具有拆分功能的ir(ⅲ)基手性金属-有机多孔材料及制备方法和应用
技术领域
1.本发明涉及一种具有拆分1,1
’‑
联-2-萘酚功能的ir(ⅲ)基手性金属-有机多孔材料及制备方法和应用,属于手性化合物拆分技术领域。
背景技术:
2.光活性1,1
’‑
联-2-萘酚及其衍生物被广泛应用在不对称催化和手性识别领域,如何获得1,1
’‑
联-2-萘酚的光学纯产品成为科学家关注的热点之一。目前获得光学纯1,1
’‑
联-2-萘酚的方法主要分为两类:一、直接合成法:由萘二酚氧化偶联反应中加入手性催化剂如(-)-鹰爪豆碱得到可产物s-1,1
’‑
联-2-萘酚;二、手性拆分法:利用手性拆分试剂如 (8s,9r)-(-)-n-苄基氯化辛可宁丁、r-甲基苄胺等,溶液条件下加热与r-1,1
’‑
联-2-萘酚形成结晶物,进一步分离水解再结晶得到r-1,1
’‑
联-2-萘酚。这些方法大部分产率低,步骤繁琐,因此开发一种新合成方法或者新材料以获得光学纯1,1
’‑
联-2-萘酚尤为重要。
3.近年来,金属-有机多孔手性材料受到科学家的青睐,将其应用于手性小分子的拆分也得到越来越多的关注。国内外超分子领域科学家在该方面做出了很多的研究工作。通过手性配体与过渡金属配位自组装,得到一系列手性多孔材料,并用于手性拆分中。但由于动力学活性的过渡金属具有灵活的配位方式,在组装过程中会产生多种异构体,组装体易消旋化;选择动力学惰性的金属离子作为节点构筑的手性螺旋体稳定性增强,手性得以长久保持,但组装过程中的中间产物容易陷入“动力学陷阱”,形成聚合物。另一方面,目前研究重心集中在调控手性组装体的内空腔以实现更优的对映选择性。作为一种多孔材料,分子堆积产生的孔道却往往受到忽视。而在实际应用过程中,堆积孔道在传质过程中起到不可忽视的作用,因此探索发掘手性堆积孔道也许能够带来意想不到的应用前景。另外,目前的金属-有机多孔材料中的手性模块仅有一种,大大限制了其拆分效果。
技术实现要素:
4.为了克服现有技术中存在的不足,本发明的目的是提供一种具有拆分1,1
’‑
联-2-萘酚功能的ir(ⅲ)基手性金属-有机多孔材料及制备方法和应用。本发明的ir(ⅲ)基手性金属-有机多孔材料具有合成简单、产率高、拆分操作简单、效率高并可循环使用等优势。
5.为了实现上述发明目的,解决现有技术中存在的问题,本发明采取的技术方案是:一种具有拆分1,1
’‑
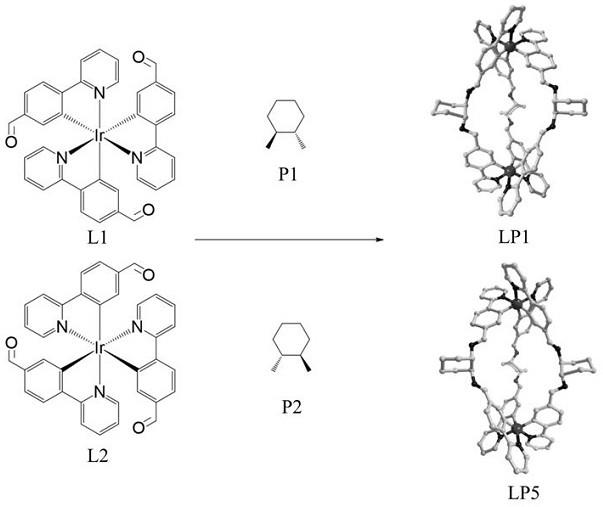
联-2-萘酚功能的ir(ⅲ)基手性金属-有机多孔材料,该材料包含l1-i-p1、 l2-i-p2两种多孔材料,所述l1-i-p1多孔材料的结构如下:
所述l2-i-p2多孔材料的结构如下:
所述的一种具有拆分1,1
’‑
联-2-萘酚功能的ir(ⅲ)基手性金属-有机多孔材料的制备方法:以手性ir(ⅲ)基配合物l1、l2作为构筑模块,分别与手性二胺p1, p2通过希夫碱反应生成多手性模块金属-有机多孔材料l1-i-p1,l2-i-p2 (i为亚胺键),其合成路线如下:l1 p1
ꢀ→ꢀ
l1-i-p1,l2 p2
ꢀ→ꢀ
l2-i-p2
所述手性ir(ⅲ)基配合物构筑模块l1,l2分别选自分子式为c
36h24
n3o3ir的三(2-苯基吡啶)合铱衍生物中的λ-fac-ir-cho(l1),δ-fac-ir-cho(l2),分别具有如下分子结构式:所述手性二胺p1选自如下分子;所述手性二胺p2选自如下分子;所述手性二胺p1最优选1s,2s-环己二胺;所述手性二胺p2最优选1r,2r-环己二胺;所述制备方法采用以下步骤:步骤1、将l1与p1或者l2与p2按照1:2~3的摩尔比投料,加入体积比为1:2~2.5的甲苯和乙腈溶液,加入催化剂量的对甲苯磺酸,在100℃~120℃条件下反应10~15 h;步骤2、反应液冷却除去溶剂,得到的固体重新溶解、扩散结晶得到l1-i-p1或l2-i-p2。
6.所述的一种具有拆分1,1
’‑
联-2-萘酚功能的ir(ⅲ)基手性金属-有机多孔材料在拆分1,1
’‑
联-2-萘酚中的应用。
7.所述材料在有机溶剂中拆分1,1
’‑
联-2-萘酚。
8.所述材料拆分1,1
’‑
联-2-萘酚的方法采用如下步骤:将l1-i-p1或者l2-i-p2加入到含有1,1
’‑
联-2-萘酚的有机溶剂中,室温搅拌至吸附饱和,过滤,滤饼有机溶剂洗涤后用萃取剂萃取,萃取液除掉溶剂得到r-1,1
’‑
联-2-萘酚或者s-1,1
’‑
联-2-萘酚。
9.所述的有机溶剂为异丙醇、乙酸乙酯、甲醇、丙酮、异丙醚中的一种或几种,优选为异丙醇、丙酮、异丙醚,更优选为异丙醇。
10.所述的萃取剂为乙醚。
11.所述材料、1,1
’‑
联-2-萘酚、有机溶剂的用量比为0.01mmol:0.005mmol:3-5ml-0.01mmol:0.05mmol:30-50ml。
12.优选的,所述材料、1,1
’‑
联-2-萘酚、有机溶剂的用量比为0.01mmol:0.01mmol:6-10ml。
13.本发明的有益效果是:具有拆分1,1
’‑
联-2-萘酚功能的ir(ⅲ)基手性金属-有机多孔材料,以手性ir(ⅲ)基配合物l1和l2作为构筑模块,分别与手性二胺p1, p2通过希夫碱反应生成手性金属-有机多孔材料,以外消旋1,1
’‑
联-2-萘酚为待拆分分子,加入溶剂,以所得多手性螺旋化合物为固体拆分剂,通过固液搅拌实现对相应单一手性分子的吸附分离。ir(ⅲ)配合物使得目标材料手性得以长久保持;手性二胺的引入,丰富了目标晶态材料孔道的手性信息,实现了与手性分子的严格匹配,大大提高了其拆分效果。与已有技术相比,本发明涉及的手性多孔材料合成简单,产率高;多种手性构筑单元的引入实现对1,1
’‑
联-2-萘酚的高效拆分(ee 高达》99%);多相条件下进行手性拆分,使得固体多孔材料在保持高选择性的同时可以简单的进行分离纯化,具有非常好的应用前景。
附图说明
14.图1是目标化合物lp1和lp5的反应式结构图。
15.图2是目标化合物lp1和lp5的核磁谱图。
16.图3是目标化合物lp1和lp5的质谱谱图。
17.图4是目标化合物lp1和lp5的圆二色谱图。
18.图5是目标化合物lp1和lp5的热重分析图。
19.图6是目标化合物lp1和lp5的磷光发射图。
20.图7是目标化合物lp1和lp5的磷光寿命图。
21.图8是目标化合物lp1、lp3 、lp4 、lp5、lp6拆分1,1
’‑
联-2-萘酚的结果图。
22.图9是目标化合物lp1和lp5的拆分轴手性分子的十次循环结果图。
具体实施方式
23.下面结合具体的实施例对本发明作进一步的说明。
24.实施例1合成目标化合物lp1:将λ-fac-ir-cho (37.0 mg , 0.05 mmol)和1s,2s-环己二胺(11.4 mg,0.10mmol) 置于50 ml三口烧瓶中,向其中加入30 ml体积比为1:2的甲苯和乙腈溶剂,搅拌使其完全溶解,随后加入15 mol%对甲苯磺酸,110 ℃氩气氛围下反应12 h,反应液冷却至室温后,减压蒸馏得到橙黄色固体,将其溶解在二氯甲烷中,乙醚扩散得晶态lp1,38.5 mg,产率90%。元素分析理论值[c
90h78
ir2n
12
·
h2o
·
(c7h8)]: h, 4.87; c, 69.93; n, 9.22。实验值h, 4.81; c, 61.68; n, 9.54. 得到的目标材料结构如图1所示,核磁谱图如图2所示,质谱谱图如图3所示。
[0025]
实施例2合成目标化合物lp3:将λ-fac-ir-cho (37.0 mg , 0.05 mmol)和(s)-(-)-二氨基丙烷(7.4mg,0.10mmol) 置于50 ml三口烧瓶中,向其中加入35 ml体积比为1:2.1的甲苯
和乙腈溶剂,搅拌使其完全溶解,随后加入15 mol%对甲苯磺酸,100 ℃氩气氛围下反应13.5 h,反应液冷却至室温后,减压蒸馏得到橙黄色固体,将其溶解在二氯甲烷中,乙醚扩散得晶态lp3,36.6 mg,产率92%。得到的目标材料lp3结构经过质谱鉴定,hrms-esi m/z calcd for c
81h67
ir2n
12 [m h]
1593.4878 found 1598.4831实施例3合成目标化合物lp4:将λ-fac-ir-cho (37.0 mg , 0.05 mmol)和(1s,2s)-1,2-二苯基乙二胺(25.4 mg,0.12mmol) 置于50 ml三口烧瓶中,向其中加入45 ml体积比为1:2.5的甲苯和乙腈溶剂,搅拌使其完全溶解,随后加入15 mol%对甲苯磺酸,120 ℃氩气氛围下反应10h,反应液冷却至室温后,减压蒸馏得到橙黄色固体,将其溶解在二氯甲烷中,乙醚扩散得晶态lp4,46.5 mg,产率93%。得到的目标材料lp4结构经过质谱鉴定,hrms-esi m/z calcd for c
114h85
ir2n
12 [m h]
2007.6298 found 2007.6200。
[0026]
实施例4合成目标化合物lp5:将δ-fac-ir-cho (37.0 mg , 0.05 mmol)和1r,2r-环己二胺 (11.4 mg,0.10mmol) 置于50 ml三口烧瓶中,向其中加入30 ml体积比为1:2的甲苯和乙腈溶剂,搅拌使其完全溶解,随后加入15 mol%对甲苯磺酸,110 ℃氩气氛围下反应12 h,反应液冷却至室温后,减压蒸馏得到橙黄色固体,将其溶解在二氯甲烷中,乙醚扩散得晶态lp5,37.3 mg,产率87%。元素分析理论值[c
90h78
ir2n
12
·
h2o
·
(c7h8)]: h, 4.87; c, 69.93; n, 9.22。实验值h, 4.97; c, 69.58; n, 9.22. 得到的目标材料结构如图1所示,核磁谱图如图2所示,质谱谱图如图3所示。
[0027]
实施例5合成目标化合物lp6:将δ-fac-ir-cho (37.0 mg , 0.05 mmol)和1r,2r-环戊二胺(15.0 mg,0.15mmol) 置于50 ml三口烧瓶中,向其中加入35 ml体积比为1:2.3的甲苯和乙腈溶剂,搅拌使其完全溶解,随后加入15 mol%对甲苯磺酸,120 ℃氩气氛围下反应10 h,反应液冷却至室温后,减压蒸馏得到橙黄色固体,将其溶解在二氯甲烷中,乙醚扩散得晶态lp6,37.6 mg,产率90%。得到的目标材料lp6结构经过质谱鉴定,hrms-esi m/z calcd for c
87h73
ir2n
12 [m h]
1671.5355 found 1671.5261。
[0028]
实施例6合成目标化合物lp7:将δ-fac-ir-cho (37.0 mg , 0.05 mmol)和(r)-(-)-二氨基丙烷(7.4mg,0.10mmol) 置于50 ml三口烧瓶中,向其中加入50 ml体积比为1:2.0的甲苯和乙腈溶剂,搅拌使其完全溶解,随后加入15 mol%对甲苯磺酸,115 ℃氩气氛围下反应12h,反应液冷却至室温后,减压蒸馏得到橙黄色固体,将其溶解在二氯甲烷中,乙醚扩散得晶态lp7,36.6mg,产率92%。得到的目标材料lp7结构经过质谱鉴定,hrms-esi m/z calcd for c
81h67
ir2n
12 [m h]
1593.4878, found 1593.4819。
[0029]
实施例7实施例1、2、3、4、5所制得的目标材料的预处理操作:lp1、lp3 、lp4 、lp5、lp6的晶体置于乙醚溶液中浸泡24小时进行客体分子的置换,期间每隔6个小时更换一次新鲜的溶剂。滤出晶体并置于80 ℃的真空干燥箱中4小时除去客体分子。预处理的晶体放置于氮气氛围中,以备后续使用。(预处理的目的是将材料中的溶剂进行去除,防止称量不准确,预处理所用溶剂、干燥时间、干燥温度可以改变,将目标材料中溶剂除去即可)
实施例8lp1、lp3 、lp4 、lp5、lp6对1,1
’‑
联-2-萘酚的拆分实验:lp1拆分1,1
’‑
联-2-萘酚:将预处理得到的 lp1(0.01mmol)置于含有1,1
’‑
联-2-萘酚(0.01mmol)的6毫升异丙醇溶剂中。室温搅拌1小时,抽滤,滤饼用异丙醇洗三次,得到的滤饼用乙醚进行萃取,收集的乙醚溶液减压蒸馏,固体异丙醇溶解,利用hplc分析ee值,结果如图8所示。(搅拌1h可以达到饱和,再长没有意义,再短可以达到99的效率,但是不能达到材料吸附饱和,异丙醇洗的目的是将滤饼上残留的反应液洗掉,萃取剂可以用其它的醚类)lp3拆分1,1
’‑
联-2-萘酚:将预处理得到的 lp3(0.01mmol)分别置于含有1,1
’‑
联-2-萘酚(0.02mmol)的15毫升乙酸乙酯溶剂中。室温搅拌1.5小时,抽滤,滤饼用乙酸乙酯洗三次,得到的滤饼用乙醚进行萃取,收集的乙醚溶液减压蒸馏,固体异丙醇溶解,利用hplc分析ee值,结果如图8所示。
[0030]
lp4拆分1,1
’‑
联-2-萘酚:将预处理得到的 lp4(0.01mmol)置于含有1,1
’‑
联-2-萘酚(0.03mmol)的20毫升丙酮溶剂中。室温搅拌2小时,抽滤,滤饼用丙酮洗三次,得到的滤饼用乙醚进行萃取,收集的乙醚溶液减压蒸馏,固体异丙醇溶解,利用hplc分析ee值,结果如图8所示。
[0031]
lp5拆分1,1
’‑
联-2-萘酚:将预处理得到的 lp5(0.01mmol)置于含有1,1
’‑
联-2-萘酚(0.01mmol)的6毫升异丙醇溶剂中。室温搅拌1小时,抽滤,滤饼用异丙醇洗三次,得到的滤饼用乙醚进行萃取,收集的乙醚溶液减压蒸馏,固体异丙醇溶解,利用hplc分析ee值。结果如图8所示。
[0032]
lp6拆分1,1
’‑
联-2-萘酚:将预处理得到的 lp6(0.01mmol)置于含有1,1
’‑
联-2-萘酚(0.05mmol)的45毫升异丙醚溶剂中。室温搅拌2小时,抽滤,滤饼用异丙醚洗三次,得到的滤饼用乙醚进行萃取,收集的乙醚溶液减压蒸馏,固体异丙醇溶解,利用hplc分析ee值,结果如图8所示。
[0033]
通过图8的拆分数据可以看出,l1-i-p1类材料更倾向于吸附r-1,1
’‑
联-2-萘酚,而l2-i-p2类材料对s-1,1
’‑
联-2-萘酚具有特异性吸附;两类材料均具有很高的拆分ee值,能够实现高效拆分1,1
’‑
联-2-萘酚。
[0034]
实施例9lp1,lp5的十次循环试验:预处理得到的 lp1(0.01mmol)和lp5(0.01mmol)分别置于含有1,1
’‑
联-2-萘酚(0.01mmol)的异丙醇溶剂中。室温搅拌半小时,抽滤,滤饼用异丙醇洗三次,得到的滤饼用乙醚进行萃取,收集的乙醚溶液减压蒸馏,固体异丙醇溶解,利用hplc分析ee值;滤饼抽滤,乙醚洗涤,干燥得到的固体再次进行拆分实验,往复十次,结果如图9。经过多次循环使用,lp1和lp5依然保持ee大于99% 的效果。这表明它们具有优异的稳定性,能够重复使用,为工业化应用奠定一定的基础。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。