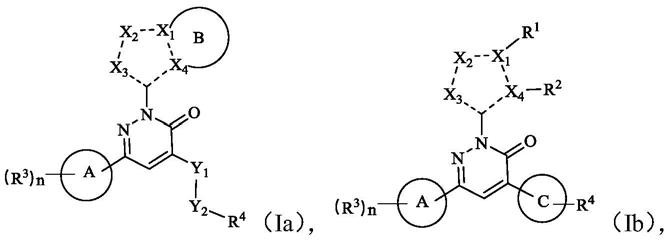
1.本发明涉及一种三唑啉酮类除草剂技术领域,具体的涉及一种磺酰唑草酮的制备方法。
背景技术:
2.三唑啉酮类除草剂磺酰唑草酮又名甲磺草胺,化学名称为n-(2,4-二氯-5-(4-二氟甲基-4,5-二氢-3-甲基-5-氧代-1h-1,2,4-三唑-1-基)苯基)甲磺酰胺。该除草剂最早由美国fmc公司开发,对一年生阔叶杂草、禾本科杂草和莎草有较好的防除效果,其具体的化学结构式如下所示:
[0003][0004]
关于磺酰唑草酮的制备方法也多有报道,如us4743291和us4818275公开了以2, 4-二氯苯胺为起始原料的磺酰唑草酮的合成方法。此外,梁凯(梁凯,徐刚,杨立荣,吴坚平,化学反应工程与工艺,2012,28(5),412-417.)和张元元(张元元,孙永辉,史跃平等,农药,2013,52(4),260-262)等也报道了以2,4-二氯苯胺为起始原料的磺酰唑草酮合成方法,这些方法反应流程基本如下式所示:
[0005][0006]
通过上述的反应方程式可知,这些方法均是以2,4-二氯苯胺为起始原料,经重氮化、还原成肼、成腙、成环、n-二氟甲基化、硝化、还原等步骤得到最终产物;其中梁凯等报道的反应总收率为30.7%,张元元等报道的反应总产率为25.3%。上述所有方法的共同点都是先合成出中间体ⅳ、该中间体结构的右侧为n-二氟甲基取代三唑啉酮环结构,然后再在n-二氟甲基取代三唑啉酮环存在的前提下、在左侧苯环的5位上进行浓硫酸、浓硝酸条件下
的混酸硝化和硝基还原等反应;然而在混酸硝化反应时会生成一定比例的硝化异构体和二硝化产物等副产物导致目标产物的产率低,并且其中的n-二氟甲基取代的三唑啉酮环在混酸和硝基还原条件等条件下存在不稳定的风险,使得采用现有的这些技术方案合成的磺酰唑草酮的成本高、收率低,而且在n-二氟甲基取代的三唑啉酮环存在的受限下、再进行左侧苯环5位上硝化和还原等反应所能采用的方法选择范围窄,不利于磺酰唑草酮这一高效除草剂生产工艺的持续改进。
技术实现要素:
[0007]
本发明针对上述现有技术的不足,提供一种磺酰唑草酮的制备方法,该方法在n
‑ꢀ
二氟甲基取代三唑啉酮环形成之前即进行苯环上的硝化和还原反应,无需将n-二氟甲基取代三唑啉酮环置于浓硫酸、浓硝酸存在的混酸硝化和硝基还原等反应环境中,从而降低副产物的产生、提高了该化合物的合成产率,降低了该化合物的生产成本;并且还避免了n-二氟甲基取代三唑啉酮环在某些硝化条件下不稳定的问题,使硝化和还原方法有更多选择,有利于开发更为先进的磺酰唑草酮生产工艺。
[0008]
为了解决上述技术问题,本发明采用的技术方案为:一种磺酰唑草酮的制备方法,该方法的具体合成路径为:
[0009][0010]
优选的,本技术上述磺酰唑草酮的制备方法,具体的合成步骤包括:
[0011]
(1)向反应容器中加入2,4-二氯苯胺和浓硫酸,冰水浴冷却;然后在0℃或以下滴加由浓硫酸和浓硝酸组成的混合物,滴加完毕后于同样温度下继续反应;反应完成后将反应混合物加入到冰水混合物中,滤出沉淀,在异丙醇/水混合溶剂中结晶得2,4-二氯-5-硝基苯胺;
[0012]
(2)向含有溶剂的反应容器中加入碱和步骤(1)方法制备的2,4-二氯-5-硝基苯胺,搅拌均匀;室温下向反应容器中滴加乙酰氯,滴加完毕后室温继续反应;反应完毕后将反应混合物加入到冰水中,分液,溶剂层洗涤、干燥,过滤,然后除去溶剂、所得粗产物在乙醇/水中结晶得到n-(2,4-二氯-5-硝基苯基)乙酰胺;
[0013]
(3)向反应容器中加入进行硝基还原的材料和步骤(2)方法制备的n-(2,4-二氯-5-硝基苯基)乙酰胺,搅拌均匀后缓慢升温至80-90℃反应;反应完毕后将反应混合物中和
到ph=7.5-8.5,萃取合并萃取剂,干燥、过滤,除去萃取剂所得粗产物在乙醇/ 水中结晶得到n-(5-氨基-2,4-二氯苯基)乙酰胺;
[0014]
(4)向反应容器中加入浓盐酸和步骤(3)方法制备的n-(5-氨基-2,4-二氯苯基) 乙酰胺,搅拌0.5-1.5小时后降温到-10℃以下,氮气保护下滴加亚硝酸钠溶于水中形成的溶液,滴加完毕后相同温度下继续反应1.5-2.5小时;-10℃或以下将氯化亚锡分批加入到上述反应混合物中,加完后搅拌反应0.5-2小时,然后升温到室温再继续反应2-4 小时;调节至ph=8-9,萃取、干燥、过滤,除去萃取溶剂后得到n-(2,4-二氯-5-肼基苯基)乙酰胺;
[0015]
(5)将盐酸和步骤(4)方法制备的n-(2,4-二氯-5-肼基苯基)乙酰胺加入到反应容器中,室温下搅拌均匀;滴加丙酮酸溶于水所形成的溶液,滴加完毕后继续搅拌反应20-40min,然后过滤获得沉淀物,然后用冰水淋洗沉淀2-5次,所得固体干燥得到2
‑ꢀ
(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸;
[0016]
(6)将步骤(5)合成的2-(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸,三乙胺,叠氮磷酸二苯酯和甲苯加入到反应容器中,搅拌均匀;缓慢加热上述的反应混合物至回流;回流4-6小时后停止反应,冷却后的反应混合物加入到氢氧化钠溶液中,分液;然后向其中的下层溶液中加入浓盐酸调节ph=5-6.5,过滤,滤饼分别用清水淋洗2-5 次,所得固体干燥得到n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑-1
‑ꢀ
基)苯基)乙酰胺;
[0017]
(7)将步骤(6)方法制备的n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1, 2,4-三唑-1-基)苯基)乙酰胺,碱,四丁基溴化铵和溶剂加入到反应容器中,搅拌均匀;升温至110-130℃,向反应混合物中通入一氯二氟甲烷气体,停止通气后,110-130℃下继续搅拌反应4-6小时;降温,减压蒸馏除去溶剂,剩余物中加入水,用乙酸乙酯萃取2-5次,合并乙酸乙酯,干燥,过滤,除去乙酸乙酯后所得粗产物在乙酸乙酯/正己烷混合溶剂中结晶得产物n-(2,4-二氯-5-(4-二氟甲基-3-甲基-5-氧代-4,5-二氢-1h-1, 2,4-三唑-1-基)苯基)乙酰胺;
[0018]
(8)将步骤(7)方法制备的n-(2,4-二氯-5-(4-二氟甲基-3-甲基-5-氧代-4, 5-二氢-1h-1,2,4-三唑-1-基)苯基)乙酰胺加入到盐酸溶液中,85-110℃反应3-6小时;反应完毕后冷至室温,调节ph=9-11,然后萃取、合并萃取剂层,干燥、过滤,除去萃取剂后得到4,5-二氢-3-甲基-1-(2,4-二氯-5-氨基苯基)-4-二氟甲基-1,2,4-三唑-5(1h)-酮;
[0019]
(9)将步骤(8)方法制备的4,5-二氢-3-甲基-1-(2,4-二氯-5-氨基苯基)-4-二氟甲基-1,2,4-三唑-5(1h)-酮,碱和n,n-二甲基甲酰胺溶于有机溶剂中,冰水浴下滴加甲基磺酰氯,滴加完毕后升温回流反应7-9小时,反应完毕后将反应混合物冷却至室温,加入水,分出有机溶剂层和水层,然后将有机溶剂层中的有机溶剂除去后得到目标产物。
[0020]
优选的,步骤(1)中所述的2,4-二氯苯胺与浓硫酸(第一次添加的浓硫酸)的添加量比值为0.1-0.2mol:100ml(即每100ml浓硫酸对应添加0.1-0.2mol的2,4-二氯苯胺);浓硫酸和浓硝酸组成的混合物中浓硫酸与浓硝酸的体积比为9-12:1。
[0021]
优选的,步骤(1)中所述的2,4-二氯苯胺与浓硫酸和浓硝酸组成的混合物的添加量比值为0.2-0.25mol:100ml(即每100ml浓硫酸和浓硝酸组成的混合物对应添加 0.2-0.25mol的2,4-二氯苯胺)。
[0022]
优选的,步骤(2)中所述的2,4-二氯-5-硝基苯胺与碱的摩尔比为:1:2-3。
[0023]
优选的,步骤(2)中所述的乙酰氯与碱的摩尔比为:1:1.8-2.2。
[0024]
优选的,步骤(2)中所述的溶剂为二氯甲烷、三氯甲烷、二氯乙烷中至少一种有机溶剂,所述的碱为三乙胺、吡啶或者二异丙基乙基胺中的一种。
[0025]
优选的,步骤(3)中所述的硝基还原的材料为浓盐酸和氯化亚锡,或者fe与醋酸,或者硫化钠或者催化加氢中的一种。
[0026]
进一步优选的,步骤(3)中所述的硝基还原的材料为浓盐酸和氯化亚锡,其中氯化亚锡与n-(5-硝基-2,4-二氯苯基)乙酰胺的摩尔比为:2.5-3.5:1。
[0027]
优选的,步骤(4)中所述的n-(5-氨基-2,4-二氯苯基)乙酰胺与亚硝酸钠和氯化亚锡的摩尔比为:1:1:2.5-3.5。
[0028]
优选的,步骤(5)中所述的盐酸的浓度为4-6mol/l。
[0029]
优选的,步骤(5)中所述的n-(2,4-二氯-5-肼基苯基)乙酰胺与4-6mol/l的盐酸的反应配比为:0.15-0.3mol:100ml。
[0030]
优选的,步骤(5)中所述的丙酮酸与水所形成的溶液中、丙酮酸与水的配比为: 0.15-0.3mol:100ml。
[0031]
优选的,步骤(6)中所述的2-(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸、三乙胺、叠氮磷酸二苯酯的摩尔用量比为1:0.8-1.2:0.8-1.2。
[0032]
优选的,步骤(6)中所述的氢氧化钠溶液的浓度为0.8-1.2mol/l。
[0033]
优选的,步骤(7)中所述的n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1,2, 4-三唑-1-基)苯基)乙酰胺、碱与四丁基溴化铵的摩尔比为1:1:0.08-0.12。
[0034]
优选的,步骤(7)中所述的碱为为碳酸钾、碳酸钠、碳酸铯中的一种,所述的溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、四乙二醇二甲醚中的一种。
[0035]
优选的,步骤(7)中所述n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1,2, 4-三唑-1-基)苯基)乙酰胺与一氯二氟甲烷气体的摩尔比为1:1.4-1.6。
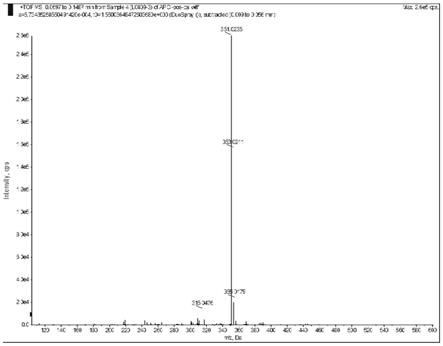
[0036]
优选的,步骤(8)中所述的n-(2,4-二氯-5-(4-二氟甲基-3-甲基-5-氧代-4,5
‑ꢀ
二氢-1h-1,2,4-三唑-1-基)苯基)乙酰胺与盐酸溶液的比值为0.08-0.12mol:200-300ml 1.0mol/l的盐酸溶液;步骤(8)中所述的调节ph采用的是2.5mol/l氢氧化钠溶液来调节,所述的萃取剂为乙酸乙酯。
[0037]
优选的,步骤(9)中所述的4,5-二氢-3-甲基-1-(2,4-二氯-5-氨基苯基)-4-二氟甲基-1,2,4-三唑-5(1h)-酮、碱与甲基磺酰氯的摩尔比为:1:1.2-1.6:1-1.5;所述的碱为吡啶、三乙胺或二乙基异丙基胺中的一种;所述的水层可以再用有机溶剂萃取1-2 次与有机溶剂层合并、然后再除去有机溶剂(上述操作可以充分的回收水层中的目标产物,提高目标产物的产率)。
[0038]
本发明的优点和有益效果:
[0039]
1.本技术以2,4-二氯苯胺为起始原料,通过混酸硝化、乙酰基保护氨基、氯化亚锡还原硝基、重氮化、还原重氮基、成腙、三唑啉酮环的成环以及三唑啉酮环n-二氟甲基化、水解脱乙酰基保护等反应来合成最终的目标产物,这些反应步骤易于实施,而且涉及到的各种原料价廉易得、也不需要改变现有方法的起始原料,反应总产率达到 32.45%,比现有技术产率高,从而可以高效、经济地合成磺酰唑草酮。
[0040]
2.本技术采用的磺酰唑草酮的制备方法,在苯环上进行硝化和硝基还原反应时,由于没有n-二氟甲基取代三唑啉酮环的存在,因此可以选择多种苯环硝化和硝基还原的方
法和条件,而不用考虑在这些方法和条件下n-二氟甲基取代三唑啉酮环的稳定性问题,从而能为开发更为先进的磺酰唑草酮合成工艺提供更多的选择和开发思路,对磺酰唑草酮的合成具有重要意义。
附图说明
[0041]
图1实施例1制备的n-(2,4-二氯-5-肼基苯基)乙酰胺的核磁共振氢谱图。
[0042]
图2实施例1制备的n-(2,4-二氯-5-肼基苯基)乙酰胺的核磁共振碳谱图。
[0043]
图3实施例1制备的n-(2,4-二氯-5-肼基苯基)乙酰胺的质谱图。
[0044]
图4实施例1制备的2-(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸的核磁共振氢谱图。
[0045]
图5实施例1制备的2-(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸的核磁共振碳谱图。
[0046]
图6实施例1制备的2-(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸的核磁共振碳谱图。
[0047]
图7实施例1制备的n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑
ꢀ‑
1-基)苯基)乙酰胺的核磁共振氢谱图。
[0048]
图8实施例1制备的n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑
ꢀ‑
1-基)苯基)乙酰胺的核磁共振碳谱图。
[0049]
图9实施例1制备的n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑-1-基)苯基)乙酰胺的质谱图。
[0050]
图10实施例1制备的n-(2,4-二氯-5-(4-二氟甲基-3-甲基-5-氧代-4,5-二氢-1h-1, 2,4-三唑-1-基)苯基)乙酰胺的核磁共振氢谱图。
[0051]
图11实施例1制备的n-(2,4-二氯-5-(4-二氟甲基-3-甲基-5-氧代-4,5-二氢-1h-1, 2,4-三唑-1-基)苯基)乙酰胺的核磁共振碳谱图。
[0052]
图12实施例1制备的n-(2,4-二氯-5-(4-二氟甲基-3-甲基-5-氧代-4,5-二氢-1h-1, 2,4-三唑-1-基)苯基)乙酰胺的质谱图。
具体实施方式
[0053]
下面通过实施例进一步详细描述本发明,但本发明不仅仅局限于以下实施例。
[0054]
本技术实施例涉及的各个物料的具体用量均采用四舍五入的方式进行定量;其中的原料不做特殊说明均为行业常规原料或者市售产品。
[0055]
实施例1
[0056]
1、向500ml圆底烧瓶内加入2,4-二氯苯胺64.80克(0.40mol)、浓硫酸300ml,冰水浴冷却;0℃下滴加由160ml浓硫酸和16ml浓硝酸组成的混合物,滴加完毕后同样温度下继续反应2小时。然后将反应混合物加入到1500ml冰水混合物中,滤出沉淀,在异丙醇/水混合溶剂(异丙醇/水混合溶剂中的二者体积比为3:1,实施例2与此相同) 中结晶得2,4-二氯-5-硝基苯胺66.24克(0.32mol),产率80%。
[0057]
2、向500ml圆底烧瓶内加入200ml二氯甲烷、62.10克(0.30mol)2,4-二氯-5
‑ꢀ
硝基苯胺和66.79克(0.66mol)三乙胺,搅拌均匀。室温下向烧瓶中滴加86.35克(0.33mol) 乙酰
氯,滴加完毕后室温继续反应5小时。反应完毕后将反应混合物加入到200ml冰水中,分液,二氯甲烷层分别用100ml饱和食盐水洗涤两次,再用100ml水洗涤一次,无水硫酸钠干燥,过滤,除去二氯甲烷后所得粗产物在乙醇/水(乙醇/水中的二者体积比为2:1,实施例2与此相同)中结晶得产物n-(2,4-二氯-5-硝基苯基)乙酰胺(n
‑ꢀ
(5-硝基-2,4-二氯苯基)乙酰胺)70.97克(0.285mol),产率95%。
[0058]
3、向500ml圆底烧瓶内加入200ml浓盐酸、113.77克(0.6mol)氯化亚锡和49.80 克(0.20mol)n-(5-硝基-2,4-二氯苯基)乙酰胺,搅拌均匀后缓慢升温至85℃反应8 小时。反应完毕后将反应混合物用氢氧化钠溶液中和到ph=8,分别用300ml乙酸乙酯萃取三次,合并乙酸乙酯,无水硫酸钠干燥,过滤,除去乙酸乙酯,所得粗产物在乙醇 /水(乙醇/水中的二者体积比为3:1,实施例2与此相同)中结晶得产物n-(5-氨基-2, 4-二氯苯基)乙酰胺42.05克(0.192mol),产率96%。
[0059]
4、向1000ml圆底烧瓶中加入n-(5-氨基-2,4-二氯苯基)乙酰胺65.72克(0.30mol) 和200ml浓盐酸,搅拌1小时后降温到-10℃,氮气保护下滴加亚硝酸钠20.7克(0.30mol) 溶于120ml水中形成的溶液,滴加完毕后相同温度下继续反应2小时。-10℃下将氯化亚锡170.65克(0.90mol)分批加入到上述反应混合物中,加完后搅拌反应1小时,然后升温到室温再继续反应3小时。加入饱和氢氧化钠溶液,调节至ph=9,二氯甲烷萃取(二氯甲烷作为萃取溶剂),无水硫酸钠干燥,过滤,除去二氯甲烷后得产物n-(2,4-二氯-5-肼基苯基)乙酰胺56.16克(0.24mol),产率80%。
[0060]
对获得的产物n-(2,4-二氯-5-肼基苯基)乙酰胺进行氢谱和碳谱的检测:1h nmr (500mhz,cdcl3)δ8.45(1h,s,-nh-co-),7.95(1h,s,-nh-nh2),7.50(1h, s,-c6h-),7.23(1h,s,-c6h-),4.12(2h,s,-nh
2-),2.23(3h,s,-ch3co-) (具体见附图1)。
[0061]
13
c nmr(500mhz,cd3od)δ170.60(-co-),108.10~142.71(-c6h
2-),22.08 (-ch
3-)(具体见附图2)。
[0062]
esi-ms c8h9cl2n3o[m hcooh]
,计算值:279.02,found 279.10(见附图3)。
[0063]
5、将n-(2,4-二氯-5-肼基苯基)乙酰胺46.82克(0.20mol)和100ml浓度为5.0mol/l 的盐酸加入到250ml圆底烧瓶中,室温下搅拌均匀。滴加17.61克(0.20mol)丙酮酸溶于100毫升水所形成的溶液,滴加过程中反应混合物逐渐变浑浊。滴加完毕后继续搅拌反应半小时,过滤沉淀,分别用70ml冰水淋洗沉淀三次,所得固体50℃下真空干燥,得到2-(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸54.72克,产率90%。
[0064]
如附图4-6所示,对获得的产物2-(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸进行谱图检测:
[0065]1h nmr(500mhz,cdcl3)δ8.03(s,1h),7.42(s,1h),2.19(s,3h),2.15 (s,3h)(图4,氢谱)。
[0066]
13
c nmr(500mhz,cd3od)δ170.66,154.09,145.69,139.50,134.27,128.92, 118.00,114.26,110.12,21.98,18.91(图5碳谱)。
[0067]
esi-ms c
11h11
cl2n3o3[m-h]-,计算值:303.0177,305.0148,实测值:302.0185, 304.0141(图6质谱)。
[0068]
6.将2-(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸60.83克(0.20mol),三乙胺20.24克(0.20mol),dppa(叠氮磷酸二苯酯)55.04克(0.20mol),甲苯120ml 加入到250ml圆
底烧瓶中,搅拌均匀。缓慢加热反应混合物至80℃时即有气体放出, 110℃时开始回流,溶液逐渐分层。回流5小时后停止反应,冷却后的反应混合物加入到150ml浓度为1.0mol/l的氢氧化钠溶液中,分液。下层溶液中加入浓盐酸调节ph=6,有大量棕黄色固体析出。过滤,滤饼分别用50ml清水淋洗三次,所得固体50℃下真空干燥得产物n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑-1-基)苯基) 乙酰胺54.36克(0.18mol),产率90%。
[0069]
如附图7-9所示,对获得的产物n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1, 2,4-三唑-1-基)苯基)乙酰胺进行谱图检测:
[0070]1h nmr(500mhz,cd3od)δ8.09(s,1h),7.70(s,1h),2.24(s,3h), 2.19(s,3h)(图7氢谱)。
[0071]
13
c nmr(500mhz,cd3od)δ170.57,154.09,145.69,132.75,130.11,128.79, 127.88,126.83,124.90,22.14,10.65(图8碳谱)。
[0072]
esi-ms c
11h10
cl2n4o2[m h]
,计算值:300.0181,302.0151,实测值:301.0278, 303.0243(图9质谱)。
[0073]
7.将n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑-1-基)苯基) 乙酰胺30.2克(0.10mol),碳酸钾13.82克(0.10mol),四丁基溴化铵3.22克(0.01mol), dmf(n,n-二甲基甲酰胺)150ml加入到250ml圆底烧瓶中,搅拌均匀。升温至120℃,向反应混合物中通入一氯二氟甲烷气体,大约通入13克(0.15mol)后停止通气,120℃下继续搅拌反应5小时。降温,减压蒸馏除去dmf,剩余物中加入150ml水,分别用 150ml乙酸乙酯萃取三次,合并乙酸乙酯,无水硫酸钠干燥,过滤,旋蒸除去乙酸乙酯后所得粗产物在乙酸乙酯/正己烷混合溶剂中结晶(乙酸乙酯/正己烷体积比为2:1,实施例2与此处相同)得产物n-(2,4-二氯-5-(4-二氟甲基-3-甲基-5-氧代-4,5-二氢-1h-1, 2,4-三唑-1-基)苯基)乙酰胺29.90克,产率85%。
[0074]
如附图10-12所示,对获得的产物n-(2,4-二氯-5-(4-二氟甲基-3-甲基-5-氧代
ꢀ‑
4,5-二氢-1h-1,2,4-三唑-1-基)苯基)乙酰胺进行谱图检测:
[0075]1h nmr(500mhz,cd3od)δ8.12(s,1h),7.73(s,1h),7.10~7.39(t,1h), 2.44(s,3h),2.19(s,3h)(图10,氢谱)。
[0076]
13
c nmr(500mhz,cd3od)δ170.60,150.36,142.71,134.62,131.88,130.22,127.68,124.91,110.55,108.10,22.08,11.17(图11,碳谱)。
[0077]
esi-ms c
12h10
cl2f2n4o2[m h]
,计算值:350.0149,352.0119,实测值:351.0235, 353.0211(图12,质谱)。
[0078]
8.将n-(2,4-二氯-5-(4-二氟甲基-3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑-1
‑ꢀ
基)苯基)乙酰胺35.11克(0.10mol)加入到250ml 1.0mol/l的盐酸溶液中,100℃反应5小时。反应完毕后冷至室温,用2.5mol/l氢氧化钠溶液调节ph=10,分别用200ml 乙酸乙酯萃取三次(每次都是200ml),合并乙酸乙酯层,无水硫酸钠干燥,过滤,减压蒸馏除去乙酸乙酯后得固体产物4,5-二氢-3-甲基-1-(2,4-二氯-5-氨基苯基)-4-二氟甲基-1,2,4-三唑-5(1h)-酮29.36克(0.095mol),产率95%。
[0079]
9.将4,5-二氢-3-甲基-1-(2,4-二氯-5-氨基苯基)-4-二氟甲基-1,2,4-三唑-5(1h)
‑ꢀ
酮30.91克(0.1mol),吡啶11.85克(0.15mol)和n,n-二甲基甲酰胺1.5ml溶于250ml 甲苯,冰水浴下滴加甲基磺酰氯13.74克(0.12mol),滴加完毕后升温回流反应8小时,反应
完毕后将反应混合物冷却至室温,加入200ml水,分出甲苯层,水层用100ml甲苯萃取一次,合并甲苯层,除去甲苯后得固体32.91克(0.085mol),产率85%。
[0080]
通过上述实施例可知,本技术的方法获得了化合物甲磺草胺,总产率为32.45%,高于现有技术的目标产物的总收率;此外,更为重要的是,本技术的制备方法还能避免硝化和硝基还原反应对n-二氟甲基取代三唑啉酮环的影响,有利于开发更多的硝化和硝基还原方法来改进该目标化合物的生产工艺。
[0081]
实施例2
[0082]
1、向500ml圆底烧瓶内加入2,4-二氯苯胺72.90克(0.45mol)、浓硫酸320ml,冰水浴冷却。0℃下滴加由170ml浓硫酸和17ml浓硝酸组成的混合物,滴加完毕后同样温度下继续反应2.5小时。将反应混合物加入到1550ml冰水混合物中,滤出沉淀,在异丙醇/水混合溶剂中结晶得2,4-二氯-5-硝基苯胺75.55克(0.365mol),产率81%。
[0083]
2、向250ml圆底烧瓶内加入加入210ml三氯甲烷、51.75克(0.25mol)2,4-二氯
ꢀ‑
5-硝基苯胺和51.35克(0.65mol)吡啶,室温下搅拌均匀。室温下向烧瓶中滴加91.58 克(0.35mol)乙酰氯,滴加完毕后室温继续反应4.5小时。反应完毕后将反应混合物加入到210ml冰水中,分液,三氯甲烷层分别用100ml饱和食盐水洗涤两次,再用110ml 水洗涤一次,无水硫酸钠干燥,过滤,除去三氯甲烷后所得粗产物在乙醇/水中结晶得产物n-(2,4-二氯-5-硝基苯基)乙酰胺(n-(5-硝基-2,4-二氯苯基)乙酰胺)58.77克 (0.236mol),产率94.5%;
[0084]
3、向500ml圆底烧瓶内加入230ml浓盐酸、132.73克(0.7mol)氯化亚锡和56.03 克(0.225mol)n-(5-硝基-2,4-二氯苯基)乙酰胺,搅拌均匀后缓慢升温至85℃反应 8.5小时。反应完毕后将反应混合物用氢氧化钠溶液中和到ph=8,分别用320ml乙酸乙酯萃取三次,合并乙酸乙酯,无水硫酸钠干燥,过滤,除去乙酸乙酯,所得粗产物在乙醇/水中结晶得产物n-(5-氨基-2,4-二氯苯基)乙酰胺46.87克(0.214mol),产率95%。
[0085]
4、向1000ml圆底烧瓶中加入n-(5-氨基-2,4-二氯苯基)乙酰胺76.65克(0.35mol) 和210ml浓盐酸,搅拌1小时后降温到-10℃,氮气保护下滴加亚硝酸钠24.15克(0.35mol) 溶于125ml水中形成的溶液,滴加完毕后相同温度下继续反应2小时。-10℃下将氯化亚锡205.73克(1.085mol)分批加入到上述反应混合物中,加完后搅拌反应1小时,然后升温到室温再继续反应3小时。加入饱和氢氧化钠溶液,调节至ph=9,二氯甲烷萃取,无水硫酸钠干燥,过滤,除去二氯甲烷后得产物n-(2,4-二氯-5-肼基苯基)乙酰胺66.46克(0.284mol),产率81%;
[0086]
5、将n-(2,4-二氯-5-肼基苯基)乙酰胺42.13克(0.18mol)和120ml浓度为4.8mol/l 的盐酸加入到250ml圆底烧瓶中,室温下搅拌均匀。滴加15.85克(0.18mol)丙酮酸溶于90毫升水所形成的溶液,滴加过程中反应混合物逐渐变浑浊。滴加完毕后继续搅拌反应25min,过滤沉淀,分别用65ml冰水淋洗沉淀三次,所得固体50℃下真空干燥,得产物2-(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸48.97克,产率89.5%;
[0087]
6、将2-(2-(5-乙酰氨基-2,4-二氯苯基)亚肼基)丙酸60.82克(0.20mol),三乙胺18.22克(0.18mol),dppa(叠氮磷酸二苯酯)49.54克(0.18mol),甲苯110ml 加入到250ml圆底烧瓶中,搅拌均匀。缓慢加热反应混合物至80℃时即有气体放出, 110℃时开始回流,溶液逐渐分层。回流5.5小时后停止反应,冷却后的反应混合物加入到145ml浓度为1.0mol/l
的氢氧化钠溶液中,分液。下层溶液中加入浓盐酸调节ph=6,有大量棕黄色固体析出。过滤,滤饼分别用50ml清水淋洗三次,所得固体50℃下真空干燥得产物n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑-1-基)苯基) 乙酰胺52.85克(0.175mol),产率87.5%。
[0088]
7、将n-(2,4-二氯-5-(3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑-1-基)苯基) 乙酰胺27.2克(0.09mol),碳酸钠9.54克(0.09mol),四丁基溴化铵3.54克(0.011mol), n,n-二甲基乙酰胺160ml加入到250ml圆底烧瓶中,搅拌均匀。升温至122℃,向反应混合物中通入一氯二氟甲烷气体,大约通入13.87克(0.16mol)后停止通气,122℃下继续搅拌反应5小时。降温,减压蒸馏除去n,n-二甲基乙酰胺,剩余物中加入160ml 水,分别用160ml乙酸乙酯萃取三次,合并乙酸乙酯,无水硫酸钠干燥,过滤,旋蒸除去乙酸乙酯后所得粗产物在乙酸乙酯/正己烷混合溶剂中结晶得产物n-(2,4-二氯-5
‑ꢀ
(4-二氟甲基-3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑-1-基)苯基)乙酰胺26.8克,产率84.5%。
[0089]
8、将n-(2,4-二氯-5-(4-二氟甲基-3-甲基-5-氧代-4,5-二氢-1h-1,2,4-三唑-1
‑ꢀ
基)苯基)乙酰胺38.62克(0.11mol)加入到270ml 1.0mol/l的盐酸溶液中,98℃反应 5.5小时;反应完毕后冷至室温,用2.5mol/l氢氧化钠溶液调节ph=10,分别用220ml 乙酸乙酯萃取三次,合并乙酸乙酯层,无水硫酸钠干燥,过滤,减压蒸馏除去乙酸乙酯后得固体产物4,5-二氢-3-甲基-1-(2,4-二氯-5-氨基苯基)-4-二氟甲基-1,2,4-三唑
ꢀ‑
5(1h)-酮32.45克(0.105mol),产率96%。
[0090]
9.将4,5-二氢-3-甲基-1-(2,4-二氯-5-氨基苯基)-4-二氟甲基-1,2,4-三唑-5(1h)
‑ꢀ
酮37.1克(0.12mol),吡啶13.43克(0.17mol)和n,n-二甲基甲酰胺1.7ml溶于260ml 甲苯,冰水浴下滴加甲基磺酰氯17.18克(0.15mol),滴加完毕后升温回流反应8.5小时,反应完毕后将反应混合物冷却至室温,加入220ml水,分出甲苯层,水层用120ml 甲苯萃取一次,合并至甲苯层中,然后减压蒸馏除去甲苯后得固体38.72克(0.101mol),产率84.6%。
[0091]
本技术公开了一种全新的磺酰唑草酮的制备方法,与现有方法相比先进性在于:该制备方法在苯环上进行硝化和硝基还原反应时,由于没有n-二氟甲基取代三唑啉酮环的存在,因此可以选择多种苯环硝化和硝基还原的方法和条件,而不用考虑在这些方法和条件下n-二氟甲基取代三唑啉酮环的稳定性问题,从而能为开发更为先进的磺酰唑草酮合成工艺提供更多的选择和思路,对磺酰唑草酮的合成具有重要意义;另外,采用本技术所述方法合成磺酰唑草酮有效提高了最终目标产物的产率,降低了磺酰唑草酮的生产成本。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。