制备2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮的方法
1.本发明涉及制备通式(i)的2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮的新方法。
2.2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮和相应的衍生物在制药和农业化学工业中作为制备例如手性亚砜的中间体是非常重要的。这类亚砜例如在作物保护中用作杀螨剂(参见例如wo2013/092350或wo2015/150348)。
3.2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮的化学合成是已知的。这可以例如通过使通式(ii)的适当n,n
’‑
二取代硫脲与通式(iii)的乙酸衍生物反应来实现(参见例如wo2013/092350;ep 985670;advances in heterocycl.chem.25,(1979)85))。原则上有许多制备通式(ii)的n,n
’‑
二取代硫脲的方法。一种简单有效的方法包括使适当取代的通式(iv)苯胺与通式(v)的异硫氰酸酯反应(wo2014/202510)。相反,也可以通过使通式(vi)的异硫氰酸芳基酯与通式(vii)的胺反应,以这种方式获得通式(ii)的n,n-二取代硫脲(jp2011/042611)。
4.因此,制备通式(i)的2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮的常见方法的特征在于,在第一步中,通式(iv)的苯胺与通式(v)的异硫氰酸酯反应,或通式(vi)的异硫氰酸芳基酯与通式(vii)的胺反应,然后例如通过过滤分离由此形成的通式(ii)的n,n
’‑
二取代硫脲。在该已知方法的第二步中,通式(ii)的n,n
’‑
二取代硫脲与通式(iii)的乙酸衍生物在碱存在下反应,形成通式(i)的2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮。
5.该方法的缺点是使用异硫氰酸酯,即通式(v)的异硫氰酸烷基酯或通式(vi)的异硫氰酸芳基酯。异硫氰酸酯通常只能通过使用危险化学品的费力方法来制备。例如,通式(v)和(vi)的异硫氰酸酯的制备是已知的,通过使通式(vii)的胺或通式(iv)的苯胺与硫光气反应(rapid communications in mass spectrometry 8(1994)737)。在这种情况下,使用硫光气是非常不利的。硫光气具有很高的毒性;腐蚀性很强;有难闻的气味;并且通常很难获得并且成本很高。制备通式(v)和(vi)的异硫氰酸酯的另一种常见方法包括使通式(vii)的胺或通式(iv)的苯胺在碱如三乙胺的存在下与二硫化碳反应,得到通式(viii)的二硫代氨基甲酸酯,随后使它们与以下试剂反应,如氯甲酸酯(j.org.chem.29(1964)3098)、甲苯磺酰氯(wo2012/129338)、光气(chem.zentralblatt 101(1930)buch 1(3),3431)、次氯酸钠(liebigs ann.chem.585(1954)230)、亚氯酸钠(de 960276)或过氧化氢(j.org.chem.62(1997)4539)。这些方法有各种缺点,如使用低沸点和高度易燃的二硫化碳或使用剧毒的光气。此外,工业过程的收率不够高。同样已知的卤代烷与硫氰酸盐反应生成硫氰酸酯,随后异构化为异硫氰酸酯的反应并不是在所有情况下都有效。
6.现有技术已知的制备2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮的方法(a)如方案(1)所示,其中x、y1、y2、w、r1、r2和r3定义如下。
7.方案(1)
[0008][0009]
鉴于上述缺点,因此迫切需要一种简化的、工业上和经济上可行的制备通式(i)的2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮的方法。用该设想的方法可获得的2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮应优选以高收率和高纯度提供。
[0010]
令人惊讶的是,已经发现通式(i)的2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮可以通过通式(viii)的2-(苯基亚氨基)-3h-1,3-噻唑烷-4-酮与通式(ix)的烷基化剂反应来制备。
[0011]
因此,本发明提供了制备通式(i)的2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮的新方法(b)
[0012][0013]
其中
[0014]
y1和y2各自独立地是氟、氯或氢,
[0015]
r1和r2各自独立地是氢、(c
1-c
12
)烷基、(c
1-c
12
)卤代烷基、氰基、卤素或硝基,和
[0016]
r3是任选取代的(c
6-c
10
)芳基、(c
1-c
12
)烷基或(c
1-c
12
)卤代烷基,其中取代基选自卤素、(c
1-c6)烷基、(c
3-c
10
)环烷基、氰基、硝基、羟基、(c
1-c6)烷氧基、(c
1-c6)卤代烷基和(c
1-c6)卤代烷氧基,特别是氟、氯、(c
1-c3)烷基、(c
3-c6)环烷基、环丙基、氰基、(c
1-c3)烷氧基、(c
1-c3)卤代烷基和(c
1-c3)卤代烷氧基,
[0017]
其特征在于通式(viii)的2-(苯基亚氨基)-3h-1,3-噻唑烷-4-酮与通式(ix)的烷基化剂反应:
[0018][0019]
其中y1、y2、r1和r2如上定义,
[0020]r3-z
[0021]
(ix),
[0022]
其中
[0023]
r3如上定义,
[0024]
和
[0025]
z是碘、溴、氯、oso2me、oso2ph、oso2(4-me-ph)、oso2cf3、oso2c2f5、oso2c3f7、oso2c4f9、oso2cf2coome、oso2cf2cooet、oso2cf2coonpr、oso2cf2cooipr或oso2cf2coonbu。
[0026]
通式(i)的2-(苯基亚氨基)-3-烷基-1,3-噻唑烷-4-酮可通过本发明的方法以良好的收率和高纯度制备。
[0027]
式(i)的化合物可以作为e-异构体或z-异构体或这些异构体的混合物存在。这由式(i)中的交叉双键表示。在本发明的一个单独的实施方案中,化合物在每种情况下都是e-异构体的形式。在本发明的另一个单独的实施方案中,化合物在每种情况下都是z-异构体的形式。在本发明的另一个单独的实施方案中,化合物是e-异构体和z-异构体的混合物的形式。在本发明的一个优选的单独实施方案中,化合物是z-异构体或e-异构体和z-异构体的混合物的形式,其中z-异构体的比例大于50%,并且越来越优选大于60%、65%、70%、75%、80%、85%、90%、95%,基于混合物中e-异构体和z-异构体的总量计。
[0028]
因为根据本发明的方法,原料通式(viii)也可以由其互变异构形式的通式(viii’)反应得到式(i)的化合物,也可以获得异构产物通式(x)的(2-[{2-苯基}(烷基)氨基]-1,3-噻唑-4(5h)-酮)
[0029][0030]
其中y1、y2、r1和r2如上定义,
[0031][0032]
其中y1、y2、r1、r2和r3如上定义。
[0033]
根据本发明的方法的特征还在于以高选择性,即以比通式(x)的化合物显著高的比例,获得通式(i)的化合物。
[0034]
在上面提到的式(i)、(viii)、(viii’)、(ix)和(x)中列出的基团y1、y2、z、r1、r2和r3的优选的、特别优选的和非常特别优选的定义在下面阐明。
[0035]
优选的是当
[0036]
y1和y2各自独立地是氟、氯或氢,
[0037]
r1和r2各自独立地是氟、氯、(c
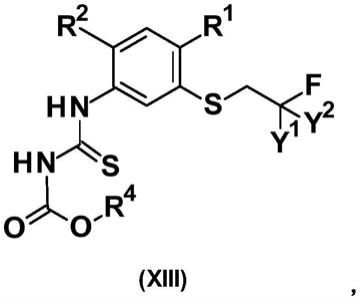
1-c3)烷基或氢,
[0038]
r3是(c
1-c6)烷基或(c
1-c6)卤代烷基,和
[0039]
z是oso2me、oso2ph、oso2(4-me-ph)、oso2cf3、oso2c2f5、oso2c3f7、oso2c4f9、oso2cf2coome、oso2cf2cooet、oso2cf2coonpr、oso2cf2cooipr或oso2cf2coonbu。
[0040]
特别优选的是当
[0041]
y1和y2各自独立地是氟或氢,
[0042]
r1和r2各自独立地是氟、氯、氢或甲基,
[0043]
r3是(c
1-c6)卤代烷基,和
[0044]
z是oso2cf3、oso2c2f5、oso2c3f7、oso2c4f9、oso2cf2coome、oso2cf2cooet、oso2cf2coonpr、oso2cf2cooipr或oso2cf2coonbu。
[0045]
非常特别优选的是当
[0046]
y1和y2是氟,
[0047]
r1和r2各自独立地是氟、氢或甲基,
[0048]
r3是(c
1-c6)氟烷基,和
[0049]
z是oso2cf3、oso2c4f9、oso2cf2coome、oso2cf2cooet、oso2cf2coonpr、oso2cf2cooipr或oso2cf2coonbu。
[0050]
最优选的是当
[0051]
y1和y2是氟,
[0052]
r1是甲基,
[0053]
r2是氟,
[0054]
r3是ch2cf3,和
[0055]
z是oso2cf3、oso2c4f9、oso2cf2coome、oso2cf2cooipr。
[0056]
本技术同样提供了通式(viii)的化合物
[0057][0058]
其中y1、y2、r1和r2如上定义。
[0059]
因此,在通式(viii)中,优选的是当
[0060]
y1和y2各自独立地是氟、氯或氢,和
[0061]
r1和r2各自独立地是氟、氯、(c
1-c3)烷基或氢。
[0062]
因此特别优选的是当
[0063]
y1和y2各自独立地是氟或氢,和
[0064]
r1和r2各自独立地是氟、氯、氢或甲基。
[0065]
因此非常特别优选的是当
[0066]
y1和y2是氟,和
[0067]
r1和r2各自独立地是氟、氢或甲基。
[0068]
因此,最优选的是当
[0069]
y1和y2是氟,
[0070]
r1是甲基,和
[0071]
r2是氟。
[0072]
通式(viii)的化合物可以例如由相应的通式(xi)的单芳基硫脲(其中y1、y2、r1和r2如上定义)通过与通式(iii)的化合物(其中x是溴、氯、oso2me、oso2ph、oso2(4-me-ph)或oso2cf3,且w是oh或基团o(c
1-c
6-烷基))反应来制备(方案2)。
[0073]
方案(2)
[0074][0075]
优选的是当x是溴或氯,且w是基团o(c
1-c
6-烷基)。非常特别优选的是当x是溴或氯,且w是基团och3或oc2h5。最优选的是当x是溴或氯,且w是基团och3。
[0076]
因此,本技术同样提供通式(xi)的化合物
[0077][0078]
其中y1、y2、r1和r2如上定义。
[0079]
因此在通式(xi)中,优选的是当
[0080]
y1和y2各自独立地是氟、氯或氢,和
[0081]
r1和r2各自独立地是氟、氯、(c
1-c3)烷基或氢。
[0082]
因此特别优选的是当
[0083]
y1和y2各自独立地是氟或氢,和
[0084]
r1和r2各自独立地是氟、氯、氢或甲基。
[0085]
因此非常特别优选的是当
[0086]
y1和y2是氟,和
[0087]
r1和r2各自独立地是氟、氢或甲基。
[0088]
因此最优选的是当
[0089]
y1和y2是氟,
[0090]
r1是甲基,和
[0091]
r2是氟。
[0092]
通式(xi)的单芳基硫脲可以通过各种方法制备。优选的方法包括使通式(iv)的苯胺与通式(xii)的异硫氰酸烷氧基羰基酯反应得到通式(xiii)的(苯基氨基硫代甲酰基)氨基甲酸烷基酯,然后在酸性或碱性条件下使通式(xiii)的化合物皂化和脱羧,得到通式(xi)的单芳基硫脲(方案3),
[0093][0094]
其中y1、y2、r1和r2如上定义,
[0095][0096]
其中r4是甲基、乙基或异丙基,
[0097][0098]
其中y1、y2、r1、r2和r4如上定义。
[0099]
对于皂化和脱羧,这是本领域技术人员熟知的。
[0100]
方案(3)
[0101][0102]
因此,本技术还提供了通式(xiii)的(苯基氨基硫代甲酰基)氨基甲酸烷基酯:
[0103][0104]
其中y1、y2、r1、r2和r4如上定义。
[0105]
因此在通式(xiii)中,优选的是当
[0106]
y1和y2各自独立地是氟、氯或氢,
[0107]
r1和r2各自独立地是氟、氯、(c
1-c3)烷基或氢,和
[0108]
r4是甲基、乙基或异丙基。
[0109]
因此特别优选的是当
[0110]
y1和y2各自独立地是氟或氢,
[0111]
r1和r2各自独立地是氟、氯、氢或甲基,和
[0112]
r4是甲基或乙基。
[0113]
因此非常特别优选的是当
[0114]
y1和y2是氟,
[0115]
r1和r2各自独立地是氟、氢或甲基,和
[0116]
r4是甲基或乙基。
[0117]
因此,最优选的是当
[0118]
y1和y2是氟,
[0119]
r1是甲基,
[0120]
r2是氟,和
[0121]
r4是甲基或乙基。
[0122]
在本发明主题的另一个实施方案中,式(xiii)的化合物的进一步特征在于它不是2-氨基-1-(3-甲氧基羰基-1-2-硫脲基)-4-(2,2,2-三氟乙硫基)苯。
[0123]
制备通式(viii)的化合物的另一种可能性包括使通式(xiv)的2-卤代-n-(苯基)乙酰胺与通式(xv)的碱金属或铵硫氰化物反应:
[0124][0125]
其中y1、y2、r1和r2如上定义
[0126]
和
[0127]
hal是氯或溴,
[0128]
mscn(xv),
[0129]
其中m是li、na、k或nh4。
[0130]
该反应如方案4所示。
[0131]
方案(4)
[0132][0133]
因此,本技术还提供了通式(xiv)的2-卤代-n-(苯基)乙酰胺
[0134][0135]
其中y1、y2、r1、r2和hal如上定义。
[0136]
因此在通式(xiv)中优选的是当
[0137]
y1和y2各自独立地是氟、氯或氢,
[0138]
r1和r2各自独立地是氟、氯、(c
1-c3)烷基或氢,和
[0139]
hal是溴或氯。
[0140]
因此特别优选的是当
[0141]
y1和y2各自独立地是氟或氢,
[0142]
r1和r2各自独立地是氟、氯、氢或甲基,和
[0143]
hal是溴或氯。
[0144]
因此非常特别优选的是当
[0145]
y1和y2是氟,
[0146]
r1和r2各自独立地是氟、氢或甲基,和
[0147]
hal是氯。
[0148]
因此最优选的是当
[0149]
y1和y2是氟,
[0150]
r1是甲基,
[0151]
r2是氟和
[0152]
hal是氯。
[0153]
通式(xiv)的2-卤代-n-(苯基)乙酰胺可通过通式(iv)的苯胺(如上所指明的)与通式(xvi)的卤代乙酰基卤化物反应获得:
[0154][0155]
其中hal和hal’各自独立地是氯或溴,特别优选氯。
[0156]
根据本发明的方法在方案5中全部示出。
[0157]
方案(5)
噁二唑-3-基、1,2,4-噁二唑-5-基、1,2,4-噻二唑-3-基、1,2,4-噻二唑-5-基、1,2,4-三唑-3-基、1,3,4-噁二唑-2-基、1,3,4-噻二唑-2-基和1,3,4-三唑-2-基;1-吡咯基、1-吡唑基、1,2,4-三唑-1-基、1-咪唑基、1,2,3-三唑-1-基、1,3,4-三唑-1-基;3-哒嗪基、4-哒嗪基、2-嘧啶基、4-嘧啶基、5-嘧啶基、2-吡嗪基、1,3,5-三嗪-2-基和1,2,4-三嗪-3-基。
[0168]
在根据本发明的方法中,通式(viii)的2-(苯基亚氨基)-3h-1,3-噻唑烷-4-酮生成式(i)的化合物的反应优选在溶剂存在下进行。在根据本发明的方法中,合适的溶剂特别是下列溶剂:乙腈、丙腈、丁腈、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、甲醇、乙醇、正丙醇、异丙醇、正丁醇、异丁醇、仲丁醇、叔丁醇、戊醇、己醇、辛醇、异辛醇、环戊醇、环己醇、乙二醇、甘油、二甲基亚砜、环丁砜。也可以使用所述溶剂的混合物。
[0169]
优选的溶剂是乙腈、丁腈、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、甲醇、乙醇、正丙醇、异丙醇、正丁醇、异丁醇、仲丁醇、叔丁醇、己醇、辛醇、异辛醇、环己醇、二甲基亚砜、环丁砜或所述溶剂的混合物。
[0170]
特别优选的溶剂是乙腈、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、二甲基亚砜或所述溶剂的混合物。
[0171]
通式(ix)的烷基化剂r
3-z优选以0.9:1至2:1的摩尔比使用,基于通式(viii)的2-(苯基亚氨基)-3h-1,3-噻唑烷-4-酮计。进一步优选摩尔比为0.95:1至1.5:1,同样在每种情况下基于通式(viii)的2-(苯基亚氨基)-3h-1,3-噻唑烷-4-酮计。
[0172]
在另一个优选的实施方案中,根据本发明的方法在碱的存在下进行。
[0173]
根据本发明的方法中使用的碱可以是有机碱和无机碱。有机碱包括例如三甲胺、三乙胺、三丁胺、乙基二异丙基胺、吡啶、2-甲基吡啶、2,3-二甲基吡啶、2,5-二甲基吡啶、2,6-二甲基吡啶、2-甲基-5-乙基吡啶、喹啉、甲醇钾、乙醇钾、叔丁醇钾、甲醇钠、乙醇钠、叔丁醇钠、乙酸钾和乙酸钠。无机碱包括例如氢氧化锂、氢氧化钾、氢氧化钠、碳酸氢钾、碳酸氢钠、碳酸钾、碳酸钠、碳酸铯、碳酸钙和碳酸镁。优选三乙胺、三丁胺、乙基二异丙基胺、2-甲基-5-乙基吡啶、甲醇钠、碳酸氢钾、碳酸氢钠、碳酸钾和碳酸钠。特别优选三乙胺、三丁胺、碳酸氢钠、碳酸氢钾、碳酸钾、碳酸钠和甲醇钠。
[0174]
在根据本发明的方法中,碱优选以0.9:1至3:1的摩尔比使用,基于通式(viii)的2-(苯基亚氨基)-3h-1,3-噻唑烷-4-酮计。进一步优选摩尔比为1:1至2:1,同样在每种情况下基于通式(viii)的2-(苯基亚氨基)-3h-1,3-噻唑烷-4-酮计。
[0175]
根据本发明的方法通常在-20℃至150℃,优选0℃至120℃,最优选5℃至80℃的温度下进行
[0176]
反应通常在标准压力下进行,但也可以在升高或降低的压力下进行。
[0177]
可以例如通过随后的过滤或提取分离所需的式(i)的化合物。这样的方法是本领域技术人员已知的。
[0178]
通过下面的实施例详细阐明本发明,但不应以它们限制本发明的方式解释这些实施例。
[0179]
制备实施例:
[0180]
实施例1:2-氯-n-{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}乙酰胺的合成
[0181][0182]
在0-5℃下,向11.96g[50mmol]的2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯胺和10.12g[100mmol]的三乙胺在100ml二氯甲烷中的溶液中滴加6.78g[60mmol]的氯乙酰氯。将混合物在0-5℃下搅拌1小时,然后在20℃下搅拌过夜。用150ml水搅拌反应混合物。分离出有机相,水相用50ml二氯甲烷萃取,合并的有机相用50ml 15%盐酸洗涤两次,然后用50ml水洗涤,用硫酸钠干燥并减压浓缩。得到15g褐色固体,根据gc(气相色谱法),其纯度为96.5%(a/a),收率为理论值的92.9%。
[0183]
熔点:128℃。
[0184]
gc/ms:m/e=315(m
,1cl,33%),239(m
-76,43%),156(100%)。
[0185]1h-nmr(600mhz,d
6-dmso):δ=2.44(s,3h),3.87(q,2h),4.4(s,2h),7.32(d,1h),8.12(d,1h),10.17(s,1h)ppm。
[0186]
19
f-nmr(565mhz,d
6-dmso):δ=-64.3(t,3f),-124.3(dd,1f)ppm。
[0187]
实施例2:({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}氨基硫代甲酰基)氨基甲酸甲酯的合成
[0188][0189]
步骤1(异硫氰酸甲氧基羰基酯的制备):在30℃下,向56.75g[0.7mol]的硫氰酸钠的300ml甲苯中加入0.4g吡啶和0.9g水。随后,在20分钟内加入56.7g[0.6mol]的氯甲酸甲酯。将混合物在30℃下搅拌2小时,冷却至20℃,滤出氯化钠。滤液用于步骤2。
[0190]
步骤2(标题化合物的制备):首先加入步骤1的滤液,并在30℃下加入119.6g[0.5mol]的2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯胺在100ml甲苯中的溶液。加入完成后,将混合物加热至80℃,并在此温度下搅拌90分钟。然后将反应混合物冷却至0℃,滤出沉淀的固体,用250ml戊烷洗涤并干燥。以此方式,获得165.5g白色固体,根据定量1h-nmr,其含量为98.1%(w/w)。因此,这相当于理论值的91.1%的收率。
[0191]
熔点:153-154℃。
[0192]1h-nmr(600mhz,d
6-dmso):δ=2.40(s,3h),3.76(s,2h),3.86(q,2h),7.28(d,1h),8.05(d,1h),11.36(s,1h),11.55(s,1h)ppm。
[0193]
19
f-nmr(565mhz,d
6-dmso):δ=-64.4(t,3f),-123.3(dd,1f)ppm。
[0194]
实施例3:({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}氨基硫代甲酰基)氨基甲酸乙酯的合成
[0195][0196]
步骤1(异硫氰酸乙氧基羰基酯的制备):在5分钟内向5.35g[0.066mol]的硫氰酸钠的50ml丙酮中加入6.51g[0.06mol]的氯甲酸乙酯。将混合物回流搅拌15分钟,冷却至20℃,滤出氯化钠。滤液用于步骤2。
[0197]
步骤2(标题化合物的制备):首先加入步骤1的滤液,并初始在20℃下在不冷却的情况下加入11.96g[0.05mol]的2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯胺在20ml丙酮中的溶液。添加完成后,将混合物回流加热1小时。然后将反应混合物冷却至20℃,加入370ml水中,滤出沉淀的固体并干燥。以此方式获得19.25g白色固体,根据hplc分析,其纯度为92.6%(a/a)。因此,这相当于理论值的96%的收率。
[0198]
熔点:126℃。
[0199]
lc/ms:m/e=371(mh
)。
[0200]1h-nmr(600mhz,d
6-dmso):δ=1.26(t,3h),2.4(s,3h),3.86(q,2h),4.22(q,2h),7.28(d,1h),8.05(d,1h),11.4(s,1h),11.5(s,1h)ppm。
[0201]
实施例4:1-{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}硫脲的合成
[0202][0203]
在约10分钟内,向装入2升反应器中的893ml的1n氢氧化钠水溶液和530ml乙醇的混合物中计量加入169.6g[0.458mol]的({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}氨基硫代甲酰基)氨基甲酸乙酯。将混合物在30分钟内加热至50℃,并在此温度下搅拌17小时。将反应混合物冷却,并在约40℃下从反应器中倒空。在20℃下,用半浓盐酸将ph值调节至6-8。抽吸滤出沉淀的固体,用水洗涤并干燥。得到130.38g标题化合物,根据定量
19
f-nmr,其含量为94.7%(w/w)。因此,这相当于理论值的90.4%的收率。
[0204]
熔点:120-122℃。
[0205]
lc/ms:m/e=299(mh
)。
[0206]1h-nmr(600mhz,d
6-dmso):δ=2.37(s,3h),3.85(q,2h),4.22(q,2h),7.22(d,1h),7.86(d,1h),9.38(s,1h)ppm。
[0207]
19
f-nmr(565mhz,d
6-dmso):δ=-64.8(t,3f),-123.5(dd,1f)ppm。
[0208]
实施例5:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-1,3-噻唑烷-4-酮的合成
[0209][0210]
在75ml乙腈中首先加入14.92g[50mmol]的1-{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}硫脲和5.33g[65mmol]的乙酸钠。在20-25℃下,滴加9.18g[55mmol]的溴乙酸乙酯。将反应混合物在20℃下搅拌20小时。然后在减压下蒸馏掉大部分乙腈,并向残余物中加入100ml水。用100ml二氯甲烷搅拌混合物。滤出沉淀的固体并干燥。以此方式获得2.60g固体,根据hplc分析,其纯度为99.3%(a/a),相当于理论值的15.3%的收率。分离出二氯甲烷相,干燥并浓缩。得到12.72g标题化合物,纯度为97.6%(a/a),相当于理论值的73.4%的收率。
[0211]
熔点:128℃。
[0212]
lc/ms:m/e=339(mh
)。
[0213]1h-nmr(600mhz,d
6-dmso):δ=2.36(s,3h),3.87(q,2h),4.03(s,2h),7.33(m,2h),11.98(s,1h)ppm。
[0214]
实施例6:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-1,3-噻唑烷-4-酮的合成
[0215][0216]
将3.16g[10mmol]的2-氯-n-{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}乙酰胺和1.14g[15mmol]的硫氰酸铵在25ml乙醇中的混合物回流加热15小时。随后,在室温下向反应混合物中加入50ml水和50ml二氯甲烷。分离出有机相,水相再次用50ml二氯甲烷萃取,合并有机相,用50ml水洗涤,用硫酸钠干燥并减压浓缩。得到3.33g的产物,根据gc/ms分析,纯度为70.8%(a/a)(理论值的70%)。
[0217]
实施例7:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0218][0219]
将138mg[0.4mmol]的(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-1,3-噻唑烷-4-酮、94.7mg[0.4mmol]的2,2,2-三氟乙基三氟甲基磺酸酯和113mg[0.82mmol]的碳酸钾在5ml乙腈中的混合物在20℃下搅拌18小时。过滤反应混合物,用5ml乙腈洗涤残余物并浓缩滤液。得到260mg固体。hplc分析显示完全转化,并且(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮与2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)的比例为79.9:20.1。
[0220]
实施例8:2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮的合成
[0221][0222]
将1.69g[5mmol]的(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-1,3-噻唑烷-4-酮、2.29g[6mmol]的2,2,2-三氟乙基1,1,2,2,3,3,4,4,4-九氟丁烷-1-磺酸酯和1.01g[10mmol]的三乙胺在50ml甲基叔丁基醚(mtbe)中的混合物在40℃下加热26小时,然后回流加热5小时。然后在室温下向反应混合物中加入20ml水。分离出有机相,用硫酸钠干燥并减压浓缩。得到3.8g粗产物,将其通过柱色谱纯化(洗脱剂环己烷/乙酸乙酯)。得到0.73g白色固体,根据hplc分析,纯度》99%。
[0223]
熔点:135℃
[0224]
lc/ms:m/e=421(mh
)。
[0225]1h-nmr(600mhz,d
6-dmso):δ=2.45(s,3h),4.02(q,2h),4.11-4.19(m,2h),4.76(m,1h),4.99(m,1h),7.49(d,1h),7.88(d,1h)ppm。
[0226]
19
f-nmr(565mhz,d
6-dmso):δ=-64.7(t,3f),-68.8(m,3f),-122.3(m,1f)ppm。
[0227]
13
c-nmr(151mhz,d
6-dmso):δ=20.3(ar-ch3),34.7(sch2),41.9(sch2co),52.9(nch2cf3),118.8(c
ar
h),123.8(nch2cf3),125.4(c
ar
n),125.9(sch2cf3),130.0(c
ar
s),132.5(c
ar
h),144.2(c
ar
me),156.8/c
ar
f),187.0(nco),187.1(n-c(=n)s)ppm。
[0228]
实施例9:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0229][0230]
将169mg[0.5mmol]的(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-1,3-噻唑烷-4-酮、191mg[0.5mmol]的2,2,2-三氟乙基1,1,2,2,3,3,4,4,4-九氟丁烷-1-磺酸酯和138mg[1mmol]的碳酸钾在5ml乙腈中的混合物在20℃下搅拌19小时。hplc分析显示完全转化,并且产物a和b的比例约为80:20。
[0231]
实施例10:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0232]
将169mg[0.5mmol]的(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-1,3-噻唑烷-4-酮、191mg[0.5mmol]的2,2,2-三氟乙基1,1,2,2,3,3,4,4,4-九氟丁烷-1-磺酸酯和101mg[1mmol]的三乙胺在5ml乙腈中的混合物在20℃下搅拌19小时。hplc分析显示转化率约82%,并且产物a和b的比例约为71:29。
[0233]
实施例11:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0234]
将169mg[0.5mmol]的(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-1,3-噻唑烷-4-酮、191mg[0.5mmol]的2,2,2-三氟乙基1,1,2,2,3,3,4,4,4-九氟丁烷-1-磺酸酯和138mg[1mmol]的碳酸钾在5ml n,n-二甲基乙酰胺中的混合物在20℃下搅拌19小时。hplc分析显示完全转化,并且产物a和b的比例约为90:10。
[0235]
实施例12:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0236]
步骤同实施例11,但用1mmol碳酸钠代替碳酸钾。hplc分析显示转化率为99%,并且产物a和b的比例约为92:8。
[0237]
实施例13:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0238]
步骤同实施例11,但用1mmol碳酸氢钠代替碳酸钾。hplc分析显示转化率为99%,并且产物a和b的比例约为92:8。
[0239]
实施例14:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0240]
步骤同实施例11,但用1mmol碳酸铯代替碳酸钾。hplc分析显示转化率为100%,并
且产物a和b的比例约为80:20。
[0241]
实施例15:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0242]
步骤同实施例11,但用1mmol三乙胺代替碳酸钾。hplc分析显示转化率为93%,并且产物a和b的比例约为91:9。
[0243]
实施例16:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0244]
步骤同实施例11,但用1mmol二异丙基乙胺代替碳酸钾。hplc分析显示转化率为92%,并且产物a和b的比例约为91:9。
[0245]
实施例17:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0246]
步骤同实施例11,但用1mmol的甲醇钠(作为30%的甲醇溶液)代替碳酸钾。hplc分析显示转化率为98%,并且产物a和b的比例约为95:5。
[0247]
实施例18:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0248]
步骤同实施例11,但使用相同量的n-甲基吡咯烷酮代替n,n-二甲基乙酰胺。hplc分析显示转化率为100%,并且产物a和b的比例约为91:9。
[0249]
实施例19:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0250]
步骤同实施例11,但使用相同量的二甲基亚砜代替n,n-二甲基乙酰胺。hplc分析显示转化率为98%,并且产物a和b的比例约为80:20。
[0251]
实施例20:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮(化合物a)和2-[{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}(2,2,2-三氟乙基)氨基]-1,3-噻唑-4(5h)-酮(化合物b)的合成
[0252]
将677mg[2mmol]的(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-1,3-噻唑烷-4-酮、544mg[2mmol]的二氟[(2,2,2-三氟乙氧基)磺酰基]乙酸甲基酯和404mg[4mmol]的三乙胺在20ml n,n-二甲基乙酰胺中的混合物在20℃下搅拌72小时。hplc分析显示转化率约为65%,并且产物a和b的比例约为91:9。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
