一种肝素酶i
技术领域
1.本发明广泛的涉及生物基因工程和发酵工程领域。在该领域内,本发明涉及肝素酶i及其编码基因,本发明进一步提供了一种使用重组载体和宿主细胞制备肝素酶i的方法。
背景技术:
2.肝素酶(heparinase)是一类作用于肝素(heparin)或者硫酸乙酰肝素(heparan sulfate)的多糖裂解酶,用于研究肝素酶及其底物多糖肝素之间的相互作用,有助于阐明多糖裂解酶的作用机制,在解析肝素等复杂粘多糖的结构及其生物学功能、解析人体内凝血和抗凝血机制、只会被低分子的抗凝血药物低分子的抗凝血药物低分子肝素和临床等多方面都有重要用途。肝素酶在许多微生物中发现,其中来自肝素黄杆菌的肝素酶主要有三种,分别为肝素酶i(ec4.2.2.7)、肝素酶ⅱ(no ec code)与肝素酶ⅲ(ec4.2.2.8)。
3.天然的肝素酶i通常是从肝素黄杆菌发酵液中提纯获得的,通常需要经过多步的色谱纯化,酶的活性损失极大,产率低,很难超过10%,而且其诱导添加物肝素钠目前只能从动物(主要是猪和牛)小肠粘膜提取,工艺复杂,成本较高,严重限制了肝素酶i的产量及应用。
技术实现要素:
4.目前现有方法制得的肝素酶i的稳定性较差,利用现有方法制得的肝素酶i以液体形式存放在4℃环境下,在很短的时间内活性即降低为原来的一半,而经过一次冻融、一次冻干,其活性只能保持为原来的45%和25%。因此,在本领域中,需要对现在有的肝素酶i及其制备方法进行改进。
5.有鉴于此,本技术的发明人进行了深入的研究,特提出本发明。
6.通过基因工程技术和分子生物学的方法,研究开发出一种稳定性强的肝素酶i以及利用重组细胞生产肝素酶i的方法。
7.本发明提供了一种肝素酶i及其编码基因,相对于原始肝素酶i,本发明提供的肝素酶i在不影响酶活的条件下,稳定性更强,本发明还提供了一种用于制备所述肝素酶i的方法。
8.为达到此发明目的,本发明采用以下技术方案:
9.在一个方面,本发明提供了一种肝素酶i,所述肝素酶i包括如seq id no:02所示的氨基酸序列,所述氨基酸序列是将原始肝素酶i(如seq id no:01)的氨基酸序列多个位置的谷氨酰胺(q)定点突变为丙氨酸(a)得到的,同时为了提高蛋白纯化效率,在肝素酶i的c端添加strepii标签序列,细胞粗提物可以通过脱硫生物素纯化柱,借助strepii标签于生物素之间的相互作用,实现目标肝素酶i的纯化。
10.本发明还提供了一种编码上述肝素酶i的核苷酸序列。
11.作为优选方案,所述核苷酸的序列如seq id no:03所示。
12.在另一个方面,本发明提供的一种重组载体,所述重组载体包含上述核苷酸序列。
13.进一步的,所述重组载体包括真核细胞重组表达载体。
14.更进一步的,所述真核细胞重组表达载体包括ppink-hc、ppiczaa、ppicz a中的任意一种;
15.作为优选方法,所述真核细胞重组表达载体为ppink-hc。
16.本发明还提供了一种宿主细胞,所述宿主细胞包含上述重组载体。
17.进一步的,所述宿主细胞为毕赤酵母或酿酒酵母中的一种;
18.更进一步的,所述宿主细胞为毕赤酵母。
19.在一个方面,本发明提供了一种肝素酶i的制备方法,所述制备方法包括以下步骤:
20.首先合成编码上述肝素酶i的核苷酸序列,随后将核苷酸序列与真核细胞重组表达载体结合,得到重组载体;
21.将重组载体转入宿主细胞,随后诱导表达,并经过纯化得到所述肝素酶i。
22.优选的,所述宿主细胞为ppink-hc、ppiczaa、ppicza中一种,进一步优选的方案,所述真核细胞重组表达载体为ppink-hc。
23.优选的,在上述制备方法中,所述核苷酸序列与真核细胞重组表达载体结合重组载体步骤按照pichiapink system试剂盒的操作说明进行操作。
24.优选的,在上述制备方法中,所述诱导表达步骤包括:将重组表达载体转化酵母菌、蛋白胨和ynb,加入水和磷酸缓冲液,分装bmmy,余下培养基加入甘油,再分装bmgy,挑取阳性转化子接种于bmgy培养基的摇瓶中,培养、离心去上清,取bmmy重悬菌体,加入装有20-25ml bmmy培养基的摇瓶中,控制起始od600约为1,继续培养,定时取样并添加甲醇,测od600和外源蛋白表达量,发酵完成后,离心收集发酵液。
25.优选的,在上述制备方法中,所述纯化步骤包括:收集菌体(3~6℃),每100ml收集的菌体用1~2ml buffer w(事先在3~6℃预冷)悬液,加入蛋白酶抑制剂,在冰水混合物上通过超声破碎细胞,得到裂解液。然后进行纯化,具体的,用buffer w清洗纯化柱,取0.5-10cvs的裂解液(3~6℃)缓慢上柱,等样品完全进柱后,用buffer w洗柱,并收集每部分的洗脱液,加入4~7倍的buffer e并在每段收集,全程在低温层析柜中操作。
26.进一步的,所述纯化柱为脱硫生物素纯化柱。
27.发明效果
28.本发明提供的肝素酶i,其包括如seq id no:02所示的氨基酸序列。本发明对原始肝素酶i的氨基酸序列(如seq id no:01所示)中可能影响肝素酶i稳定性的蛋白酶酶切位点进行突变,具体地,将天然肝素酶i(如seq id no:01)的氨基酸序列多个位置的谷氨酰胺(q)定点突变为丙氨酸(a)。上述点突变后得到的肝素酶i,酶活没有明显的下降,且在30度条件下,与原始的肝素酶i相比,酶活稳定性有明显提高。
附图说明
29.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前
提下,还可以根据这些附图获得其他的附图。其中:
30.图1为本发明实施例4提供的纯化电泳鉴定结果;
31.图2为本发明实施例6提供的酶活稳定性分析。
具体实施方式
32.下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
33.本发明提供一种肝素酶i,具体的,是将原始肝素酶i的氨基酸序列中42、102、209三个位点的谷氨酰胺(q)定点突变为丙氨酸(a),所述肝素酶i包括如seq id no:02所示的氨基酸序列。
34.所述原始的肝素酶i氨基酸序列如seq id no:01所示,来自于ncbi数据库中已经公布的肝素酶i的序列,ncbi数据库网址为:https://www.ncbi.nlm.nih.gov/protein/acb38160.1。
35.肝素酶i可以切断肝素和硫酸乙酰肝素在葡萄糖胺和
○‑
硫酸艾杜糖醛酸之间的连键,产物主要是二糖,也能切断肝素分子中的抗凝血酶ⅲ结合五糖位点,对肝素类物质具有很好的降解作用,但原始肝素酶i的稳定性很差,通常情况下,得到纯化的肝素酶i需要多步操作,在这个过程中,酶的活性损失巨大,不到10%的产率低限制了肝素酶i的应用。虽然可以在纯化过程中使用某些物质提高酶的使用寿命,但要从根本上解决酶的不稳定性难题还要从其结构和催化机制上进一步研究。
36.原始肝素酶i氨基酸序列中42、102、209三个位点为在肝素酶i结构中处于蛋白外侧负电荷聚集区以及结构中较为薄弱的位置,当肝素酶i被表达出来后,肝素酶i可能会遭受蛋白酶的攻击而被降解,尤其是在肝素酶纯化过程中,菌体被裂解后,释放出大量蛋白酶,肝素酶i极易被降解,影响肝素酶i稳定性。因此本发明分别对42、102、209这三个位点做了逐一的氨基酸替换以及复合突变,具体的,将三个位点的谷氨酰胺(q)定点突变为丙氨酸(a),能够显著提高肝素酶i的稳定性。同时,通过活性测定,证明对该位点进行突变改造不会影响肝素酶i的活性,并且,相对于原始肝素酶i具有更好的稳定性。
37.本发明提供一种编码上述肝素酶i的核苷酸序列,编码上述肝素酶i的核苷酸序列为能够表达出上述肝素酶i的任意一种核苷酸序列,作为一种优选实施方式中,所述核苷酸序列如seq id no:03所示。
38.本发明提供一种重组载体,所述重组载体包含上述核苷酸序列,所述重组载体用于肝素酶i的表达。
39.所述重组载体是指能够插入外源性dna序列,并进行自主复制的一段dna序列,而有意识地修饰的氨基酸序列或核苷酸序列,通常包括原核表达载体和真核细胞重组表达载体。
40.本发明提供的重组载体可以是编码所述肝素酶i的任何重组载体,在本发明的一种优选实施方式中,所述重组载体为真核细胞重组表达载体,包括但不限于ppic9k、ppiczaa、pgapzaa、ppic3.5k、ppink-hc、ppiczaa、ppicza。
41.所述真核细胞重组表达载体可以产生糖基化的可溶性表达的肝素酶i,而原核表
达载体表达的肝素酶i,不能对蛋白进行糖基化,其蛋白大部分为无活性的包涵体形式,无法纯化获得有活性的肝素酶i。因此,相比于原核表达载体,真核细胞重组表达载体得到的肝素酶i具有更高的活性。
42.在本发明中,作为一种优选的实施方案,所述真核细胞重组表达载体包括ppink-hc、ppiczaa、ppicza中一种,进一步优选的方案,所述真核细胞重组表达载体为ppink-hc。
43.本发明提供一种宿主细胞,所述宿主细胞包含上述重组载体,用于肝素酶i的表达。
44.本发明提供的宿主细胞可以是用于表达所述肝素酶i的任何宿主细胞,包括但不限于单细胞的原核生物、真核生物生物体、在细胞培养物中生长时来自高等植物或者动物的单细胞。
45.作为单细胞原核生物的实例,可以举出大肠杆菌、枯草芽孢杆菌、放线菌等。
46.作为真核生物生物体的实例,可以举出酵母、曲霉等。
47.作为本发明的优选实施方式,所述宿主细胞为毕赤酵母或酿酒酵母中的一种,进一步优选的方案,所述宿主细胞为毕赤酵母。
48.毕赤酵母作为宿主细胞表达外源蛋白是一种新的高效的表达系统,它含有特有的强有力的醇氧化酶基因启动子,用甲醇可严格地调控外源基因的表达,且外源蛋白基因遗传稳定,其以高拷贝数整合到毕赤酵母基因组中,不易丢失并能够得到高表达菌株,毕赤酵母作为真核表达系统,具有真核生物的亚细胞结构,也具有糖基化、脂肪酰化、蛋白磷酸化等翻译后修饰加工功能。毕赤酵母的培养成本很低,所用发酵培养基十分廉价,一般碳源为甘油或葡萄糖及甲醇,其余为无机盐,培养基中不含蛋白,有利于下游产品分离纯化,产物易分离。
49.本发明提供一种肝素酶i的制备方法,所述制备方法包括以下步骤:首先合成编码本文中涉及的肝素酶i的核苷酸序列,随后将核苷酸序列与真核细胞重组表达载体结合,得到重组载体;将重组载体转入宿主细胞,随后诱导表达,再经纯化得到肝素酶i。
50.在本发明一种优选的实施方案中,在上述制备方法中,所述合成编码本文中涉及的肝素酶i的核苷酸序列步骤如下:
51.首先将原始肝素酶i的氨基酸序列逆序翻译为核苷酸序列,优选的,得到的核苷酸序列应为所选宿主细胞所偏爱的,本发明中,所翻译的核苷酸序列为毕赤酵母所偏爱,然后将翻译得到的核苷酸序列进行定点突变,突变后得到目标肝素酶i的核苷酸序列。
52.在本发明一种优选的实施方案中,所述肝素酶i的核苷酸序列与真核细胞重组表达载体结合,得到重组载体步骤如下:
53.先将重组表达载体上连接点的酶切位点断开,然后将肝素酶i核苷酸与断开后的表达载体连接,构建出所述重组表达载体。
54.在本发明一种优选的实施方案中,在上述制备方法中,所述宿主细胞为ppink-hc、ppiczaa、ppicza中一种,进一步优选的方案,所述真核细胞重组表达载体为ppink-hc。
55.在本发明一种优选的实施方案中,在上述制备方法中,所述核苷酸序列与真核细胞重组表达载体结合重组载体步骤按照pichiapink system试剂盒的操作说明进行操作。
56.在本发明一种优选的实施方案中,在上述制备方法中,所述诱导表达过程步骤如下:
57.将重组表达载体转化酵母菌、蛋白胨和ynb,加入水和磷酸缓冲液,分装bmmy,余下培养基加入甘油,再分装bmgy,挑取阳性转化子接种于bmgy培养基的摇瓶中,培养、离心去上清,取bmmy重悬菌体,加入装有20-25ml bmmy培养基的摇瓶中,控制起始od600约为1,继续培养,定时取样并添加甲醇,测od600和外源蛋白表达量,发酵完成后,离心收集发酵液。
58.所述ynb培养基又称无氨基酵母氮培养基,用于酵母菌的发酵培养。
59.所述bmmy培养基即诱导表达培养基,用于甲醇诱导毕赤酵母重组菌株分泌表达目的蛋白。
60.所述bmgy培养基即高密度菌体培养基,用于蛋白表达前毕赤酵母重组菌株的培养,以获得产生保密度菌体。
61.所述蛋白胨即为将肉、酪素或明胶用酸或蛋白酶水解后干燥而成的外观呈淡黄色的粉剂。
62.在本发明一种优选的实施方案中,在上述制备方法中,所述纯化步骤如下:
63.收集菌体(3~6℃),每100ml收集的菌体用1~2ml buffer w(事先在3~6℃预冷)悬液,加入蛋白酶抑制剂,在冰水混合物上通过超声破碎细胞,得到裂解液。然后进行纯化,具体的,用buffer w清洗纯化柱,取0.5-10cvs的裂解液(3~6℃)缓慢上柱,等样品完全进柱后,用buffer w洗柱,并收集每部分的洗脱液,加入4~7倍的buffer e并在每段收集,全程在低温层析柜中操作。
64.所述buffer w即结合缓冲液,当粗蛋白上样时,可将目标蛋白吸附在纯化柱上。
65.所述buffer e即洗脱缓冲液,用于将目标蛋白从纯化柱上洗脱下来进行收集。
66.作为优选的实施方式,所述纯化操作步骤全程在4℃条件下进行。
67.作为优选的实施方式,所述buffer e为6倍。
68.所述纯化柱可以使用本领域常用的用于蛋白纯化的各种类型的纯化柱,包括但不限于亲和类型、离子交换类型、疏水作用类型、尺寸排阻类型。作为本发明的优选方案,所述纯化柱为脱硫生物素纯化柱,其为亲和纯化柱中的链霉亲和素突变体填料的纯化柱,为了提高蛋白纯化效率,本发明在所述肝素酶i的c端添加strepii标签序列,借助strepii标签与脱硫生物素纯化柱中生物素之间的相互作用,实现目标蛋白肝素酶i的更好的纯化效果。
69.有益效果:
70.本发明提供的肝素酶i,在不影响酶活的情况下,与原始的肝素酶i相比,在30度条件下的酶活稳定性有明显提高,酶活半衰期提高将近一倍,稳定性更强,本发明提供的一种肝素酶i的制备方法,生产效率高、经济性强,同时对环境友好,有效缓解了现有肝素酶i产品制备工艺复杂、产量低、环境污染大且价格昂贵的问题。
71.本发明的以下实施例仅用来说明实现本发明的具体实施方式,这些实施方式不应理解为对本发明的限制。其他的任何在未背离本发明的精神实质与原理下作出的改变、修饰、替代、组合、简化,均视为等效的置换方式,落在本发明的保护范围之内。
72.实施例
73.下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径购买得到。
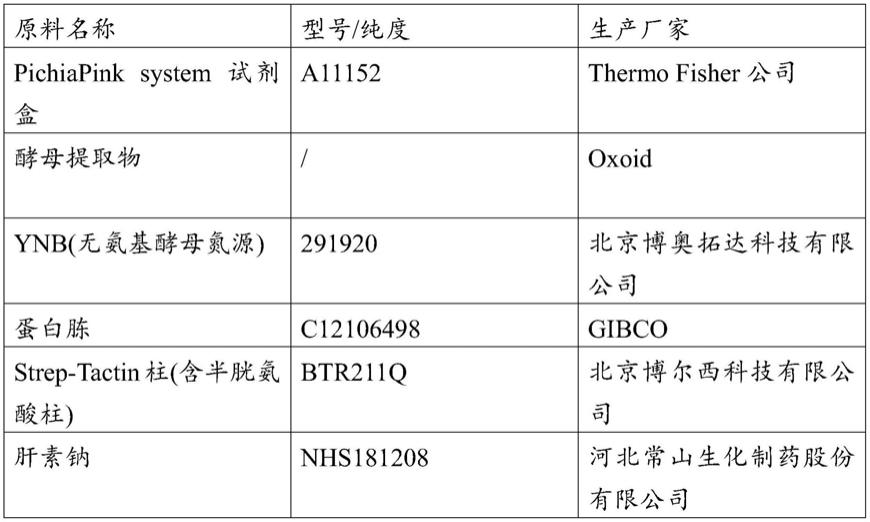
74.表1所使用的实验原料
[0075][0076]
实施例1改进肝素酶i的核苷酸序列优化并构建表达载体
[0077]
(a)原始肝素酶i的氨基酸序列是来自于ncbi数据库中已经公布的肝素酶i的序列,ncbi数据库网址为:https://www.ncbi.nlm.nih.gov/protein/acb38160.1。
[0078]
(b)由苏州金唯智生物技术有限公司,按照毕赤酵母密码子偏爱性数据表中毕赤酵母的密码子使用偏好,将该蛋白质序列逆翻译为dna序列,使该dna序列的密码子均为毕赤酵母偏爱的。
[0079]
(c)对潜在的蛋白酶切位点进行突变改造,将改造后的序列分别进行全序列合成,得到突变体核苷酸序列。
[0080]
(d)在ppink-hc重组表达载体上,选ecori和ecorv两个酶切位点断开表达载体,将突变体核苷酸序列与断开后的表达载体连接,构建hepi-ppink-hc重组表达载体。
[0081]
实施例2感受态制备及电转化
[0082]
按照pichiapink system试剂盒的操作说明进行操作。
[0083]
实施例3诱导表达
[0084]
一、培养基配置:
[0085]
(1)bmgy液体培养基:1%(w/v)酵母提取物,2%(w/v)蛋白胨,1.34%(w/v)ynb培养基,1%(w/v)甘油,10%(v/v)1m ph 6.0的磷酸缓冲液。115℃灭菌20min;
[0086]
(2)bmmy液体培养基:1%(w/v)酵母提取物,2%(w/v)蛋白胨,1.34%(w/v)ynb培养基,1%(v/v)甲醇,10%(v/v)1m ph 6.0的磷酸缓冲液。115℃灭菌20min。
[0087]
(3)ynb培养基:称取134gynb粉末,超纯水溶解定容至1l,0.45um无菌滤膜过滤除菌,4℃保存备用。
[0088]
二、诱导表达:称量酵母提取物、蛋白胨和ynb,加入水和磷酸缓冲液,分装bmmy,余下培养基加入甘油,再分装bmgy即可。
[0089]
三、发酵过程:
[0090]
挑取三株阳性转化子接种于装有5-10ml bmgy培养基的50ml摇瓶中,30℃、250rpm条件下培养至od600(600nm时测定的光密度值)=5(16-18小时),3000g、3min离心去上清,
取1ml bmmy重悬菌体,加入装有25ml bmmy培养基的250ml摇瓶中,控制起始od600约为1,然后在30℃、250rpm条件下培养,每24小时取样(~200ul)和添加1%甲醇,测od600和外源蛋白表达量,发酵96小时后停止发酵,离心收集发酵液。
[0091]
实施例4纯化
[0092]
一、纯化前细胞破碎裂解液的处理
[0093]
buffer w溶液配比:20mm na2hpo4,0.28m nacl,6mm kcl,ph7.4
[0094]
buffer e溶液配比:20mm na2hpo4,0.28m nacl,6mm kcl,2.5mm脱硫生物素,ph7.4
[0095]
收集菌体(4500g,15min,4℃),每100ml收集的菌体用1ml buffer w(事先在4℃预冷)悬液,加入蛋白酶抑制剂,在冰水混合物上通过超声破碎细胞,得到裂解液。
[0096]
二、纯化及鉴定
[0097]
用2cvs的buffer w清洗strep-tactin柱(含半胱氨酸柱),取0.5-10cvs的裂解液(4℃)缓慢上柱,等样品完全进柱后,用5cvs的buffer w洗柱,并收集每部分的洗脱液,加入6倍的0.5cvs的buffer e并在每段(0.5cvs)收集。收集每一部分并各取20μl sds-page鉴定,融合标签蛋白通常在2
nd
和5
th
部分。全程在4℃低温层析柜中操作。
[0098]
经sds-page电泳分析,结果如图1所示,表明该方法能够成功获得纯度在90%以上的肝素酶i蛋白,分子量与预期一致。
[0099]
实施例5蛋白活性分析
[0100]
底物为肝素钠,使用紫外可见分光光度计(上海凌光技术有限公司golds54),测定吸光度随时间的变化曲线。扫描波长为232nm,时间为3min。取反应缓冲液(20mm tris(三羟甲基氨基甲烷),200mm nacl,充分溶解后用6m的盐酸调节ph=7.4,4℃保存),与一定量的酶溶液共约1000μl,500μl底物溶液(17mm tris,44mm nacl,3.5mm cacl2,25g/l肝素钠,充分搅拌后用6m的盐酸调节ph=7.0,4℃保存)于石英比色皿,混匀后立即放入分光光度计进行扫描(混匀前反应缓冲液与底物溶液均在30℃水浴中预热至恒温,至少30min),扫描时间70s,取40-60s的数据,结束后计算曲线的斜率k(min-1),则肝素酶的酶活(iu/l)计算公式及推导过程如下:
[0101]
根据比尔定律,吸光度a=εc,其中ε=3800m-1.cm-1,故1500μl反应体系中酶的总活力为15/38k(min-1)iu,若1500μl反应体系中加入酶溶液体积为v(μl),则所加酶溶液的酶活计算如下:
[0102][0103]
本发明选取的六个突变位点分别为2、16、42、102、209、272,做了逐一的氨基酸替换以及复合突变,对酶活性及稳定性的数据进行测试。在肝素酶i结构中,根据肝素酶i的结构信息以及蛋白酶降解位点的基本特征,这六个位点均处于蛋白外侧负电荷聚集区以及结构中较为薄弱的位置,对酶活及稳定性可能具有较大的影响。
[0104]
在相同的发酵、破碎及纯化条件下,突变体的酶活性数据见表2,由表2可知,q2a、q16a、q272a的突变体酶活相对于原始肝素酶i具有明显的下降,这三种突变意义不大;而q42a、q102a、q209a、q42a&q102a、q42a&q102a&q209a的突变体酶活下降都较轻微,其中q42a&q102a&q209a的突变体的酶活最接近原始的肝素酶i的酶活,下降最少。
[0105]
表2:酶活测定结果
[0106]
突变位点酶活性(iu/l)未突变1203.66q42a942.22q102a1035.12q209a901.99q2a398.33q16a455.32q272a711.02q42a、q102a1087.14q42a、q102a、q209a1132.05
[0107]
实施例6热稳定性分析
[0108]
将纯化后的原始序列的肝素酶i和本发明进行q42a&q102a&q209a突变后肝素酶i分别置于冰上,立即检测酶活,以此时的时间记为0,酶活值作为100%。随后将该酶置于30℃温浴,每隔10min进行取样测定酶活,记录此时酶活相对于0时刻的酶活值的比例。检测时间直到肝素酶i到达半衰期停止。比较不同的酶的稳定性时,则根据该酶在相同浓度、相同溶液条件以及相同温浴条件下的失活速率来进行判断。(酶活测定时均平行测3次,取平均值后作为该酶在该时刻的酶活)
[0109]
分析结果见图2,由图2可以看出,在相同温浴条件下,相比于原始序列的肝素酶i,本发明提供的肝素酶i的热稳定性显著上升:肝素酶i酶活半衰期由原来的30min提高到60min左右,升高将近一倍,表明这几个位点的谷氨酰胺的突变,有效提高了肝素酶i在30度℃的温度下的热稳定性。
[0110]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
[0111]
[0112][0113]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。