)是由t(9,22)染色体相互易位形成的染色体22中的遗传异常,导致称为bcr-abl1的融合基因。该融合基因包含来自染色体9的abl1基因和bcr基因的一部分。abl1蛋白的酪氨酸激酶活性通常受到严格调节,然而,融合基因中的bcr结构域导致abl1激酶的组成型激活。然而,bcr-abl和c-abl的结合结构域是相同的。c-abl的激活涉及多种疾病,特别是癌症。例如,bcr-abl突变的存在与慢性髓性白血病(chronic myeloid leukaemia,cml)强相关。其也在急性淋巴细胞白血病(acute lymphocytic leukaemia,all)和急性淋巴母细胞白血病(acute lymphoblastic leukaemia,all)的一些情况中被发现。尼洛替尼(nilotinib)和帕纳替尼(ponatinib)二者均为已用于治疗慢性髓性白血病(cml)和急性淋巴细胞白血病(all)的c-abl抑制剂。可通过c-abl抑制治疗的白血病的范围包括慢性髓性白血病(cml)、急性淋巴母细胞白血病(all)、急性髓性白血病(acute myelogenous leukaemia,aml)、混合表型急性白血病(mixed-phenotype acute leukaemia,mpal)及其中枢神经系统(central nervous system,cns)转移。c-abl的激活也涉及神经退行性疾病。神经退行性疾病可被神经元的进行性变性和最终死亡表征。具体的神经退行性疾病包括肌萎缩侧索硬化(amyotrophic lateral sclerosis,als)和帕金森病(parkinson’s disease,pd)。als是由运动神经元的进行性变性引起的致命的神经退行性疾病。据报道,c-abl信号传导激活有助于神经元凋亡,并且c-abl抑制剂可防止运动神经元死亡[rojas et al.frontiers in cellular neuroscience,2015,9,203;imamura et al.science translational medicine,2017]。帕金森病(pd)是由黑质致密部(substantia nigra pars compacta)中多巴胺能神经元的选择性损失引起的进行性神经退行性病症。据报道,c-abl在患有pd的患者的脑中被激活,并且c-abl抑制可防止多巴胺神经元损失[pagan et al.pharmacology research&perspectives,2019;karuppagounder et al.scientific reports,2014,4,4874]。c-abl的激活还涉及宽范围的其它疾病,包括但不限于朊病毒病、病毒感染、糖尿
fibrosis in mice.respiration 2011,82,273-87)。尽管在这些研究中,作者聚焦于与pdgfr有关的机制的意义,但有趣的是rhee et a1.(respiration.2011,82,273-87)的研究,比伊马替尼更强效的c-abl抑制剂尼洛替尼显示出优异的治疗性抗纤维化作用,因此支持c-abl抑制剂用于治疗具有肺部炎症的人疾病的治疗适用性。在另一项研究中,将小鼠暴露于高氧提高了发动蛋白2磷酸化和活性氧物质产生和肺渗漏所需的abl1激活(singleton et al.dynamin 2and c-abl are novel regulators of hyperoxia-mediated nadph oxidase activation and reactive oxygen species production in caveolin-enriched microdomains of the endothelium j.biol.chem.2009,284,34964-75)。鉴于以上,对可用于治疗和预防其中c-abl的抑制是有益的医学病症,例如神经退行性疾病(即als和pd)和癌症(尤其是白血病)的新化合物的需求尚未满足。
附图说明
图1示出了显示帕纳替尼在与重组cyp1a1和人肝胞质溶胶一起孵育之后形成谷胱甘肽加合物(在4.33分钟处的峰)的uv色谱图。认为该加合物(至少部分地)是与帕纳替尼相关的毒性的原因。图2示出了显示实施例25在与重组cyp1a1和人肝胞质溶胶一起孵育之后不形成谷胱甘肽加合物的uv色谱图。图3示出了在存在als患者iastrocyte的情况下,示例性化合物在挽救运动神经元存活方面的作用。图4示出了示例性化合物在降低在过表达α-突触核蛋白的rencell vm神经元细胞中的α-突触核蛋白水平方面的作用。
技术实现要素:
出乎意料地,已发现式(i)化合物可抑制c-abl并且因此治疗或预防上述医学病症。此外,与已知化合物相比,其具有导致用作药物的潜力提高的某些有益的特性。这可依据其效力、c
max
下的游离脑水平、溶解度、选择性分布(例如激酶选择性)、低herg抑制活性、安全性特性和/或其它显著的药代动力学特性。因此,本发明涉及式(i)化合物,或其可药用盐、溶剂合物、水合物、互变异构体、光学异构体、n-氧化物和/或前药,其中a为未经取代的吡啶基;b为经取代的5元杂芳基;并且r1是h或选自以下:(i)c
1-c6烷基、c
2-c6烯基和c
2-c6炔基,其各自任选地经一个或更多个独立地选自-nrarb、-orc、卤素和氧代的取代基取代;以及(ii)c
6-c
10
芳基、c
1-c9杂芳基、c
1-c9杂环,其各自任选地经一个或更多个独立地选
自卤素和c
1-c6烷基的取代基取代,其中该c
1-c6烷基任选地经一个或更多个卤素原子取代,其中ra和rb各自独立地选自h和c
1-c6烷基,其中该c
1-c6烷基任选地经一个或更多个卤素原子取代,或者ra和rb可与其所连接的氮原子一起形成5或6元饱和、部分饱和或不饱和的环;并且rc各自独立地选自h和c
1-c6烷基,其中该c
1-c6烷基任选地经一个或更多个卤素原子取代。这些化合物是本发明的化合物。高度优选地,r1是h。在这种情况下,本发明的化合物具有式(ii)。本文中使用的术语“吡啶基”是吡啶的一价基团。吡啶基可以是经邻位、间位或对位取代的,即基团a可以是以下三种基团之一。优选基团a的吡啶基是经间位或对位取代的,即以下基团之一。最优选地,基团a是:不希望受理论的束缚,本发明的化合物的出乎意料的有益特性可部分归因于吡啶基。出乎意料地发现,包含未经取代的吡啶基的化合物具有提高的血脑屏障穿透,这使得其特别地可用于治疗某些疾病和病症。值得注意的是,血脑屏障穿透是不可预测的并且是凭经验建立的。克服与将治疗剂递送至脑的特定区域相关的挑战对大多数脑病症的治疗提出了主要挑战。本文中使用的“5元杂芳基”是芳族单环烃环,其中至少一个,例如1、2、3或4个环原子是杂原子。可用作基团b的5元杂芳基的一些实例包括但不限于经取代的吡咯、吡唑、咪唑、三唑、四唑、异唑、二唑和噻唑。优选的5元杂芳基是经取代的吡唑、咪唑、三唑、四唑、异唑、二唑或噻唑,更优选的是经取代的吡唑、三唑或咪唑基团,最优选的是经取代的吡唑或咪唑基团。5元杂芳基的一些优选实例包括:
或其互变异构体,并且每个基团是经取代的。更优选地,其包含或其互变异构体,并且每个基团是经取代的。在本发明的优选特征中,基团b的5元杂芳基经一个或更多个独立地选自以下的取代基取代:(i)c
1-c6烷基、c
2-c6烯基、c
2-c6炔基和c
1-c6烷氧基,其各自任选地经一个或更多个独立地选自-nrdre、-orf、卤素和氧代的取代基取代;(ii)卤素、-cn、-c(o)nrgrh、-nrgrh、-c(o)ori、-c(o)ri和-ori;以及(iii)c
6-c
10
芳基、c
1-c9杂芳基和c
1-c9杂环,其各自任选地经一个或更多个独立地选自卤素和c
1-c6烷基的取代基取代,其中该c
1-c6烷基任选地经一个或更多个卤素原子取代,其中rd、re、rg和rh独立地选自h和c
1-c6烷基,其中该c
1-c6烷基任选地经一个或更多个卤素原子取代,或者rd和re、和/或rg和rh可与其所连接的氮原子一起形成5或6元饱和、部分饱和或不饱和的环,其中环任选地经一个或更多个选自卤素和c
1-c3烷基的基团取代,其中该c
1-c3烷基任选地经一个或更多个卤素原子取代;并且其中rf和ri独立地选自h和c
1-c6烷基,其中该c
1-c6烷基任选地经一个或更多个卤素原子取代。在更优选的特征中,基团b的5元杂芳基经一个或更多个独立地选自以下的取代基取代:(i)任选地经一个或更多个选自-nrdre、-orf、卤素和氧代的取代基取代的c
1-c6烷基;(ii)卤素、-c(o)nrgrh、-c(o)ori;以及(iii)-ori。在更优选的特征中,基团b的5元杂芳基经一个或更多个独立地选自以下的取代基取代:(i)任选地经一个或更多个选自卤素和-orf的取代基取代的c
1-c6烷基;(ii)卤素、-c(o)ori;以及(iii)-ori。不希望受理论的束缚,特别有利的可以是包含疏水基团作为在基团b的5元杂芳基上的取代基,因为这可提高与c-abl的疏水口袋的相互作用,从而提高结合亲和力。最优选地基团b的5元杂芳基经c
1-c6烷基、异丙基或叔丁基取代,更优选地经异丙基或叔丁基取代,
最优选地经叔丁基取代。取代基优选位于相对于与化合物其余部分的连接点的3-位或4-位,如由以下两个实例中的叔丁基所示。除了本发明的上述优选特征之外,亚烷基可与基团b的5元杂芳基的两个相邻原子连接以形成与基团b的5元杂芳基稠合的5、6或7元(优选5或6元)不饱和、部分饱和或饱和的环。优选地,其为与基团b的5元杂芳基稠合的部分饱和或饱和的环。该稠合双环环体系是基团b的5元杂芳基的特定方面。在这种情况下,“亚烷基”是c3、c4或c5烷基的直链双基,其中每个基团位于烷基链的每个末端。基团b的稠合双环环体系任选地具有亚烷基的独立地被替换为杂原子的一个或两个碳原子。当杂原子是氮时,则所述氮可经c
1-c6烷基或-c(o)o-(c
1-c6烷基)取代,其中c
1-c6烷基任选地经一个或更多个卤素原子取代。当杂原子是硫时,则所述硫可形成亚硫酰或磺酰基团,例如在基团b的以下两个实例中的。基团b的稠合双环环体系的亚烷基的碳原子可任选地经一个或更多个独立地选自以下的取代基取代:卤素、-c(o)o-(c
1-c6烷基)、c
1-c6烷基和氧代,优选c
1-c6烷基和氧代,其中所述c
1-c6烷基任选地独立地经一个或更多个卤素原子取代。此外,与基团b的稠合双环环体系的亚烷基的同一碳连接的两个氢原子可任选地被替换为碳原子,所述碳原子与其所连接的碳原子一起形成c
3-c6环状烷基(即形成螺基序),其中所述环状烷基任选地经一个或更多个卤素原子取代,和/或一个碳被替换为杂原子,优选被替换为o或n。当化合物包含基团b的稠合双环环体系(例如上述那些)时,最优选的是亚烷基与5元杂芳基形成部分饱和或饱和的环。基团b的一些示例性稠合双环环体系包括:
或其互变异构体,其各自可以是任选地经取代的,如上文所概述。在其中亚烷基的一个或更多个碳原子已被杂原子(例如n、o或s)替代的情况下,合适的一些实例包括:或其互变异构体,其各自可以是任选地经取代的,如上文所概述。上述螺基序的合适的一些实例发现于基团b的以下实例中:
或其互变异构体,其各自可以是任选地经取代的,如上文所概述。不希望受理论的束缚,当亚烷基连接在基团b的5元杂芳基的2-位和3-位时,如在以下基团中:亚烷基可具有提高的与c-abl的疏水口袋的相互作用,从而提高结合亲和力。当亚烷基连接在基团b的5元杂芳基的3-位和4-位时,如在以下中:特别有利的可以是在亚烷基上包含另外的疏水取代基以提高与疏水口袋的相互作用并从而提高对c-abl的结合亲和力。优选地,疏水基团位于桥头原子的α-位(如下所示)。疏水基团优选为c
1-c6烷基,更优选为二-c
1-c6烷基(即在同一原子上取代的两个c
1-c6烷基),最优选形成偕二甲基,例如在基团b的以下实例中的。特别优选的是基团b不包含任何n-h基团。换言之,优选的是基团b中的所有氮原子是经三取代的,例如其可以是叔胺。为了避免疑问,这不包含在基团b的氮原子与可药用盐(例如hcl)之间形成的n-h键。不包含n-h作为基团b的一部分之化合物可具有提高的与c-abl的疏水口袋的相互作用,从而进一步提高结合亲和力。更优选地,基团b的5元杂芳基经一个或更多个独立地选自以下的取代基取代:(i)任选地经一个或更多个选自卤素和-orf的取代基取代的c
1-c6烷基;
(ii)卤素、-c(o)ori;以及(iii)-ori,和/或其中亚烷基与基团b的5元杂芳基的两个相邻原子连接以形成5或6元部分饱和或饱和的环,任选地,其中亚烷基的一个或更多个碳原子经一个或更多个独立地选自卤素、c
1-c6烷基和氧代的取代基取代,其中所述烷基任选地独立地经一个或更多个卤素原子取代;和/或任选地,其中与亚烷基的同一碳连接的两个氢原子被替换为碳原子,该碳原子与其所连接的碳原子一起形成c
3-c6环状烷基,其中所述环状烷基任选地经一个或更多个卤素原子取代。在本发明的一个特别优选的特征中,该化合物是式(i)化合物,优选式(ii)化合物,其中a是b选自并且经一个或更多个独立地选自以下的取代基取代:(i)任选地经一个或更多个选自卤素和-orf的取代基取代的c
1-c6烷基;(ii)卤素、-c(o)ori;以及(iii)-ori和/或其中亚烷基与基团b的5元杂芳基的两个相邻原子连接以形成5、6元部分饱和或饱和的环,其与基团b的5元杂芳基稠合,任选地,其中亚烷基的一个或更多个碳原子经一个或更多个独立地选自卤素、c
1-c6烷基和氧代的取代基取代,其中所述烷基任选地独立地经一个或更多个卤素原子取代;和/或任选地,其中与亚烷基的同一碳连接的两个氢原子被替换为碳原子,该碳原子与其所连接的碳原子一起形成c
3-c6环状烷基,其中所述环状烷基任选地经一个或更多个卤素原子取代。在一个方面中,本发明的化合物包含上述基团b的稠合双环环体系。这些特定的化合物可限定为式(i)化合物,优选式(ii)化合物,其中基团b选自任选地经取代的基团(v)和任选地经取代的基团(w)
其中x和y各自独立地选自c、s、o和n,至少一个x是n;至少两个x是c;至少两个y是c;n=1、2或3,优选1或2;其中x各自任选地独立地经一个或更多个选自以下的取代基取代:卤素、-cn、-c(o)oh、-c(o)o-(c
1-c6烷基)和c
1-c6烷基,优选卤素、-c(o)oh、-c(o)o-(c
1-c6烷基)和c
1-c6烷基,最优选c
1-c6烷基,其中所述烷基任选地经一个或更多个卤素原子取代;并且y各自任选地独立地经一个或更多个选自以下的取代基取代:卤素、-c(o)oh、-c(o)o-(c
1-c6烷基)、c
1-c6烷基和氧代,优选卤素、-c(o)o-(c
1-c6烷基)、c
1-c6烷基和氧代,最优选c
1-c6烷基和氧代,其中所述烷基任选地经一个或更多个卤素原子取代;和/或任选地,其中与同一y连接的两个氢原子被替换为碳原子,该碳原子与其所连接的碳原子一起形成c
3-c6环状烷基,其中所述环状烷基任选地经一个或更多个卤素原子取代。在该特定的方面中,优选本发明的化合物选自式(ii-v)或(ii-w),其中x和y如上所限定。基团b的稠合双环环体系的一些优选实例是以下列出的那些,或其互变异构体,其中每个基团是任选地经取代的,如上所概述。基团b的稠合双环环体系的这些特定的实例优选形成式(ii-v)化合物和式(ii-w)化合物。
基团b的稠合双环环体系的尤其优选的一些实例是以下列出的那些,或其互变异构体,其中每个基团是任选地经取代的,如上所概述。基团b的稠合双环环体系的这些特定的实例尤其优选形成式(ii-v)和(ii-w)的化合物。
本发明的特定化合物是以下列出的那些。
·
4-甲基-n-{4h,5h,6h,7h-吡唑并[1,5-a]吡啶-2-基}-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-(2-甲基-4,5,6,7-四氢-2h-吲唑-3-基)-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[1-(丙烷-2-基)-1h-1,2,3-三唑-4-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-3-[2-(吡啶-3-基)乙炔基]-n-[1-(2,2,2-三氟乙基)-1h-吡唑-3-基]苯甲酰胺;
·
4-甲基-3-[2-(吡啶-3-基)乙炔基]-n-[1-(3,3,3-三氟丙基)-1h-吡唑-4-基]苯甲酰胺;
·
n-(1-叔丁基-1h-吡唑-3-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(5-环丙基-1-甲基-1h-吡唑-3-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[5-甲基-1-(2,2,2-三氟乙基)-1h-吡唑-3-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(1-环丁基-1h-吡唑-4-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(1-叔丁基-1h-吡唑-4-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(3-叔丁基-1-甲基-1h-吡唑-5-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(2,2-二甲基丙基)-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(环丙基甲基)-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[5-(二氟甲基)-1-甲基-1h-吡唑-3-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[5-甲基-1-(2-甲基丙基)-1h-吡唑-4-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(2,2-二氟环丙基)-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(1-环丙基乙基)-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(1-环丁基-5-甲基-1h-吡唑-4-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[1-(丙烷-2-基)-1h-1,2,4-三唑-3-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[1-(2-甲基丙基)-1h-吡唑-4-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[5-(二氟甲氧基)-1-甲基-1h-吡唑-3-基]-4-甲基-3-[2-(吡啶-3-基)乙炔
基]苯甲酰胺;
·
n-[1-(2,2-二氟乙基)-5-甲基-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[5-甲基-1-(丙烷-2-基)-1h-吡唑-4-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(1-异丙基吡唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-(1-异丙基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-(4,4-二甲基-5,6-二氢吡咯并[1,2-b]吡唑-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-[1-(环丙基甲基)咪唑-4-基]-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
4-甲基-3-[2-(3-吡啶基)乙炔基]-n-[1-(2,2,2-三氟乙基)咪唑-4-基]苯甲酰胺;
·
4-甲基-3-[2-(3-吡啶基)乙炔基]-n-[5-(三氟甲基)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-2-基]苯甲酰胺;
·
4-甲基-3-[2-(3-吡啶基)乙炔基]-n-螺[6,7-二氢吡咯并[1,2-a]咪唑-5,1
’‑
环丙烷]-2-基-苯甲酰胺;
·
4-甲基-n-(1-丙基咪唑-4-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-(1-环丙基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
4-甲基-3-[2-(3-吡啶基)乙炔基]-n-[5-(三氟甲基)-5,6,7,8-四氢咪唑并[1,2-a]19yridine-2-基]苯甲酰胺;
·
n-(4-叔丁基-1,3-唑-2-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[4-(丙烷-2-基)-1,3-唑-2-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[2-(丙烷-2-基)-2h-1,2,3,4-四唑-5-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(1-叔丁基-1h-1,2,4-三唑-3-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(1-叔丁基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-(5,5-二甲基-6,8-二氢咪唑并[2,1-c][1,4]嗪-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-(6,6-二甲基-5,7-二氢吡咯并[1,2-c]咪唑-1-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
4-甲基-3-[2-(3-吡啶基)乙炔基]-n-螺[5,6-二氢吡咯并[1,2-b]吡唑-4,1
’‑
环丁烷]-2-基-苯甲酰胺;
·
n-(1-环戊基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-[1-(二氟甲基)-5-甲基-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(5,5-二甲基-6,7-二氢吡咯并[1,2-a]咪唑-2-基)-4-甲基-3-[2-(3-吡啶
基)乙炔基]苯甲酰胺;
·
4-甲基-n-[4-(2-甲基丙基)-1,3-唑-2-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[3-(丙烷-2-基)-1,2,4-二唑-5-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(丁烷-2-基)-1h-吡唑-3-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-3-[2-(吡啶-3-基)乙炔基]-n-[1-(2,2,2-三氟乙基)-1h-吡唑-4-基]苯甲酰胺;
·
n-{6,6-二甲基-5h,6h,7h-吡唑并[3,2-b][1,3]嗪-3-基}-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-(1-丙基-1h-吡唑-4-基)-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(3-环丁基-1-甲基-1h-吡唑-5-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[3-甲氧基-1-(2,2,2-三氟乙基)-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(2,2-二氟乙基)-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(2,2-二氟乙基)-5-甲基-1h-吡唑-3-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[5-甲基-1-(丙烷-2-基)-1h-吡唑-3-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(5-异丙基-6,7-二氢-5h-吡咯并[1,2-a]咪唑-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-(5-异丙基-6,8-二氢-5h-咪唑并[2,1-c][1,4]嗪-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
4-甲基-n-[4-甲基-1-(丙烷-2-基)-1h-吡唑-3-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[5-(二氟甲基)-1-甲基-1h-1,2,3-三唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-(5-甲基-6,7-二氢-5h-吡咯并[1,2-c]咪唑-1-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
4-甲基-n-[1-甲基-3-(2-甲基丙基)-1h-吡唑-5-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(1-环丙基-1h-吡唑-4-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-3-[2-(3-吡啶基)乙炔基]-n-螺[5,6-二氢吡咯并[1,2-b]吡唑-4,1
’‑
环丙烷]-2-基-苯甲酰胺;
·
n-(1-异丁基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-(4-乙基-5,6-二氢-4h-吡咯并[1,2-b]吡唑-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
4-甲基-n-[1-(丙烷-2-基)-1h-吡唑-3-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-(5-甲基-5,6,7,8-四氢咪唑并[1,2-a]吡啶-2-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-[1-(环丁基甲基)-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(1-环丁基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-[3-(环丙基甲基)-1-甲基-1h-吡唑-5-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[3-甲基-1-(2,2,2-三氟乙基)-1h-吡唑-4-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(2,2-二氟乙基)-3-甲基-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(2,2-二氟环丙基)-1h-吡唑-3-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[3-甲基-1-(2-甲基丙基)-1h-吡唑-4-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(2,2-二氟乙基)-1h-吡唑-3-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[4-甲基-1-(2,2,2-三氟乙基)-1h-吡唑-3-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(1-环丁基-3-甲基-1h-吡唑-4-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(2-氟乙基)咪唑-4-基]-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
4-甲基-n-[3-甲基-1-(丙烷-2-基)-1h-吡唑-4-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[1-(2-甲基丙基)-1h-吡唑-3-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-{5-甲基-4-氧代-4h,5h,6h,7h-吡唑并[1,5-a]吡嗪-2-基}-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-(5-甲基-6,7-二氢-5h-吡咯并[1,2-a]咪唑-2-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-[1,4-二甲基-3-(三氟甲基)-1h-吡唑-5-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(3-环丙基-1-甲基-1h-吡唑-5-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[1-甲基-3-(丙烷-2-基)-1h-1,2,4-三唑-5-基]-3-[2-(吡啶-3-基)
乙炔基]苯甲酰胺;
·
n-[1-(二氟甲基)-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(2,2-二氟乙基)-4-甲基-1h-吡唑-3-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-(4-氧代-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
4-甲基-n-(5-甲基-6,8-二氢-5h-咪唑并[2,1-c][1,4]嗪-2-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-[4-氯-1-(丙烷-2-基)-1h-吡唑-3-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-(3-环丙基-1-乙基-1h-吡唑-5-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-[5-甲基-1-(2-甲基丙基)-1h-吡唑-3-基]-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(1-甲氧基丙烷-2-基)-1h-吡唑-3-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
n-[1-(环丁基甲基)咪唑-4-基]-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
4-甲基-3-[2-(3-吡啶基)乙炔基]-n-螺[6,7-二氢-5h-吡唑并[1,5-a]吡啶-4,1
’‑
环丙烷]-2-基-苯甲酰胺;
·
n-(6,6-二甲基-5,8-二氢咪唑并[2,1-c][1,4]嗪-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-(4,4-二氟-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺;
·
n-[1-(2-氟乙基)-1h-吡唑-4-基]-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-n-{4h,5h,6h,7h-吡唑并[1,5-a]吡啶-3-基}-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;
·
4-甲基-3-[2-(吡啶-3-基)乙炔基]-n-(4,5,6,7-四氢-1,2-苯并唑-3-基)苯甲酰胺;
·
4-甲基-3-[2-(吡啶-3-基)乙炔基]-n-(4,5,6,7-四氢-2,1-苯并唑-3-基)苯甲酰胺;
·
n-(5,5-二氟-4,5,6,7-四氢-1,2-苯并唑-3-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺;或
·
n-(5-叔丁基-1,3,4-二唑-2-基)-4-甲基-3-[2-(吡啶-3-基)乙炔基]苯甲酰胺。本发明的化合物可包括同位素标记和/或同位素富集形式的化合物。本文中本发
明的化合物可在构成这样的化合物的一个或更多个原子处包含非天然比例的原子同位素。可并入到所公开化合物中的同位素的一些实例包括氢、碳、氮、氧、磷、硫、氯的同位素,例如2h、3h、
11
c、
13
c、
14
c、
13
n、
15
o、
17
o、
32
p、
35
s、
18
f、
36
cl。本发明的化合物可原样使用,或在适当时作为其药理学上可接受的盐(酸或碱加成盐)使用。下文提及的药理学上可接受的加成盐意指包含化合物能够形成的具有治疗活性的无毒酸和碱加成盐形式。通过用合适的酸处理碱形式,可将具有碱性特性的化合物转化为其药理学上可接受的酸加成盐。一些示例性酸包括无机酸,例如氯化氢、溴化氢、碘化氢、硫酸、磷酸;以及有机酸,例如甲酸、乙酸、丙酸、羟基乙酸、乳酸、丙酮酸、乙醇酸、马来酸、丙二酸、草酸、苯磺酸、甲苯磺酸、甲磺酸、三氟乙酸、富马酸、琥珀酸、苹果酸、酒石酸、柠檬酸、水杨酸、对氨基水杨酸、扑酸(pamoic acid)、苯甲酸、抗坏血酸等。一些示例性碱加成盐形式是钠盐、钾盐、钙盐,以及具有可药用胺的盐,例如如氨、烷基胺、苄星(benzathine)和氨基酸,例如如精氨酸和赖氨酸。本文中使用的术语加成盐还包含化合物及其盐能够形成的溶剂合物,例如水合物、醇化物(alcoholate)等。在整个本公开内容中,给定的化学式或命名还将涵盖其所有可药用盐、溶剂合物、水合物、n-氧化物和/或前药形式。应当理解,本发明的化合物包括化合物式的任何和所有水合物和/或溶剂合物。应理解,在化合物的多种物理形式中,某些官能团,例如羟基、氨基等基团与水和/或多种溶剂形成复合物和/或配位化合物。因此,上式应理解为包含并代表那些多种水合物和/或溶剂合物。本发明的化合物还包含互变异构形式。互变异构形式由单键与相邻双键的交换以及伴随的质子迁移而产生。互变异构形式包括质子移变互变异构体,其为具有相同经验式和总电荷的异构质子化状态。示例性质子移变互变异构体包括酮-烯醇对、酰胺-亚胺酸对、内酰胺-内酰亚胺对、酰胺-亚胺酸对、烯胺-亚胺对和其中质子可占据杂环体系的两个或更多个位置的环状形式,例如1h-和3h-咪唑、1h、2h-和4h-1,2,4-三唑、1h-和2h-异吲哚以及1h-和2h-吡唑。互变异构形式可通过适当的取代处于平衡中或空间锁定成一种形式。本文中所述的化合物可以是不对称的(例如,具有一个或更多个立构中心)。除非另外指明,否则意指所有立体异构体,例如对映体和非对映体。包含不对称的取代的碳原子的本发明的化合物可以以光学活性或外消旋形式分离。如何由光学活性起始物料制备光学活性形式的方法是本领域已知的,例如通过拆分外消旋混合物或通过立体选择性合成。烯烃、c=n双键等的许多几何异构体也可存在于本文中所述的化合物中,并且所有这样的稳定的异构体均在本发明中考虑。描述了本发明化合物的顺式-和反式-几何异构体,并且可将其作为异构体的混合物或作为经分离的异构体形式进行分离。在包含不对称碳原子的化合物的情况下,本发明涉及d型、l型和d,l混合物,并且在存在多于一个不对称碳原子的情况下,本发明还涉及非对映体形式。包含不对称碳原子且通常作为外消旋体产生的那些本发明化合物可以以已知方式例如使用光学活性酸分离成光学活性异构体。然而,也可从一开始就使用光学活性起始物料,随后获得相应的光学活性或非对映体化合物作为终产物。术语“前药”是指可在生理条件下或通过溶剂解转化为本发明的生物活性化合物的化合物。当向有此需要的对象施用时,前药可以是无活性的,但在体内转化为本发明的活性化合物。前药通常在体内快速转化以产生本发明的母体化合物,例如通过在血液中水解。
前药化合物通常在哺乳动物生物体中提供溶解度、组织相容性或延迟释放的优点(参见silverman,r.b.,the organic chemistry ofdrug design and drug action,第2版,elsevier academic press(2004),第498至549页)。本发明化合物的前药可通过以将修饰在常规操作中或在体内裂解成本发明母体化合物这样的方式对本发明化合物中存在的官能团,例如羟基、氨基或巯基进行修饰来制备。前药的一些实例包括但不限于羟基官能团的乙酸酯、甲酸酯和琥珀酸酯衍生物或者氨基官能团的氨基甲酸苯酯衍生物。本发明的另一个目的涉及用于治疗的本发明的化合物。本发明的化合物可用作c-abl的抑制剂。因此,其可用于治疗或预防其中c-abl的抑制是有益的医学病症(病症或疾病)。因此,提供了用于治疗或预防对c-abl抑制有响应的疾病或病症的方法,其包括向对象施用治疗有效量的本发明的化合物。虽然本发明的化合物可适合于预防一系列疾病和病症,但优选其用于治疗所述疾病和病症。因此,优选该方法用于治疗疾病或病症,并因此该方法包括向有此需要的对象施用治疗有效量的本发明的化合物。本文中使用的术语“治疗”可包括预防指定的障碍或病症,或者一旦障碍已确立就减轻或消除该障碍。术语“预防”是指预防指定的障碍或病症。通过c-abl抑制可治疗或可预防的一系列疾病和病症是公知的。因此,本发明的化合物可用于治疗或预防该一系列疾病或病症。这包括神经退行性病症、癌症、朊病毒病、病毒感染、糖尿病、炎性疾病例如肺纤维化、或者骨骼或肌营养不良。优选地,疾病是神经退行性病症或癌症。可治疗或可预防的神经退行性病症包括但不限于阿尔茨海默病(alzheimer disease)、唐氏综合征(down’s syndrome)、额颞痴呆、进行性核上性麻痹、皮克病(pick’s disease)、尼曼-皮克病(niemann-pick disease)、帕金森病(parkinson’s disease)、亨廷顿病(huntington’s disease,hd)、齿状核红核苍白球丘脑下核萎缩(dentatorubropallidoluysian atrophy)、肯尼迪病(kennedy’s disease)、和脊髓小脑性共济失调、脆性x(雷特(rett’s))综合征、脆性xe智力低下、弗里德赖希共济失调(friedreich’s ataxia)、强直性肌营养不良、脊髓小脑性共济失调8型和脊髓小脑性共济失调12型、亚历山大病(alexander disease)、阿尔珀斯病(alper’s disease)、肌萎缩侧索硬化(als)、共济失调毛细血管扩张症、巴藤病(batten disease)、卡纳万病(canavan disease)、科凯恩综合征(cockayne syndrome)、皮质基底节变性、克罗伊茨费尔特-雅各布病(creutzfeldt-jakob disease)、缺血性卒中、克拉伯病(krabbe disease)、路易体痴呆(lewy body dementia)、多发性硬化、多系统萎缩、佩利措伊斯-梅茨巴赫病(pelizaeus-merzbacher disease)、皮克病、原发性侧索硬化、雷弗素姆氏病(refsum’s disease)、山德霍夫病(sandhoffdisease)、希尔德病(schilder’s disease)、脊髓损伤、脊髓性肌萎缩症、斯蒂尔-理查森-奥尔谢夫斯基病(steele-richardson-olszewski disease)和脊髓痨。在可治疗或可预防的神经退行性病症中,最值得注意的是肌萎缩侧索硬化(als)和帕金森病。最优选地,神经退行性病症是als。可治疗或可预防的癌症包括但不限于白血病。在可治疗或可预防的癌症中,最值得注意的是慢性髓性白血病(cml)、急性淋巴母细胞白血病(all)、急性髓性白血病(aml)和混合表型急性白血病(mpal)、或其任何中枢神经系统(cns)转移。最优选地,癌症是cml或all。
因此,本发明包括本发明化合物在制备用于治疗或预防疾病或病症(例如上述神经退行性病症和癌症)的药物中的用途。本发明还涉及用于治疗疾病或病症(例如上述神经退行性病症和癌症)的本发明的化合物。本文中描述的方法包括其中对象被确定为需要特定的所述治疗的那些。确定需要这样的治疗的对象可以是在对象或健康护理专业人员的判断中,并且可以是主观的(例如意见)或客观的(例如可通过测试或诊断方法测量)。在另一些方面中,本文中的方法包括还包括监测对治疗施用有响应的对象的那些。这样的监测可包括对对象组织、流体、标本、细胞、蛋白质、化学标志物、遗传物质等的定期取样,作为治疗方案的标志物或指示物。在另一些方法中,通过评估用于这样的治疗的适合的相关标志物或指示物,将对象预筛选或确定为需要这样的治疗。本发明提供了监测治疗进展的方法。该方法包括在患有或易患有本文中描述的病症或其症状的对象中确定诊断标志物(标志物)(例如,本文中描述的由本文中的化合物调节的任何靶标或细胞类型)的水平或诊断测量(例如,筛选、测定)的步骤,其中已向对象施用了足以治疗疾病或其症状的治疗量的本文中的化合物。可将该方法中确定的标志物水平与健康正常对照中或其他患病患者中的标志物已知水平进行比较以确立对象的疾病状态。在一些优选实施方案中,在晚于确定第一水平的时间点确定对象中标志物的第二水平,并比较这两个水平以监测疾病进程或治疗效力。在某些优选实施方案中,对象中标志物的治疗前水平在开始根据本发明的治疗之前确定;然后可将该标志物的治疗前水平与治疗开始之后对象中的标志物水平进行比较,以确定治疗的效力。对象中的标志物或标志物活性的水平可至少一次确定。标志物水平的比较,例如,与先前或随后从同一患者、另一患者或正常对象获得的标志物水平的另一测量的比较,可用于确定根据本发明的治疗是否具有期望的作用,并从而允许适当地调节剂量水平。标志物水平的确定可使用本领域已知的或本文中所述的任何合适的取样/表达测定方法进行。优选地,首先从对象中移出组织或流体样品。合适的样品的一些实例包括血液、尿、组织、口腔或颊细胞以及包含根部的毛发样品。另一些合适的样品将是本领域技术人员已知的。样品中蛋白质水平和/或mrna水平(例如标志物水平)的确定可使用本领域已知的任何合适的技术进行,包括但不限于酶免疫测定、is elisa、放射性标记/测定技术、印迹/化学发光方法、实时pcr等。对于临床应用,本文中公开的化合物被配制成用于多种施用模式的药物组合物(或制剂)。应当理解,本发明的化合物可与生理学上可接受的载体、赋形剂和/或稀释剂(即这些中的一种、两种或所有三种)一起施用。本文中公开的药物组合物可通过任何合适的途径施用,优选通过经口、经直肠、经鼻、表面(包括口含和舌下)、舌下、经皮、鞘内、经黏膜或肠胃外(包括皮下、肌内、静脉内和皮内)施用。其它制剂可方便地以单位剂型(例如,片剂和持续释放的胶囊剂)和以脂质体存在,并且可通过药学领域公知的任何方法来制备。药物制剂通常通过使活性物质或其可药用盐与常规的可药用载体、稀释剂或赋形剂混合来制备。赋形剂的一些实例是水、明胶、阿拉伯树胶、乳糖、微晶纤维素、淀粉、羟基乙酸淀粉钠、磷酸氢钙、硬脂酸镁、滑石、胶体二氧化硅等。这样的制剂还可包含其他药理学活性剂和常规添加剂,例如稳定剂、润湿剂、乳化剂、矫味剂、缓冲剂等。通常来说,活性化合物的量为按制剂的重量计0.1至95%,优选地为按用于肠胃外用制剂的重量计0.2至20%,并且更优选地为
按用于经口施用制剂的重量计1至50%。制剂还可通过已知方法,例如制粒、压制、微囊化、喷涂等来制备。制剂可通过常规方法以片剂、胶囊剂、颗粒剂、散剂、糖浆剂、混悬剂、栓剂或注射剂的剂型来制备。液体制剂可通过将活性物质溶解或混悬在水或其它适合的载剂中来制备。片剂和颗粒剂可以以常规方式进行包衣。为了长时间维持治疗有效的血浆浓度,可将本文中公开的化合物并入到缓慢释放制剂中。特定化合物的剂量水平和剂量频率将根据多种因素而改变,所述因素包括所使用的特定化合物的效力、该化合物的代谢稳定性和作用时长、患者的年龄、体重、一般健康状况、性别、饮食、施用的模式和时间、排泄率、药物组合、待治疗病症的严重程度,以及正进行治疗的患者。每日剂量可例如为约0.001mg至约100mg/千克体重,以例如每次约0.01mg至约25mg的剂量单次或多次施用。通常,这样的剂量是经口给予的,但也可选择肠胃外施用。定义术语“未经取代的”意指其所指的基团不具有取代不同基团的氢原子。例如,“未经取代的吡啶基”是指吡啶的一价基团,其除了其与化合物的其余部分连接的点之外仅有与环连接的氢。术语“杂原子”意指o、n或s。通常,优选地5元杂芳基基团b中的一个或更多个杂原子为氮。“任选的”或“任选地”意指随后描述的事件或情况可以但不必发生,并且该描述包括其中事件或情况发生的情况以及其中事件或情况未发生的情况。术语“c
1-c6烷基”表示具有1至6个碳原子,即1、2、3、4、5或6个碳原子的直链、支链或者环状或部分环状的烷基。对于包含环状部分的“c
1-c6烷基”,其应由3至6个碳原子形成。对于“c
1-c6烷基”范围的部分,考虑了其所有亚基,例如c
1-c5烷基、c
1-c4烷基、c
1-c3烷基、c
1-c2烷基、c1烷基、c
2-c6烷基、c
2-c5烷基、c
2-c4烷基、c
2-c3烷基、c2烷基、c
3-c6烷基、c
3-c5烷基、c
3-c4烷基、c3烷基、c
4-c6烷基、c
4-c5烷基、c4烷基、c
5-c6烷基、c5烷基和c6烷基。“c
1-c6烷基”的一些实例包括甲基、乙基、正丙基、异丙基、环丙基、正丁基、异丁基、仲丁基、叔丁基、环丁基、环丙基甲基、以及直链、支链或者环状或部分环状戊基和己基等。当术语表示范围,例如在c
1-c6烷基的定义中的“1至6个碳原子”时,每个整数被认为是公开的,即1、2、3、4、5和6。术语“c
2-c6烯基”表示具有至少一个碳-碳双键并且具有2至6个碳原子的直链、支链或者环状或部分环状的烷基。烯基可包含由3至6个碳原子形成的环。对于“c
2-c6烯基”范围的部分,考虑了其所有亚基,例如c
2-c5烯基、c
2-c4烯基、c
2-c3烯基、c2烯基、c
3-c6烯基、c
3-c5烯基、c
3-c4烯基、c3烯基、c
4-c6烯基、c
4-c5烯基、c4烯基、c
5-c6烯基、c5烯基和c6烯基。“c
2-c6烯基”的一些实例包括2-丙烯基、2-丁烯基、3-丁烯基、2-甲基-2-丙烯基、2-己烯基、5-己烯基、2,3-二甲基-2-丁烯基。术语“c
2-c6炔基”表示具有至少一个碳-碳三键并且具有2至6个碳原子的直链、支链或者环状或部分环状的烷基。炔基可包含由3至6个碳原子形成的环。对于“c
2-c6炔基”范围的部分,考虑了其所有亚基,例如c
2-c5炔基、c
2-c4炔基、c
2-c3炔基、c2炔基、c
3-c6炔基、c
3-c5炔基、c
3-c4炔基、c3炔基、c
4-c6炔基、c
4-c5炔基、c4炔基、c
5-c6炔基、c5炔基和c6炔基。“c
2-c6炔基”的一些实例包括2-丙炔基、2-丁炔基、3-丁炔基、2-戊炔基、3-甲基-4-戊炔基、2-己炔基、5-己炔基等。
术语“c
1-c6烷氧基”表示-o-(c
1-c6烷基),其中c
1-c6烷基如上文所定义并且通过氧原子与化合物的其余部分连接。“c
1-c6烷氧基”的一些实例包括甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、仲丁氧基、叔丁氧基以及直链和支链戊氧基和己氧基。术语“卤代”意指卤素原子,并且优选f、cl、br和i,更优选f和cl,并且最优选f。术语“氧代”表示与氧原子的双键(=o)。这通常形成酮基或醛基。术语“c
6-c
10
芳基”表示包含6至10个环原子的芳族单环或稠合双环烃环。“c
6-c
10
芳基”的一些实例包括苯基、茚基、萘基和萘。术语“c
1-c9杂芳基”表示具有5至10个环原子的芳族单环或稠合双环杂芳环体系,其中1至9个环原子为碳并且一个或更多个环原子选自氮、硫和氧。“c
1-c9杂芳基”的一些实例包括呋喃基、吡咯基、噻吩基、唑基、异唑基、咪唑基、噻唑基、异噻唑基、吡啶基、嘧啶基、四唑基、喹唑啉基、吲哚基、二氢吲哚基、异吲哚基、异二氢吲哚基、吡唑基、哒嗪基、吡嗪基、喹啉基、喹喔啉基、噻二唑基、苯并呋喃基、2,3-二氢苯并呋喃基、1,3-苯并二氧杂环戊烯基、1,4-苯并二氧杂环己烯基(1,4-benzodioxinyl)、2,3-二氢-1,4-苯并二氧杂环己烯基、苯并噻唑基、苯并咪唑基、苯并噻二唑基、苯并三唑基和色满基。术语“c
1-c9杂环”表示具有5至10个环原子的非芳族单环或稠合双环环体系,所述环原子包含1至9个碳原子并且一个或更多个环原子选自氮、硫和氧。当存在时,硫原子可以是氧化形式(即s=o的双基或o=s=o的双基)。环体系可以是完全饱和的或部分不饱和的。“c
1-c9杂环”的一些实例包括哌啶基、四氢吡喃基、四氢呋喃基、氧杂环丁烷基、氮杂基(azepinyl)、氮杂环丁烷基、吡咯烷基、吗啉基、咪唑啉基、咪唑烷基、硫代吗啉基、吡喃基、二烷基、哌嗪基、高哌嗪基和5,6-二氢-4h-1,3-嗪-2-基。“有效量”是指赋予所治疗对象治疗作用的本发明的化合物的量。治疗作用可以是客观的(即,通过一些测试或标志物可测量的)或主观的(即,对象给出对作用的指示或感觉到作用)。本文中使用的术语“施用”或其变化形式意指用于本文中公开的化合物的施用途径。一些示例性施用途径包括但不限于经口、静脉内、腹膜内、动脉内和肌内。优选的施用途径可取决于多种因素变化,例如包含本文中公开的化合物的药物组合物的组分、潜在或实际疾病的部位和疾病的严重程度。术语“对象”和“患者”在本文中可互换使用。它们是指可受疾病或病症折磨或易于患有疾病或病症但是可患有或可未患有疾病或病症的人或其他哺乳动物(例如,小鼠、大鼠、兔、狗、猫、牛、猪、绵羊、马或灵长类)。优选对象是人。本发明的化合物可通过名称或化学结构来公开。如果化合物的名称与其相关化学结构之间存在差异,则以化学结构为准。现将通过以下非限制性实施例进一步说明本发明。以下具体实施例应被解释为仅是说明性的,并且不以任何方式限制本公开内容的其余部分。无需进一步阐述,认为本领域技术人员可基于本文中所述以其最大程度地利用本发明。本文中引用的所有参考文献和出版物通过引用整体并入于此。本发明化合物的制备本文中公开的式(i)化合物可通过常规方法或以与常规方法类似的方法来制备。
3300和agilent lc\msd g6130a、g6120b质谱仪的agilent 1200系列lc/msd系统;具有dad\elsd alltech 3300和agilent lc\msd g6120b质谱仪的agilent technologies 1260infinity lc/msd系统;或具有dad\elsd g7102a 1290infinity ii和agilent lc\msd g6120b质谱仪的agilent technologies 1260infinity ii lc/msd系统。方法clc-ms phenomenex kinetex xb-c18,1.7μm,2.1
×
50mm,40℃,0.8ml/分钟,在1.2分钟内在水( 0.1%tfa)中的5%至100%mecn( 0.085%tfa),保持0.2分钟,再平衡0.6分钟,200至300nm。方法dagilent 1100(四元泵)xbridge-c18,5μm,4.6
×
50mm,25℃,2ml/分钟,5μl注射液,在水( 10mm甲酸铵)中的5%mecn,3.5分钟内梯度5至95%,保持1分钟,200至400nm。方法ewaters aquity系统csh-c18,1.7μm,2.1
×
50mm,40℃,0.5μl注射液,0.4ml/分钟。在水( 0.1%甲酸 5%mecn)中的0%mecn( 0.1%甲酸 5%水)持续0.2分钟,3.3分钟内0至100%,保持1分钟,200至400nm。方法fwaters aquity系统beh-c18,1.7μm,2.1
×
50mm,40℃,0.5μl注射液,0.4ml/分钟。在水( 0.1%nh3 5%mecn)中的0%mecn( 0.1%nh3水溶液 5%水)持续0.2分钟,3.3分钟内0至100%,保持1分钟,再平衡1.0分钟,200至400nm。方法gwaters aquity系统beh-c18,1.7μm,2.1
′
50mm,40℃,0.5μl注射液,0.4ml/分钟。在水( 0.1%nh3 5%mecn)中的50%mecn( 0.1%nh3水溶液 5%水)持续0.2分钟,1.8分钟内50%至100%,保持2.5分钟,200至400nm。方法hphenomenex kinetex xb-c18,1.7μm,2.1
×
100mm,40℃,0.5ml/分钟,在水( 0.1%tfa)中的5%mecn( 0.085%tfa)持续0.7分钟,8.0分钟内5%至100%,保持0.3分钟,再平衡1.0分钟。200至300nm。方法iphenomenex kinetex xb-c18,1.7μm,2.1
×
50mm,40℃,0.8ml/分钟,在水( 0.1%tfa)中的5%mecn( 0.085%tfa)持续0.7分钟,3.0分钟内5%至100%,保持0.3分钟,再平衡1.0分钟。200至300nm。方法jphenomenex kinetex xb-c18,1.7μm,2.1
×
50mm,40℃,0.8ml/分钟,在水( 0.1%tfa)中的5%mecn( 0.085%tfa)持续1.0分钟,3.0分钟内5%至100%,保持0.2分钟,再平衡0.8分钟。200至300nm中间体14-甲基-3-(吡啶-3-基乙炔基)苯甲酸甲酯
向脱气的在etoac(87ml)中的3-碘-4-甲基苯甲酸甲酯(5.5g,20mmol,1当量)、3-乙炔基吡啶(2.1g,20mmol,1当量)和三乙胺(6.1g,60mmol,3当量)的溶液添加碘化铜(i)(190mg,1mmol,0.05当量)和pd(pph3)2cl2(702mg,1mmol,0.05当量),并将反应物在室温下在氮气下搅拌3小时。通过过滤除去固体并将溶液在真空中浓缩,将粗物质通过正相色谱etoac/庚烷(1∶1)进行纯化,以得到为浅黄色固体的4-甲基-3-(吡啶-3-基乙炔基)苯甲酸甲酯(4.3g,85%)。uplc(方法a)3.44分钟,99%,[m h]
=252.2。中间体24-甲基-3-(吡啶-3-基乙炔基)苯甲酸向在thf(40ml)中的中间体1(2.80g,11.0mmol,1.0当量)的溶液添加在水(10ml)中的氢氧化锂一水合物(0.69g,16.5mmol,1.5当量)的溶液,并将反应物在室温下在氮气下搅拌超过72小时。通过添加2m hcl(8.5ml)将反应物酸化至ph约5并搅拌1小时。通过过滤收集固体,用叔丁基甲基醚(2
×
30ml)洗涤,并在真空中干燥,以得到为灰白色固体的4-甲基-3-(吡啶-3-基乙炔基)苯甲酸(2.00g,77%)。uplc(方法a)1.97分钟,98%,[m h]
=238.2。中间体31-异丙基-4-硝基-1h-咪唑向在dmf(35ml)中的4-硝基-1h-咪唑(0.80g,7.1mmol,1.0当量)的溶液添加k2co3(1.96g,14.2mmol,2.0当量)和2-碘丙烷(0.85ml,8.5mmol,1.2当量),并将反应物在50℃下搅拌过夜。将反应混合物冷却至室温,用h2o(150ml)淬灭并用etoac(150ml)稀释。分离各相并用etoac(2
×
100ml)萃取水相。将合并的有机物干燥(mgso4)并在真空中浓缩(与甲苯共沸)。将粗物质通过正相色谱和叔丁基甲基醚/庚烷(1∶1)随后通过100%etoac进行纯化以分离为黄色油状物的主要产物,其在静置时结晶。将晶体与甲苯(
×
2)共沸,以得到为浅黄色针状结晶的1-异丙基-4-硝基-1h-咪唑(2.76g,84%)。uplc(方法a)1.96分钟,100%,[m h]
=156.1。中间体4
1-异丙基-1h-咪唑-4-胺盐酸盐向在甲醇(50ml)中的异丙基-4-硝基-1h-咪唑(930mg,6mmol,1当量)的溶液添加钯碳(palladium on carbon)(100mg,10%w/w),并将反应物在室温下在氢气氛(1atm/14psi)下搅拌过夜。通过硅藻土垫过滤混悬液,并向所得溶液添加2m hcl(3ml)并在真空下浓缩混合物,以得到1-异丙基-1h-咪唑-4-胺盐酸盐(假定定量)。uplc(方法a)1.36分钟,85%,[m h]
=126.1。中间体56-羟基-4,4-二甲基-3-氧代己腈将在thf(29ml)中的双(三甲基甲硅烷基)氨基锂(1m,在thf中)(38.5ml,38.5mmol,2.2当量)的溶液在氮气下冷却至-78℃,随后逐滴添加在thf(20ml)中的3,3-二甲基二氢呋喃-2(3h)-酮(2.0g,27.5mmol,1.0当量)和mecn(1.83ml,35.0mmol,2.0当量)的溶液。将反应物在-78℃下搅拌15分钟,随后温热至室温并另外搅拌2小时。将反应物用饱和nh4c1水溶液淬灭,用etoac稀释,分离各相并用etoac萃取水相。将合并的有机物用饱和nacl洗涤,干燥(mgso4)并在真空中浓缩,以得到为油状物的6-羟基-4,4-二甲基-3-氧代己腈粗物质(假定定量)。中间体63-(3-氨基-1h-吡唑-5-基)-3-甲基丁-1-醇向在meoh(25ml)中的中间体5(2.72g,17.5mmol,1.0当量)的溶液添加水合肼(1.64ml,26.3mmol,1.5当量),并将反应物在60℃下加热64小时。将反应物冷却至室温并使co2鼓泡通过混合物1小时。将反应物从灰白色沉淀中倾析,并将溶液在真空中浓缩。将所得剩余物通过正相色谱1至10%meoh/dcm(用kmno4染色)进行纯化,以得到为澄清油状物的3-(3-氨基-1h-吡唑-5-基)-3-甲基丁-1-醇(1.18g,40%)。lcms(方法d)1.36分钟,100%,[m h]
=170.10。中间体74,4-二甲基-4h,5h,6h-吡咯并[1,2-b]吡唑-2-胺
向在thf(38ml)中的中间体6(1.18g,7.0mmol,1当量)的溶液逐滴添加socl2(2.53ml,34.9mmol,5当量),并将反应物在室温下搅拌1小时。将反应物倒入冰上的28%nh4oh溶液中并用dcm稀释。分离各相,用dcm萃取水相并将合并的有机物用饱和nacl洗涤、干燥(mgso4)并在真空中浓缩。通过正相色谱1至10%meoh/dcm纯化粗物质,以得到为黄色油状物的4,4-二甲基-4h,5h,6h-吡咯并[1,2-b]吡唑-2-胺(334mg,29%产率)。lcms(方法d)1.87分钟,90%,[m h]
=152.10。中间体82-[2-氧代-5-(三氟甲基)吡咯烷-1-基]乙酸甲酯在恒定搅拌的情况下在n2气氛下在0℃下向在无水thf(20ml)中的5-(三氟甲基)吡咯烷-2-酮(500mg,3.3mmol,1.0当量)的混悬液中以小部分地添加60%nah(157mg,3.9mmol,1.2当量)。使反应物温热至室温并搅拌30分钟,随后添加2-溴乙酸甲酯(550mg,3.6mmol,1.1当量)并另外在室温下搅拌18小时。将反应物用饱和nh4cl(5ml)淬灭并在真空中除去挥发物。将剩余物在etoac(25ml)与h2o(15ml)之间分配,并将有机层干燥(mgso4)并在真空中浓缩。将剩余物通过正相色谱30%至100%叔丁基甲基醚/庚烷进行纯化,以得到为无色油状物的2-[2-氧代-5-(三氟甲基)吡咯烷-1-基]乙酸甲酯(400mg,54%)。1h-nmr(400mhz,cdcl3)δ
h 4.64(d,j=18.2hz,1h),4.28-4.20(m,1h),3.78(d,j=18.2hz,1h),3.74(s,3h),2.64-2.50(m,1h),2.48-2.31(m,2h),2.24-2.13(m,1h)ppm,19f-nmr(373mhz,氯仿-d)δ
f-75.4(d,j=7.0hz,3f)ppm。中间体92-(2-氧代-5-(三氟甲基)吡咯烷-1-基)乙酰胺将2-(2-氧代-5-(三氟甲基)吡咯烷-1-基)乙酸甲酯(400mg,1.8mmol,1.0当量)溶解在nh3(7m在meoh中,5ml)中并在密封管中在室温下搅拌4小时。将反应物在真空中浓缩,以得到为白色固体的2-(2-氧代-5-(三氟甲基)吡咯烷-1-基)乙酰胺(374mg,100%)。1h-nmr(400mhz,cdcl3)δ
h 5.78(s,1h),5.42(s,1h),4.42(d,j=17.0hz,1h),4.33-4.22(m,1h),3.79(d,j=16.3hz,1h),2.65-2.50(m,1h),2.49-2.32(m,2h),2.24-2.14(m,1h)ppm。中间体105-(三氟甲基)-6,7-二氢-3h-吡咯并[1,2-a]咪唑-2(5h)-酮
向在mecn(10ml)中的中间体9(400mg,1.9mmol,1当量)的混悬液添加三溴氧化磷(v)(phosphorus(v)oxybromide)(1.64g,5.7mmol,3当量)并将反应物在70℃下在n2下加热2小时。在真空中除去挥发物,并将剩余物与h2o(20ml)一起搅拌,用固体k2co3碱化,用etoac(3
×
20ml)萃取,干燥(mgso4)并在真空中浓缩。将剩余物通过正相色谱0.2∶2∶98 nh3∶meoh∶dcm进行纯化,以得到为无色油状物的5-(三氟甲基)-6,7-二氢-3h-吡咯并[1,2-a]咪唑-2(5h)-酮(240mg,50%)。1h-nmr(400mhz,cdcl3)(d,j=17.6hz,1h),4.21(m,1h),3.90(d,j=17.6hz,1h),2.65-2.50(m,1h),2.50-2.32(m,2h),2.32-2.20(m,1h),19f-nmr(373mhz,氯仿-d)δ
f-75.2(d,j=6.3hz,3f)ppm。中间体112-溴-5-(三氟甲基)-6,7-二氢-5h-吡咯并[1,2-a]咪唑向在mecn(4ml)中的中间体10(300mg,1.6mmol,1当量)的混悬液添加三溴氧化磷(v)(1.34g,4.7mmol,3当量),并将反应物在密封管中在100℃下加热18小时。在真空中除去挥发物,并将剩余物与h2o(20ml)一起搅拌,用固体k2co3碱化,用etoac(3
×
20ml)萃取,干燥(mgso4)并在真空中浓缩。将剩余物通过正相色谱0.2∶2∶98 nh3∶meoh∶dcm进行纯化,以得到为无色油状物的2-溴-5-(三氟甲基)-6,7-二氢-5h-吡咯并[1,2-a]咪唑(160mg,24%)。uplc(方法a)2.61分钟,89%,[m h]
=255.0、257.0。中间体124-甲基-3-(吡啶-3-基乙炔基)苯甲酰胺向在dmf(20ml)中的4-甲基-3-(吡啶-3-基乙炔基)苯甲酸(1.50g,6.3mmol,1.0当量)、氯化铵(1.01g,19.0mmol,3.0当量)和三乙胺(2.67ml,19.6mmol,3.1当量)的溶液添加(1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶3-氧化物六氟磷酸盐/酯(3.12g,8.2mmol,1.3当量),将反应物在室温下搅拌2小时。将反应混合物与h2o(100ml)和叔丁基甲基醚(100ml)一起搅拌,并通过过滤收集固体并在真空中干燥,以得到为无色固体的4-甲基-3-(吡啶-3-基乙炔基)苯甲酰胺(1.0g,67%)。uplc(方法a)2.59分钟,100%,[m h]
=237.2。中间体132-(5-氧代-4-氮杂螺[2.4]庚烷-4-基)乙酸甲酯
在n2下且在约15℃(冷水浴)下,向在thf(8ml)中的nah(60%在油中,216mg,5.40mmol,1.2当量)的混悬液逐滴添加在thf(5ml)中的4-氮杂螺[2.4]庚-5-酮(500mg,4.50mmol,1.0当量)的溶液,并将反应物在15℃下搅拌30分钟。向反应物逐滴添加在thf(1.5ml)中的溴乙酸甲酯(516μl,5.40mmol,1.2当量)的溶液并将反应物在室温下搅拌1小时。将反应物用饱和nh4cl(60ml)淬灭,用etoac(30ml)稀释,分离各相并用etoac(3
×
30 ml)萃取水相。将合并的有机物用盐水(50ml)洗涤,干燥(na2so4)并在真空中浓缩,以得到为淡黄色-橙色油状物的2-(5-氧代-4-氮杂螺[2.4]庚烷-4-基)乙酸甲酯(824mg,假定定量)。1h nmr(400mhz,cdcl3)δ
h 3.72(s,3h),3.70(s,2h),2.58(t,j=8.2hz,2h),2.14(t,j=8.2hz,2h),0.80(t,j=6.7hz,2h),0.61(t,j=6.7hz,2h)ppm。中间体142-(5-氧代-4-氮杂螺[2.4]庚烷-4-基)乙酰胺将在nh3(7m在meoh中,10ml,15当量)中的中间体13(821mg,4.48mmol,1当量)的溶液在60℃下加热70小时。将反应物冷却至室温并在真空中浓缩,以得到为浅黄色黏性剩余物的2-(5-氧代-4-氮杂螺[2.4]庚烷-4-基)乙酰胺(753mg,假定定量)。1h nmr(400mhz,dmso-d6)δ
h 7.23(br.s,1h),7.03(br.s,1h),3.41(s,2h),2.38(t,j=8.2hz,2h),2.05(t,j=8.2hz,2h),0.82(t,j=6.5hz,2h),0.51(t,j=6.5hz,2h)ppm。中间体152
’‑
溴-6’,7
’‑
二氢螺[环丙烷-1,5
’‑
吡咯并[1,2-a]咪唑]向在mecn(2ml)中的中间体14(400mg,2.38mmol,1当量)的混悬液添加三溴氧化磷(v)(2.73g,9.51mmol,4当量),并将反应物在85℃下加热3.5小时。将反应物冷却至室温,倒入h2o(50ml)中并用dcm(2
×
40ml)萃取。将水层用k2co3碱化(至ph=9),用dcm(40ml)萃取并将合并的有机物用k2co3(3%wt/wt水溶液,100ml)、盐水(150ml)洗涤,干燥(na2so4)并在真空中浓缩。将剩余物通过正相色谱(干装载的)在0至15%meoh/dcm中进行纯化,以得到为棕色固体的2
’‑
溴-6’,7
’‑
二氢螺[环丙烷-1,5
’‑
吡咯并[1,2-a]咪唑](94mg,19%)。uplc(方法e)2.05分钟,100%,[m h]
=213.0、215.0。中间体16甲基4-溴-1-环丙基-1h-咪唑
在氮气下向在dce(100ml)中的4-溴-1h-咪唑(2.00g,13.6mmol,1当量)的溶液添加环丙基硼酸(2.34g,27.2mmol,2当量)、乙酸铜(ii)(copper(ii)acetate)(2.47g,13.6mmol,1当量)、2,2
’‑
联吡啶(2.13g,13.6mmol,1当量)和碳酸钾(3.76g,27.2mmol,2当量),并将混悬液在70℃下加热3小时。将反应物冷却至室温并倒入h2o(100ml)中。分离各相,并用1m hcl溶液(100ml)和饱和na2co3(100ml)洗涤有机相,用k2co3碱化水相(至ph=10)并用dcm(100ml)和dcm/异丙醇(9∶1,2
×
100ml)萃取。将合并的有机物用盐水洗涤,干燥(mgso4)并在真空中浓缩。将剩余物通过正相色谱(干装载的)10%至50%etoac/dcm进行纯化,以得到为浅黄色油状物的甲基4-溴-1-环丙基-1h-咪唑(195mg,7%)。uplc(方法f)2.26分钟,100%,[m h]
=187.0、189.0。中间体173-乙炔基咪唑并[1,2-a]吡啶将乙炔基三甲基硅烷(1.41ml,10.20mmol,2.0当量)和n-环己基环己胺(1.21ml,6.09mmol,1.2当量)添加至在mecn(10ml)中的3-溴咪唑并[1,2-a]吡啶(1.00g,5.08mmol,1.0当量)、双(三苯基膦)二氯化钯(ii)(89.1mg,127μmol,0.025当量)和碘化铜(i)(33.8mg,178μmol,0.035当量)的溶液,将溶液用n2鼓泡10分钟,并随后加热至80℃持续5分钟以形成非常黏稠的浆料。将反应物冷却至室温,用mecn(40ml)稀释并在80℃下再加热3小时。随后将反应混合物在真空中浓缩并将剩余物在dcm(100ml)和h2o(100ml)之间分配。将水层用dcm(100ml)萃取并合并有机层,干燥(mgso4)并在真空中浓缩。将剩余物溶解在meoh(20ml)中,添加k2co3(701mg,5.08mmol)并将反应物在室温下搅拌30分钟。将反应混合物过滤并在真空中浓缩。将剩余物通过柱色谱(正相,[24g],redisep硅胶,35至60μm(230至400目),35ml/分钟,梯度为在异己烷中的0%至100%etoac)进行纯化。将产物在真空烘箱中在50℃下干燥3小时,以得到为棕色固体的3-乙炔基咪唑并[1,2-a]吡啶(473mg,66%)。lc-ms(方法c)0.50分钟,[m h]
=143.0。中间体18n-(5-叔丁基异唑-3-基)-3-碘代-4-甲基-苯甲酰胺
将dipea(1.32ml,7.63mmol,2当量)添加至在dcm(20ml)中的3-碘-4-甲基-苯甲酸(1.00g,3.82mmol,1当量)、3-氨基-5-叔丁基异唑(535mg,3.82mmol,1当量)和hatu(1.45g,3.82mmol,1当量)的溶液,并将反应物在回流下搅拌96小时。将混合物在dcm(50ml)与h2o(50ml)之间分配并用dcm(50ml)萃取水层。将合并的有机物干燥(mgso4)并在真空中浓缩。将剩余物通过柱色谱(正相,[40g],redisep硅胶,35至60μm(230至400目),40ml/分钟,梯度为在异己烷中的0%至100%etoac)进行纯化。将产物在真空烘箱中在60℃下干燥2小时,以得到为白色固体的n-(5-叔丁基异唑-3-基)-3-碘代-4-甲基-苯甲酰胺(767mg,51%)。lc-ms(方法c)1.47分钟,[m h]
=385.2。中间体191-(环丁基甲基)-4-碘代-咪唑在n2下在室温下,向在dmf(9ml)中的碘代咪唑(0.90g,4.6mmol,1.0当量)的溶液添加cs2co3(4.54g,13.9mmol,3.0当量),随后添加溴甲基环丁烷(0.63ml,5.5mmol,1.2当量),并将所得混悬液加热至60℃持续1小时。随后将白色混悬液冷却至室温并在真空中除去挥发物。向剩余物添加h2o(50ml)和etoac(25ml),分离各相并用etoac(2
×
25ml)萃取水相。将合并的有机物用盐水(50ml)洗涤,用na2so4干燥并在真空中浓缩。将粗制混合物与来自100mg规模反应的粗制品合并,并将合并的剩余物通过柱色谱(biotage isolera,在10cv内100%etoac至etoac∶dcm,1∶1)进行纯化,以得到为无色油状物的标题产物(819mg,61%)。结构由noesy确定。uplc(方法a)2.88分钟,100%,[m h]
=263.0。中间体203-[1-(3-羟基丙基)环丙基]-3-氧代-丙腈将用无水thf(20ml)稀释的lhmds(1.5m在thf中,11.6ml,17.3mmol)冷却至-78℃并逐滴添加mecn(0.82ml,15.8mmol)。将所得溶液搅拌1小时,随后逐滴添加在无水thf(5ml)中的5-氧杂螺[2.5]辛-4-酮(995mg,7.9mmol)的溶液。将反应物在-78℃下搅拌2小时,并随后温热至室温,在此情况下通过将反应混合物添加至饱和nh4cl溶液(130ml)中来淬灭反应并用dcm(3
×
60ml)萃取。将合并的有机物干燥(mgso4),过滤并在真空中浓缩,以
得到为红色/棕色油状物的粗产物(1.63g,》100%),其不经进一步纯化用于下一步。中间体213-[1-(3-氨基-1h-吡唑-5-基)环丙基]丙-1-醇在高压釜(150ml)中,将一水合肼(1.2ml,25.0mmol)逐滴添加至在meoh(25ml)中的中间体20(1.39g,8.3mmol)的溶液。随后将所得反应混合物在120℃下加热18小时。随后将混合物冷却至室温,随后在10分钟的时期内添加干冰。倾析澄清溶液,并在真空中除去溶剂,以得到为橙色油状物的粗产物(1.60g)。将其通过柱色谱在二氧化硅(meoh/dcm,1∶99至1∶9)上进行纯化,以得到为橙色油状物的标题化合物(1.10g,73%)。uplc(方法a)1.72分钟,94.6%,[m h]
=182.1。中间体225-[1-(3-氯丙基)环丙基]-1h-吡唑-3-胺在室温下将亚硫酰氯(150μl,1.99mmol)添加至在1,2-二氯乙烷(5.0ml)中的中间体21(300mg,1.66mmol)的溶液。随后将反应混合物在90℃下加热1小时。将反应物冷却至室温并与在1.17mmol规模下进行的相同反应合并。向混合物添加碳酸钾水溶液(2m、15ml)并分离层。用dcm(2
×
20ml)萃取水相并将合并的有机物干燥(na2so4)并在真空中浓缩,以得到为棕色油状物的粗制品标题化合物(474mg),其不经进一步纯化用于下一步。中间体23螺[6,7-二氢-5h-吡唑并[1,5-a]吡啶-4,1
’‑
环丙烷]-2-胺将在mecn(20ml)中的中间体22(474mg,2.37mmol)和碳酸钾(656mg,4.75mmol)在80℃下加热过夜。将反应物冷却至室温,并在真空中除去溶剂。向所得剩余物添加h2o(20ml)和dcm(20ml)并分离各相。用dcm(2
×
20ml)洗涤水相并将合并的有机物在减压下干燥,以得到粗产物。将其通过柱色谱在二氧化硅(meoh/dcm,1∶99至1∶9)上进行纯化,以得到为棕色固体的标题产物(260mg,67%),其不经进一步纯化用于下一步。uplc(方法a)2.29分钟,81.4%,[m h]
=164.1。中间体24
2-氯-n-(2-羟基-2-甲基-丙基)乙酰胺在0℃下向在dcm(80ml)和2m naoh(39.3ml,78.5mmol)中的1-氨基-2-甲基-丙-2-醇(5.00g,56.1mmol)逐滴添加氯乙酰氯(5.4ml,67.3mmol)。将反应物在室温下搅拌2小时,分离层,用mgso4干燥有机层并在真空中除去溶剂,以得到为无色油状物的2-氯-n-(2-羟基-2-甲基-丙基)乙酰胺(5.60g,60%)。1h nmr(400mhz,cdcl3):δ
h 7.02(s,1h),4.13(m,3h),3.35(d,j=6.1hz,2h),1.30-1.25(m,6h)ppm。中间体256,6-二甲基吗啉-3-酮在0℃下在n2下向在ipa(40ml)中的中间体24(5.60g,33.8mmol)添加叔丁氧基钾(7.59g,67.6mmol)并且在16小时内将反应混合物温热至室温。将反应用hcl溶液(2m)中和至ph 7并在减压下除去有机溶剂。用dcm(3
×
20ml)萃取所得的水相。将合并的有机物用mgso4干燥并在减压下除去溶剂,以得到为黄色油状物的粗产物(1.99g)。将其通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,在etoac中的0%至10%meoh)进行纯化,以得到为结晶白色固体的6,6-二甲基吗啉-3-酮(832mg,19.1%)。1h nmr(400mhz,cdcl3):δ
h 6.39(s,1h),4.18(s,2h),3.25(d,j=2.4hz,2h),1.33(s,6h)ppm。中间体262-(2,2-二甲基-5-氧代-吗啉-4-基)乙酸甲酯在室温下在n2下向在thf(25ml)中的中间体25(832mg,6.44mmol)的搅拌溶液分批添加nah(在矿物油中的60%,258mg,6.44mmol)。将反应物在室温下搅拌40分钟,随后在室温下逐滴添加2-溴乙酸甲酯(608μl,6.44mmol)并将反应物在室温下搅拌16小时。将反应物小心地用水(20ml)稀释并分离层。用etoac(3
×
10ml)萃取水相。将合并的有机物用盐水(20ml)洗涤,干燥(mgso4)并在真空中浓缩,以得到粗产物(1.20g),其为粉色油状物。将剩余物通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,在庚烷中的70%至100%etoac)进行纯化,以得到为无色油状物的标题化合物(726mg,56%)。1h nmr(400mhz,cdcl3):δ
h 422(s,2h),4.15(s,2h),3.75(s,3h),3.28(s,2h),1.36(s,6h)ppm。中间体272-(2,2-二甲基-5-氧代-吗啉-4-基)乙酸
在室温下向在thf(20ml)中的中间体26(726mg,3.61mmol)的搅拌溶液添加tmsok(669mg,4.69mmol),随后搅拌16小时。将反应物用tbme(30ml)稀释,在真空下过滤并弃去滤液。将滤饼溶解在水(10ml)中,使用2m hcl酸化至ph 2,通过添加固体nacl使饱和并用etoac(8
×
10ml)萃取。将合并的有机物干燥(mgso4)并在真空中浓缩,以得到为白色固体的标题化合物(378mg,56%)。1h nmr(400mhz,cdcl3):δ
h 4.24(s,2h),4.17(s,2h),3.31(s,2h),1.36(s,6h)ppm。中间体282-(2,2-二甲基-5-氧代-吗啉-4-基)乙酰胺在室温下向在1,4-二氧六环(10ml)中的中间体27(378mg,2.02mmol)的搅拌溶液添加吡啶(81μl,1.01mmol)、碳酸铵(194mg,2.02mmol)和boc2o(617mg,2.83mmol),并将反应物在室温下搅拌16小时。将反应物在真空中浓缩并将剩余物通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,在etoac中的0%至20%meoh)进行纯化,以得到为白色固体的标题化合物(349mg,92.8%)。1h nmr(400mhz,cdcl3):δ
h 6.20(s,1h),5.38(s,1h),4.22(s,2h),4.02(s,2h),3.35(s,2h),1.34(s,6h)ppm。中间体292-溴-6,6-二甲基-5,8-二氢咪唑并[2,1-c][1,4]嗪向包含mecn(4.0ml)中的中间体28(160mg,0.86mmol)的微波小瓶添加pobr3(1.48g,5.16mmol)。随后将小瓶在biotage引发器中加热至120℃持续35分钟。随后在0℃下将反应物逐滴添加至h2o(10ml)和dcm(10ml)的搅拌溶液。随后添加固体k2co3以使水层碱化至ph10。分离有机层并用4∶1 dcm∶ipa(5
×
10ml)萃取水层。将合并的有机层用mgso4干燥,过滤并浓缩,以得到粗产物(310mg)。将粗剩余物通过硅胶柱色谱(在庚烷中的0至60%etoac)进行纯化,以得到为棕色油状物的标题化合物(35.0mg,17.6%)。其不经进一步纯化用于下一步。uplc(方法a)2.44分钟,28.3%,[m h]
=231.0/233.0。中间体301-(4-乙氧基-4-氧代-丁基)吡唑-3,5-二甲酸二乙酯
根据文献程序(pfizer ep1241170a2(2002),其内容并入本文),向在mecn(100ml)中的二乙基-3,5-吡唑二羧酸盐/酯(10.00g,47.1mmol,1.0当量)的溶液添加k2co3(6.51g,47.1mmol,1.0当量),随后添加4-溴丁酸乙酯(9.19g,47.1mmol,1.0当量),并将混合物在80℃下加热3.5小时。将所得白色混悬液冷却至室温并使其静置过夜。在真空中除去溶剂并将剩余物与nh4cl水溶液(100ml)和etoac(100ml)合并,并分离各相。用etoac(1
×
50ml)萃取水层并将合并的有机物用nahco3水溶液(100ml)、nh4cl水溶液(100ml)洗涤,干燥(na2so4),过滤并在真空中除去溶剂,以得到为浅黄色油状物的标题化合物(15.77g,定量)。lc-ms(方法d)2.89分钟,98%,[m h]
=327.2。中间体314-氧代-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2,5-二甲酸二乙酯在5分钟内向在甲苯(100ml)中的中间体30(10.0g,30.6mmol,1当量)的溶液逐滴添加在thf中的t-buok(1.6m在thf中,21.1ml,33.7mmol,1.1当量)的溶液,在此期间温度稳定升至30℃,产生在淡橙色溶液中的沉淀。将混合物在室温下搅拌25分钟,并随后在90℃下搅拌3小时,以产生黏稠浆料。随后使混合物冷却至室温并搅拌过夜。将浆料用etoac(150ml)稀释,倒入nh4cl水溶液(200ml)中并添加2m hcl(50ml)以酸化水层。分离层并用etoac(2
×
80ml)萃取水相。将合并的有机物用nh4cl水溶液(2
×
200ml)洗涤,干燥(na2so4)并过滤。在真空中除去溶剂,以得到为浅黄色固体的标题化合物(7.72g,90%)。uplc(方法a)1.87分钟,99%,[m h]
=281.2。中间体324-氧代-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-羧酸将中间体31(5.38g,19.20mmol)和浓hcl/h2o(2∶1,90ml)的混悬液在100℃下加热6小时,冷却至室温并在真空中除去溶剂。将黄色剩余物溶解在mecn∶thf(1∶4)中并在真空中除去溶剂,并重复该过程(
×
1),并随后用mecn重复,以得到为浅黄色固体的标题化合物(3.44g,99%)。uplc(方法e)1.55分钟,97%,未观察到电离。中间体33
4-氧代-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-羧酸甲酯向在dmf(25ml)中的4-氧代-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-羧酸(1.83g,10.1mmol,1.0当量)的溶液添加k2co3(2.81g,20.3mmol,2.0当量)以形成混悬液。向该混悬液添加碘甲烷(0.76ml,12.2mmol,1.2当量)并在室温下继续搅拌过夜。向所得黑色混合物添加nh4cl水溶液(30ml),并用etoac(3
×
30ml)萃取混合物。将合并的有机物用硫代硫酸钠水溶液(30ml)、nahco3水溶液(30ml)、nh4cl水溶液(2
×
30ml)洗涤,干燥(na2so4),过滤并在真空中除去溶剂,以得到为灰白色固体的标题化合物(1.36g,69%)。中间体344,4-二氟-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-羧酸甲酯在10ml微波小瓶中设置两个相同的反应:向在dce(3.5ml)中的中间体33(0.67g,3.4mmol,1当量)的溶液逐滴添加dast(4.5ml,34.3mmol,10当量)并将混合物在室温下搅拌2至3分钟,密封并随后在室温下搅拌5天。将两个微波小瓶开盖并用dcm(10ml)稀释每个小瓶的内容物。在10分钟内将反应混合物各自吸移到搅拌的nahco3水溶液(100ml)中。添加另外的dcm(20ml)并将混合物搅拌1至2小时。分离各相并将合并的有机物用nahco3水溶液(2
×
100ml)洗涤,干燥(mgso4),过滤并在真空中除去溶剂。将剩余物通过柱色谱在二氧化硅上(100%dcm至etoac/dcm,1∶99)进行纯化,以得到为黄色油状物的标题化合物(650mg,44%)。uplc(方法a)2.58分钟,84%,[m h]
=217.1。中间体354,4-二氟-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-羧酸向在thf(10ml)中的中间体34(735mg,3.40mmol,1当量)的溶液添加lioh(163mg,6.80mmol,2当量)和h2o(0.5ml),并将溶液在室温搅拌过夜。使用2m hcl将所得黄色溶液酸化至ph 5并在减压下部分地除去溶剂。将油状剩余物用h2o(6ml)和2m hcl(1ml)稀释,并将所得的黏稠白色沉淀进行超声处理,过滤,在滤纸上用h2o(2
×
10ml)洗涤,并在真空中干燥,以得到为白色粉末的标题化合物(551mg,80%)。uplc(方法e)2.37分钟,92%,[m h]
=203.1。中间体36n-(4,4-二氟-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-基)氨基甲酸苄酯
向在甲苯(10ml)和et3n(233μl,1.67mmol,2当量)中的中间体35(169mg,0.84mmol,1当量)的混悬液添加苯甲醇(434μl,4.18mmol,5当量)和dppa(359μl,1.67mmol,2当量),并将反应物加热至90℃过夜。将所得橙色反应混合物用etoac(40ml)稀释,用h2o(10ml)、饱和nahco3水溶液(10ml)和盐水(10ml)洗涤,干燥(na2so4),过滤并浓缩至橙色油状物。将粗油状物通过柱色谱(甲苯/etoac,9∶1至4∶1至1∶1)进行纯化,以得到为灰白色固体的所期望产物(145mg,56%)。uplc(方法a)3.20分钟,80%,[m h]
=308。中间体374,4-二氟-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-胺向在meoh(10ml)中的中间体36(145mg,0.47mmol,1当量)的混悬液添加10%pd/c(50mg)并将反应物在1atm h2下在室温下搅拌16小时。随后将混合物通过硅藻土过滤,并将滤液在减压下浓缩,以得到为棕色油状物的标题化合物(78mg,95%),其不经进一步纯化用于下一步。uplc(方法a)2.10分钟,72%,[m h]
=174。中间体383-乙炔基-4-甲基-苯甲酸甲酯将在mecn(50ml)中的3-碘代-4-甲基苯甲酸甲酯(6.00g,21.7mmol)、乙炔基三甲基硅烷(7.52ml,54.3mmol)和et3n(9.09ml,65.2mmol)的溶液用n2脱气15分钟。添加cui(207mg,1.09mmol)和pd(pph3)2cl2(763mg,1.09mmol)并将混合物用n2脱气5分钟,随后在n2下搅拌45分钟。将反应混合物在真空中浓缩,将样品再溶解在meoh(50ml)中,添加k2co3(3.00g,21.7mmol)并将混合物在室温下搅拌1小时。在15分钟之后添加另一份k2co3(3.00g,21.7mmol)。将反应混合物在真空中浓缩并将样品再溶解在etoac(75ml)和水(75ml)中。用etoac(2
×
35ml)萃取水相,并将合并的有机相用盐水(50ml)洗涤并在真空中浓缩。将样品通过柱色谱(c18反相,[(86g)],redisep c18-来源的二氧化硅,40至63μm(230至400目),60ml/分钟,梯度为10%至100%meoh,在10%meoh/h2o中)并在真空烘箱中在60℃下干燥18小时,以得到为深黑色固体的3-乙炔基-4-甲基-苯甲酸甲酯(1.40g,37%)。uplc(方法j)2.76分钟,100%。中间体393-乙炔基-4-甲基-苯甲酸
将中间体39(700mg,4.0mmol)和lioh一水合物(512mg,11.9mmol)溶解在thf∶h2o(10ml,1∶1)中,并将反应物在室温下搅拌2小时。添加另一份lioh一水合物(512mg,11.9mmol),并将反应物在室温下搅拌3天。在真空中除去thf,并将剩余物用1m hcl酸化。将产物用etoac(4
×
50ml)萃取,干燥(mgso4)并在真空中浓缩,以得到为浅棕色固体的3-乙炔基-4-甲基-苯甲酸(200mg,29%)。lc-ms(方法i)2.27分钟,未观察到电离。中间体403-乙炔基-4-甲基-n-[1-(2-吗啉代乙基)吡唑-3-基]苯甲酰胺将1-[2-(吗啉-4-基)乙基]-1h-吡唑-3-胺(251mg,1.28mmol)、中间体39(200mg,1.16mmol,93%纯)、dipea(202μl,1.16mmol)和hatu(574mg,1.51mmol)溶解在dcm(40ml)中,并将反应物在室温下搅拌18小时。将混合物用饱和nahco3水溶液(30ml)洗涤,干燥(mgso4)并在真空中浓缩。将剩余物通过柱色谱(正相,[24g],redisep硅胶,35至60μm(230至400目),35ml/分钟,梯度为在异己烷中的0%至100%etoac,随后在dcm中的0%至20%meoh[剩余物装载在dcm中])进行纯化,以得到为浅棕色固体的3-乙炔基-4-甲基-n-[1-(2-吗啉代乙基)吡唑-3-基]苯甲酰胺(377mg,92%)。lc-ms(方法i)2.04分钟,[m h]
=339.2。中间体416-氟异喹啉-3-胺向meoh(15ml)添加2,2-二乙氧基乙腈(3.00g,23.2mmol)和naome的甲醇溶液(0.25g,4.7mmol在1ml meoh中),并将反应物在室温下搅拌24小时。随后添加4-氟苄胺(2.39ml,20.9mmol)并将反应物在室温下另外搅拌24小时。将反应混合物在真空中浓缩并将容器冷却至0℃,随后添加浓硫酸(15.0ml)并在室温下另外搅拌24小时。使用4m koh将反应中和至ph 7,将产物使用dcm(3
×
150ml)萃取,干燥(mgso4)并在真空中浓缩。将剩余物通过柱色谱(正相,[80g],redisep硅胶,35至60μm(230至400目),60ml/分钟,梯度为在异己烷中的0%至100%etoac[剩余物装载在dcm中])进行纯化,以得到为棕色固体的6-氟异喹啉-3-胺(292mg,7.8%)。lc-ms(方法i)1.38分钟,[m h]
=163.0。中间体426-氟-4-碘代-异喹啉-3-胺
将中间体41(292mg,1.66mmol,92%纯)和nis(347mg,1.54mmol)溶解在meoh(75ml)中并将该反应在室温下搅拌3小时。随后添加另一份nis(187mg,0.83mmol)并将反应物在室温下搅拌另外18小时。在真空中除去溶剂并将剩余物通过柱色谱(正相,[40g],redisep硅胶,35至60μm(230至400目),40ml/分钟,梯度为在异己烷中的0%至100%etoac[剩余物装载在dcm中])进行纯化,以得到为棕色固体的6-氟-4-碘代-异喹啉-3-胺(296mg,57%)。lc-ms(方法i)2.15分钟,[m h]
=289。中间体434-甲基-3-(2-三甲基甲硅烷基乙炔基)苯甲酸甲酯将在etoac(700ml)中的3-碘代-4-甲基苯甲酸甲酯(50.0g,0.18mol)、et3n(55.0g,0.54mol)和乙炔基三甲基硅烷(23.1g,0.24mol)的溶液使用减压/n2的三个循环来脱气。添加双(三苯基膦)二氯化钯(ii)(1.27g,1.81mmol)和cui(0.35g,1.81mmol)并将反应物在室温下在n2下搅拌2小时。将反应物在真空中变得干燥并将深棕色固体通过柱色谱(100%庚烷至庚烷/etoac,9二1)进行纯化,以得到为浅黄色固体的标题化合物(44.6g,95.1%)。uplc(方法a)4.26分钟,95%,未检出质量离子(mass ion)。中间体443-(2-咪唑并[1,2-b]哒嗪-3-基乙炔基)-4-甲基-苯甲酸甲酯将在微波管中的在mecn(4.0ml)中的中间体43(318mg,1.29mmol)、3-溴咪唑并[1,2-b]哒嗪(307mg,1.55mmol)、三乙胺(0.54ml,3.87mmol)、碘化铜(i)(25mg,0.13mmol)、双(三苯基膦)二氯化钯(ii)(91mg,0.13mmol)和氟化铯(392mg,2.58mmol)的混合物用n2脱气2分钟并在微波中在100℃下加热2小时。将反应混合物与另外两批反应物(1.29和0.81mmol)合并,在此情况下向混合物添加dcm(50ml)和h2o(50ml)。分离各层并用dcm(2
×
50ml)萃取水相。将合并的有机物浓缩并将粗产物在硅胶(dcm/meoh,99∶1至9∶1)上进行纯化并用mecn(15ml)进行重结晶,以得到为黄色固体的标题化合物(238mg,24%)。uplc(方法a),3.21分钟,99%,[m h]
=292.2。中间体453-(2-咪唑并[1,2-b]哒嗪-3-基乙炔基)-4-甲基-苯甲酸
在室温下向在meoh(10ml)和thf(10ml)中的中间体44(238mg,0.82mmol)的溶液添加在h2o(2.0ml)中的lioh.h2o(103mg,2.45mmol),并将所得混合物在室温下搅拌过夜。随后在真空中除去溶剂并向剩余物添加h2o(10ml),其由hcl水溶液酸化至ph 1至2。在真空中除去溶剂,以得到为棕色固体的标题化合物(320mg),其不经进一步纯化用于下一步。uplc(方法a),1.87分钟,99%,[m h]
=278.2。中间体466
’‑
硝基螺[1,3-二硫戊环-2,1
’‑
二氢化茚]将乙烷-1,2-二硫醇(1.68g,17.9mmol,1.10当量)、6-硝基茚满-1-酮(2.88g,16.2mmol,1.00当量)、对甲苯磺酸(0.56g,3.3mmol,0.20当量)和甲苯(15ml)在dean stark条件下在100℃下加热24小时。随后将反应物冷却至室温并在真空下除去甲苯。将剩余物通过柱色谱在硅胶(etoac/庚烷,1∶9)上进行纯化,以得到为黄色油状物的标题产物(4.05g,98%)。uplc(方法a)1.71分钟,100%,未观察到电离。中间体472-溴-1,1-二氟-6-硝基-茚满在-70℃下向在无水dcm(90ml)中的1,3-二溴-5,5-二甲基-咪唑烷-2,4-二酮(18.1g,63.2mmol)的溶液逐滴添加hf-吡啶(18.8ml,70%)并将反应物在-70℃下搅拌30分钟,随后逐滴添加在dcm(10ml)中的中间体46(4.00g,15.8mmol)的溶液并另外搅拌4小时。随后将混合物温热至室温并搅拌过夜。向深棕色混合物添加naoh(2m,50ml)和nahso3(3m,5.0ml)并分离各相。用dcm(2
×
80ml)萃取水相并将合并的有机物在真空中浓缩,以得到为橙色油状物的粗产物(4.02g)。将其在硅胶(etoac/庚烷,1∶9至3∶7)上进行纯化,以得到为黄色油状物的2-溴-1,1-二氟-6-硝基-茚满(3.63g),其不经进一步纯化用于下一步。uplc(方法g),1.30分钟,80%,未电离。中间体481,1-二氟-6-硝基-茚
在室温下将dbu(3.3ml,22.2mmol)添加至在无水dcm(50ml)中的中间体47(3.63g,13.1mmol)的溶液持续2小时。随后向混合物添加hcl水溶液(50ml,2m)并分离各相。用dcm(2
×
50ml)洗涤水相并将合并的有机物在真空中浓缩,以得到为紫色固体的粗产物(3.01g)。将粗产物在二氧化硅(etoac/庚烷,1∶9至3∶7)上进行纯化,以产生为黄色固体的1,1-二氟-6-硝基-1h-茚(2.62g),其不经进一步纯化用于下一步。uplc(方法g)0.99分钟,83%,未电离。中间体491,1-二氟-6-硝基-茚满在0℃下,将一水合肼(2.58ml,53.2mmol)逐滴添加至在mecn(60ml)中的中间体48(2.62g,13.3mmol)及2-硝基苯磺酰氯(5.89g,26.6mmol)的溶液,以得到黄色混悬液。使反应混合物温热至室温,导致形成澄清溶液,将其在室温下搅拌72小时。在30℃下在真空中除去mecn,并将h2o(80ml)添加至反应混合物中以溶解沉淀。将粗产物用etoac(3
×
50ml)萃取并将合并的有机物在真空中浓缩。将粗产物在二氧化硅上使用etoac/庚烷(1∶9至3∶7)进行纯化,以得到为黄色油状物的1,1-二氟-6-硝基-茚满(2.00g,76%产率),其在静置时固化。uplc(方法g),1.12分钟,99.6%,未电离。中间体503-溴-1,1-二氟-6-硝基-茚满将在四氯化碳(30ml)中的中间体49(500mg,2.51mmol)、aibn(41mg,0.25mmol)和nbs(536mg,3.01mmol)在回流(90℃)下加热过夜。随后对另一批材料重复该反应,并在冷却至室温之后合并这两个反应。向粗制混合物添加硅胶(10.0g)并在真空中除去溶剂。将粗产物在二氧化硅(etoac/庚烷,3∶97)上进行纯化,以得到为黄色油状物的3-溴-1,1-二氟-6-硝基-茚满(788mg,56%),其通过uplc包含16%起始材料并且不经进一步纯化即使用。uplc(方法g)1.39分钟,83%,未电离。中间体511-(3,3-二氟-5-硝基-茚满-1-基)-4-甲基-哌嗪在室温下,将1-甲基哌嗪(568mg,5.7mmol)添加至在dmf(10ml)中的中间体50(788mg,2.8mmol)和碳酸钾(783mg,5.7mmol)中,并将所得混合物在室温下搅拌4小时。随后向反应物添加h2o(100ml)和etoac(3
×
50ml)并在真空中除去来自合并的有机物的溶剂,以
得到紫色油状物。将其在二氧化硅(meoh/dcm,1∶99至1∶9)上进行纯化,以得到为深绿色固体的标题产物(100mg,12%)。uplc(方法a)3.07分钟,78%,[m h]
=298.2。中间体523,3-二氟-1-(4-甲基哌嗪-1-基)茚满-5-胺将pd/c(15.0mg,10%wt)添加至在ipa(5ml)中的中间体51(100mg,0.34mmol)中。将所得反应混合物在室温下在1atm下氢化3小时。将反应混合物通过celite过滤并用ipa(5ml)洗涤。在真空中除去溶剂,以得到为灰色固体的粗制3,3-二氟-1-(4-甲基哌嗪-1-基)茚满-5-胺(103mg),其直接用于下一步。uplc(方法a)2.31分钟,67%,[m h]
=268.2。中间体532-(3,3-二甲基-5-氧代-吗啉-4-基)乙酸甲酯在室温下向在无水thf(70ml)中的5,5-二甲基吗啉-3-酮(2.00g,15.5mmol)的经n2吹扫的搅拌溶液分批添加nah(60%在油中)(619mg,15.5mmol),并将反应物在室温下搅拌50分钟。逐滴添加2-溴乙酸甲酯(1.46ml,15.5mmol)并将反应物搅拌另外4小时,随后向反应物逐滴添加水(5.0ml)并随后将反应物倒入水(40ml)中。使用etoac(6
×
10ml)萃取所得溶液,将合并的有机物用盐水(30ml)洗涤,用mgso4干燥并浓缩,以得到为浑浊油状物的粗产物(4.0g)。将粗产物通过硅胶柱色谱(在庚烷中的20%至90%etoac)进行纯化,以得到为无色油状物的2-(3,3-二甲基-5-氧代-吗啉-4-基)乙酸甲酯(1.60g,46%)。1h nmr(400mhz,cdcl3):δ
h 4.24(s,2h),4.04(s,2h),3.75(s,3h),3.66(s,2h),1.26(s,6h)ppm。中间体542-(3,3-二甲基-2-氧代-1-哌啶基)乙酰胺将中间体53(2.40g,11.9mmol)在7n nh3/meoh(100ml)中的溶液在密封的parr氢化器容器中在100℃下搅拌过夜。将反应物浓缩,分装在两个微波小瓶中,每个小瓶用7n nh3/meoh(10ml)稀释并在biotage引发器中加热至110℃持续1.5小时。将反应物浓缩并通过硅胶柱色谱(在etoac中的0至20%meoh)进行纯化,以得到为无色油状物的所期望的产物(119mg,5%)。中间体55
2-溴-8,8-二甲基-6,7-二氢-5h-咪唑并[1,2-a]吡啶向各自包含mecn(5.0ml)中的中间体54(243mg,1.31mmol)的两个微波小瓶添加pobr3(2.25g,7.83mmol),并将两个小瓶在biotage引发器中加热至120℃持续35分钟。随后在0℃下将两个反应物逐滴添加至h2o(40ml)和dcm(40ml)的搅拌溶液。随后将溶液通过添加固体k2co3小心地碱化至ph 10。用1∶9 ipa∶dcm(7
×
10ml)萃取两相溶液,将合并的有机层用盐水(30ml)洗涤,用mgso4干燥,过滤并浓缩至棕色剩余物。通过硅胶色谱(在庚烷中的40%至100%etoac)进行纯化,得到为棕色结晶固体的标题化合物(215mg,36%)。uplc(方法a)2.41分钟,17%,[m h]
=233.0/231.0。中间体566,6-二甲基-5,7-二氢吡咯并[1,2-c]咪唑将一水合肼(0.13ml,2.76mmol)添加至溶解在二甘醇(5.0ml,0.55mmol)中的6,6-二甲基-5h-吡咯并[1,2-c]咪唑-7-酮(83.0mg,0.55mmol)的搅拌溶液。将所得溶液使用biotage引发器微波反应器在180℃下加热60分钟。随后使反应物冷却至室温,并将烧瓶开启,随后将koh(217mg,3.87mmol)小心地添加至混合物。将烧瓶重新密封并将所得混悬液使用biotage引发器微波反应器在220℃下加热120分钟。将反应混合物用稀hcl水溶液(2m)酸化至ph 5,并在真空中除去溶剂。将所得粗剩余物用dcm/meoh(1∶1)研磨,过滤并在真空中除去溶剂,以得到包含显著deg的标题化合物(40mg,53%),其直接用于下一步。中间体571,3-二碘代-6,6-二甲基-5,7-二氢吡咯并[1,2-c]咪唑在室温下将中间体56(17.0mg,0.07mmol)和n-碘代琥珀酰亚胺(35.2mg,0.16mmol)溶解在dmf(0.7ml)中并将反应物在70℃下加热2小时。将混合物冷却至室温,用水(2ml)稀释,用dcm(3
×
5ml)萃取,并分离各层。将合并的有机物用mgso4干燥,过滤并在真空中浓缩,以得到粗产物(29.0mg),将其通过柱色谱(手动柱,正相,p60硅胶40至63μm/230至400目,[二氧化硅/粗制品=30/1],在dcm中的0至5%meoh)进行纯化,以得到为浅黄色固体的标题化合物(11mg,40%)。lc-ms(方法d)2.82分钟,37%,[m h]
=388.7。
中间体581-碘代-6,6-二甲基-5,7-二氢吡咯并[1,2-c]咪唑向在etoh(5.0ml)中的中间体57(50.0mg,0.13mmol)的搅拌溶液添加在h2o(5.0ml)中的na2so3(81.2mg,0.64mmol)的溶液,并将溶液在60℃下搅拌35分钟。使反应物冷却至室温并在真空中除去挥发物。用etoac(3
×
5ml)萃取水相,将合并的有机相用na2so4干燥,过滤并在真空中浓缩,以得到1-碘代-6,6-二甲基-5,7-二氢吡咯并[1,2-c]咪唑(54mg),将其以粗制品的形式用于下一步。uplc(方法a)2.77分钟,86%,[m h]
=263.0。中间体592-(5-羟基-6-氧杂螺[3.4]辛烷-5-基)乙腈在-78℃下向在无水thf(20ml)中的lhmds(1m在thf中,14.7ml,14.7mmol,2.2当量)的溶液逐滴添加在thf(6.6ml)中的起始内酯(840mg,6.7mmol,1.0当量)和mecn(0.69ml,13.3mmol,2.0当量)的溶液。将反应物在-78℃下搅拌30分钟并且随后在室温下持续2小时。向反应混合物添加饱和nh4cl水溶液(10ml)并将反应物在室温下在n2下搅拌5分钟。将反应混合物在饱和nh4cl水溶液(20ml)与dcm(20ml)之间分配并分离各相。用dcm(3
×
50ml)洗涤水相,并将合并的有机物用na2so4干燥并在真空中除去溶剂。将粗剩余物通过柱色谱(silicycle 40g,在庚烷中的etoac,在15cv内0至40%)进行纯化,以得到为无色油状物的所期望的产物(640mg,57%)。uplc(方法a)1.85分钟,98%,[m-h]-=166.1。中间体602-[1-(3-氨基-1h-吡唑-5-基)环丁基]乙醇将中间体59(490mg,2.93mmol)溶解在etoh(5.0ml)中并添加一水合肼(213μl,4.38mmol)。将所得溶液在60℃下搅拌3天。将反应冷却至室温并使co2鼓泡通过1小时。将反应物浓缩并添加meoh(10ml),并滤出所得白色固体。浓缩滤液以得到为棕色油状物的粗制标题产物(526mg)。其不经进一步纯化用于下一步。中间体61螺[5,6-二氢吡咯并[1,2-b]吡唑-4,1
’‑
环丁烷]-2-胺
向在无水thf(10ml)中的中间体60(使用的粗制品,354mg,1.95mmol)的溶液逐滴添加socl2(708μl,9.77mmol),并将所得混合物在室温下搅拌3小时。将反应物小心地倒入nh4oh(28%水溶液)和冰的1∶1混合物(总共60ml)中,并用dcm(3
×
50ml)萃取水相三次。将合并的有机相用盐水(60ml)洗涤,用na2so4干燥并浓缩至干燥,以得到为棕色油状物的粗制螺[5,6-二氢吡咯并[1,2-b]吡唑-4,1
’‑
环丁烷]-2-胺(200mg,24%),其不经进一步纯化即使用。lc-ms(方法d)2.38分钟,38.2%,[m h]
=264.0。中间体621-环戊基-4-碘代-咪唑在室温下向在dmf(10ml)中的4-碘代-1h-咪唑(1.00g,5.2mmol)和cs2co3(5.07g,15.5mmol)的经n2吹扫的搅拌溶液逐滴添加环戊基溴(663μl,6.2mmol)并将反应物在60℃下搅拌16小时。随后将温度提高至80℃并将反应物另外搅拌3小时。在真空中除去溶剂并将粗制固体用h2o(40ml)和etoac(30ml)稀释,随后超声处理。随后分离有机层并使用etoac(4
×
10ml)萃取水层。将合并的有机物用盐水(30ml)洗涤,干燥(mgso4)并浓缩至棕色/橙色油状物(1.41g)。将粗制品通过硅胶色谱(在庚烷中的20%至70%etoac)进行纯化,以得到1-环戊基-4-碘代-咪唑(874mg,65%)。1h nmr(400mhz,cdcl3:δh 7.41(s,1h),7.02(s,1h),4.47-4.40(m,1h),2.20-2.14(m,2h),1.86-1.68(m,6h)ppm。中间体632-(2,2-二甲基-5-氧代-吡咯烷-1-基)乙酸甲酯向在无水thf(300ml)中的5,5-二甲基吡咯烷-2-酮(10.0g,88mmol)的冰冷溶液添加溴乙酸甲酯(10.0ml,106mmol),随后分批添加nah(3.9g,97mmol),并将反应物在0℃下在n2下搅拌1.5小时。随后将反应物小心地倒入饱和nh4cl水溶液(300ml)中并用etoac(3
×
100ml)萃取。将合并的有机相用na2so4干燥,过滤并在真空中浓缩,以得到为黄色油状物的2-(2,2-二甲基-5-氧代-吡咯烷-1-基)乙酸甲酯(21.2g),其以粗制品的形式用于反应的下一步。1h nmr(400mhz,cdcl3)δ3.90(s,2h),3.72(s,3h),2.44(t,j=8hz,2h),1.93(t,j=8hz,2h),1.20(s,6h)ppm。中间体642-(2,2-二甲基-5-氧代-吡咯烷-1-基)乙酰胺
将中间体63(32.7g,177mmol)溶解在nh3(7n在meoh中,200ml)中并转移到hasteloid高压容器(bomb)中。将反应容器密封并加热至60℃过夜。使反应物冷却至室温并在真空中除去溶剂,以通过1h nmr得到sm和产物(7∶3)的混合物(35.9g,119%),其为黄色油状物。将粗制混合物溶解在nh3(7n在meoh中,600ml)中并转移至1l高压容器中,密封并加热至80℃过夜。使反应物冷却至室温并在真空中除去溶剂,以得到2-(2,2-二甲基-5-氧代-吡咯烷-1-基)乙酸甲酯(33.0g),其以粗制品的形式用于下一步。中间体652-溴-5,5-二甲基-6,7-二氢吡咯并[1,2-a]咪唑将该反应分成8个不同的批次,全部以相同的方式进行。向mw小瓶添加粗制中间体64(2.0g,11.8mmol)、pobr3(10.1g,35.3mmol)和mecn(10ml)。将反应混合物密封并加热至70℃过夜。使8个批次均冷却至室温并倒入水(1l)中。将水相用k2co3(120g)碱化至ph 10并用etoac(3
×
1l)萃取。将合并的有机相用na2so4干燥,过滤并浓缩至干燥。将剩余物通过np色谱(二氧化硅,etoac/dcm 0至50%)进行纯化,以得到为黄色油状物的2-溴-5,5-二甲基-6,7-二氢吡咯并[1,2-a]咪唑(11.6g,56%),其在静置时结晶。uplc(方法a)2.60分钟,99%,[m h]
=215.1。中间体662-(2-异丙基-5-氧代-吡咯烷-1-基)乙酸甲酯在n2下向在无水thf(15ml)中的5-异丙基吡咯烷-2-酮(500mg,3.9mmol,1.0当量)的冷却(0℃)混悬液中分批添加nah(在矿物油中的60%,142mg,5.9mmol,1.5当量)并将混合物在室温下搅拌30分钟,随后添加2-溴乙酸甲酯(751mg,4.9mmol,1.3当量)并将混合物在室温下搅拌18小时。将反应物用饱和nh4cl(5ml)淬灭并在真空中除去挥发物。将剩余物在etoac(25ml)与h2o(15ml)之间分配,并将有机相用mgso4干燥并在真空中浓缩。将剩余物通过正相色谱(100%叔丁基甲基醚)进行纯化,以得到为无色油状物的标题化合物(350mg,45%)。1h-nmr(396mhz,氯仿-d)δ4.48(d,j=17.6hz,1h),3.74-3.66(m,4h),3.57(d,j=17.6hz,1h),2.38(t,j=8.5hz,2h),2.04-1.92(m,2h),1.80-1.71(m,1h),0.91(d,j=6.7hz,3h),0.76(d,j=6.7hz,3h)ppm。中间体67
2-(2-异丙基-5-氧代-吡咯烷-1-基)乙酰胺将中间体66(350mg,1.76mmol,1当量)溶解在7n甲醇nh3(5ml)中并在密封管中在室温下搅拌48小时。将反应物在真空中浓缩,以得到为无色胶状物的标题化合物(320mg,99%)。1h-nmr(396mhz,氯仿-d)δ6.53(s,1h),5.40(s,1h),3.94(d,j=15.1hz,1h),3.82(d,j=15.1hz,1h),3.68-3.64(m,1h),2.41-2.37(m,2h),2,18-2-07(m,1h),2.06-1.95(m,1h),1.87-1.75(m,1h),0.92(d,j=7.3hz,3h),0.77(d,j=6.7hz,3h)ppm。中间体682-溴-5-异丙基-6,7-二氢-5h-吡咯并[1,2-a]咪唑将中间体67(310mg,1.68mmol,1当量)和三溴氧化磷(v)(1.93g,6.73mmol,4当量)密封在管中并在100℃下加热1小时。将反应冷却至室温并倒入水(25ml)中,将不溶性物质通过过滤除去并且将溶液用k2co3碱化,用etoac(2
×
20ml)萃取,将合并的有机物干燥(mgso4)并在真空中浓缩,以得到为棕色胶状物的标题化合物(310mg,80%)。uplc(方法a):2.84分钟,80%,[m h]
=229.0/231.0。中间体692-溴-5-异丙基-6,8-二氢-5h-咪唑并[2,1-c][1,4]嗪在5分钟内向在thf(15ml)中的5-异丙基吗啉-3-酮(1.25g,8.7mmol,1.0当量)的溶液分批添加nah(在矿物油中的60%,0.45g,11.4mmol,1.3当量),并将反应物在室温下搅拌10分钟,随后在10分钟内逐滴添加在thf(10ml)中的碘乙酰胺(1.78g,9.6mmol,1.1当量)的溶液。将反应物在室温下搅拌2小时,随后添加另一份碘乙酰胺(0.32g,1.9mmol,0.2当量)并在室温下另外搅拌2小时。添加另一份nah(0.25g,1.7mmol,0.2当量),并将反应混合物在室温下搅拌另外的16小时,通过添加3滴水淬灭并且随后在真空中浓缩,以得到浅橙色泡沫状固体。将其混悬在mecn(20ml)中,添加三溴化磷酰基(7.22g,25.2mmol,3.0当量)并将反应混合物在95℃下加热3小时。随后将反应混合物在真空中浓缩并在饱和nahco3溶液(100ml)与dcm(200ml)之间分配。将有机层在真空中浓缩,吸收在二氧化硅上并通过正相色谱0至30%etoac/dcm进行纯化,以得到为橙棕色油状剩余物的标题化合物(282mg,14%),
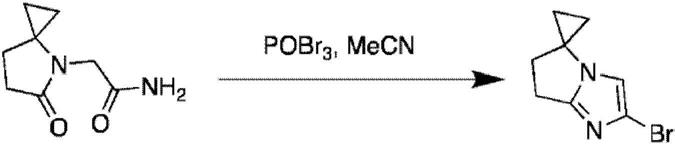
其直接用于下一步。uplc(方法a):2.73分钟,60%,[m h]
=245.0/247.0。中间体704-(1h-咪唑-4-基)丁-3-炔-2-醇将在mecn(3ml)中的4-碘代-1h-咪唑(190mg,0.98mmol,1.00当量)、cui(9mg,0.05mmol,0.05当量)、pd(ph3p)2cl2(34mg,0.05mmol,0.05当量)和丁-3-炔-2-醇(70mg,1.00mmol,2.00当量)的混悬液用n2脱气,随后添加三乙胺(410μl,2.94mmol,3.00当量)。将反应物在100℃下在密封管中加热3小时,并随后在真空中浓缩。将粗物质溶解在h2o(10ml)中,过滤并通过离子交换(dowex w50x)进行纯化,用水洗涤并用20%nh3/h2o洗脱,以得到为棕色胶状物的标题化合物(610mg,87%)。uplc(方法a):0.91分钟,89%,[m h]
=137.1。中间体714-(1h-咪唑-4-基)丁烷-2-醇将在meoh(25ml)中的中间体70(610mg,4.48mmol,1当量)的溶液分到5个小瓶中。向每个小瓶分批添加10%pd/c(40mg)和甲酸铵(566mg,8.96mmol,10当量)。将反应物在55℃下加热3小时,并随后在回流下1小时。将反应物合并,通过硅藻土垫过滤并真空浓缩。将粗物质溶解于h2o中,通过离子交换(dowex w50x)纯化,用水洗涤并用20%nh3/h2o洗脱以得到为褐色胶状物的标题化合物(410mg,65%)。uplc(方法a):0.89分钟,88.7%,[m h]
=141.1。中间体724-(3-氯丁基)-1h-咪唑将在亚硫酰氯(5ml)中的中间体71(410mg,2.92mmol,1当量)的溶液在80℃下加热5分钟,冷却至室温并将反应物真空浓缩以得到标题化合物(464mg 100%,2.92mmol)。uplc(方法a):2.36分钟,83%,[m h] =159.1/161.1。中间体735-甲基-6,7-二氢-5h-吡咯并[1,2-c]咪唑向在dmf(15ml)中的中间体72(0.46g,2.9mmol,1当量)的溶液添加k2co3(2.02g,14.6mmol,5当量)并将混合物在100℃下加热过夜。将反应物真空浓缩,将粗物质溶解于etoac(2
×
15ml)中,将固体通过过滤去除并在真空中浓缩溶液以得到为胶状物的标题化合物(410mg,假定定量)。uplc(方法a):1.95分钟,83%,[m h]
=123.1。中间体74
1,3-二碘代-5-甲基-6,7-二氢-5h-吡咯并[1,2-c]咪唑向在dmf(10ml)中的中间体73(100mg,0.8mmol,1.0当量)的溶液添加n-碘代琥珀酰亚胺(405mg,1.8mmol,2.2当量)并将反应物在75℃在n2下加热2小时。将反应物真空浓缩,用dcm(10ml)和h2o(10ml)稀释,分离各相,并将水相用dcm(2
×
10ml)萃取。将合并的有机物干燥(mgso4)并真空浓缩。将粗物质通过正相色谱0.5∶5∶95 nh3/meoh/dcm纯化以得到为褐色胶状物的标题化合物(157mg,51%)。uplc(方法a):2.98分钟,22%,[m h]
=375.0。中间体751-碘代-5-甲基-6,7-二氢-5h-吡咯并[1,2-c]咪唑向在etoh(10ml)中的中间体74(150mg,0.4mmol,1当量)的溶液添加在h2o(10ml)中的亚硫酸钠(253mg,2.0mmol,5当量)的溶液且将反应物在60℃下加热15分钟。真空去除etoh,并将水相用etoac(3
×
10ml)萃取,将合并的有机物干燥(mgso4),并真空去除溶剂以得到为浅褐色胶状物的标题化合物(75mg,75%)。uplc(方法a):2.52分钟,54%,[m h]
=249.1。中间体763-[1-(2-羟基乙基)环丙基]-3-氧代-丙腈向冷却至-78℃的在thf(22ml)中的lhmds(在thf中的1m,9.8mmol,2.2当量)的溶液逐滴添加在thf(2ml)中的5-氧杂螺[2.4]庚烷-4-酮(500mg,4.5mmol,1.0当量)和mecn(0.47ml,8.9mmol,2.0当量)的溶液,并将反应物在-78℃下搅拌15分钟,并随后在1小时内温热至室温。将反应物通过逐滴添加饱和nh4cl水溶液(20ml)淬灭并用etoac(3
×
30ml)洗涤。将合并的有机物用mgso4干燥并真空浓缩以得到为无色油状物的标题化合物(468mg,69%),所述油状物不经进一步纯化即用于下一步。1h-nmr(396mhz,cdcl3)δ3.92-3.87(m,2h),3.65(t,j=5.4hz,2h),1.88(t,j=5.4hz,2h),1.32-1.27(m,2h),0.83-0.79(m:2h)ppm.中间体772-[1-(3-氨基-1h-吡唑-5-基)环丙基]乙醇向在etoh(4ml)中的中间体76(468mg,3.1mmol,1.0当量)的溶液添加水合肼
(4.45ml,4.6mmol,1.5当量),并将反应物在密封小瓶中加热至90℃持续5小时。然后将反应物真空浓缩,并将粗剩余物通过正相柱色谱,0至15%meoh/dcm纯化以得到为淡黄色胶状物的标题化合物(340mg,73%),将其直接用于下一步骤。中间体78螺[5,6-二氢吡咯并[1,2-b]吡唑-4,1
′‑
环丙烷]-2-胺在1分钟内向在thf(11ml)中的中间体77(340mg,2.0mmol)的搅拌溶液逐滴添加socl2(0.74ml,10.2mmol),并将反应物在室温下搅拌45分钟。将反应物缓慢倒入nh4oh(h2o中的35%溶液)和冰(8g)的搅拌溶液(nh3oh体积=5/3
×
thf体积,冰质量=nh3oh体积/2.5)并搅拌5分钟。将溶液用dcm(3
×
20ml)萃取,将合并的有机物用mgso4干燥并在真空中浓缩。将粗物质通过正相柱色谱(biotage isolera,25g,siliasep硅胶40至63μm/230至400目,剩余物[装载到dcm中],在dcm中的0至10%甲醇纯化,以得到为无色玻璃状物的螺[5,6-二氢吡咯并[1,2-b]吡唑-4,1
′‑
环丙烷]-2-胺(46.0mg,15%)。1h-nmr(396mhz,氯仿-d4)δ4.95(s,1h),4.13-4.02(m,2h),3.52(br s,2h),2.51-2.43(m,2h),1.00-0.95(m,2h),0.92-0.86(m,2h)ppm.中间体794-碘代-1-异丁基-咪唑向在dmf(10ml)中的碘代咪唑(1.0g,5.2mmol,1.0当量)的溶液添加碳酸铯(5.0g,15.5mmol,3.0当量)和1-碘代-2-甲基丙烷(710μl,6.2mmol,1.2当量)并将混合物在60℃下加热2小时。添加另一份1-碘代-2-甲基丙烷(237μl,2.1mmol,0.4当量)并继续加热另外3小时。然后将反应物冷却至室温,并真空去除挥发物。将剩余物用水(50ml)和etoac(25ml)稀释,分离各相,并将水相用etoac(2
×
25ml)萃取。将合并的有机物用盐水(50ml)洗涤、干燥(na2so4)并真空去除溶剂。将剩余物通过正相色谱,0至20%etoac/dcm纯化以得到为黄色油状物的标题化合物(687mg,53%)。uplc(方法a)2.74分钟,99%,[m h]
=251.0。中间体802-(3-乙基-2-羟基-四氢呋喃-2-基)乙腈将在无水thf(20ml)中的lhmds(在thf中的1m,17.3ml,17.3mmol)的溶液冷却至-78℃并逐滴添加mecn(821μl,15.8mmol)。将所得溶液搅拌30分钟,然后逐滴添加在无水thf(5.0ml)中的3-乙基四氢呋喃-2-酮(900mg,7.9mmol)的溶液,并将反应物在-78℃下搅拌5
小时。将反应物通过套管转移至含有meoh(100ml)的圆底烧瓶中,并将所得溶液使用1m hcl小心地中和至ph 8。将混合物用dcm(3
×
100ml)萃取并将合并的有机物用盐水(200ml)洗涤,用na2so4干燥并真空浓缩以得到为浅褐色油状物的粗产物。将粗剩余物通过柱色谱(biotage isolera,正相,25g,siliasep硅胶40至63μm/230至400目,装载到dcm中的剩余物,在庚烷中的0至40%etoac,不具有uv活性的产物)纯化以得到为浅黄色油状物的2-(3-乙基-2-羟基-四氢呋喃-2-基)乙腈(702mg,57%),所述油状物不经进一步纯化即使用。中间体813-(3-氨基-1h-吡唑-5-基)戊烷-1-醇向在etoh(19ml)中的中间体80(600mg,3.87mmol)的溶液添加nh2nh2.h2o(282μl,5.80mmol),并将所得溶液在60℃下搅拌20小时。将反应物冷却至室温并将co2(干冰)鼓泡通过1小时。将反应混合物倾析并将滤液浓缩成粗制黄色油状物(575mg)。将所述物质通过柱色谱(biotage isolera,正相,40g,siliasep硅胶40至63μm,230至400目,[剩余物装载在dcm(0.5ml)中]在dcm中的0至10%meoh纯化以得到为浅粉色胶状物的3-(3-氨基-1h-吡唑-5-基)戊-1-醇(260mg,33%)。uplc(方法a)1.56分钟,84%,[m-h]-=168.1。中间体824-乙基-5,6-二氢-4h-吡咯并[1,2-b]吡唑-2-胺向在thf(7.7ml)中的中间体81(260mg,1.54mmol)的溶液逐滴添加socl2(0.56ml,7.68mmol)并将反应物在室温下在n2下搅拌。在4小时之后,向反应物添加另一份socl2(0.56ml,7.68mmol),在室温下再搅拌1小时。然后将反应物真空浓缩并将剩余物溶解于dcm(20ml)中并在回流下加热2小时。将反应物冷却至室温,添加socl2(0.56ml,7.68mmol)并将混合物在室温下搅拌2小时,然后真空去除溶剂。将粗物质直接用于下一步骤。中间体864-甲基-n-(6-甲基-2-吡啶基)苯磺酰胺向在吡啶(5ml)中的6-甲基吡啶-2-胺(1.00g,9.3mmol,1.0当量)的溶液添加对甲苯磺酰氯(2.29g,12.2mmol,1.3当量)且将反应物在80℃下搅拌4小时,接着在室温下搅拌过夜。将反应物用h2o(25ml)淬灭,用dcm(20ml)稀释,分离各相并将水相用dcm(3
×
10ml)萃取。将合并的有机物用磷酸盐缓冲液(ph 7,3
×
10ml)萃取、干燥(na2so4)并真空浓缩。将粗物质通过正相色谱用2%至10%etoac/庚烷洗脱进行纯化以得到为白色黏性油状物的标题化合物(1.60g,66%)。uplc(方法a):2.17分钟,100%,[m h]
=263.1。中间体872-[(6e)-2-甲基-6-(对甲苯磺酰基亚氨基)-1-吡啶基]乙酰胺
向在dmf(2ml)中的中间体86(867mg,3.31mmol,1.0当量)的溶液添加n,n-二异丙基乙胺(0.63ml,3.64mmol,1.1当量)并将反应物在室温下搅拌15分钟,然后添加在dmf(1.5ml)中的碘代乙酰胺(672mg,3.64mmol,1.1当量)的溶液,并将反应物在室温下搅拌过夜。将反应物真空浓缩,将油混悬于水(8ml)中,并将固体通过过滤收集,并从叔丁基甲基醚研磨(通过超声处理辅助,
×
3)以得到为褐色固体的标题化合物(668mg,63%)。uplc(方法a):2.18分钟,88%,[m h]
=320.2。中间体882,2,2-三氟-n-(5-甲基咪唑并[1,2-a]吡啶-2-基)乙酰胺向在dcm(2ml)中的中间体87(0.67g,2.1mmol,1.0当量)的混悬液添加三氟乙酸酐(1.37ml,9.9mmol,4.7当量)并将反应物在室温下搅拌过夜。将反应物真空浓缩,溶解于dcm(10ml)中并用饱和nahco3水溶液(3
×
10ml)和磷酸盐缓冲液(ph 7,3
×
10ml)洗涤,将有机物干燥(na2so4)并真空浓缩。将粗物质通过正相色谱3%至10%丙酮/dcm纯化以得到为浅黄色固体的标题化合物(315mg,62%)。uplc(方法a):1.43分钟,99%,[m h]
=244。中间体892,2,2-三氟-n-(5-甲基-5,6,7,8-四氢咪唑并[1,2-a]吡啶-2-基)乙酰胺在高压釜中制备在meoh(8ml)中的中间体88(200mg,0.82mmol,1当量)的溶液。添加rh(在氧化铝上5%,1.69g,0.82mmol,1当量)。将高压釜用n2(
×
3)吹扫并再填充,并随后用h2(8atm)吹扫并装载。将反应物在室温下搅拌18小时,真空浓缩并将粗物质通过正相色谱(10%丙酮/dcm)纯化以得到为白色固体的标题化合物(136mg,55%)。uplc(方法a):2.28分钟,97%,[m h]
=248.2。中间体905-甲基-5,6,7,8-四氢咪唑并[1,2-a]吡啶-2-胺将在thf(0.50ml)和meoh(0.50ml)中的中间体89(136mg,0.45mmol,1当量)和naoh
(180mg,4.49mmol,10当量)的混悬液通过微波辐照在80℃下加热1小时。将反应物真空浓缩并添加h2o(1ml)。通过逐滴添加2m hcl将ph调节至ph 7,并将所得溶液冷冻干燥过夜,并直接用于下一步骤,假定定量。uplc(方法a):1.84分钟,85%,[m h]
=152.1。中间体942-(3-甲基-5-氧代-吗啉-4-基)乙酰胺在n2下,在约15℃(冷水浴)下,向在thf(4ml)中的氢化钠(在矿物油中的60%,267.5mg,6.7mmol,1.1当量)的混悬液逐滴添加在thf(4ml)中的5-甲基吗啉-3-酮(700.0mg,6.1mmol,1.0当量)的溶液,并将反应混合物在15℃下搅拌30分钟。然后向溶液逐滴添加溴乙酸甲酯(640μl,6.7mmol,1.1当量),并将所述反应物在室温下再搅拌1小时。将反应物用饱和nh4cl(60ml)淬灭,用etoac(30ml)稀释,分离各相并将水相用etoac(2
×
30ml)萃取。将合并的有机物干燥(na2so4)并真空浓缩。将粗剩余物通过正相色谱,etoac/dcm 1∶1纯化以得到为浅黄色油状物的2-(3-甲基-5-氧代-吗啉-4-基)乙酸甲酯(858mg,75%)。1h nmr(cdcl3,400mhz)δ
h 4.49(m,1h),4.23(m,2h),3.94(m,1h),3.84-3.75(m,4h),3.69(m,1h),3.59-3.51(m,1h),1.29(m,3h)ppm.将在nh3(meoh中的7m,5ml,15.0当量)中的2-(3-甲基-5-氧代-吗啉-4-基)乙酸甲酯(429.0mg,2.3mmol,1.0当量)的溶液在密封小瓶中加热至60℃持续63小时。将反应混合物冷却至室温并真空浓缩。将剩余物用dcm/叔丁基甲基醚(约10ml,3∶7)研磨以得到为乳白色胶状剩余物的标题化合物(270mg,34%)。1h nmr(400mhz,dmso-d6)δ
h ppm,7.33(br.s,1h),7.05(br.s,1h),4.12(d,j=16.3hz,1h),4.04(d,j=1.2hz,2h),3.85(dd,j=11.5,3.6hz,1h),3.66-3.58(m,2h),3.53-3.47(m,1h),1.16(d,j=6.7hz,3h)ppm.中间体952-溴-5-甲基-6,8-二氢-5h-咪唑并[2,1-c][1,4]嗪向在mecn(1ml)中的中间体94(270.0mg,1.57mmol,1当量)的混悬液添加三溴氧化磷(v)(2.25g,7.84mmol,5当量),并将小瓶密封并在90℃下加热3小时。将反应物冷却至室温,倒入h2o(40ml)中,用dcm(40ml)稀释并分离各相。将水相用dcm(30ml)萃取,用固体k2co3中和(至ph 9)并进一步用dcm(30ml)萃取。将合并的有机物用水(40ml)稀释并用固体k2co3碱化(至ph 9)。分离各相,将有机相干燥(na2so4)并真空去除溶剂。将粗剩余物通过正相色谱(干装载的)0至100%etoac/dcm然后0至15%meoh/dcm纯化,并将所得剩余物用丙酮(2ml)研磨以得到为橙色固体的标题化合物(100mg,29%)。uplc(方法e)2.02分钟,100%,[m h]
=217.0/219.0。
中间体99n-(4-氧代-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-基)氨基甲酸叔丁酯向在无水甲苯(20ml)中的中间体98(1.54g,8.5mmol,1.0当量)的混悬液添加三乙胺(1.43ml,10.2mmol,1.2当量),然后添加无水t-buoh(10.0ml,104.0mmol,12.0当量)。向溶液添加二苯基磷酰叠氮化物(2.21ml,10.2mmol,1.2当量)并将反应物在80℃下搅拌过夜。将反应物冷却至室温,用etoac(45ml)和h2o(45ml)稀释,分离各相,并将水相用etoac(25ml)萃取。将合并的有机物用饱和nahco3水溶液(30ml)、h2o(30ml)洗涤、干燥(na2so4)并真空浓缩。将剩余物通过短硅胶垫0至10%etoac/dcm纯化以得到为白色固体的标题化合物(1.17g,54%)。uplc(方法a)2.68分钟,88%,[m-h]-=250.2。中间体1002-氨基-6,7-二氢-sh-吡唑并[1,5-a]吡啶-4-酮向在二氧六环(4ml)中的中间体99(190mg,0.76mmol,1当量)的溶液添加hcl(4ml,二氧六环中的4m),随后从玻璃移液管添加一滴水,并将反应物在室温下搅拌过夜。真空去除溶剂,并将固体混悬在dcm(10ml)中。添加饱和nahco3水溶液(10ml)并搅拌直至形成溶液,分离各相并将水相用dcm(2
×
5ml)萃取。将合并的有机物干燥(相分离器)并真空浓缩以得到为黄色油状物的标题化合物(81mg,71%)。uplc(方法a)0.80分钟,56%,[m h]
=152.1。中间体1012-(2-甲基-5-氧代-吡咯烷-1-基)乙酸甲酯向无水thf(60ml)的冰冷溶液分批添加氢化钠(在矿物油中的60%,2.22g,55.4mmol,1.1当量)。在10分钟内向该灰色浆液逐滴添加无水thf(20ml)中的5-甲基吡咯烷-2-酮(5.00g,50.4mmol,1.0当量)的溶液,保持温度低于7℃。再过5分钟之后,添加另外的thf(40ml)并将反应物再搅拌45分钟,然后添加thf(10ml)中的溴乙酸甲酯(5.7ml,60.5mmol,1.2当量)。使反应物温热至室温并搅拌1.5小时。将反应物倒入饱和nh4cl水溶液中(100ml),用etoac(3
×
80ml)萃取,用水(2
×
100ml)、nh4cl水溶液(2
×
100ml)洗涤,用na2so4干燥,过滤并在减压下浓缩以得到为无色油状物的标题化合物(4.81g,56%)。uplc(方法a):1.86分钟,66%,[m h]
=172.1。中间体1022-(2-甲基-5-氧代-吡咯烷-1-基)乙酰胺
将在氨(甲醇中的7m,40ml,13当量)中的中间体101(3.80g,22.2mmol,1当量)的溶液在室温下搅拌48小时。将反应混合物真空浓缩并再溶解于meoh(40ml)和mecn(10ml)中并用庚烷洗涤。将meoh/mecn层真空浓缩,并将剩余物用mecn(30ml)研磨,并在5℃下储存3至4小时。将溶剂倾析相对于不溶性杂质分离并在减压下蒸发以得到为无色油状物的标题化合物(2.16g,62%)。1h nmr(dmso-d6,396mhz):δ7.29(br s,1h),7.01(br s,1h)3.88(m,1h),3.63(m,1h),3.51(m,1h),2.08-2.21(m,3h),1.47(m,1h),1.07(d,j=6.1hz,3h)ppm,中间体1032-溴-5-甲基-6,7-二氢-5h-吡咯并[1,2-a]咪唑将中间体102(2.00g,12.8mmol,1当量)和pobr3(7.34g,25.6mmol,2当量)在80℃下在密封小瓶中加热3小时,并随后将反应物冷却至室温过夜。将混合物吸收在dcm/h2o(3
×
10ml)中,分离各相,将水相用k2co3(3至4g)碱化并用dcm(2
×
30ml)萃取。将合并的有机物用k2co3水溶液(20ml)和h2o(20ml)洗涤、干燥(na2so4)并真空浓缩。将剩余物通过短硅胶垫(0至50%dcm/etoac)纯化以得到为琥珀色油状物的标题化合物(115g,45%)。其在静置时结晶。uplc(方法a):2.42分钟,96%,[m h]
=201.1/203.1。中间体104a和104b1-(2-氟乙基)-5-碘代-咪唑(中间体104a)和1-(2-氟乙基)-4-碘代-咪唑(中间体104b)向在dmf(9.5ml)中的4-碘代-1h-咪唑(0.93g,4.77mmol,1.0当量)的溶液添加碳酸铯(4.66g,14.31mmol,3.0当量)和1-氟-2-碘代-乙烷(1.00g,5.72mmol,1.2当量)。将反应物在室温下搅拌3小时,真空浓缩并将剩余物溶解于etoac(100ml)中。将有机层用水(3
×
50ml)、盐水(50ml)洗涤,干燥(mgso4)并真空浓缩。将剩余物通过正相色谱20%etoac/dcm纯化以得到为无色油状物的不可分离的异构体混合物(749mg,65%)。uplc(方法a):2.03分钟,77%,[m h]
=240.9;uplc(方法a):2.07分钟,22%,未观察到质量离子。中间体1056-(三氟甲基)哌啶-2-酮
将起始吡啶酮(1.50g,9.20mmol,1当量)溶解于无水meoh(90ml)中并置于高压容器中。添加pto2(300mg,20%wt)。将反应容器密封并用h2(3
×
)吹扫气氛。将反应物在室温下在10巴的h2下搅拌16小时。将反应混合物通过dicalite过滤并用meoh(约200ml)充分洗涤。将滤液浓缩至干燥以得到为无色固体的标题化合物(1.49g,97%)。1h nmr(dmso,400mhz)δ
h 8.01(bs,1h),4.02-4.09(m,1h),2.13-2.21(m,2h),1.85-1.94(m,1h),1.57-1.78(m,3h)ppm.19f nmr(dmso,376mhz)δ
f-75.34(d,j=8.65hz)ppm.中间体1062-[2-氧代-6-(三氟甲基)-1-哌啶基]乙酸甲酯在n2下向在无水thf(50ml)中的中间体105的吡啶酮和哌啶酮(约67%哌啶酮)的混合物(2.40g,14.4mmol,1.0当量)添加溴乙酸甲酯(1.63ml,17.2mmol,1.2当量)。将混合物在冰浴中冷却至0℃并分批添加nah(在矿物油中的60%,632mg,15.8mmol,1.1当量)。将反应物在0℃下搅拌30分钟并在室温下搅拌1.5小时。将反应物小心地倒入饱和nh4cl水溶液(100ml)中并用etoac(3
×
75ml)萃取。将合并的有机相用na2so4干燥,过滤并浓缩至干燥以得到黄色油状物。将剩余物通过np色谱(hept/etoac 10%至75%)纯化以得到为无色油状物的标题化合物(2.21g,64%)。1h nmr(cdcl3,400mhz)δ
h 4.79(d,j=17.6hz,1h),4.00-3.92(m,1h),3.71-3.75(m,4h),2.54-2.49(m,2h),2.17-1.99(m,3h),1.88-1.82(m,1h)ppm.19f nmr(cdcl3,376mhz)δ
f 78.2(d,j=6.7hz)ppm.中间体1072-[2-氧代-6-(三氟甲基)-1-哌啶基]乙酰胺将中间体106(2.21g,9.23mmol,1当量)溶解于7n甲醇氨(30ml)中并将溶液分成2
×
15ml的批次,将其各自在密封管中以mw加热至60℃持续24小时。使两个小瓶冷却至室温,合并且浓缩至干燥以得到为灰白色固体的标题化合物(1.91g,92%)。1h nmr(dmso,400mhz)δ
h 7.28(bs,1h),6.98(bs,1h),4.34(d,j=16.3hz,1h),4.23-4.29(m,1h),3.49(d,j=16.3hz,1h),229-2.33(m,2h),1.91-2.09(m,2h),1.71-1.83(m,2h)ppm.19f nmr(dmso,376mhz)δ
f-70.50(d,j=7.8hz)ppm.中间体1082-溴-5-(三氟甲基)-5,6,7,8-四氢咪唑并[1,2-a]吡啶
庚烷和1%et3n洗脱进行纯化。将产物级分合并,再溶解在ch3cn/h2o中并冷冻干燥过夜。分离出为浅黄色固体的3-溴-4,4-二氟-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-胺(248mg,78%、)。uplc(方法f)2.45分钟和2.63分钟,66.6%和25.5%,es :252.0/254.0[m(79br/81br) h] 中间体1124,4-二氟-3-甲基-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-胺向在无水1,4-二氧六环(6.0ml)中的三甲基环硼氧烷(trimethylboroxine)(293mg,2.33mmol)和中间体111(195mg,0.77mmol)添加碳酸钾(250mg,1.81mmol)和pd(pph3)4(90mg,0.08mmol),并将混合物脱气(真空/氮气吹扫
×
3)并在90℃下加热过夜。将反应混合物冷却至室温,用etoac(15ml)稀释并过滤以去除无机物。真空去除溶剂以得到粗剩余物。第二反应平行进行。向在无水(先前经过脱气)1,4-二氧六环(2.5ml)中的三甲基环硼氧烷(75mg,0.60mmol)和中间体111(50mg,0.20mmol)添加碳酸钾(55mg,0.40mmol)和pd(pph3)4(23mg,0.02mmol),并将混合物在90℃下加热过夜。添加另外的pd(pph3)4(23mg,0.02mmol)、三甲基环硼氧烷(75mg,0.60mmol)和1,4-二氧六环(1.0ml)并在90℃下继续反应。将反应物冷却并过滤并用etoac洗涤。蒸发溶剂并将剩余物溶解于甲醇(3.0ml)中。将混合物使用scx-2(2g)滤筒用甲醇(3cv)洗脱,然后用在meoh(3cv)中的4.5m nh3洗脱产物进行纯化。将溶剂蒸发并通过uplc显示所得剩余物(20mg)含有作为与起始材料和脱甲基杂质的混合物的所期望产物。将来自两个反应的粗剩余物合并用于纯化。将合并的产物吸附到二氧化硅上并通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,)dcm中的0%至3%meoh纯化。将所获得混合物(214mg)通过scx-2(5g)滤筒,用甲醇(3cv)洗脱以去除opph3,然后用在meoh(3cv)中的4.5m nh3洗脱以将产物作为与起始材料的混合物洗脱以得到黄色油状物(130mg)。将该黄色油状物溶解于dcm(1.0ml)中,并通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,)在dcm中的0%至2%meoh纯化。合并级分以得到两批粗产物。通过uplc,分离的物质a(60mg)含有期望的产物(57%)、opph3(27%)和脱甲基副产物(9%)。分离的物质b(76mg)含有产物(40%)、opph3(16%)和溴起始材料(25%)。将两个不纯的批次直接用于下一步骤。uplc(方法e)2.13分钟,57%,es
:188.1[m h]
。中间体1134,4-二氟-3-碘代-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-胺向在dcm(15ml)中的中间体37(317mg,1.83mmol)的溶液添加n-碘代琥珀酰亚胺(618mg,2.75mmol)并且将反应物在室温下在氮气下搅拌72小时。在真空中蒸发挥发物并将
剩余物通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,dcm中的4%丙酮,含有1%三乙胺)纯化。将含有产物的级分浓缩,并将剩余物通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,在dcm中的2%丙酮,含有1%三乙胺)进一步纯化以得到为黄色固体的4,4-二氟-3-碘代-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-胺(113mg,15%)。uplc(方法a)2.55分钟,72%,es
:300.0[m h]
。中间体1144,4-二氟-3-乙烯基-6,7-二氢-5h-吡唑并[1’5-a]吡啶-2-胺将在1,4-二氧六环(4.0ml)和水(1.0ml)的脱气混合物中的中间体113(113mg,0.38mmol)、2-乙烯基硼酸频哪醇酯(0.08ml,0.45mmol)、pd(pph3)4(43.7mg,0.04mmol)、k2co3(157mg,1.13mmol)的混悬液在密封小瓶中在mw辐照下加热至150℃持续20分钟。将反应混合物用水(10ml)淬灭并用etoac(3
×
10ml)萃取。将合并的有机物用盐水(15ml)洗涤,用na2so4干燥并真空浓缩以得到粗产物。将其通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,在庚烷中的10%至50%etoac)纯化以得到为浅黄色固体的4,4-二氟-3-乙烯基-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-胺(71.0mg,71%产率)。uplc(方法a)2.51分钟,75%,es
:200.1[m h]
。中间体1153-乙基-4,4-二氟-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-胺向不锈钢容器中的在meoh(5.0ml)中的中间体114(71.0mg,0.30mmol)的溶液添加pd/c(10%纯度,32.2mg,0.03mmol)。将容器密封并在氢气气氛(4巴)下在室温下搅拌过夜。将混合物通过dicalite垫过滤,用甲醇洗脱。真空去除溶剂以得到为白色固体的3-乙基-4,4-二氟-6,7-二氢-5h-吡唑并[1,5`-a]吡啶-2-胺(54.0mg,57%)。uplc(方法a)2.52分钟,64%,es
:202.1[m h]
。中间体1161-[2-(4-甲基哌嗪-1-基)乙基]-5-(三氟甲基)吡唑-3-胺向在dmf(10ml)中的5-(三氟甲基)-1h-吡唑-3-胺(380mg,2.52mmol)的溶液添加碳酸钾(1.11g,8.05mmol)并将混合物在室温下搅拌0.25小时。然后向其添加1-(2-氯乙基)-4-甲基-哌嗪;将二盐酸盐(711mg,3.02mmol)和反应混合物在室温下搅拌72小时。然后将反应物在55℃下加热24小时。将反应物浓缩并在h2o(40ml)和tbme(2
×
40ml)之间分配。
将合并的有机层用水(2
×
10ml)洗涤,用mgso4干燥并浓缩以得到油状剩余物。将合并的水层用dcm(3
×
20ml)萃取,并将这些层合并、用mgso4干燥并浓缩以得到油状剩余物。将原始tbme萃取物通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,[干装载的剩余物],在dcm中的3%至5%meoh,含有1%氨水)纯化以得到为浅橙色固体的不期望的区域异构体2-[2-(4-甲基哌嗪-1-基)乙基]-5-(三氟甲基)吡唑-3-胺(122mg,15.9%)。合并混合的产物级分以得到93mg褐色油状剩余物,将其与来自前述水层的dcm萃取物合并。将该剩余物通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,[干装载的剩余物],在dcm中的3%至5%meoh,含有1%氨水)纯化以得到具有所期望区域异构体的1-[2-(4-甲基哌嗪-1-基)乙基]-5-(三氟甲基)吡唑-3-胺级分的一些分离物,但并不纯净。将混合的产物级分浓缩以得到为灰白色固体的1-[2-(4-甲基哌嗪-1-基)乙基]-5-(三氟甲基)吡唑-3-胺(114mg,6.6%),含有作为显著杂质的2-[2-(4-甲基哌嗪-1-基)乙基]-5-(三氟甲基)吡唑-3-胺。uplc(方法a)2.23分钟,40.3%,es
:278.1[m h]
。中间体1176-氨基吡啶-2-碳酰肼向在meoh(2.0ml)中的6-氨基吡啶-2-羧酸甲酯(152mg,1.00mmol)的溶液添加一水合肼(0.15ml,3.00mmol)。将混合物在78℃下在氮气下加热3小时。将混合物冷却至室温并去除溶剂。将所得固体用etoac和tbme的混合物(1∶3,15ml)洗涤,并随后用tbme(10ml)洗涤。将所得固体风干以得到为灰白色固体的6-氨基吡啶-2-碳酰肼(117mg,77%),其不经进一步纯化即用于下一步骤中。中间体118n,6-双[(e)-二甲基氨基亚甲基氨基]吡啶-2-甲酰胺将中间体117(117mg,0.77mmol)和dmf-dma(5.0ml,37.60mmol)的搅拌混合物在110℃下加热24小时。将混合物冷却至室温并真空去除挥发物。将所得固体混悬于tbme中。将混合物置于超声浴中5分钟,去除溶剂并干燥固体以得到n,6-双[(e)-二甲基氨基亚甲基氨基]吡啶-2-甲酰胺(220mg,102%),其不经进一步纯化即以粗制品的形式用于下一步骤。uplc(方法a)1.97分钟,93.2%,es
:263.1[m h]
。中间体1196-(4-异丙基-1,2,4-三唑-3-基)吡啶-2-胺向在mecn(1.0ml)和acoh(0.25ml,4.44mmol)的混合物中的中间体118(220mg,
0.78mmol)的搅拌溶液添加异丙基胺(0.34ml,3.91mmol)。将混合物在100℃下在氮气下搅拌16小时。将反应混合物用水淬灭并用2m naoh水溶液将ph调节至8。将混合物用etoac(3
×
15ml)萃取并将合并的有机物用盐水(10ml)洗涤,用na2so4干燥并浓缩以得到为黄色油状物的6-(4-异丙基-1,2,4-三唑-3-基)吡啶-2-胺(50mg,22%),将其直接用于下一步骤。uplc(方法a)1.86分钟,68.8%,es
:204.1[m h]
。中间体120[3-[2-(3-吡啶基)乙炔基]苯甲酰基]氧基钾向在thf(10ml)中的3-[2-(3-吡啶基)乙炔基]苯甲酸甲酯(237mg,1.00mmol)的溶液添加kotms(385mg,3.00mmol)。将反应物在室温下在氮气下搅拌过夜。真空蒸发挥发物。将所得固体混悬于tbme(20ml)中并置于超声浴中5分钟。去除溶剂并干燥固体以得到为浅黄色固体的[3-[2-(3-吡啶基)乙炔基]苯甲酰基]氧基钾(288mg,110%),其不经进一步纯化即用于下一步骤。uplc(方法a)1.79分钟,100%,es
:224.1[m-k 2h]
。中间体1213-[2-(3-吡啶基)乙炔基]苯甲酰氯向在dcm(5.0ml)中的中间体120(288mg,0.99mmol)的混悬液添加草酰氯(0.11ml,1.29mmol)和几滴dmf。将反应物在室温下在氮气下搅拌过夜。将另一份草酰氯(0.09ml,0.99mmol)添加至反应混合物并将反应物在室温下在氮气下搅拌过夜。将另一份草酰氯(0.09ml,0.99mmol)添加至反应混合物并将反应物在室温下在氮气下搅拌2小时。真空蒸发挥发物。向反应混合物添加甲苯(10ml)并真空蒸发挥发物。将剩余物溶解于dcm(10ml)中并过滤。将固体用dcm(10ml)洗涤并合并有机相并真空浓缩以得到3-[2-(3-吡啶基)乙炔基]苯甲酰氯和3-[2-(3-吡啶基)乙炔基]苯甲酸的混合物。向混悬在dcm(5.0ml)中的该混合物添加草酰氯(0.11ml,1.29mmol)。将反应物在室温下在氮气下搅拌2小时。真空蒸发挥发物。向混合物添加甲苯(10ml)并真空蒸发挥发物。将剩余物溶解于dcm(10ml)中并过滤。将固体用dcm(10ml)洗涤并将有机相合并并在真空中浓缩以得到为橙色油状物的3-[2-(3-吡啶基)乙炔基]苯甲酰氯(230mg,92%),其以粗制品的形式用于下一步骤中。uplc(方法a)3.23分钟,95.8%,es
:238.1[mh-cl ome]
。中间体1224-氟-3-[2-(3-吡啶基)乙炔基]苯甲酸甲酯将3-溴-4-氟-苯甲酸甲酯(1.00g,4.30mmol)、3-乙炔基吡啶(665mg,6.45mmol)、
三乙胺(1.82ml,13.1mmol)和etoac(20ml)添加至双颈圆底烧瓶,用n2脱气15分钟,然后添加cui(26.8mg,0.14mmol)和pd(pph3)2cl2(85.0mg,0.12mmol)并将所得混合物在室温下在n2下搅拌过夜。使反应混合物冷却至室温,通过硅藻土垫过滤并浓缩。将剩余物通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,[二氧化硅/粗制品=40/1,剩余物装载在dcm中],在庚烷中的0%至20%etoac进行纯化,以得到为浅黄色固体的4-氟-3-[2-(3-吡啶基)乙炔基]苯甲酸甲酯(577mg,52%)。uplc(方法a)3.30分钟,99.0%,es
:256.1[m h]
。中间体1234-吡唑-1-基-3-[2-(3-吡啶基)乙炔基]苯甲酸甲酯向rbf中装入甲基中间体122(208mg,0.81mm0l)、cs2co3(797mg,2.44mmol),吡唑(56mg,0.81mmol)和dmf(15ml)并在n2气氛下加热至80℃持续0.5小时。使反应混合物冷却至室温,部分去除溶剂,将剩余物用水(15ml)和etoac(20ml)稀释并分离各层。将水层用etoac(3
×
30ml)萃取,将合并的有机物用na2so4干燥,过滤并浓缩以得到米色固体。将剩余物通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,[二氧化硅/粗制品=20/1,剩余物装载在dcm中],在庚烷中的0%至30%etoac进行纯化,以得到为无色固体的4-吡唑-1-基-3-[2-(3-吡啶基)乙炔基]苯甲酸甲酯(177mg,65%)。uplc(方法a)3.13分钟,91%,es
:304.1[m h]
。中间体1244-吡唑-1-基-3-[2-(3-吡啶基)乙炔基]苯甲酸向在thf∶meoh∶水(2.0ml∶1.0ml∶1.0ml)中的中间体123(177mg,0.58mmol)的溶液添加lioh.h2o(40mg,0.95mmol)并将混合物在室温下在n2气氛下搅拌过夜。将反应混合物部分浓缩,添加水(5.0ml)并使用2n hcl将溶液酸化至ph 4。将所得沉淀物收集在玻璃料上并用mtbe(20ml)洗涤,并干燥以得到4-吡唑-1-基-3-[2-(3-吡啶基)乙炔基]苯甲酸(149mg,87.8%)。uplc(方法a)1.74分钟,99.5%,es
:290.1[m h]
。一般程序a将中间体2(1.1mmol)和在dmso中的n-羟基苯并三唑的溶液(100g/l,2ml,1.5mmol)置于小瓶中并添加胺(1mmol;添加“起始胺”)。如果胺是盐酸盐,则还添加et3n(1mmol)。将反应混合物在振荡器中搅拌30分钟,并添加edc(1.2mmol)。
在装载所有试剂后,将小瓶密封并在振荡器中搅拌1小时。如果形成澄清溶液,则将小瓶在室温放置24小时。否则,将反应混合物在超声浴中保持24小时(应避免强烈加热)。如果观察到反应混合物显著增稠使得搅拌无效,则一次性添加0.2ml dmso。通过lc-ms(方法b)分析粗制反应混合物,并随后进行色谱纯化。一般程序b将中间体2(0.10mmol,1.0当量)、无水mecn(0.5ml)、胺(0.10mmol,1.0当量)和dipea*(0.25mmol,2.5当量)置于小瓶中。将反应混合物搅拌30分钟,并添加吡啶盐(0.12mmol,1.2当量)。将小瓶密封并将反应混合物在100℃下加热6小时。冷却至室温之后蒸发混合物。将剩余物溶解于dmso中,过滤,并将溶液进行色谱纯化。通过lc-ms(方法b)分析产物。*如果胺作为盐购买,则向反应混合物添加另外量的dipea,产生游离碱。一般程序c将在dmf(1ml)中的中间体2(0.1mmol,1.0当量)和cdi(1.0当量)置于小瓶中。将反应混合物在搅拌下于50℃下加热1小时。然后添加胺(1.0当量)和在thf中的naotbu*的溶液(2.0当量)。将小瓶密封并将反应混合物在60℃下加热6小时。冷却至室温之后,将混合物用过量乙酸淬灭并蒸发。将剩余物溶解于dmso中,过滤,并将溶液进行色谱纯化。通过lc-ms(方法b)分析产物。*如果胺作为盐购买,则向反应混合物添加另外量的naotbu产生游离碱。一般程序d在室温下向中间体2(0.11mmol,1.1当量),n-甲基咪唑(2.2当量)和mecn(0.7ml)小心添加甲磺酰氯(1.1当量)(观察到少量放热)。将反应混合物搅拌0.5小时以使其冷却回室温。然后添加胺(1.0当量),将小瓶密封,并将反应混合物在振荡器中在室温下搅拌1小时并在100℃下加热2小时。在冷却至室温之后,真空蒸发溶剂,将剩余物溶于dmso中,过滤,并将溶液进行色谱纯化。通过lc-ms(方法b)分析产物。实施例1至23实施例1至23根据一般程序a制备。
实施例24n-(1-异丙基吡唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺
在约2分钟向在thf(40ml)中的1-异丙基-1h-吡唑-4-胺(1.00g,8.0mmol,1.0当量)和4-甲基-3-(吡啶-3-基乙炔基)苯甲酸甲酯(2.01g,8.0mmol,1.0当量)的冰冷溶液逐滴添加ktobu(在thf中的20%wt溶液,5.17ml,8.8mmol,1.1当量),并将所得溶液在0℃下搅拌30分钟,然后使其温热至室温1小时。将反应混合物通过添加h2o(250ml)淬灭,并随后真空浓缩以去除约20ml thf。然后将其用水(100ml)稀释并用叔丁基甲基醚(2
×
200ml)萃取水相。将合并的有机物用饱和盐水(150ml)洗涤,用mgso4干燥并真空浓缩以得到紫色/褐色油状固体。将其与另外的粗产物(来自相同的220mg规模的反应)合并,吸收到二氧化硅上并通过正相色谱,etoac∶庚烷(1∶1)至100%etoac纯化,并将所得剩余物用叔丁基甲基醚研磨以得到为无色固体的n-(1-异丙基吡唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(1.28g,37%)。uplc-ms(方法a)3.11分钟,100%,[m h]
=345.3。实施例25n-(1-异丙基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺向在四氢呋喃(75ml)中的中间体2(1.42g,6mmol,1当量)的混悬液添加三乙胺(1.82g,18mmol,3当量)并将混悬液搅拌10分钟,之后添加中间体4(假定6mmol,1当量),随后添加苯并三唑-1-基-氧基三吡咯烷基六氟磷酸盐/酯(3.12g,6mmol,1当量)并将反应物在室温下在氮气下搅拌18小时。将反应物通过倒入h2o(80ml)中淬灭并用dcm(80ml)稀释,分离各相并将有机相干燥(mgso4)并真空浓缩。将粗物质通过正相色谱(meoh∶dcm,1∶19)纯化,并随后通过正相色谱(meoh∶dcm,1∶39)再纯化。将该物质从etoac(40ml)中结晶以得到两批(878mg,350mg)。将第二批(350mg)溶解于dcm(10ml)中,并用h2o(10ml)洗涤、干燥(mgso4)并真空浓缩,并随后在整个周末期间将所述物质从etoac(7ml)中结晶。将所得固体用etoac(0℃,2ml)洗涤以得到n-(1-异丙基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(124mg&878mg,48%)。uplc(方法a)2.75分钟,99.7%,[m h]
=345.2。实施例26n-(4,4-二甲基-5,6-二氢吡咯并[1,2-b]吡唑-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺
将在thf(4.6ml)中的中间体1(150mg,0.60mmol,1.0当量)和中间体7(108mg,0.72mmol,1.2当量)的溶液冷却至0℃,此后逐滴添加kotbu(在thf中的20%wt溶液)(0.45ml,0.72mmol,1.2当量)并在0℃下搅拌1.5小时。将反应物用盐水淬灭,分离各相并将水层用etoac(
×
3)萃取,将合并的有机物干燥(mgso4)并真空浓缩。将粗物质通过反相色谱5%至80%mecn/h2o(0.1%nh3改性剂)纯化以得到为灰白色固体的n-(4,4-二甲基-5,6-二氢吡咯并[1,2-b]吡唑-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(36mg,16%)。uplc(方法a)3.36分钟,98.91,[m h]
=371.30。实施例27n-[1-(环丙基甲基)咪唑-4-基]-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺向在thf(8ml)中的中间体2(169mg,0.71mmol,1.0当量)的混悬液添加三乙胺(398μl,2.86mmol,4.0当量)并将反应物在室温下搅拌10分钟,然后添加1-(环丙基甲基)-1h-咪唑-4-胺二盐酸盐(150mg,0.71mmol,1.0当量)和(苯并三唑-1-基-氧基三吡咯烷基六氟磷酸盐/酯)(409mg,0.79mmol,1.1当量)并再在室温下搅拌2小时。添加另外的1-(环丙基甲基)-1h-咪唑-4-胺二盐酸盐(15mg,0.07mmol,0.1当量)和三乙胺(100μl,0.71mmol,1.0当量)并将反应物再搅拌1小时,然后用h2o(40ml)淬灭并用dcm(3
×
30ml)萃取。将合并的有机物用1m氨(40ml)、h2o(40ml)和盐水(40ml)洗涤、干燥(相分离器)并真空浓缩。使粗物质吸附在二氧化硅上并通过正相色谱10%至40%丙酮/dcm纯化,并将所得固体从etoac中结晶以得到为无色结晶固体的n-[1-(环丙基甲基)咪唑-4-基]-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(130mg,51%)。uplc(方法a)3.05分钟,98%,[m h]
=357.2。实施例284-甲基-3-[2-(3-吡啶基)乙炔基]-n-[1-(2,2,2-三氟乙基)咪唑-4-基]苯甲酰胺
[0001]
向在thf(5ml)中的中间体2(118mg,0.50mmol,1.0当量)的混悬液添加三乙胺(207μl,1.49mmol,3.0当量)并将反应物在室温下搅拌10分钟,然后添加1-(2,2,2-三氟乙基)-1h-咪唑-4-胺盐酸盐(100mg,0.50mmol,1.0当量)和(苯并三唑-1-基-氧基三吡咯烷基六氟磷酸盐/酯)(284mg,0.55mmol,1.1当量)并再在室温下搅拌7小时。然后向反应物添加1-(2,2,2-三氟乙基)-1h-咪唑-4-胺盐酸盐(10mg,0.05mmol,0.1当量)和三乙胺(69μl,0.50mmol,1当量),并将反应物在室温下再搅拌18小时。将反应物用水(40ml)淬灭,用dcm(3
×
20ml)萃取并将合并的有机物用1m氨(40ml)、水(40ml)和盐水(40ml)洗涤、干燥(相分离
器)并真空浓缩。将粗物质通过正相色谱用4%meoh/dcm纯化,并将固体从热etoac中结晶过夜。将所得晶体用etoac(
×
2)洗涤并真空干燥以得到为含有吡咯烷膦氧化物之无色固体的标题化合物。将母液吸收在二氧化硅上并通过正相色谱用dcm/丙酮9/1至6/4纯化。收集正确的级分并使其干燥以得到为无色固体的4-甲基-3-[2-(3-吡啶基)乙炔基]-n-[1-(2,2,2-三氟乙基)咪唑-4-基]苯甲酰胺(64mg,34%)。uplc(方法a)3.04分钟,98%,[m h]
=385.2。实施例294-甲基-3-[2-(3-吡啶基)乙炔基]-n-[5-(三氟甲基)-6,7-二氢-5h-吡咯并[1,2-a]咪唑-2-基]苯甲酰胺将2-溴-5-(三氟甲基)-6,7-二氢-5h-吡咯并[1,2-a]咪唑中间体11(160mg,0.63mmol,1.0当量)、中间体12(148mg,0.63mmol,1.0当量)、碳酸铯(306mg,0.94mmol,1.5当量)、碘化亚铜(i)(120mg,0.63mmol,1.0当量)和n,n
′‑
二甲基亚乙基二胺(55mg,0.63mmol,1.0当量)合并于二氧六环(4ml)及dmso(1ml)中,将反应物用n2脱气5分钟且随后在密封管中在100℃下加热4小时。将混合物冷却、在减压下蒸发并将剩余物在h2o(10ml)和etoac(2
×
15ml)之间分配。将合并的有机物干燥(mgso4)、真空浓缩并将剩余物通过正相色谱(干装载的)0.2∶2∶98至0.5∶5∶95 nh3∶meoh∶dcm纯化,并用乙醚研磨,以得到为白色固体的标题化合物(49mg,19%)。uplc(方法a):3.12分钟,100%,[m h]
=411,[m-h]-409。实施例304-甲基-3-[2-(3-吡啶基)乙炔基]-n-螺[6,7-二氢吡咯并[1,2-a]咪唑-5,1
′‑
环丙烷]-2-基-苯甲酰胺将在脱气dmso(1.5ml)和脱气二氧六环(3.5ml)中的中间体15(90mg,0.42mmol,1.0当量)的溶液进一步用氮气脱气约10分钟。向溶液添加碳酸铯(208mg,0.63mmol,1.5当量)、碘化亚铜(i)(81mg,0.42mmol,1.0当量)、n,n
′‑
二甲基亚乙基二胺(55μl,0.51mmol,1.2当量)和中间体12(100mg,0.42mmol,1.0当量),并将混合物加热至95℃持续18小时。将反应物冷却至室温并真空去除挥发物。将剩余物用h2o(20ml)和dcm/异丙醇(9∶1,70ml)稀释,用(15%溶液)nh4oh碱化(至ph=10),分离各相并将水相用dcm/异丙醇(9∶1,2
×
70ml)
a]吡啶-2-基]苯甲酰胺向在1,4-二氧六环/dmso(12ml,3∶1)中的中间体108(250mg,0.93mmol,1.0当量)、碳酸铯(454mg,1.39mmol,1.5当量)、碘化亚铜(i)(177mg,0.93mmol,1.0当量)和反式n,n
′‑
二甲基环己烷-1,2-二胺(0.18ml,1.11mmol,1.2当量)的脱气溶液添加中间体12(220mg,0.93mmol,1.0当量),并将反应物在90℃下加热2小时。然后向反应物添加另一份的碘化亚铜(i)(0.5当量)和反式n,n
′‑
二甲基环己烷-1,2-二胺(0.5当量),并在90℃下继续加热另外1小时。添加另一份碘化亚铜(0.2当量)和反式n,n
′‑
二甲基环己烷-1,2-二胺(0.2当量),并将反应物在90℃下加热另外3小时。将反应混合物在h2o(50ml)和tbme(50ml)之间分配并将水层用2-methf(3
×
30ml)萃取。将合并的有机物真空浓缩以得到褐色油状剩余物。将其通过柱色谱(手动柱,正相,p60硅胶40至63μm/230至400目,dcm中的0至2%meoh)纯化以得到120mg褐色油状固体。将其通过柱色谱(biotage isolera,反相,12g,hp-sphere c18 ultra,25μm,在h2o中的35%至60%mecn,两种洗脱剂均含有0.1体积%nh3)进一步纯化。将产物级分真空浓缩以去除溶剂,得到混悬液,将其过滤并用1ml mecn研磨以得到为无色固体的4-甲基-3-[2-(3-吡啶基)乙炔基]-n-[5-(三氟甲基)-5,6,7,8-四氢咪唑并[1,2-a]吡啶-2-基]苯甲酰胺(42mg,11%)。uplc(方法a)3.21分钟,100%,[m h]
=425.2,[m-h]-=423.2。下表中的实施例使用其中列出的一般程序制备。用于每个实施例的胺在随后的表中提及。
实施例38n-(1-叔丁基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺
向微波小瓶中的中间体12(263mg,1.11mmol)添加cs2co3(905mg,2.78mmol)和cui(212mg,1.11mmol),并将气氛吹扫并用n2(
×
3)替代。添加1,4-二氧六环(7.0ml)并将混合物脱气5分钟,然后添加在dmso(1.4ml)中的1-叔丁基-4-碘代-咪唑和1-叔丁基-5-碘代-咪唑的不可分离混合物(中间体110)(278mg,1.11mmol)的溶液,随后添加反式-n,n
′‑
二甲基环己烷-1,2-二胺(175μl,1.11mmol),并将反应物密封并加热至100℃持续2小时。使反应物冷却至室温,用dcm(15ml)稀释并用饱和nh4cl水溶液(10ml)洗涤。将水相用dcm(3
×
10ml)萃取三次。将合并的有机相用na2so4干燥,过滤并浓缩至干燥。将剩余物通过rp色谱([biotage系统,30gc18筒,装载于dmso中]0.1%nh3/mecn5%至80%)纯化并将产物冻干以获得为白色固体的n-(1-叔丁基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(135mg,33%)。uplc(方法a)3.15分钟,99%,[m h]
=359.3。结构通过noesy分析证实,在咪唑和tbu上的2-和5-质子之间具有明显的相互作用。实施例39n-(5,5-二甲基-6,8-二氢咪唑并[2,1-c][1,4]嗪-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺在室温下在n2下向在1,4-二%氧六环(9.0ml)和dmso(3.0ml)中的中间体55(307mg,1.33mmol)和cs2co3(649mg,1.99mmol)的脱气搅拌溶液分批添加碘化亚铜(i)(253mg,1.33mmol)并且逐滴添加反式n,n
′‑
二甲基环己烷-1,2-二胺(251μl,1.59mmol)。然后添加中间体12(314mg,1.33mmol)并将反应物在90℃下搅拌2小时。将反应物冷却、浓缩、用饱和nh4cl(20ml)稀释并用1∶4 ipa∶dcm(7
×
10ml)萃取。将合并的有机层用mgso4干燥,过滤并浓缩成粗制蓝色/黑色油状物(5.50g)。使粗物质通过二氧化硅塞(在etoac中的0至20%meoh)以得到绿色/褐色油状物(910mg)。将其通过反相柱色谱(biotage isolera,反相,[60g],hp-sphere c18 ultra,25μm[装载于dmso中的剩余物],在h2o中的0至45%mecn,两种洗脱剂均含有0.1体积%nh3)进一步纯化以得到为米色固体的标题化合物(130mg,25%)。uplc(方法a)3.01分钟,100%,[m h]
=387.3。实施例40n-(6,6-二甲基-5,7-二氢吡咯并[1,2-c]咪唑-1-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺在微波小瓶添加中间体12(45.1mg,0.19mmol)、cs2co3(155.0mg,0.48mmol)和cui
(36.3mg,0.19mmol),并将气氛吹扫并用n2(
×
3)替代。添加1,4-二氧六环(1.5ml)并将混合物脱气5分钟,然后添加在dmso(0.4ml)中的中间体58(50.0mg,0.19mmol)的溶液,然后添加反式n,n
′‑
二甲基环己烷-1,2-二胺(30.1μl,0.19mmol)。将反应物密封并加热至100℃持续2小时。使反应物冷却至室温,用dcm(15ml)稀释并用饱和nh4cl水溶液(10ml)洗涤。将水相用dcm(3
×
10ml)萃取并将合并的有机物用na2so4干燥,过滤并浓缩至干燥。将剩余物通过反相色谱([biotage系统,30g c18滤筒,装载于dmso中]0.1%nh3/mecn 5%至80%)纯化。将物质通过制备型hplc(phenomenex luna c18,5μm,mecn/h2o65/35等度,5分钟运行)进一步纯化,并将产物冷冻干燥以得到n-(6,6-二甲基-5,7-二氢吡咯并[1,2-c]咪唑-1-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(5.7mg,8.0%)。uplc(方法a)3.11分钟,99.3%,[m h]
=371.3。实施例414-甲基-3-[2-(3.吡啶基)乙炔基]-n.螺[5,6-二氢吡咯并[1,2-b]吡唑-4,1
′‑
环丁烷]-2-基-苯甲酰胺向在dmf(3.0ml)中的中间体2(145mg,0.61mmol)和中间体61(使用粗物质,假定50%,200mg,0.61mmol)的溶液添加pybop(478mg,0.92mmol),然后添加dipea(320μl,1.84mmol)。将反应混合物在室温下搅拌1小时,然后浓缩并通过柱色谱(biotage isolera,反相,30g,hp-sphere c18 ultra,25μm[剩余物装载于dmf中],h2o中的0%至70%meoh,两种洗脱剂均含有0.1体积%nh3)纯化,以得到含有芳族杂质的粗产物(35.0mg),通过1h nmr观察。将粗制混合物用tbme/mecn(9∶1)研磨,以得到4-甲基-3-[2-(3-吡啶基)乙炔基]-n-螺[5,6-二氢吡咯并[1,2-b]吡唑-4,1
′‑
环丁烷]-2-基-苯甲酰胺(14.7mg,6%)。uplc(方法a)3.40分钟,100%,[m h]
=383.3。实施例42n-(1-环戊基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺在室温下将在1,4-二氧六环(9.0ml)和dmso(3.0ml)中的中间体62(300mg,1.15mmol)和碳酸铯(560mg,1.72mmol)的经n2吹扫的搅拌溶液脱气15分钟,然后分批添加碘化亚铜(i)(220mg,1.14mmol)并逐滴添加反式n,n
′‑
二甲基环己烷-1,2-二胺(220μl,1.37mmol)。然后添加中间体12(270mg,1.14mmol)并将反应物在90℃下搅拌6小时。然后将反应物用碘化亚铜(i)(110mg,0.57mmol)和反式n,n
′‑
二甲基环己烷-1,2-二胺(110μl,0.69mmol)再处理并再搅拌16小时。将反应物冷却、浓缩并将所得剩余物用饱和nh4cl(10ml)稀释并用1∶3 ipa∶dcm(5
×
10ml)萃取。将合并的有机层用mgso4干燥,过滤并浓缩以
得到为蓝色/黑色油状物的粗产物(3.01g)。使粗产物通过二氧化硅塞(用etoac中的0至20%meoh洗脱),以得到半纯化的产物(1.30g)。通过反相色谱(biotage isolera,反相,[30g],hp-sphere c18 ultra,25μm[装载于dmso中的剩余物],在h2o的10%至70%mecn,两种洗脱剂均含有0.1体积%nh3)纯化,以得到为黄色固体的经纯化产物(45mg)。将产物用tbme(10ml)研磨并过滤,以得到为白色固体的标题化合物(25mg,6%)。uplc(方法a)3.24分钟,99%,[m h]
=371.3。实施例44n-(5,5-二甲基-6,7-二氢吡咯并[1,2-a]咪唑-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺在n2气氛下向中间体12(1.33g,5.63mmol)、cui(1.07g,5.63mmol)和cs2co3(4.58g,14.1mmol)添加1,4-二氧六环(56ml)并将混合物脱气5分钟,然后添加在dmso(14ml)中的中间体65(1.21g,5.63mmol)的溶液,然后添加反式n,n
′‑
二甲基环己烷-1,2-二胺(0.89ml,5.63mmol)。将反应物加热至100℃持续2小时,使其冷却至室温并通过硅藻土过滤。将滤液真空浓缩,用dcm(300ml)稀释,用nh4cl饱和水溶液(3
×
50ml)洗涤,用na2so4干燥,过滤并真空浓缩。将剩余物通过rp色谱([biotage系统,c18 30g滤筒,装载于dmso中]0.1%nh3水溶液/mecn10%至80%)纯化并且将所期望的产物冷冻干燥,以得到为无色固体的n-(5,5-二甲基-6,7-二氢吡咯并[1,2-a]咪唑-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(812mg,38%)。uplc(方法a)3.11分钟,99%,[m h]
=371.3。实施例56n-(5-异丙基-6,7-二氢-5h-吡咯并[1,2-a]咪唑-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺将中间体68(150mg,0.65mmol,1.0当量)、中间体12(155mg,0.65mmol,1.0当量)、碘化亚铜(i)(125mg,0.65mmol,1.0当量)、n,n
′‑
二甲基亚乙基二胺(58mg,0.65mmol,1.0当量)和碳酸铯(320mg,0.98mmol,1.5当量)混悬于1,4-二氧六环(3ml)和dmso(1ml)中,用n2脱气、密封且在100℃下加热4小时。添加另一份的碘化亚铜(i)(125mg,0.65mmol,1.0当量)和n,n
′‑
二甲基亚乙基二胺(58mg,0.65mmol,1.0当量)并将反应物加热至100℃持续3小时。将反应物冷却至室温并真空去除挥发物。将剩余物在mecn(25ml)中搅拌并过滤。将固体在2m hcl(25ml)中搅拌,过滤,将滤液用固体k2co3碱化,用etoac(2
×
25ml)萃取、干燥(mgso4)并真空浓缩。将剩余物用叔丁基甲基醚研磨,然后用etoac再研磨,并将固体从etoac中重结晶以得到为白色固体的标题化合物(11mg,4%)。uplc(方法a):3.26分钟,99%,[m h] =385.3。
实施例57n-(5-异丙基-6,8-二氢-5h-咪唑并[2,1-c][1,4]嗪-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺将在二氧六环∶dmso(3ml,2∶1)中的中间体69(约20%w/w,282mg,0.23mmol,1.0当量)、中间体12(82mg,0.35mmol,1.5当量),cui(66mg,0.35mmol,1.5当量)、n,n
′‑
二甲基亚乙基二胺(37μl,0.35mmol,1.5当量)和碳酸铯(150mg,0.46mmol,2.0当量)的混合物在95℃下在密封小瓶中加热16小时。添加另外1当量的cui和n,n
′‑
二甲基亚乙基二胺,并将反应物在密封小瓶中在110℃下加热3小时。将反应物真空浓缩并在h2o(20ml)和叔丁基甲基醚(20ml)之间分配,并过滤固体物质。将固体和有机层合并,吸附到二氧化硅上并通过正相色谱1%至3%meoh/dcm,然后通过反相色谱20%至80%meoh/h2o(0.1%nh3改性剂)纯化,以得到标题化合物(4mg,4%),在冷冻干燥之后为橙色固体。uplc(方法a):3.15分钟,100%,[m h]
=401.3。实施例604-甲基-n-(5-甲基-6,7-二氢-5h-吡咯并[1,2-c]咪唑-1-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺将在二氧六环(6ml)中的中间体12(70mg,0.3mmol,1当量)、中间体75(73mg,0.3mmol,1当量)、cui(56mg,0.3mmol,1当量)、n,n
′‑
二甲基亚乙基二胺(26mg,0.3mmol,1当量)和碳酸铯(193mg,0.6mmol,2当量)的混悬液用n2脱气且在密封小瓶中在100℃下加热72小时。将反应物冷却至室温并真空浓缩。将剩余物溶解于0.5∶5∶95 nh3/meoh/dcm中,通过硅藻土垫过滤并真空浓缩。将剩余物溶解于etoac中,通过过滤去除固体并将滤液真空浓缩。将粗物质通过正相色谱0.2∶2∶98至0.5∶5∶95 nh3/meoh/dcm纯化,以得到为白色固体的标题化合物(16mg,15%)。lcms(方法d):2.50分钟,98%,[m h]
=357.1。实施例634-甲基-3-[2-(3-吡啶基)乙炔基]-n-螺[5,6-二氢吡咯并[1,2-b]吡唑-4,1
′‑
环丙烷]-2-基-苯甲酰胺
向在dmf(2.0ml)中的中间体2(73.0mg,0.31mmol)和中间体78(46.0mg,0.31mmol)的溶液添加pybop(241.0mg,0.46mmol),然后添加dipea(161μl,0.92mmol)。将所得溶液在室温下搅拌1小时,并随后真空浓缩。将粗剩余物通过柱色谱(biotage isolera,反相,12g,hp-sphere c18 ultra,25μm[装载于dmso中的剩余物],在h2o中的5%至60%mecn,两种洗脱剂均含有0.1体积%nh3)纯化。将所得油状物在ch3cn中(用超声处理)研磨。将所得固体过滤并真空干燥,以得到为灰白色固体的4-甲基-3-[2-(3-吡啶基)乙炔基]-n-螺[5,6-二氢吡咯并[1,2-b]吡唑-4,1
′‑
环丙烷]-2-基-苯甲酰胺(50.8mg,44%)。uplc(方法a)3.17分钟,99%,[m h]
=369.3。实施例64n-(1-异丁基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺在n2气氛下,向中间体12(180mg,0.76mmol,1.0当量)、碳酸铯(373mg,1.14mmol,1.5当量)和碘化亚铜(i)(145mg,0.76mmol,1.0当量)添加二氧六环(8ml),并使n2鼓泡通过反应混合物5分钟。向混合物添加在dmso(2ml)中的4-碘代-1-异丁基-咪唑中间体79(191mg,0.76mmol,1.0当量)的溶液,然后添加n,n
′‑
二甲基亚乙基二胺(99μl,0.92mmol,1.2当量)。将小瓶密封并在95℃下加热过夜。将反应物冷却至室温,用dcm(50ml)和饱和nh4cl水溶液(20ml)稀释,分离各相,并将水相用dcm(3
×
30ml)萃取。将合并的有机物用na2so4干燥并真空浓缩。将剩余物通过正相色谱(kp-nh)0至30%丙酮/甲苯纯化,并将所得固体在mecn(1ml)中研磨以得到为白色固体的标题化合物(50mg,18%)。uplc(方法a)3.19分钟,100%,[m h]
=359.3。实施例65n-(4-乙基-5,6-二氢-4h-吡咯并[1,2-b]吡唑-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺向在thf(6.6ml)中的中间体82(300mg,1.98mmol)、中间体2(471mg,1.98mmol)和tea(1.11ml,7.94mmol)的溶液添加hatu(1.13g,2.98mmol),并将反应物在室温下在n2下搅拌过夜。将反应物用盐水(10ml)淬灭、用etoac(10ml)稀释并分离各相。将水相用etoac(2
×
20ml)萃取,将合并的有机物用h2o(10ml)洗涤、用mgso4干燥并真空浓缩。将粗物质通过柱色谱(biotage isolera,反相,[60g],hp-sphere c18 ultra,25μm[装载于dmso中的剩余物],
5%至100%mecn/h2o,两种洗脱剂均含有0.1体积%nh3)纯化并通过柱色谱(biotage isolera,正相,[25g],kp-nh 40至63μm/230至400目,装载于dcm中的剩余物,0至50%etoac/庚烷)再纯化,以得到为无色固体的n-(4-乙基-5,6-二氢-4h-吡咯并[1,2-b]吡唑-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(62.7mg,8.5%)。lcms(方法d)2.78分钟,100%,[m h]
=371.1。实施例674-甲基-n-(5-甲基-5,6,7,8-四氢咪唑并[1,2-a]吡啶-2-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺将在dmf(1.5ml)中的中间体2(106mg,0.45mmol,1.0当量)、苯并三唑-1-基-氧基三吡咯烷基六氟磷酸盐/酯(257mg,0.49mmol,1.1当量)和n,n-二异丙基乙基胺(120μl,0.67mmol,1.5当量)的混悬液在室温下搅拌5分钟,并将所得溶液转移至在dmf(1.5ml)中的中间体90(68mg,0.45mmol,1.0当量)的混悬液中,并将反应混合物在室温下搅拌18小时。将反应物通过倒入h2o(10ml)中淬灭,用dcm(10ml)稀释,分离各相并用dcm(3
×
10ml)萃取水相。将合并的有机物干燥(na2so4)并真空浓缩。将粗物质通过正相色谱(kp-nh)50%丙酮/dcm纯化,随后通过在15%至100%mecn/h2o(0.1%nh3改性剂)中的反相色谱纯化,以得到为浅黄色固体的标题化合物(7.3mg,4%)。uplc(方法a):3.10分钟,96%,[m h]
=371。实施例69n-(1-环丁基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺在n2下向在dmf(2ml)中的中间体2(200mg,0.84mmol)的混悬液添加dipea(294μl,1.69mmol)。向所得澄清溶液添加1-丙基膦酸环酐(1.00ml,1.68mmol),并将反应物在室温下搅拌5分钟,然后添加1-环丁基咪唑-4-胺盐酸盐(220mg,1.26mmol)和dipea(294μl,1.69mmol)为dmf溶液(2ml)。将反应物在60℃下加热24小时。将反应物冷却至室温并用dcm(10ml)稀释、用磷酸盐缓冲液(ph7,3
×
5.0ml)洗涤、干燥(na2so4)并真空浓缩。将剩余物通过柱色谱(biotage isolera,反相,12g,hp-sphere c18 ultra,25μm,h2o中的10%至100%mecn,两种洗脱剂均含有0.1体积%nh3)纯化,以得到n-(1-环丁基咪唑-4-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(28.7mg,96.3%),在冷冻干燥之后为白色固体。uplc(方法g)3.10分钟,96.9%,[m h]
=357.3。实施例78n-[1-(2-氟乙基)咪唑-4-基]-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺
在n2下向在1,4-二氧六环(8ml)中的中间体12(196mg,0.83mmol,1.0当量)、碘化亚铜(i)(159mg,0.83mmol,1.0当量)和碳酸铯(407mg,1.25mmol,1.5当量)的脱气混悬液添加在dmso(2ml)中的中间体104a及104b的不可分离异构体混合物(200mg,0.83mmol,1.0当量)的溶液,然后添加n,n
′‑
二甲基亚乙基二胺(0.11ml,1.00mmol,1.2当量)并将整个混合物再脱气5分钟,然后在95℃下加热3小时。将反应物冷却至室温,用dcm(50ml)和饱和nh4cl水溶液(50ml)稀释,分离各相,并将水相用dcm(2
×
50ml)萃取。将合并的有机物用盐水(50ml)洗涤、干燥(mgso4)并真空浓缩。将剩余物通过正相色谱,5%meoh/etoac纯化,并将所得固体用etoac(5ml)研磨以得到为无色固体的标题化合物(84mg,29%)。uplc(方法a):2.82分钟,100%,[m h]
=349.2。实施例824-甲基-n-(5-甲基-6,7-氢-5h-吡咯并[1,2-a]咪唑-2-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺在小瓶中合并中间体103(40mg,0.20mmol,1.00当量)、碳酸铯(130mg,0.40mmol,2.00当量)和碘化亚铜(i)(2mg,0.01mmol,0.05当量),接着合并在无水二氧六环(0.5ml)中的n,n
′‑
二甲基亚乙基二胺(4mg,0.04mmol,0.20当量)的溶液和在无水二氧六环(2ml)中的中间体12(47mg,0.20mmol,1.00当量)的溶液,并将小瓶密封并在100℃下加热过夜。将另一份的碘化亚铜(i)(2mg,0.01mmol,0.05当量)和n,n
′‑
二甲基亚乙基二胺(4mg,0.04mmol,0.20当量)添加到反应物,将其加热另外24小时。然后将反应物冷却至室温,用etoac(10ml)、h2o(2ml)和35%氨水溶液(1ml)稀释,并分离各相。将水相用etoac(15ml)萃取,将合并的有机物用水/氨溶液(35%水溶液)、14∶1(15ml)和饱和nh4cl(10ml)洗涤,干燥(na2so4)并真空浓缩。将粗物质通过反相色谱20%至95%mecn/h2o(0.1%nh3改性剂)纯化以得到为白色固体的标题化合物(14mg,20%)。uplc(方法a):2.99分钟,98%,[m h]
=357.3。实施例884-甲基-n-(4-氧代-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺
向在dcm(4ml)中的中间体100(80mg,0.53mmol,1.0当量)和中间体2(126mg,0.53mmol,1.0当量)的混悬液添加(1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶3-氧化物六氟磷酸盐/酯(242mg,0.63mmol,1.2当量)、n,n-二异丙基乙基胺(0.28ml,1.59mmol,3.0当量)和dmf(4ml),并将反应物在室温下搅拌72小时。将混合物通过添加饱和nh4cl水溶液(20ml)淬灭,并用dcm(3
×
15ml)萃取。将合并的有机物用饱和nh4cl水溶液(2
×
20ml)洗涤、干燥(na2so4,随后相分离器)并真空浓缩。将粗剩余物通过反相色谱20%至95%mecn/h2o(0.1%nh3改性剂)纯化,以得到标题化合物(67mg,34%),在冷冻干燥之后为白色固体。uplc(方法a)3.23分钟,97%,[m h]
=371.2。实施例894-甲基-n-(5-甲基-6,8-二氢-sh-咪唑并[2,1-c][1,4]嗪-2-基)-3-[2-(3-吡啶基)乙炔基]苯甲酰胺向含有碘化亚铜(i)(17.6mg,0.09mmol,0.2当量)和碳酸铯(300.2mg,0.92mmol,2.0当量)的小瓶添加n,n-二甲基亚乙基二胺(40μl,0.37mmol,0.8当量)、中间体95(100.0mg,0.46mmol,1.0当量)和脱气二氧六环(0.25ml),接着一次性添加中间体12(108.9mg,0.46mmol,1.0当量),用脱气二氧六环(0.25ml)冲洗烧瓶,将反应物密封并在100℃下通过微波辐照加热1小时。将另一份碘化亚铜(i)(17.6mg,0.09mmol,0.2当量)和n,n-二甲基亚乙基二胺(40μl,0.37mmol,0.8当量)与二氧六环(0.25ml)和dmso(0.25ml)一起添加,并将反应物在120℃下通过微波辐照加热3小时。将反应物用水(20ml)和etoac(20ml)稀释并用15%nh4oh溶液碱化至ph 10。分离各相,将水相用etoac(2
×
20ml)萃取,并将合并的有机物用盐水(50ml)洗涤、干燥(na2so4)并真空浓缩。将粗剩余物通过反相色谱10%至70%mecn/h2o(0.1%nh3改性剂)纯化,以得到标题化合物(9.5mg,5%),在冷冻干燥之后为米色固体。uplc(方法a)2.89分钟,95%,[m h]
=373.3。实施例94n-[1-(环丁基甲基)咪唑-4-基]-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺
[0002]
在微波小瓶中添加中间体12(180mg,0.76mmol,1.0当量),cs2co3(373mg,1.14mmol,1.5当量)和cui(145mg,0.76mmol,1.0当量),并将气氛吹扫并用n2(
×
3)替代。向混合物添加二氧六环(8ml),并将n2鼓泡通过反应混合物5分钟,然后添加在dmso(2ml)中的中间体19(200mg,0.76mmol,1.0当量)的溶液,接着添加dmeda(99μl,0.92mmol,1.2当量),并将反应容器密封并加热至95℃过夜。使反应物冷却至室温并用dcm(100ml)和饱和nh4cl水溶液(50ml)稀释。分离各相并将水相用dcm(3
×
30ml)萃取。将合并的有机物用na2so4干燥,真空浓缩并将剩余物通过反相柱色谱(biotage isolera,mecn∶h2o(0.1%nh3),10cv内1∶19至4∶1)纯化,以得到标题化合物(30.8mg,11%),冷冻干燥之后为白色固体。uplc(方法a)3.26分钟,99%,[m h]
=371.3。实施例954-甲基-3-[2-(3-吡啶基)乙炔基]-n-螺[6,7-二氢-5h-吡唑并[1,5-a]吡啶-4,1
′‑
环丙烷]-2-基-苯甲酰胺在室温下向在dmf(2.0ml)中的4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酸中间体2(116mg,0.49mmol)、中间体23(80mg,0.49mmol)和n,n-二异丙基乙基胺(256μl,1.47mmol)的溶液添加(苯并三唑-1-基氧基)三吡咯烷基六氟磷酸盐/酯(383mg,0.74mmol),并将反应物在室温下搅拌4小时。将反应溶液装载到反相滤筒上并通过反相柱色谱(biotage isolera,反相,30g,hp-sphere c18 ultra,25μm,在h2o中的5%至80%mecn,两种洗脱剂均含有0.1体积%nh3)纯化。将含有产物的级分真空浓缩并冷冻干燥过夜,以得到为灰白色固体的标题产物(50.8mg,26.9%)。uplc(方法a)3.53分钟,99.3%,[m h]
=383.3。实施例96n-(6,6-二甲基-5,8-二氢咪唑并[2,1-c][1,4]嗪-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺将在1,4-二氧六环(2.4ml)和dmso(0.6ml)中的中间体29(70.0mg,0.30mmol)、cs2co3(148.0mg,0.45mmol)和中间体12(71.6mg,0.30mmol)的n2吹扫的搅拌溶液脱气10分钟。然后添加cui(57.7mg,0.30mmol)和反式n,n
′‑
二甲基环己烷-1,2-二胺(57.3μl,0.36mmol)并将反应物在95℃下搅拌2小时。将反应物再搅拌1.5小时,并随后真空浓缩。将
油状物溶解于饱和nh4cl溶液(10ml)中并用dcm∶ipa,4∶1(6
×
5ml)萃取。将合并的有机物干燥(mgso4)并真空浓缩,以得到粗制深绿色油状物(890mg)。将剩余物通过柱色谱(biotage isolera,反相,30g,hp-sphere c18 ultra,25μm,在h2o中的10%至70%mecn,两种洗脱剂均含有0.1体积%nh3)纯化,以得到为灰白色固体的标题化合物(10.8mg,9.0%)。uplc(方法a)3.06分钟,97%,[m h]
=387.3。实施例97n-(4,4-二氟-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺向在dmf(3ml)中的中间体37(106mg,0.61mmol,1当量)的溶液添加中间体2(145mg,0.61mmol,1当量)、et3n(256μl,1.84mmol,3当量)和pybop(318mg,0.61mmol,1当量),并将反应物在室温下搅拌过夜。将所得橙色反应混合物真空浓缩,并将所得油状物溶解于dcm(30ml)中,用h2o(10ml)洗涤,并将有机物干燥(na2so4)、过滤,并真空去除溶剂以得到为橙色油状物的粗产物。将其通过biotage isolera(30g c18柱)mecn∶h2o,1∶19(3cv)然后mecn∶h2o,35∶65至80∶20(25cv内)与0.1%nh3和流量=25ml/分钟纯化,以得到为白色固体的标题化合物(95mg,40%)。uplc(方法a)3.28分钟,98%,[m h]
=393。实施例104n-(4,4-二氟-3-甲基-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺在室温下向在无水thf(3.0ml)中的中间体112(60.0mg,0.32mmol)和中间体1(101.0mg,0.40mmol)的溶液逐滴添加叔丁氧基钾(0.71ml,1.21mmol,1.7m于thf中)。在室温下搅拌所得橙色混悬液1.5小时。向反应混合物添加nh4cl水溶液(10ml),将产物用etoac(2
×
15ml)萃取并用水(15ml)洗涤,用mgso4干燥,过滤,吸收到二氧化硅上并通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,)用dcm-etoac 1∶1洗脱进行纯化。分离为白色固体的产物n-(4,4-二氟-3-甲基-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(23.9mg,18%)。uplc(方法e)3.17分钟,99%,[m h]
=407.2。实施例105n-(3-乙基-4,4-二氟-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-基)-4-甲基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺
向在thf(5.0ml)中的中间体1(66.8mg,0.27mmol)和中间体115(54.0mg,0.24mmol)的溶液逐滴添加kotbu(在thf中的1.7m溶液,0.57ml,0.97mmol)并且将混合物在氮气下在室温下搅拌0.5小时。将反应物用水(10ml)淬灭并用etoac(3
×
10ml)萃取。将合并的有机物用盐水(10ml)洗涤,用na2so4干燥并浓缩以得到粗产物。将粗产物通过柱色谱(biotage isolera,反相,40g,siliasep,在h2o中的20%至80%mecn,两种洗脱剂均含有0.1体积%nh3)进一步纯化。将含有产物的级分浓缩,并将剩余物通过柱色谱(手动柱,正相,硅胶40至63μm/230至400目,在庚烷中的20%至80%etoac)进一步纯化,以得到所期望产物n-(3-乙基-4,4-二氟-6,7-二氢-5h-吡唑并[1,5-a]吡啶-2-基)-4-甲基-3-[2.(3-吡啶基)乙炔基]苯甲酰胺(26.0mg,25%),冷冻干燥之后为无色固体。uplc(方法a)3.42分钟,97.5%,es
:421.2[m h]
。实施例1064-甲基-n-[1-[2-(4-甲基哌嗪-1-基)乙基]-5-(三氟甲基)吡唑-3-基]-3-[2-(3-吡啶基)乙炔基]苯甲酰胺向在thf(4.0ml)中的中间体1(113mg,0.45mmol)和中间体116(100mg,0.36mmol)的溶液逐滴添加kotbu(在thf中的20%w/w溶液,0.80ml,1.44mmol),并将混合物在室温下搅拌0.5小时。将反应混合物通过添加水(20ml)和饱和盐水溶液(20ml)淬灭并用tbme(2
×
30ml)萃取。将合并的有机层用10%k2co3溶液(20ml)洗涤、用mgso4干燥并浓缩,以得到橙色油状剩余物。将剩余物通过柱色谱(biotage isolera,反相,12g,sfar c18d duo30μm,在h2o中的55%至100%mecn,两种洗脱剂均含有0.1体积%nh3)纯化,以得到4-甲基-n-[1-[2-(4-甲基哌嗪-1-基)乙基]-5-(三氟甲基)吡唑-3-基]-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(23.0mg,13%),冷冻干燥之后为无色固体。uplc(方法a)3.34分钟,99.8%,es
:497.3[m h]
。比较例1n-(5-叔丁基异唑-3-基)-3-(2-咪唑并[1,2-a]吡啶-3-基乙炔基)-4-甲基-苯甲酰胺
将在mecn(7.0ml)中的中间体18(280mg,0.70mmol,1当量)、中间体17(100mg,0.7mmol,1当量),双(三苯基膦)二氯化钯(ii)(12.3mg,17.6μmol,0.025当量)、碘化亚铜(i)(4.7mg,24.6μmol,0.035当量)和tea(118μl,844μmol,1.2当量)的溶液在回流下加热2小时。将反应混合物过滤并将滤饼用mecn(20ml)和dcm(20ml)洗涤并将合并的有机物真空浓缩。将剩余物通过柱色谱(正相,[24g],redisep硅胶,35至60μm(230至400目),35ml/分钟,梯度在异己烷中的0%至100%etoac)纯化。将产物在真空烘箱中在50℃下干燥过夜,以得到为黄色固体的n-(5-叔丁基异唑-3-基)-3-(2-咪唑并[1,2-a]吡啶-3-基乙炔基)-4-甲基-苯甲酰胺(179mg,64%)。lcms(方法c)5.50分钟,100%,[m h]
=399.1。比较例23-(咪唑并[1,2-b]哒嗪-3-基乙炔基)-4-甲基-n-[4-[(4-甲基-1-哌嗪基)甲基]-3-(三氟甲基)苯基]苯甲酰胺(帕纳替尼(ponatinib),cas:943319-70-8)帕纳替尼商购自ak scientific,inc.。比较例34-甲基-n-[3-(4-甲基咪唑-1-基)-5-(三氟甲基)苯基]-3-[(4-吡啶-3-基嘧啶-2-基)氨基]苯甲酰胺(尼洛替尼(nilotinib),cas:641571-10-0)尼洛替尼商购自medchem tronica。比较例43-[2-(3-氨基-6-氟-4-异喹啉基)乙炔基]-4-甲基-n-[1-(2-吗啉乙基)吡唑-3-基]苯甲酰胺将中间体42(185.0mg,0.59mmol,91.7%纯)、中间体40(160.0mg,0.45mmol,96%纯)、et3n(75.9μl,0.54mmol)、双(三苯基膦)二氯化钯(ii)(8.0mg,11.3μmol)和cui(3.0mg,15.9μmol)添加至mecn(5.0ml),并将反应物在82℃下回流加热3小时。将混合物通过硅藻土过滤并真空浓缩。将剩余物通过反相hplc(ace-5aq,100
×
21.2mm,5μm,25ml/分钟,梯度为在10%meoh/水中的0%至100%(在7分钟内)meoh然后是100%(3分钟)meoh)纯化。然后将剩余物通过反相hplc(phenomenex synergi hydro-rp 80a axia,100
×
21.2mm,4μm,25ml/分钟,梯度为在10%meoh/水中的20%至100%(在7分钟内)meoh然后是100%(3分钟)meoh)[1%甲酸])再纯化。将物质通过用饱和nahco3水溶液处理脱盐并萃取到dcm中并真空浓缩至干燥。然后将剩余物通过反相hplc(ace-5aq,100
×
21.2mm,5μm,25ml/分钟,
梯度0%至100%(在7分钟内),然后在10%meoh/水中的100%(3分钟)meoh)再纯化,然后在真空烘箱中在50℃下干燥过夜,以得到标题化合物(15.5mg,6.8%)为黄色固体。lc-ms(方法h)4.57分钟,98%,[m h]
=498.5。比较例5n-[3,3-二氟-1-(4-甲基哌嗪-1-基)茚满-5-基]-3-(2-咪唑并[1,2-b]哒嗪-3-基乙炔基)-4-甲基-苯甲酰胺向在2-methf(10ml)和dmf(5ml)中的中间体52(80mg,0.30mmol)和中间体45(119mg,0.30mmol,70%纯度)的溶液添加吡啶(100μl,1.20mmol),然后添加1-丙基膦酸环酐(400μl,0.60mmol)。然后将反应混合物在70℃下加热过夜。分别地,向在2-methf(3ml)和dmf(1ml)中的中间体52(23mg,0.09mmol)和中间体45(34mg,0.09mmol,70%纯度)的溶液添加吡啶(30μl,0.34mmol),然后添加1-丙基膦酸环酐(100μl,0.17mmol)。然后将反应混合物在70℃下加热过夜。然后将两种反应物冷却至室温并合并。向所得混合物添加etoac(50ml),然后添加k2co3水溶液(2m,50ml),并分离各相。将水相用etoac(2
×
50ml)萃取并将合并的有机物用盐水(50ml)洗涤、干燥(na2so4)并真空浓缩,以得到为褐色油状物的粗产物。将粗产物在二氧化硅上使用dcm/nh3(meoh中的7m)(99∶1至95∶5)纯化,通过柱色谱(手动柱,正相,kp-nh二氧化硅,dcm中的10%至20%丙酮)进一步纯化,并使用柱色谱(biotage isolera,反相,4g,hp-sphere c18 ultra,25μm[剩余物装载于dmso中],在h2o中的10%至80%thf,两种洗脱剂均含有0.1体积%nh3)再纯化,并将物质冷冻干燥72小时,以得到为灰白色固体的标题化合物(26mg,13%)。uplc(方法a)3.24分钟,99.5%,[m h]
=527.3。比较例6n-[6-(4-异丙基-1,2,4-三唑-3-基)-2-吡啶基]-3-[2-(3-吡啶基)乙炔基]苯甲酰胺向在dcm(3.0ml)中的中间体119(50mg,0.25mmol)和三乙胺(40μl,0.32mmol)的混合物逐滴添加在dcm(2.0ml)中的中间体121(119mg,0.49mmol)的溶液,并将反应混合物在室温下在氮气下搅拌过夜。将反应混合物用水(10ml)淬灭并用dcm(3
×
10ml)萃取。将合并的有机物用盐水(15ml)洗涤、用na2so4干燥并真空浓缩。将剩余物通过柱色谱(biotage isolera,反相,25g,siliasep,在h2o中的20%至80%mecn,两种洗脱剂均含有0.1体积%nh3)纯化,以得到为灰白色固体的所期望的产物n-[6-(4-异丙基-1,2,4-三唑-3-基)-2-吡啶基]-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(8.0mg,7.9%)。uplc(方法a)2.97分钟,99.3%,es
:409.3[m h]
。比较例7n.(5.乙基.1,3,4-噻二唑-2-基)-4-吡唑-1-基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺
在n2气氛下向在dmf(2.0ml)中的中间体124(79.0mg,0.25mmol)和2-氨基-5-乙基-1,3,4-噻二唑(38.0mg,0.29mmol)的溶液添加hatu(120.0mg,0.32mmol)和三乙胺(0.10ml,0.72mmol),并将反应物在室温下搅拌过夜。将反应物用饱和nahco3水溶液(10ml)淬灭,并用etoac(30ml)稀释,分离各相并将水相用etoac(3
×
30ml)萃取。将合并的有机物用na2so4干燥,过滤并浓缩。将粗物质通过阳离子交换树脂scx-2(非封端的丙基磺酸官能化的二氧化硅,50μm,1g)纯化,将剩余物装载于dcm中,用meoh洗涤并用nh3(meoh中的2m)洗脱。将所得固体用水:mbte研磨,收集在玻璃料上并干燥以得到为灰白色固体的n-(5-乙基-1,3,4-噻二唑-2-基)-4-吡唑-1-基-3-[2-(3-吡啶基)乙炔基]苯甲酰胺(41mg,41%)。uplc(方法a)2.02分钟,98.9%,es
:401.2[m h]
。生物学数据ba/f3细胞滴度glo测定细胞滴度-glo发光细胞生存力测定是基于存在的atp的量化的确定培养物中活细胞数的同质性方法。简言之,修饰il-3依赖性ba/f3细胞以表达bcr-abl。转化激酶的活性克服了il3对细胞增殖和存活的依赖性。特异性抑制激酶活性的测试化合物导致程序性细胞死亡,这可以通过添加细胞滴度-glo试剂来测量。在该测定中,表达bcr-abl的ba/f3细胞(高级细胞动力学)或亲本ba/f3(对照)细胞在含有10%fbs、1
×
glutamax和750ng/ml嘌呤霉素的rpmi 1640中以5
×
104/ml制备。将测试化合物使用tecan d300e以10μm的最高最终测定浓度分配到384孔板中,剂量归一化为50μl体积中的0.1%dmso。向制备的384孔板的每个孔添加50μl细胞,并将板以1000rpm旋转1分钟,然后在37℃、5%co2下孵育48小时。在48小时之后,向板的每个孔添加15μl细胞滴度glo试剂。在室温孵育60分钟之后,在pherastar fs读取仪上读取发光。在ba/f3细胞滴度glo测定中测试本发明的示例性化合物,并且pic50数据示于下表中。将pic
50
数据计算为-log
10
(ic
50
,以摩尔计)那些数据表明本发明的化合物可以抑制c-abl。
体内cns渗透测定将300至350g雄性spraguedawley大鼠(charlesriver,uk)在12小时光/暗循环下分组圈养,n=3,可随意获得食物和水。在给药前一天17:00取出所有食物。在给药当天,将动物称重,标记尾巴并通过经口管饲给药体积为5ml/kg中的3mg/kg化合物。在给药之后30分钟、1小时和4小时通过腹膜内施用戊巴比妥(pentobarbital)处死动物。通过心脏穿刺抽取死后血液,并在在冰上短暂储存在k2 edta血液管中,然后在4℃下以14,000g旋转4分钟。将血浆取出至96孔板中,置于干冰上并储存在-80℃下。将脑快速解剖并置于干冰上,然后储存在-80℃下。在将测试化合物(经口)给药于雄性sprague-dawley大鼠之后,在三个时间点处死动物。在心脏放血之后通过离心血液分级从全血中分离血浆并分离全脑。将样品储存在冰上并转移至-80℃下的生物分析实验室储存。如下详述进行血浆和脑样品的生物分析。方法在工业标准文献的指导下制备。
2,3
血浆生物分析使用10mm dmso储备液(stock)在稀释剂(mecn∶h2o,1;1)中在10至100,0000ng/ml的范围内制备测试化合物的添加溶液(spiking solution)。通过将2.5μl校准添加溶液添加到25μl对照血浆中以在1至10000ng/ml的范围内的已知浓度制备在对照雄性sprague-dawley大鼠血浆中的校准线。将实验样品解冻至室温,并使用蛋白质沉淀(在室温用含有25ng/ml甲磺丁脲作为内标的400μl mecn搅拌至少5分钟)沿着校准线萃取25μl等分样品。通过在4℃下以4000rpm离心5分钟将蛋白质沉淀物与萃取的测试化合物分离。将所得上清液用相关稀释剂(例如在h2o中的0.1%甲酸或1∶1 meoh∶h2o)以1∶2的比率稀释。通过uplc-ms/ms在api6500 qtrap或waters tqs质谱仪上使用对测试化合物特异的先前优化的分析mrm(multiple reaction monitoring,多反应监测)方法分析样品。
在对照校准线的两个复制品分析样品之后,确定分离样品中测试化合物的浓度,在使用适当回归和加权的样品组之前和之后注射。校准线仅包含20%预期测试浓度范围内的样品,并且任何超出校准线限制的样品均将被视为低于或高于量化限制(lloq/aloq)。脑生物分析使用10mm dmso储备液在稀释剂(1∶1 mecn∶h2o)中在10至100,0000ng/ml的范围内制备测试化合物的添加溶液。通过将2.5μl校准添加溶液添加到25μl对照匀浆中,以在3至30000ng/g的范围内的已知浓度制备在对照雄性sprague-dawley大鼠脑匀浆中的校准线。为了制备对照和实验脑匀浆,将脑解冻、称重并以每克脑2ml的比率添加一定体积的稀释剂(h2o)。使用precellys evolution和ck147ml小陶瓷珠匀浆管通过珠-搅拌器匀浆进行脑的匀浆。使用蛋白质沉淀(在室温下用含有25ng/ml甲磺丁脲作为内标的400μlmecn搅拌至少5分钟)沿着校准线萃取25μl实验样品的等分样品。通过在4℃下以4000rpm离心5分钟将蛋白质沉淀物与萃取的测试化合物分离。将所得上清液用相关稀释剂(例如在h2o中的0.1%甲酸或1∶1 meoh∶h2o)以1∶2的比率稀释。通过uplc-ms/ms在api6500q trap或waters tqs质谱仪上使用对测试化合物具有特异性的先前优化的分析mrm(多反应监测)方法分析样品。在对照校准线的两个复制品分析样品之后,确定分离样品中测试化合物的浓度,在使用适当回归和加权的样品组之前和之后注射。校准线仅包含20%预期测试浓度范围内的样品,并且任何超出校准线限值的样品均将被视为低于或高于量化限制(lloq/aloq)。确定脑与血浆比率和游离脑浓度。通过将每个时间点脑中的浓度除以血浆中的浓度来计算总cns外显率。平均脑与血浆比率(br∶pl)通过使这些比率平均化(定义使用哪些时间点)来计算。游离药物假说表明只有未结合的化合物能够与药理学作用相互作用并引起药理学作用。因此,期望化合物具有高游离脑浓度。为了计算各基质中的游离浓度,将确定的浓度乘以%游离值,%游离值通过使用快速平衡透析法5的血浆蛋白结合和脑组织结合研究确定。然后将这些值转化为摩尔浓度以给出在每个时间点的纳米摩尔游离结果。7k
pu,u
或k
p,脑
计算为在脑中未结合的游离药物级分与血浆中未结合的游离药物的比率。
4,5,6,7
下表显示本发明化合物在c
max
下的游离脑水平。与比较例相比,本发明的实施例在c
max
下具有极大改善的游离脑水平。
通过肝外cyp1a1形成帕纳替尼的反应性代谢物包括肝毒性在内的严重副作用导致帕纳替尼从市场上短暂撤出。据假设,肝毒性可能是由帕纳替尼的反应性代谢物的形成导致的,这由delin及其同事(2017)1通过肝外cyp1a1介导的环氧化物形成证实。方法对测试化合物形成反应性代谢物的潜力的研究使用de lin et al详述的适合方法进行。简言之,在存在和不存在5mm谷胱甘肽的情况下,在存在重组人p450酶1a1(100nm)的情况下,在含有5mm mgcl2的100mm磷酸盐缓冲液(ph 7.4)中,在37℃下将测试化合物与混合性别人肝细胞溶胶(1mg/ml总蛋白浓度)预孵育5分钟(n=2;50um基质浓度)。在预孵育期结束时,将2mm nadph添加至每个样品并且将混合物在37℃下孵育60分钟。添加nadph和60分钟孵育期之后立即将每种样品的等分样品与mecn(含有内标)混合以终止反应并沉淀蛋白质。与对照bactosome一起孵育也同时进行以证实代谢激活的需要。将样品混合、离心,并在适当稀释之后使用高分辨率质谱仪通过lc-ms分析上清液以进行代谢物鉴定和表征分析。得到的迹线在图1和2中示出。结果
[0003]
在nadph和gsh的存在下,与人肝胞质溶胶和重组cyp1a1孵育之后,帕纳替尼形成显著的直接谷胱甘肽加合物。该代谢物在与对照bactosome孵育之后不形成,表明需要通过cyp1a1的代谢激活(假定通过生物转化为环氧化物)。另外,nadph和gsh都是出现谷胱甘肽加合物形成所需的。在nadph和gsh存在下,将实施例25与人肝胞质溶胶和重组cyp1a1一起孵育之后,没有观察到直接谷胱甘肽缀合的产物,表明实施例25具有显著降低的在体内形成反应性代谢物的风险。两种非常少量的代谢物被鉴定为氧化和谷胱甘肽缀合的产物,然而这些是非常少量的并因此不可能是显著的。这些结果表明,本发明的化合物不形成潜在毒性的反应性代谢物,例如对于帕纳替尼观察到的那些。人iastrocyte-鼠hb9-gfp 运动神经元共培养物材料和方法如前所述,inpc(诱导的神经元祖细胞(induced neuronal progenitor cell))来源于als患者的成纤维细胞(meyer et al.20148)。通过在iasrocyte培养基中培养至少5天使inpc分化成iastrocyte。在hb9运动神经元特异性启动子的存在下表达绿色荧光蛋白(green fluorescent protein,gfp)的鼠运动神经元(从现在开始称为hb9-gfp )通过胚状体(embryoid body,eb)从鼠胚胎干细胞分化,如前所述(haidet-phillips et a1.20119,wichterle et al.200210)。共培养程序:
第0天-inpc分裂和mesc分裂inpc和mesc在同一天分别分裂到iastrocyte培养基和meb培养基中,使得当一起接种在共培养物中时iastrocyte和运动神经元都分化7天。第3天-培养基改变iastrocyte改变iastrocyte上的培养基,并且如果从inpc接种之后3天汇合90%至100%则使用accutase分裂。将iastrocyte在iastrocyte培养基中再放置2天,直到接种到384孔板上。第5天-iastrocyte接种在pbs中以1∶400稀释纤连蛋白,并且每孔添加5μl。将板与纤连蛋白在室温下孵育至少5分钟。将培养基与iastrocyte分离并用pbs洗涤。每10cm板添加1ml accutase,并在37℃下孵育4分钟。轻敲板以去除任何残留的iastrocyte。将iastrocyte重悬于iastro培养基中,并以200
×
g离心4分钟。去除上清液,轻弹falcon以涡旋细胞,并将细胞重悬于适当量的iastrocyte培养基中。使用血细胞计数器计数细胞,并将细胞稀释至适当的稀释度用于接种。将1至2,000个iastrocyte接种在纤连蛋白包被的384孔板上的35μl培养基中。将384孔板以1,760
×
rpm离心60秒,使用pk120(alc)离心(在神经遗传学中)以收集培养基和细胞至孔的底部。将板放置24小时,使iastrocyte贴壁到板上。第6天-药物处理使用echo550液体处理器(labcyte)将药物在100%药物级dmso中递送至iastrocyte培养基。使用pk120(alc)离心机以1,760
×
rpm将384孔板离心60秒。第7天-eb解离和鼠gfp 运动神经元接种将eb的2个板收集在50ml管中并以200
×
g离心4分钟。离心之后将eb与上清液分离,并用10ml pbs洗涤,然后以200
×
g再次离心4分钟,并去除pbs洗液。对于每个50ml管,添加2.75ml eb解离缓冲液,并随后添加250μl 200u/ml(10
×
)木瓜蛋白酶。使用p1000移液管相对于falcon侧将溶液轻轻上下移液10次(不直接移液eb沉淀)。将50ml管置于37℃水浴中并孵育5分钟。每3分钟取出管并轻轻摇动。在5分钟之后,添加另外的2ml eb解离液和100μl 200u/ml(10
×
)木瓜蛋白酶,然后用p1000移液管将其再次移液5次,并再返回水浴中另外5分钟。如前所述将其放回水浴中,并孵育不超过15至20分钟。以300
×
g离心5分钟。每个50ml管制备2.7ml eb解离液,向其添加300μl fbs和150μl 0.5mg/ml dna酶i。从解离的eb中去除上清液,并添加3ml fbs/dna酶i混合物,用p1000移液管上下移液约5次。向含有解离的eb的falcon底部非常缓慢地添加5ml fbs。去除上清液,并将细胞非常温和地重悬于约3ml mn培养基(如果沉淀较大,则使用更多)中,并通过40μm过滤器过滤。添加1ml额外的mn培养基以洗涤过滤器。将2500个鼠hb9-gfp 运动神经元接种在每孔中预处理的iastrocyte上部的10μl运动神经元培养基中。使用pk120(alc)离心机以1,760
×
rpm将384孔板离心60秒。第8天每孔添加15μl运动神经元培养基。使用incell分析仪2000(ge healthcare)对hb9-gfp 运动神经元成像-共培养的第1天。第9天使用incell分析仪2000(ge healthcare)对hb9-gfp 运动神经元成像-共培养的第2天(在这一天成像是任选的)。
第10天使用incell分析仪2000(ge healthcare)对hb9-gfp 运动神经元成像-共培养的第3天。运动神经元生存力评估:使用columbus分析仪软件计数72小时之后存活的活运动神经元(限定为具有至少1个轴突的gfp 运动神经元)的数目。结果图3中的结果显示实施例10、17、24、25和26在与患者来源的iastrocyte共培养中拯救运动神经元存活。该作用是剂量依赖性的,最大响应接近阳性对照(1μm尼洛替尼)的最大响应并且最小响应接近阴性对照(dmso)的最小响应。一些化合物(例如24、25和17)在非常高的浓度下显示出效力降低,这可能是由于该范围内的化合物毒性。这些化合物在该模型中的效力表明了本发明化合物在治疗als中的效用。α-突触核蛋白聚集的调节评估化合物调节α-突触核蛋白聚集的能力。将rencell vm神经元细胞用α-突触核蛋白编码的腺病毒转导,并在24小时之后添加化合物。72小时之后,更新化合物。另外的72小时之后,将细胞固定(总共6天的化合物处理)。添加两种不同的α-突触核蛋白抗体(syn205和mjf-14),在in cell 2200上以10
×
放大倍数进行成像。通过高内涵算法量化免疫反应性。将化合物数据归一化为阴性对照(0.1%dmso)和阳性对照(10μm ku0063794;cas 938440-64-3)。结果图4中的结果显示,实施例24、25和26剂量依赖性地降低rencell vm神经元细胞中存在的α-突触核蛋白的量。该降低与两种抗体一致,并且所有化合物显示出1至10μm范围内的ic
50
,其中100%抑制是等同于10μm ku0063794的降低并且0%是等同于0.1%dmso的降低。这些化合物在该模型中的效力表明了本发明化合物在治疗帕金森病中的效用。体外安全组数据huvec细胞生存力(cell viability,cv)测定程序将细胞以10,000个细胞/孔接种在96孔板中的huvec特异性细胞培养基中。将细胞铺板并在37℃下培养过夜(16至24小时)。过夜培养之后,洗涤细胞并添加测定培养基。施用测试化合物并与细胞孵育指定的时间段,之后通过alamarblue方法测量细胞生存力。在8个浓度(默认为0.03、0.1、0.3、1、3、10、30和100μm)下一式两份测试化合物以确定ic50。最终dmso浓度为1%。在测试化合物和alamarblue孵育之后记录荧光读数。激发和发射波长分别为530nm和590nm。通过将在测试化合物存在下的读数与载剂对照进行比较来计算对照的百分比。随后,通过从100中减去对照活性百分比来计算百分比抑制。使用hill方程通过浓度-响应曲线的非线性回归分析确定ic50值(引起对照值的半数最大抑制的浓度)。用于该测定的参考化合物是星形孢菌素(staurosporine)。huvec管形成测定程序将huvec(人脐静脉内皮细胞,1.5
×
104/孔,atcc crl-1730)置于先前制备的96孔
板中。然后将测试物质和/或载剂在37℃下在5%co2的气氛下在生长培养基中以0.4%dmso的终浓度添加至每个孔。18小时孵育期之后,通过共聚焦高内涵成像系统评估内皮细胞管的形态,其类似毛细血管状网络。从每张照片测量并相对于载剂对照组确定总的管长度的破坏(抗血管生成)。将最小抑制浓度(minimum inhibitory concentration,mic,≥30%)记录为显著响应并确定gi50(gi=生长抑制)。以30、3、0.3、0.03和0.003μm(或μg/ml)一式三份筛选化合物。本实验中使用的参考化合物是预期gi50为15μm的苏拉明(suramin)。下表显示了本发明化合物和比较例的huvec数据。心肌细胞测定(crl)程序实验程序所有测量均在生理温度下在组织培养箱(37℃;5%co2)中进行。将测试样品、阳性对照和载剂添加至无菌组织培养罩。在化合物添加之前至少10小时,将sc-hcm暴露于新鲜培养基。在化合物添加之前,获得场电位和阻抗信号(基线)的一分钟记录。以2
×
浓度添加测试样品、阳性对照和载剂。在测试品添加之后0.5、1、2、24和48小时获得一分钟记录。在药物施用时,将培养基添加至独立96孔板,其在组织培养箱中孵育48小时。将该培养基用作ctni释放测定的空白。在48小时暴露记录之后,将每孔150μl离心以去除残渣。收集上清液并冷冻用于随后使用elisa板根据制造商的说明(abcam,cat#ab200016)的ctni含量分析。将数据存储在查尔斯河实验室(charles river laboratories)计算机网络上用于离线分析。使用rtca cardio软件进行数据采集。数据分析使用rtca cardio软件、microsoft excel和matlab脚本进行数据分析。数据以平均值
±
sem报告。量化以下场电位参数:钠尖峰振幅、钠尖峰速率、场电位持续时间(field potential duration,fpd)、bazett校正场电位持续时间(bazett corrected field potential duration,fpdcb)和节律障碍事件(如果存在的话)。对以下阻抗参数进行量化:细胞指数、峰振幅,从峰到50%振幅的驰豫(rel50)和颤动持续时间(twitch duration)。钠尖峰(sodium spike)是由去极化波(正峰)的传播和钠通道(负峰)的局部激活
device)在稳定表达人ether
‑á‑
go-go相关基因的cho细胞中测定herg离子通道筛选。获得细胞膜与经处理硅表面之间的千兆欧姆密封(gigaohm seal),并将特定的外部和内部缓冲溶液施加到细胞上,然后记录。在基线载剂处理之后,以递增的浓度(8pt crc)施加化合物,以2分钟记录测量对herg尾电流振幅的作用。
[0338]
下表示出了本发明化合物和比较例的herg ic50数据。实施例herg ic50(μm)实施例97>10实施例25>10实施例39>10比较例2>10比较例33.8安全组数据讨论
[0339]
帕纳替尼(比较例2)与临床严重不良事件相关,并以黑盒警告动脉闭塞事件销售。使用人脐静脉内皮细胞(human umbilical vein endothelial cell,huvec)的体外血管生成模型先前已用于研究蛋白酪氨酸激酶抑制剂的血管不良事件
11
。使用该模型,显示帕纳替尼降低huvec生存力并抑制huvec管形成。为了评价本发明化合物相对于帕纳替尼的抗血管生成活性,在huvec中进行管形成和生存力测定。上表中所示的数据表明帕纳替尼对管形成具有更显著的抑制作用,以表明比本发明化合物大至少10倍的效力的gi50。与本发明化合物相比,帕纳替尼还显示对细胞生存力约10倍(或更大)的作用。因此,根据该模型,预期本发明化合物将具有比帕纳替尼显著更低的抗血管生成活性。
[0340]
帕纳替尼(比较例2)和尼洛替尼(比较例3)二者在临床上都与严重的心血管不良事件相关。人诱导的多能干细胞来源心肌细胞已在文献中用于模拟tki(如帕纳替尼和尼洛替尼)的心脏毒性作用
12
。根据文献,发现帕纳替尼能够诱导如上表中的许多参数所示的结构性心脏毒性,包括肌钙蛋白(ctni)分泌提高(其中ctni是心脏损伤的已知标志物)。通过比较,相对于载剂,本发明的化合物在10μm(48小时)没有显示出任何显著的ctni释放。用10μm的帕纳替尼处理之后48小时的阻抗测量表明,由于对心脏细胞健康的损害或有害作用(其导致细胞变得静止(缺乏活性)),无法测量帕纳替尼的钠尖峰振幅。对于尼洛替尼,观察到对钠尖峰振幅的最大测量作用(如先前在文献中报道的),观察到另外的显著的药物性心律失常标志物,包括触发性活动和阻抗不稳定性
13
。对于尼洛替尼,在48小时之后在10μm观察到钠尖峰振幅的显著降低(96.2%vs.对照)。实施例39仅给出43%的降低,而实施例97对钠尖峰振幅没有显著作用(2.2%vs.对照)。
[0341]
从上表中的数据还可以看出,与尼洛替尼相比,本发明化合物具有降低的herg抑制活性。
[0342]
安全组数据清楚地表明,与帕纳替尼和尼洛替尼相比,本发明化合物具有提高的安全性谱。高分辨率质谱数据
参考文献1.de lin et al.(2017)novel pathways of ponatinib disposition catalyzed by cyp1a1 involving generation of potentially toxic metabolites.j pharmacol exp ther 363:12-192.food and drug administration.(2018)bioanalytical method validation:guidance for industry.3.whitmire m,ammerman j,de lisio p,killmer j,kyle d(2011)lc-ms/ms bioanalysis method development,validation,and sample analysis:points to consider when conducting nonclinical and clinical studies in accordance with current regulatory guidances.j anal bioanal techniques s4:001.doi:104172/2155-9872.s4-0014.summerfield s,stevens aj,cutler l,del carmen osuna m,hammond b,tang sp,hersey a,spalding dj,and jeffrey p(2006)improving the in vitro prediction of in vivo cns penetration:integrating permeability,pgp efflux and free fractions in blood and brain.j pharmacolexp ther 316:1282-12905.waters,nigel j.et al,.(2008)validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding.journal of pharmaceutical sciences:97(10)4586-45956.kalvass jc,maurer ts,pollack gm(2007)use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs:comparison of unbound concentration ratios to in vivo p-glycoprotein effluxratios.drug metab dispos 35:660-666.7.friden m,gupta a,antonsson m,bredberg u,hammadund-udenaes m(2007)in vitro methods for estimating unbound drug concentrations in the brain interstitial and intracellular fluids.drug metab dispos 35:1711-1719.8.meyer,k.,ferraiuolo,l.,miranda,c.j.,likhite,s.,mcelroy,s.,renusch,s.,ditsworth,d.,lagier-tourenne,c.,smith,r.a.,ravits,j.,burghes,a.h.,shaw,p.j.,cleveland,d.w.,kolb,s.j.,and kaspar,b.k.(2014)direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic als.pnas 111,829-832.
9.haidet-phillips,a.m.,hester,m.e.,miranda,c.j.,meyer,k.,braun,l.,frakes,a.,song,s.,likhite,s.,murtha,m.j.,foust,k.d.,rao,m.,eagle,a.,kammesheidt,a.,christensen,a.,mendell,j.r.,burghes,a.h.m.,and kaspar,b.k.(2011)astrocytes from familial and sporadic als patients are toxic to motor neurons.nature biotechnology 29,824-828.10.wichterle,h.,lieberam,l.,porter,j.a.and jessell,t.m.(2002)directed differentiation of embryonic stem cells into motor neurons.cell 110,385-397.11.leukemia&lymphoma,2017,vol.58,no.6,1455-1467.12.toxicological sciences,143(1),2015,147-155.13.toxicology&applied pharmacology,272(1),2013,245-255.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。