1.本发明涉及一种碳八馏分选择加氢除苯乙炔催化剂的制备方法,特别是涉及一种抗结焦性能优异的裂解汽油碳八馏分选择加氢除苯乙炔催化剂的制备。
背景技术:
2.苯乙烯(st)是生产聚苯乙烯、abs树脂和丁苯橡胶的重要单体,单靠乙苯脱氢生产苯乙烯的技术很难满足市场对苯乙烯的需求,并且乙苯脱氢法存在生产成本高的缺点。从裂解制乙烯副产碳八馏分中抽提生产苯乙烯,正在成为一条颇具吸引力的新的增产苯乙烯的途径。
3.裂解汽油是乙烯工业的副产物,产量约为乙烯产能的60-70%,利用其中的碳八馏分抽提回收苯乙烯,一套1000kt/a乙烯装置可获取24-42kt/a的苯乙烯,同时可回收混合二甲苯,使裂解碳八馏分从燃料价值升级到化学价值,且其生产成本约为传统乙苯脱氢生产苯乙烯的1/2,具有很强的市场竞争力;同时由于碳八馏分的分离,减轻了裂解汽油后续加氢装置的负荷,也减少了氢气消耗,又避免了裂解汽油加氢催化剂因苯乙烯聚合而引起的中毒。
4.从裂解汽油中回收苯乙烯的方案,目前采用的是萃取蒸馏法,但是碳八馏分中含有4000~15000μg
·
g-1
的苯乙炔(pa),而st与pa的化学结构相似,两者与萃取蒸馏溶剂之间的相互作用也相似,因此通过现有的萃取精馏工艺条件不能实现st与pa的有效分离。这些苯乙炔的存在不仅会增加sm阴离子聚合时的催化剂消耗量,对链长和聚合速度也有影响,而且会影响聚合产品的色泽、气味以及综合性能。所以在对乙烯裂解碳八进行抽提苯乙烯之前,必须对苯乙炔进行选择性加氢,而碳八馏分中含有30~50%的苯乙烯,因此在对苯乙炔进行加氢时,应该尽量降低苯乙烯的加氢损失。加氢产品中苯乙炔含量越低、苯乙烯损失率越小,是考核催化剂的重要指标,也是决定了从裂解汽油碳八馏分中回收苯乙烯工艺的效益。因此开发出一种适用于裂解碳八馏分选择性加氢脱除苯乙炔的催化剂成为该项技术的关键。
5.选择加氢技术在精细化工及聚合物行业有着重要应用,林德拉催化剂无疑是选择加氢反应最著名的催化剂。钯通常被认为是对炔烃选择加氢反应拥有最佳的加氢活性和选择性。
6.专利cn 102649063b公开了一种苯乙烯存在下苯乙炔选择性加氢催化剂,采用氧化铝和氧化硅的复合型载体,活性组分为镍,添加0.05~10w%的一种或多种稀土元素,以及0.01~6w%的选自元素周期表中ib、iib、vib或viib至少一种元素或其氧化物。但是催化剂存在苯乙炔选择性差,苯乙烯损失率高。
7.专利cn1238239a公开了一种双烯烃选择加氢催化剂,该催化剂在δ、θ、α混合相氧化铝载体上负载钯或钯和金,活性组分钯在载体上的质量分数0.05~0.5%,促进剂金在载体上的质量分数0.005~0.05%。载体的制备方法是由薄水铝石粉末经捏合、成型、干燥后,在900~1100℃下烧结2~8h而成。该催化剂在实际应用过程中易结焦阻塞催化剂孔道。
8.专利cn103785858a公开了一种非晶态纳米钯铑合金的制备方法及其催化应用,采用间歇的操作方式能够控制苯乙炔选择还原生成苯乙烯,但是苯乙烯的选择性并不高,且没有考虑长时间的苯乙烯选择性。同样pd-au双金属催化剂(phys.chem.chem.phys.,2017,19,6164-6168)较pd催化剂表现出更优的选择性,但是随着时间延长,苯乙烯的选择性明显下降,长期稳定性不佳,不适合工业中间歇或半间歇的操作,必然导致产品稳定性不佳的问题。
9.但钯催化剂在催化不饱和烃选择加氢过程中易发生不饱和烃的聚合,生成分子量较宽的低聚物,俗称“绿油”。绿油吸附在催化剂表面,并进一步形成结焦,阻塞催化剂孔道,使反应物不能扩散到催化剂活性中心表面,从而导致催化剂活性下降,影响催化剂稳定性和使用寿命。如何降低钯催化剂结焦,成为评价钯系催化剂优良的重要指标。
10.专利cn1736589报道了一种采用完全吸附浸渍法制备的pd/γ-al2o3选择加氢催化剂,催化剂在使用过程中绿油生成量较大。专利cn200810114744.0了发明了一种不饱和烃选择加氢催化剂及其制备方法。该催化剂以氧化铝为载体,以钯为活性组分,通过加入稀土和碱土金属和氟提高催化剂抗杂质和抗结焦性能,但其催化剂选择性并不理想。
11.以上方法制备的催化剂均采用孔径单一分布的催化剂,在固定床反应过程中,受到内扩散的影响,催化剂的选择性较差。具有双峰孔分布的载体,在保证催化剂高活性的同时,大孔的存在可以减少内扩散的影响,提高催化剂选择性。专利zl971187339公开了一种加氢催化剂,载体是一种蜂窝型载体,为大孔径载体,有效的提高了催化剂的选择性。cn1129606公开了一种烃类转化催化剂,其载体催化剂包括氧化铝、氧化镍、氧化铁等,该催化剂中包括两种孔,一种用于提高催化反应表面,另一种有利于扩散。专利cn101433842提供的一种加氢催化剂,其特征是催化剂具有双峰孔分布,小孔部分最可几半径为2~50nm,大孔部分最可几半径为50~250nm,由于催化剂为双峰孔分布,具有良好的加氢活性的同时,又有好的选择性。
12.催化剂的结焦是影响催化剂使用寿命的重要因素。催化剂的活性、选择性和使用寿命构成了催化剂的总体性能,以上所列出方法或对提高催化剂活性、选择性提出了较好的途径,却并没有解决催化剂容易结焦的问题,或者解决了催化剂易生成绿油和结焦的问题,却没有解决选择性的问题。具有大孔结构的载体虽然可以提高选择性,但是因聚合和链增长反应生成的较大分子也容易积留在载体大孔中,造成催化剂结焦失活,影响催化剂使用寿命。
13.专利zl201310114077.7公开了一种加氢催化剂,该催化剂中所述的活性组分有pd、ag、ni,其中pd、ag采用水溶液浸渍法负载,ni是采用w/o微乳液浸渍法负载的。采用该方法后,pd、ag与ni位于不同孔径的孔道中,反应生成的绿油在大孔中饱和加氢,催化剂结焦量降低。但ni的还原温度往往要达到500℃左右,在该温度下还原态的pd原子极易聚集,使催化剂活性大幅度下降,需要大幅度增加活性组分等量以补偿活性损失,但又会引起选择性的下降。
技术实现要素:
14.本发明的目的是提出一种碳八馏分选择加氢除苯乙炔催化剂的制备方法,特别是一种活性高、抗结焦性能好的碳八馏分选择加氢除苯乙炔催化剂的制备方法。
15.一种碳八馏分选择加氢除苯乙炔催化剂的制备方法,所述的催化剂的特征在于载体为氧化铝或主要是氧化铝,并具有双峰孔分布结构,其中小孔的孔径范围为10~25nm,大孔的孔径50~250nm,催化剂至少含有pd、li、ni、cu,每次负载活性组分后均要在60~150℃下干燥1~6小时,在300~700℃下焙烧2~8h,其中ni、cu是以微乳液方式负载,使得ni、cu分布在载体的大孔中;li采用溶液法负载,pd采用溶液法和微乳液法两种方式负载,其中大部分pd采用溶液法负载,少部分pd采用微乳液法负载。
16.采用本发明的方法制得的催化剂,苯乙炔的选择加氢反应发生在pd、li组成的主活性中心,ni和cu以微乳液的形式浸渍在载体的大孔中,反应中生成的胶质在cu与ni组成的活性中心上发生饱和加氢。
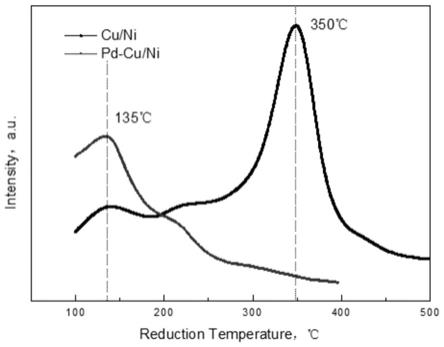
17.本发明人发现,将cu与ni一起负载后,可以大幅度降低ni的还原温度,因为要将nio完全还原,一般需要还原温度达到450℃,甚至到500℃,而这个温度对pd来说太高了,会引起pd的团聚,而加入cu后,形成cu/ni特定结构,则其还原温度与纯ni的还原温度相比可以降低100℃以上,达到350℃,从而缓解还原过程导致pd的团聚。
18.发明人意外发现,将少量pd负载在ni/cu合金的表面,还可以大幅度的降低ni的还原温度,可以达到200℃以下,最低到150℃。
19.对加氢反应而言,一般在催化剂应用前首先需要对加氢催化剂进行还原,保证活性组分以金属态存在,才能使催化剂具有加氢活性。因为催化剂制备过程中,活化是一个高位焙烧过程,在该过程中,金属盐分解为金属氧化物,氧化物会形成团簇,这种团簇一般是纳米尺寸的。不同的氧化物由于化学特性的不同,需要在不同的温度下进行还原。但对纳米尺寸的金属而言,200℃左右度是一个重要临界温度,超过该温度,金属粒子会有十分显著的产生聚集。因此,降低活性组分的还原温度,对加氢催化剂而言,有十分重要的意义。
20.发明人发现:苯乙炔的选择加氢反应发生在pd、li组成的主活性中心,反应中生成的胶质等大分子,容易进入催化剂的大孔中。如在催化剂的大孔中负载了ni、cu组分,其中ni具有饱和加氢功能,胶质组分会在ni-cu组成的活性中心上发生饱和加氢反应。由于双键被加氢饱和,胶质组分不能再发生聚合反应或聚合反应速率大大降低,其链增长反应终止或延缓,不能形成大分子量化合物,容易被物料带出反应器,因此催化剂的表面的结焦程度会大大降低,催化剂的再生周期及运行寿命得到大幅延长。
21.本发明中控制ni/cu合金定位于催化剂大孔的方法是,ni/cu以微乳液的形式负载,控制微乳液的粒径大于载体小孔孔径,而小于大孔的最大孔径。ni和cu金属盐包含在微乳液中,由于空间阻力的原因,难于进入尺寸较小的载体孔道中,因此ni和cu主要集中在大孔中。
22.在用溶液法负载钯的过程中,由于小孔的虹吸作用,含有钯的溶液,更快地进入小孔中,钯是以氯钯酸离子的形式存在,由于该离子可以与载体表面的羟基形成化学键,使钯被快速的靶定,因此溶液进入孔道的速率越快,负载的速度就越快。所以在以溶液法浸渍pd的过程中,更容易负载于小孔中。
23.本发明中,pd的负载采用溶液法和微乳液法两种方式负载,即大部分pd采用溶液法负载,少部分pd以微乳液方式负载,微乳液方式负载时控制微乳液粒径大于小孔最大孔径,小于大孔最大孔径。使得该部分pd分布在载体的大孔中,微乳液法负载的pd的量是ni cu含量的1/100~1/200,微乳液负载pd的步骤是在微乳液负载ni和cu步骤后。
24.本发明采用的载体要求具有双峰孔分布结构,特别是要有孔径在50~250nm的大孔,小孔的孔径为10~25nm。载体为氧化铝或主要是氧化铝,al2o3晶型最好为α、θ或其混合晶型。催化剂载体中氧化铝最好在80%以上,载体中还可含有其它金属氧化物如氧化镁,氧化钛等。
25.该催化剂在制备过程中ni/cu负载以微乳液形态进行浸渍。pd负载以溶液法及微乳液两种方法浸渍,li的负载则以溶液饱和浸渍方法进行。
26.本发明并不特别限定微乳液方式负载ni、cu、pd的过程,只要是能形成大于小孔最大孔径,小于大孔最大孔径的微乳液粒径,都能使得ni、cu、pd分布在载体的大孔中。
27.本发明还推荐了一种方法,该微乳液方式负载过程包括:将前驱体盐溶于水中,加入油相、表面活性剂和助表面活性剂,充分搅拌形成微乳液,其中油相为烷烃或环烷烃,表面活性剂为离子型表面活性剂和\或非离子型表面活性剂,助表面活性剂为有机醇。
28.油相、表面活性剂和助表面活性剂的种类和加入量本发明也不特别限定,可根据前驱体盐、载体的孔结构来确定油相、表面活性剂和助表面活性剂的种类和加入量。
29.本发明推荐的油相为饱和烷烃或环烷烃,优选c6~c8饱和烷烃或环烷烃,优选环己烷、正己烷;表面活性剂为离子型表面活性剂和\或非离子型表面活性剂,优选非离子型表面活性剂,更优选是聚乙二醇辛基苯基醚或十六烷基三甲基溴化铵;助表面活性剂为有机醇,优选c4~c6醇类,更优选正丁醇和\或正戊醇。
30.本发明微乳液中,推荐的水相和油相重量比为1~2,表面活性剂和油相重量比为0.4~0.7,表面活性剂和助表面活性剂的重量比为1~1.2,控制微乳液粒径大于25nm小于250nm,即微乳液粒径大于小孔的最大孔径而小于大孔的最大孔径。优选的条件是表面活性剂和助表面活性剂重量比1~1.2,水相和油相重量比是1.5~1.7,表面活性剂和油相重量比是0.5~0.6,可控制微乳液粒径大于70nm小于80nm,更有利于活性组分的负载,制备的催化剂中活性组分尤其是ni和cu的分布更均匀。
31.pd的溶液法负载在微乳液负载ni与cu步骤前或步骤后均可,pd的微乳液负载在微乳液负载ni与cu步骤之后,li的饱和浸渍负载在pd的溶液法负载步骤之后。在采用两个微乳液法的两个负载过程中,微乳液的粒径可以相同,也可以不同,最好相同。
32.本发明还提供了具体的一种碳八馏分选择加氢除苯乙炔催化剂的制备方法,包含以下步骤:
33.(1)将pd配制成活性组分浸渍液,调ph为1.5~2.5,载体加入pd活性组分浸渍液中,浸渍吸附0.5~4h后,60~150℃干燥1~6小时,300~700℃条件下焙烧1~6h,得到半成品催化剂a。
34.(2)将ni和cu的前驱体盐溶于水中,加入计量好的油相、表面活性剂和助表面活性剂,充分搅拌形成微乳液。控制微乳液粒径大于载体小孔最大孔径,小于大孔最大孔径。将半成品催化剂a加入到制好的微乳液中浸渍0.5~4小时后,滤除余液,在60~150℃下干燥1~6小时,在300~700℃下焙烧1~6h,得到半成品催化剂b。
35.(3)li的负载以饱和浸渍方法进行,即配制的li盐的溶液是载体饱和吸水率的80~110%。将半成品催化剂b浸渍在所配制的溶液中,摇晃均匀,老化0.5~4h,在60~150℃干燥1~6小时,300~700℃焙烧,时间为1~6小时。得到半成品催化剂c。
36.(4)将pd的前驱体盐溶于水中,加入计量好的油相、表面活性剂和助表面活性剂,
充分搅拌形成微乳液。控制微乳液粒径大于载体小孔孔径、小于载体的大孔孔径。将半成品催化剂c加入到制好的微乳液中浸渍0.5~4小时后,滤除余液,在60~150℃下干燥1~6小时,在300~700℃下焙烧1~6h,得到所要的催化剂。
37.上述步骤(1)中的载体为氧化铝或主要是氧化铝,al2o3晶型最好为α、θ或其混合晶型。催化剂载体中氧化铝最好在80%以上,载体中还可含有其它金属氧化物如氧化镁、氧化钛等。
38.上述步骤(1)中的载体可以是球形、圆柱形、三叶草形、四叶草形等。
39.上述步骤(1)、(2)、(3)(4)中所述的ni、cu、li和pd的前驱体盐为可溶性盐,可以是其硝酸盐、氯化盐或者其他可溶性盐。
40.在两个采用微乳液法的负载过程中,w/o微乳液的粒径可以相同,也可以不同,最好相同。
41.微乳液粒径大于小孔的孔径而小于大孔的孔径。由于空间阻力的原因,乳液粒子难于进入尺寸较小的载体孔道中,主要进入载体的大孔中。
42.使用本发明方法得到的催化剂具有以下特性:在加氢反应开始时,由于钯的加氢活性高,而且分布在小孔中,因而苯乙炔的选择性加氢反应主要发生在小孔中。随着催化剂运行时间的延长,催化剂表面生成了一部分分子量较大的副产物,这些物质由于分子尺寸较大,较多的进入大孔中,而且停留时间较长,会在镍催化剂的作用下,发生双键的加氢反应,而生成不含孤立双键的芳香烃,不再生成分子量更大的物质。使用本发明方法得到的催化剂,能大幅降低催化剂的还原温度,最低可降至150~200℃。
43.本发明人还发现,使用本制备方法的催化剂进行碳八馏分选择加氢除苯乙炔的反应,即使反应原料中含较多苯乙炔、苯乙烯,催化剂胶质生成量大幅增加,催化剂的结焦量没有明显上升,选择加氢活性和选择性也没有明显的下降趋势。
附图说明
44.图1为微乳液法负载cu/ni与pd-cu/ni制备得到样品的程序升温还原结果(tpr)。
具体实施方式
45.以下对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和过程,但本发明的保护范围不限于下述的实施例,下列实施例中未注明具体条件的实验方法,通常按照常规条件。
46.分析方法:
47.本发明催化剂在制备过程中用到以下表征方法:动态光散射粒径分析仪,在m286572动态光散射分析仪上分析ni-cu合金的微乳液粒径分布;采用美国micrometrics公司生产的tristar 3000型物理吸附仪进行载体的n2物理吸附测试,分别用bet公式和bjh方程计算样品的比表面积、孔径分布和孔体积;采用美国麦克公司9510型压汞仪测定载体的比表面积和孔结构;在a240fs原子吸收光谱仪上,测定催化剂中pd、li、ni和cu的含量。
48.所采用的十六烷基三甲基溴化铵(简称ctab)、聚乙二醇辛基苯基醚(简称triton x-100)、十二烷基硫酸钠(简称sds),下文均用简称表示。
49.下面通过实施例进一步说明本发明,但并不认为本发明仅局限于此。
50.实施例1
51.催化剂载体:
52.采用市售双峰孔分布球形氧化铝载体,直径为4mm。经过950℃焙烧4h后,双峰孔径分布范围在10~15nm和50~150nm,吸水率63%,比表面积为155m2/g。称取该载体100g。
53.催化剂制备:
54.(1)称取硝酸镍、氯化铜溶于43ml去离子水中,加环己烷43g,加triton x-100 30g,加正丁醇30g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动30min,滤除余液,用去离子水洗剂。在80℃下干燥6小时,在400℃下焙烧6h。称为半成品催化剂a。
55.(2)将氯化钯配制成活性组分浸渍液,调ph为2.0,再将半成品催化剂a浸渍到已配制的pd盐溶液中,浸渍30min后,80℃干燥6小时,500℃条件下焙烧4小时。得到半成品催化剂b。
56.(3)称取硝酸锂配制成溶液,将步骤(2)制备的半成品催化剂b浸渍于所配制的含锂的硝酸锂溶液中,摇动,待溶液全部吸收后,130℃干燥3小时,在500℃焙烧6小时,得到半成品催化剂c。
57.(4)称取氯化钯溶于43ml去离子水中,加环己烷43g,加triton x-100 30g,加正丁醇30g,充分搅拌形成微乳液,将步骤(3)制备的半成品催化剂c放置在制备的微乳液中,摇动30min,滤除余液,用去离子水洗剂。在80℃下干燥6小时,在400℃下焙烧6h。得到所要的催化剂。
58.动态光散射测定制备的微乳液的粒径是56nm。
59.催化剂的还原:
60.使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在200℃温度,还原处理8h。
61.实施例2
62.载体:
63.采用市售双峰孔分布球形氧化铝载体,直径为3mm。经过970℃焙烧4h后,双峰孔径分布范围在10~20nm和55~150nm,吸水率63%,比表面积为140m2/g。称取该载体100g。
64.催化剂制备:
65.(1)称取硝酸镍、氯化铜溶于56ml去离子水中,加正己烷47g,加ctab33g,加正戊醇28g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动90min,滤除余液,在100℃下干燥5小时,在500℃下焙烧4h。称为半成品催化剂d。
66.(2)将氯化钯配制成活性组分浸渍液,调ph为1.8,再将半成品催化剂d浸渍到已配制的pd盐溶液中,浸渍60min后,100℃干燥5小时,400℃条件下焙烧6小时。得到半成品催化剂e。
67.(3)称取碳酸锂配制成溶液,将步骤(2)制备的半成品催化剂e浸渍于所配制的含锂的碳酸锂溶液中,摇动,待溶液全部吸收后,140℃干燥2小时,在500℃焙烧4小时,得到半成品催化剂f。
68.(4)称取氯化钯溶于56ml去离子水中,加正己烷47g,加ctab33g,加正戊醇28g,充分搅拌形成微乳液,将步骤(3)制备的半成品催化剂f浸渍到所制备的微乳液中,摇动
90min,滤除余液,在100℃下干燥5小时,在500℃下焙烧4h。得到所要的催化剂。
69.动态光散射测定制备的微乳液的粒径是65nm。
70.催化剂的还原:
71.使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在150℃,还原处理8h。
72.实施例3
73.载体:
74.采用市售双峰孔分布球形氧化铝载体,直径为4mm。经过980℃焙烧4h后,双峰孔径分布范围在10~20nm和55~190nm,吸水率62%,比表面积为130m2/g。称取该载体100g。
75.催化剂制备:
76.(1)称取硝酸镍、氯化铜溶于59ml去离子水中,加环己烷45g,加sds 27g,加正丁醇25g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动240min,滤除余液,在120℃下干燥3小时,在600℃下焙烧2h。称为半成品催化剂g。
77.(2)称取硝酸钯溶于去离子水中,调ph为2,再将半成品催化剂g浸渍到已配制的pd盐溶液中,浸渍90min后,120℃干燥4小时,500℃条件下焙烧4小时。得到半成品催化剂h。
78.(3)称取硝酸锂配制成溶液,将步骤(2)制备的半成品催化剂h浸渍于所配制的含锂的硝酸锂溶液中,摇动,待溶液全部吸收后,150℃干燥2小时,在500℃焙烧6小时,得到半成品催化剂j。
79.(4)称取硝酸钯溶于59ml去离子水中,加环己烷45g,加sds 27g,加正丁醇25g,充分搅拌形成微乳液,将步骤(3)制备的半成品催化剂j浸渍到所制备的微乳液中,摇动240min,滤除余液,在120℃下干燥3小时,在600℃下焙烧2h。得到所要的催化剂。
80.动态光散射测定制备的微乳液的粒径是70nm。
81.催化剂的还原:
82.使用前放置于固定床反应装置中,用纯氢在150℃温度,还原处理8h。
83.实施例4
84.载体:
85.采用市售双峰孔分布球形氧化铝-氧化钛载体,氧化钛质量分数为20%,直径为3mm。经过1020℃焙烧4h后,双峰孔径分布范围在30~40nm和60~230nm,吸水率61%,比表面积为100m2/g。称取该载体100g。
86.催化剂制备:
87.(1)称取氯化镍、硝酸铜溶于73ml去离子水中,加正己烷43g,加sds 17g,加正戊醇15g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动180min,滤除余液,在80℃下干燥4小时,在600℃下焙烧2h。称为半成品催化剂k。
88.(2)称取氯化钯溶于73ml去离子水中,加正己烷43g,加sds 17g,加正戊醇15g,充分搅拌形成微乳液,将半成品催化剂k浸渍到所制备的微乳液中,摇动180min,滤除余液,在80℃下干燥4小时,在600℃下焙烧2h。称为半成品催化剂m。
89.(3)称取氯化钯溶于去离子水中,调ph为2.0,再将半成品催化剂m浸渍到已配制的pd盐溶液中,浸渍120min后,130℃干燥3小时,600℃条件下焙烧2小时。得到半成品催化剂n。
90.(4)称取硝酸锂配制成溶液,将步骤(2)制备的半成品催化剂n浸渍于所配制的含锂的硝酸锂溶液中,摇动,待溶液全部吸收后,100℃干燥4小时,在600℃焙烧2小时,得到所要的催化剂。
91.动态光散射测定制备的微乳液的粒径是78nm。
92.催化剂的还原:
93.使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在200℃温度,还原处理8h。
94.实施例5
95.载体:
96.采用市售双峰孔分布球形氧化铝-氧化镁载体,氧化镁质量分数为3%,直径为3mm。经过1000℃焙烧4h后,双峰孔径分布范围在15~20nm和60~200nm,吸水率60%,比表面积为100m2/g。称取该载体100g。
97.催化剂制备:
98.(1)称取氯化钯溶于去离子水中,调ph为2.5,再将载体浸渍到已配制的pd盐溶液中,浸渍120min后,130℃干燥3小时,600℃条件下焙烧2小时,得到半成品催化剂o。
99.(2)称取硝酸锂配制成溶液,将步骤(2)制备的半成品催化剂o浸渍于所配制的含锂的硝酸锂溶液中,摇动,待溶液全部吸收后,100℃干燥4小时,在300℃焙烧8小时,得到半成品催化剂p。
100.(3)称取氯化镍、硝酸铜溶于68ml去离子水中,加正己烷40g,加triton x-100 20g,加正己醇18g,充分搅拌形成微乳液,将步骤(2)制备的半成品催化剂p浸渍到所制备的微乳液中,摇动180min,滤除余液,在70℃下干燥6小时,在600℃下焙烧2h,称为半成品催化剂q。
101.(4)称取氯化钯溶于68ml去离子水中,加正己烷40g,加triton x-10020g,加正己醇18g,充分搅拌形成微乳液,将制备的半成品催化剂q浸渍到所制备的微乳液中,摇动180min,滤除余液,在70℃下干燥6小时,在600℃下焙烧2h。得到所要的催化剂。
102.动态光散射测定制备的微乳液的粒径是80nm。
103.催化剂的还原:
104.使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在180℃温度,还原处理8h。
105.实施例6
106.载体:
107.采用市售双峰孔分布球形氧化铝-氧化镁载体,氧化镁质量分数为3%,直径为3mm。经过1000℃焙烧4h后,双峰孔径分布范围在15~20nm和60~200nm,吸水率60%,比表面积为110m2/g。称取该载体100g。
108.催化剂制备:
109.(1)称取氯化镍、硝酸铜溶于69ml去离子水中,加正己烷46g,加sds23g,加正己醇20g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动180min,滤除余液,在70℃下干燥6小时,在600℃下焙烧2h。称为半成品催化剂r。
110.(2)称取氯化钯溶于去离子水中,调ph为1.8,再将半成品催化剂r浸渍到已配制的
pd盐溶液中,浸渍120min后,130℃干燥3小时,600℃条件下焙烧2小时。得到半成品催化剂s。
111.(3)称取碳酸锂配制成溶液,将步骤(2)制备的半成品催化剂s浸渍于所配制的含锂的碳酸锂溶液中,摇动,待溶液全部吸收后,100℃干燥4小时,600℃焙烧2小时,得到半成品催化剂u。
112.(4)称取氯化钯溶于69ml去离子水中,加正己烷46g,加sds23g,加正己醇20g,充分搅拌形成微乳液,将步骤(3)制备的半成品催化剂u浸渍到所制备的微乳液中,摇动180min,滤除余液,在70℃下干燥6小时,在600℃下焙烧2h。得到所要的催化剂。
113.动态光散射测定制备的微乳液的粒径是76nm。
114.催化剂的还原:
115.使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在200℃温度,还原处理8h。
116.实施例7
117.载体:
118.采用市售双峰孔分布球形氧化铝载体,直径为3mm。经过1040℃焙烧4h后,双峰孔径分布范围在20~25nm和70~250nm,吸水率63%,比表面积为85m2/g。称取该载体100g。
119.催化剂制备:
120.(1)称取氯化镍、硝酸铜溶于72ml去离子水中,加正己烷38g,加ctab 19g,加正戊醇16g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动90min,滤除余液,在80℃下干燥5小时,在500℃下焙烧4h,称为半成品催化剂v。
121.(2)称取氯化钯溶于去离子水中,调ph为1.8,再将半成品催化剂v浸渍到已配制的pd盐溶液中,浸渍60min后,100℃干燥5小时,400℃条件下焙烧6小时,得到半成品催化剂w。
122.(3)称取硝酸锂配制成溶液,将步骤(2)制备的半成品催化剂w浸渍于所配制的含锂的硝酸锂溶液中,摇动,待溶液全部吸收后,140℃干燥2小时,在500℃焙烧4小时,得到半成品催化剂x。
123.(4)称取氯化钯溶于72ml去离子水中,加正己烷38g,加ctab19g,加正戊醇16g,充分搅拌形成微乳液,将步骤(3)制备的半成品催化剂x浸渍到所制备的微乳液中,摇动90min,滤除余液,在80℃下干燥5小时,在500℃下焙烧4h得到所要的催化剂。
124.动态光散射测定制备的微乳液的粒径是92nm。
125.催化剂的还原:
126.使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在150℃温度,还原处理8h。
127.表1实施例催化剂各组分含量
[0128][0129]
对比例1
[0130]
使用与实施例1相同的载体,催化剂制备条件与实施例1相同,区别之处在于对比例1不负载cu。
[0131]
催化剂制备:
[0132]
(1)称取硝酸镍溶于43ml去离子水中,加环己烷43g,加triton x-100 30g,加正丁醇30g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动30min,滤除余液,用去离子水洗剂。在80℃下干燥6小时,在400℃下焙烧6h。称为半成品催化剂a1。
[0133]
(2)将氯化钯配制成活性组分浸渍液,调ph为2.0,再将半成品催化剂a1浸渍到已配制的pd盐溶液中,浸渍30min后,80℃干燥6小时,500℃条件下焙烧4小时。得到半成品催化剂b1。
[0134]
(3)称取硝酸锂配制成溶液,将步骤(2)制备的半成品催化剂b1浸渍于所配制的配制含锂的硝酸锂溶液中,摇动,待溶液全部吸收后,130℃干燥3小时,在500℃焙烧6小时,得到半成品催化剂c1。
[0135]
(4)称取氯化钯溶于43ml去离子水中,加环己烷43g,加triton x-100 30g,加正丁醇30g,充分搅拌形成微乳液,将步骤(3)制备的半成品催化剂c1放置在制备的微乳液中,摇动30min,滤除余液,用去离子水洗剂。在80℃下干燥6小时,在400℃下焙烧6h。得到所要的催化剂。
[0136]
动态光散射测定制备的微乳液的粒径是56nm。
[0137]
催化剂的还原:
[0138]
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在200℃,还原处理8h。
[0139]
对比例2
[0140]
催化剂制备过程与对比例1相同,区别之处在于对比例2催化剂还原温度是350℃。
[0141]
对比例3
[0142]
使用与实施例2相同的载体,催化剂制备条件与实施例2相同,区别之处在于对比例3中cu是溶液法负载的。
[0143]
催化剂制备:
[0144]
(1)称取氯化镍溶于56ml去离子水中,加正己烷47g,加ctab33g,加正戊醇28g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动90min,滤除余液,在100℃下干燥5小时,在500℃下焙烧4h。称为半成品催化剂d1。
[0145]
(2)称取氯化钯溶于去离子水中,调ph为1.8,再将半成品催化剂d1浸渍到已配制的pd盐溶液中,浸渍60min后,100℃干燥5小时,400℃条件下焙烧6小时。得到半成品催化剂e1。
[0146]
(3)称取碳酸锂、氯化铜配制成溶液,将步骤(2)制备的半成品催化剂e1浸渍于所配制的配制溶液中,摇动,待溶液全部吸收后,140℃干燥2小时,在500℃焙烧4小时,得到半成品催化剂f1。
[0147]
(4)称取氯化钯溶于56ml去离子水中,加正己烷47g,加ctab33g,加正戊醇28g,充分搅拌形成微乳液,将步骤(3)制备的半成品催化剂f1浸渍到所制备的微乳液中,摇动90min,滤除余液,在100℃下干燥5小时,在500℃下焙烧4h。得到所要的催化剂。
[0148]
动态光散射测定制备的微乳液的粒径是65nm。
[0149]
催化剂的还原:
[0150]
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在350℃温度,还原处理8h。
[0151]
对比例4
[0152]
催化剂制备条件与对比例3相同,区别之处在于对比例4催化剂还原温度为250℃。
[0153]
对比例5
[0154]
使用与实施例3相同的载体,催化剂制备条件与实施例3相同,不同之处在于取消了微乳液法负载pd这一步骤。
[0155]
催化剂制备:
[0156]
(1)称取硝酸镍、氯化铜溶于59ml去离子水中,加环己烷45g,加sds 27g,加正丁醇25g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动240min,滤除余液,在120℃下干燥3小时,在600℃下焙烧2h。称为半成品催化剂g1。
[0157]
(2)称取硝酸钯溶于去离子水中,调ph为2,再将半成品催化剂g1浸渍到已配制的pd盐溶液中,浸渍90min后,120℃干燥4小时,500℃条件下焙烧4小时。得到半成品催化剂h1。
[0158]
(3)称取硝酸锂配制成溶液,将步骤(2)制备的半成品催化剂h1浸渍于所配制的配制含锂的硝酸锂溶液中,摇动,待溶液全部吸收后,150℃干燥2小时,在400℃焙烧6小时,得到所要的催化剂。
[0159]
动态光散射测定制备的微乳液的粒径是70nm。
[0160]
催化剂的还原:
[0161]
使用前放置于固定床反应装置中,用纯氢在150℃温度,还原处理8h。
[0162]
对比例6
[0163]
催化剂制备条件与对比例5相同,不同的是催化剂还原温度为350℃。
[0164]
对比例7
[0165]
载体及制备条件与实施例4相同,不同之处在于对比例中无ni。
[0166]
(1)称取硝酸铜溶73ml去离子水中,加正己烷43g,加sds 17g,加正戊醇15g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动180min,滤除余液,在80℃下干燥4小时,在600℃下焙烧2h。称为半成品催化剂k1。
[0167]
(2)称取氯化钯溶于73ml去离子水中,加正己烷43g,加sds 17g,加正戊醇15g,充
分搅拌形成微乳液,将半成品催化剂k1浸渍到所制备的微乳液中,摇动180min,滤除余液,在80℃下干燥4小时,在600℃下焙烧2h。称为半成品催化剂m1。
[0168]
(3)称取氯化钯溶于去离子水中,调ph为2.0,再将半成品催化剂m1浸渍到已配制的pd盐溶液中,浸渍120min后,130℃干燥3小时,600℃条件下焙烧2小时。得到半成品催化剂n1。
[0169]
(4)称取硝酸锂配制成溶液,将步骤(3)制备的半成品催化剂n1浸渍于所配制的含锂的硝酸锂溶液中,摇动,待溶液全部吸收后,100℃干燥4小时,在600℃焙烧2小时,得到所要的催化剂。
[0170]
动态光散射测定制备的微乳液的粒径是78nm。
[0171]
催化剂的还原:
[0172]
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在200℃温度,还原处理8h。
[0173]
对比例8
[0174]
催化剂制备条件与实施例5相同,不同之处在于制备步骤(3)和(4)顺序对调。
[0175]
催化剂制备:
[0176]
(1)称取氯化钯溶于去离子水中,调ph为2.5,再将载体浸渍到已配制的pd盐溶液中,浸渍120min后,130℃干燥3小时,600℃条件下焙烧2小时,得到半成品催化剂o1。
[0177]
(2)称取硝酸锂配制成溶液,将步骤(1)制备的半成品催化剂o1浸渍于所配制的含锂的硝酸锂溶液中,摇动,待溶液全部吸收后,100℃干燥4小时,在300℃焙烧8小时,得到半成品催化剂p1。
[0178]
(3)称取氯化钯溶于68ml去离子水中,加正己烷40g,加triton x-100 20g,加正己醇18g,充分搅拌形成微乳液,将制备的半成品催化剂p1浸渍到所制备的微乳液中,摇动180min,滤除余液,在70℃下干燥6小时,在600℃下焙烧2h,称为半成品催化剂q1。
[0179]
(4)称取氯化镍、硝酸铜溶于68ml去离子水中,加正己烷40g,加triton x-100 20g,加正己醇18g,充分搅拌形成微乳液,将步骤(3)制备的半成品催化剂q1浸渍到所制备的微乳液中,摇动180min,滤除余液,在70℃下干燥6小时,在600℃下焙烧2h。得到所要的催化剂。
[0180]
动态光散射测定制备的微乳液的粒径是80nm。
[0181]
催化剂的还原:
[0182]
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在200℃温度,还原处理8h。
[0183]
对比例9
[0184]
催化剂制备条件与实施例6相同,不同的是催化剂还原温度为500℃。
[0185]
催化剂的还原:
[0186]
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在500℃温度,还原处理8h。
[0187]
对比例10
[0188]
与实施例7相比,不同之处在于催化剂负载时步骤(2)和(3)对调。
[0189]
催化剂制备:
[0190]
(1)称取氯化镍、硝酸铜溶于72ml去离子水中,加正己烷38g,加ctab 19g,加正戊醇16g,充分搅拌形成微乳液,将称取的100g高温焙烧过的载体浸渍到所制备的微乳液中,摇动90min,滤除余液,在80℃下干燥5小时,在500℃下焙烧4h,称为半成品催化剂v1。
[0191]
(2)称取硝酸锂配制成溶液,将步骤(1)制备的半成品催化剂v1浸渍于所配制的含锂的硝酸锂溶液中,摇动,待溶液全部吸收后,140℃干燥2小时,在500℃焙烧4小时,得到半成品催化剂w1。
[0192]
(3)称取氯化钯溶于去离子水中,调ph为1.8,再将半成品催化剂w1浸渍到已配制的pd盐溶液中,浸渍60min后,100℃干燥5小时,400℃条件下焙烧6小时,得到半成品催化剂x1。
[0193]
(4)称取氯化钯溶于72ml去离子水中,加正己烷38g,加ctab19g,加正戊醇16g,充分搅拌形成微乳液,将步骤(3)制备的半成品催化剂x1浸渍到所制备的微乳液中,摇动90min,滤除余液,在80℃下干燥5小时,在500℃下焙烧4h得到所要的催化剂。
[0194]
动态光散射测定步骤(1)制备的微乳液乳液的粒径是92nm。
[0195]
催化剂的还原:
[0196]
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在150℃温度,还原处理8h。
[0197]
催化剂应用于碳八馏分选择加氢除苯乙炔反应的性能
[0198]
试验所用裂解汽油碳八馏分来自兰州石化公司裂解汽油装置,其性质见表2、表3。
[0199]
表2裂解汽油碳八馏分性质
[0200][0201]
表3高含量苯乙炔裂解汽油碳八馏分
[0202][0203]
采用固定床反应器对制备的催化剂进行性能评价,催化剂装填量为100ml,反应物料空速2h-1
,操作压力0.3mpa,氢油比20:1,反应器入口温度30℃。
[0204]
催化剂评价结果见表4。催化剂1、2、3、4、5、6、7分别来自实施例1、2、3、4、5、6、7所制备的催化剂,对比催化剂1、2、3、4、5、6、7、8、9、10分别来源于对比例1、2、3、4、5、6、7、8、9、10所制备的催化剂。
[0205]
表4催化剂评价结果
[0206][0207]
注:苯乙炔转化率=(原料中苯乙炔含量﹣加氢产品中苯乙炔含量)/原料中苯乙炔含量;苯乙烯损失=原料中苯乙烯含量﹣加氢产品中苯乙烯含量。
[0208]
由表4催化剂评价结果对比可以看出:
[0209]
与实施例1相比,对比例1没有负载cu,还原温度为200℃,虽然初始产品中苯乙炔转化率及苯乙烯损失与相应的实施例相比基本相同。但1000小时后,则明显低于实施例。表明cu的负载或降低催化剂还原温度对提高抗结焦性能是重要的。或可能在200℃的还原温度下,未生成具有饱和加氢功能的活性中心。
[0210]
对比例2中还原温度达到350℃,从反应结果看,1000小时后的结焦量明显低于对比例1,但初始的苯乙炔转化率及苯乙烯损失却明显低于实施例1和对比例1,表明350℃的还原温度,对苯乙炔选择加氢的活性中心产生了不良影响。
[0211]
对比例3、4与实施例2相比,cu的负载采用溶液法,cu的分布主要在小孔中,没有起到有效降低还原温度的作用,在较高的还原温度致使pd产生团聚,催化剂的初始活性、选择性要低于实施例2。对比例3与对比例4相比,对比例3的还原温度高,所以其初始活性、选择性更差,但由于温度高时ni-pd活性中心得到较好的还原,所以其抗结焦性能更好。
[0212]
对比例5、6与实施例3相比,pd全部采用的是溶液法负载,pd进入了小孔;没有采用微乳液法使pd进入大孔。对比例5在150℃对催化剂进行还原,ni-cu没有还原,1000小时后结焦量很大,催化剂性能下降幅度大。对比例6在350℃还原,ni-cu得到较好的还原,催化剂性能保持较好,结焦量较对比例5明显降低,但高温还原使pd形成团聚,活性下降较快。
[0213]
对比例7与实施例4相比,不负载ni,由于对胶质的饱和加氢作用降低,1000小时后催化剂的结焦量大,性能下降幅度大。
[0214]
对比例8中,微乳液法先负载pd,后负载ni-cu,导致pd被覆盖,不能起到提高还原效率的作用,在180℃温度下ni-cu不能有效还原,1000小时后,与实施例5相比,催化剂性能下降较多。
[0215]
对比例9的催化剂与实施例6制备条件相同,只是催化剂的还原温度不同。可以看出,在500℃还原的催化剂其初始苯乙炔加氢活性、选择性均有不同程度的下降,而且1000小时后结焦量也较多,这可能是因为催化剂活性中心尺寸变大,加氢二聚反应更加剧烈的原因。
[0216]
对比例10与实施例7相比,li的负载是在溶液法pd的负载前,可以看出,其选择性一直明显较低。评价原料采用高含量苯乙炔裂解汽油碳八馏分,可以发现,在较高含量的苯乙炔评价实验中,实施例6的结焦量没有明显增加。
[0217]
当然,本发明还可有其它多种实施例,在不背离本发明精神及其实质的情况下,熟悉本领域的技术人员可根据本发明作出各种相应的改变和变形,但这些相应的改变和变形都应属于本发明权利要求的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。