1.本发明属于中药化现代领域,尤其涉及一种黑骨藤标准汤剂的制备方法及其质量检测方法。
背景技术:
2.黑骨藤为萝藦科(asclepiadaceae)杠柳属(periploca)植物黑龙骨(periplocaforrestiischltr.)的干燥根或全株。黑骨藤全株入药,具有通经、活血、解毒、祛风的功效,主治风湿关节痛、跌打损伤、月经不调等症,是民间广泛应用于治疗闭合性软组织损伤、风湿与类风湿等疾病的民族药。
3.标准汤剂又称为成为标准煎液,是一种传统的临床广泛使用的用药形式,标准汤剂系遵循中医药理论,按照临床汤剂煎煮规范化煎煮,固液分离,经适当浓缩制备或经适宜方法干燥制得,作为衡量中药配方颗粒是否与临床汤剂基本一致的标准参照物;是评价不同厂家产品质量、疗效的参照物。因此,中药标准汤剂质量标准能为所源于中药饮片水煎剂的产品的质量标准的制定提供基础。
4.目前关于黑骨藤有不少的质量标准研究报道;但是关于黑骨藤标准汤剂的制备方法以质量检测方法鲜有报道。
5.公开号为cn109828064a的专提供了一种黑骨藤有效部位群的hplc指纹图建立方法,包括以下步骤:(1)混合对照品溶液的制备:取新绿原酸、绿原酸、隐绿原酸和异绿原酸c标准品,用甲醇定容为混合对照品溶液;(2)供试品溶液这样制备:精密称定黑骨藤粗粉100.0g,用70%乙醇回流提取三次,将三次滤液合并,抽滤,浓缩至180-220ml,用蒸馏水定容至1000ml,得样品溶液;精密称取100gd101大孔吸附树脂装入层析柱,蒸馏水冲洗至层析柱下端流出液体清澈无色后,将1000ml样品溶液上样进行吸附,待吸附完全后以1200ml蒸馏水洗脱,然后以2500ml70%乙醇洗脱,收集层析柱流出的70%乙醇部分,浓缩,烘干,研磨,得黑骨藤有效部位群粉末;精密称取黑骨藤有效部位群粉末50.0mg,用70%甲醇定容至10ml,过0.45μm微孔滤膜,取续滤液,得供试品溶液;(3)指纹图谱的制作:色谱条件:色谱柱:xtimatec18,4.6
×
250mm,5μm;流动相:乙腈为a相,0.1%磷酸水溶液为b相;梯度洗脱,检测波长为360nm,流速为1ml
·
min-1,柱温38℃,进样量10μl;记录色谱图得到黑骨藤有效部位群的hplc指纹图谱;(4)标准指纹谱图的确认。该方法样品采用层析柱处理,工艺复杂,效率较低,不适宜大生产使用。
6.公开号为cn110702813a的专利公开了一种苗药黑骨藤hplc指纹图谱研究及多成分含量测定方法;采用hplc法建立不同产地黑骨藤指纹图谱,并测定其中4种咖啡酰基奎宁酸类化学成分含量;色谱柱为xtimate c18(4.6mmx250mm,5μm);以0.1%磷酸水溶液(a)-乙腈(b)为流动相,梯度洗脱;流速1ml/min;柱温30℃;进样量10μl,检测波长为330nm;使用中药色谱指纹图谱相似度评价系统对样品进行相似度分析。该方法明确不同产地的苗药黑骨藤中该4种被测成分的含量还是有明显差异的,各成分含量与产地有一定的关联,猜想与其所生长的环境、气候、湿度、土壤等有一定的关系,为后期苗药黑骨藤抗风湿关节炎质量标
准研究奠定基础,仅适用于对黑骨藤药材品质评价提供依据。
7.文献《黑骨藤指纹图谱及谱效关系探索性研究》(贵州师范大学.2016)中提供了黑骨藤指纹图谱的测定方法:供试品溶液的制备精密称定过40目筛黑骨藤粉末2.0g,置于具塞锥形瓶中,精密加入80%乙醇30ml,称定重量,回流提取2.0h,待冷却之后再次称定重量,以80%乙醇补足超声过程中损失的重量,过滤,浓缩,得黑骨藤浸膏,进样时复溶于80%乙醇水溶液中,过0.45μm微孔滤膜,得供试品。色谱条件:phecda c18(4.6mm
×
250mm,5μm);以0.5%乙酸水溶液为流动相a,以乙腈为流动相b梯度洗脱;流速1m l/mn;检测波长330nm;柱温30℃;进样量10μm。
8.上述专利或文献公开的指纹图谱检测方法,存在样品处理方法方法复杂、共有峰成分来源不清、没有对特征峰进行指认、重复性以及耐用性考察不全面等问题,不能发挥有效地控制黑骨藤水煎剂产品质量。
技术实现要素:
9.为了解决现有技术中存在的上述技术问题,本发明提供了黑骨藤标准汤剂的制备方法及其质量检测方法,具体是通过以下技术方案实现的。
10.黑骨藤标准汤剂的制备方法,包括以下过程:取黑骨藤药材饮片,加10-14倍量水浸泡30-60min后,武火煮沸后,文火煎煮30-60min,过滤;药渣再加入10-14倍量水,武火煮沸后,文火煎煮20-40min;然后将两次获得的滤液合并,减压低温浓缩,冷冻干燥,得到黑骨藤标准汤剂。
11.优选地,所述减压低温浓缩是在65
±
5℃下浓缩至相对密度为1.02~1.03g/ml。
12.优选地,所述冷冻干燥分为三个阶段:先在-45℃下预冻4-5h;然后在-30℃~0℃下升化干燥12-14h;再在0℃~25℃下解析干燥7-9h。
13.本发明的另一目的在于提供一种黑骨藤标准汤剂的质量检测方所述检测方法由出膏率、含量测定、特征图谱项目组成,所述特征图谱是对黑骨藤标准汤剂的成分,以乙腈为流动相a,以0.05-0.15%磷酸溶液为流动相b,进行梯度洗脱的hplc法指纹图谱表征过程;
14.所述含量测定是对黑骨藤标准汤剂中的绿原酸含量进行测定,以乙腈:0.5%甲酸水溶液=8-10:90-92为流动相的hplc法。
15.所述出膏率是将黑骨藤浓缩液冷冻干燥制成黑骨藤标准汤剂冻干粉的出膏率。
16.优选地,所述特征图谱表征方法如下:
17.色谱条件与系统适用性试验:inertsil ods-3 2.1
×
100mm 2μm色谱柱;以乙腈为流动相a,以0.05-0.15%磷酸溶液为流动相b,进行梯度洗脱;柱温为25-35℃;流速为0.25-0.35ml/min;检测波长为280nm;理论板数按新绿原酸计算应不低于5000;
18.所述梯度洗脱条件为:
19.0-5min,5%
→
8%a,95%
→
92%b;
20.5-12min,8%
→
10%a,92%
→
90%b;
21.12-18min,10%
→
15%a 90%
→
85%b;
22.18-38min,15%
→
20%a 85%
→
80%b;
23.38-39min,20%
→
5%a,80%
→
95%b;
24.39-45min 5%
→
5%a 95%
→
95%b;
25.参照物溶液的制备:称取新绿原酸标准品,加50%甲醇制成每1ml含新绿原酸0.5mg,即得新绿原酸标准品溶液;
26.供试品溶液的制备:精密称取黑骨藤标准汤剂,按0.1g:20ml的固液比加入50%-75%甲醇,称定重量;加热回流提取或者超声提取15-45min,放冷,再称定重量,用相应浓度的甲醇补足减失的重量,摇匀,滤过,取续滤液,即可;
27.测定法:分别精密吸取对照品溶液与供试品溶液各1μl,注入液相色谱仪,测定,记录色谱图,即得。
28.更优选地,所述特征图谱表征方法中,以乙腈为流动相a,以0.1%磷酸溶液为流动相b,进行梯度洗脱;柱温为30℃,流速为0.3ml/min;所述供试品溶液的制备:精密称取0.1g黑骨藤标准汤剂,按20ml 75%甲醇,称定重量;超声提取30min,放冷,再称定重量,用75%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即可。
29.优选地,所述含量测定方法如下:
30.色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂,以乙腈:0.5%甲酸水溶液=8-10:90-92为流动相;0.9~1.1ml/min流速进行洗脱;柱温25℃~35℃;检测波长为327nm;理论板数按绿原酸计算应不低于5000;
31.对照品溶液的制备:取绿原酸标准品,精密称定,加流动相制成每1ml含80ug的溶液;
32.供试品溶液的制备:取黑骨藤标准汤剂粉碎,按0.1g:25ml的固液比加入50-75%甲醇,称定重量;提取25-40min,放冷,再称定重量,用相应浓度的甲醇补足减失重量,摇匀,滤过,取滤液过微孔滤膜,即得;
33.测定法:分别精密吸取对照品溶液与供试品溶液10μl,注入液相色谱仪,测定,即得。
34.更优选地,所述含量测定方法中,流动相为乙腈:0.5%甲酸水溶液=9:91;十八烷基硅烷键合硅胶柱为welch xtimate c18色谱柱,规格为4.6mm
×
250mm,粒径5μm;1.0ml/min流速进行洗脱;柱温30℃;所述供试品溶液的制备:取黑骨藤标准汤剂粉碎,称取0.1g,加入50%甲醇25ml,称定重量;超声提取30min,放冷,再称定重量,用50%甲醇补足减失重量,摇匀,滤过,取续滤液过0.22μm微孔滤膜,即得。
35.优选地,所述的黑古藤标准汤剂的质量检测方法,其检测方法如下:
36.出膏率:
37.将黑骨藤标准汤剂浓缩液冷冻干燥制成黑骨藤标准汤剂冻干粉后,计算出膏率;
38.含量测定:
39.色谱条件与系统适用性试验:welch xtimate c18色谱柱,规格为4.6mm
×
250mm,粒径5μm;以乙腈:0.5%甲酸水溶液=9:91为流动相;1.0ml/min进行梯度洗脱;柱温30℃;检测波长为327nm;理论板数按绿原酸计算应不低于5000;
40.对照品溶液的制备:取绿原酸标准品,精密称定,加流动相制成每1ml含80ug的溶液;
41.供试品溶液的制备:取黑骨藤标准汤剂粉碎,称取0.1g,加入50%甲醇25ml,称定重量;超声提取30min,放冷,再称定重量,用50%甲醇补足减失重量,摇匀,滤过,取续滤液
过0.22μm微孔滤膜,即得;
42.测定法:分别精密吸取对照品溶液与供试品溶液10μl,注入液相色谱仪,测定;
43.特征图谱:
44.色谱条件与系统适用性试验:inertsil ods-3 2.1
×
100mm 2μm色谱柱;以乙腈为流动相a,以0.1%磷酸溶液为流动相b,进行梯度洗脱;柱温为30℃;流速为每分钟0.30ml;检测波长为280nm。理论板数按新绿原酸计算应不低于5000;
45.所述梯度洗脱条件为:
46.0-5min,5%
→
8%a,95%
→
92%b;
47.5-12min,8%
→
10%a,92%
→
90%b;
48.12-18min,10%
→
15%a 90%
→
85%b;
49.18-38min,15%
→
20%a 85%
→
80%b;
50.38-39min,20%
→
5%a,80%
→
95%b;
51.39-45min 5%
→
5%a 95%
→
95%b。
52.参照物溶液的制备:称取新绿原酸标准品,加50%甲醇制成每1ml含新绿原酸0.5mg,即得新绿原酸标准品溶液;
53.供试品溶液的制备:精密称取0.1g黑骨藤标准汤剂,加入20ml75%甲醇,称定重量;超声提取30min,放冷,再称定重量,用75%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即可;
54.测定法:分别精密吸取对照品溶液与供试品溶液各1μl,注入液相色谱仪,测定,记录色谱图。
55.本发明的另一目的在于提供一种黑骨藤标准汤剂特征图谱的建立方法,特征图谱建立方法如下:
56.色谱条件与系统适用性试验:inertsil ods-3 2.1
×
100mm 2μm色谱柱;以乙腈为流动相a,以0.05-0.15%磷酸溶液为流动相b,进行梯度洗脱;柱温为25-35℃;流速为0.25-0.35ml/min;检测波长为280nm;理论板数按新绿原酸计算应不低于5000;
57.所述梯度洗脱条件为:
58.0-5min,5%
→
8%a,95%
→
92%b;
59.5-12min,8%
→
10%a,92%
→
90%b;
60.12-18min,10%
→
15%a 90%
→
85%b;
61.18-38min,15%
→
20%a 85%
→
80%b;
62.38-39min,20%
→
5%a,80%
→
95%b;
63.39-45min 5%
→
5%a 95%
→
95%b;
64.参照物溶液的制备:称取新绿原酸标准品,加50%甲醇制成每1ml含新绿原酸0.5mg,即得新绿原酸标准品溶液;
65.供试品溶液的制备:精密称取黑骨藤标准汤剂,按0.1g:20ml的固液比加入50%-75%甲醇,称定重量;加热回流提取或者超声提取15-45min,放冷,再称定重量,用相应浓度的甲醇补足减失的重量,摇匀,滤过,取续滤液,即可;
66.测定法:分别精密吸取对照品溶液与供试品溶液各1μl,注入液相色谱仪,测定,记录色谱图,导入中药色谱指纹图谱相似度评价系统分析,确定共有峰,得到黑骨藤标注汤剂
的特征图谱。
67.本发明的有益效果在于:
68.1、本发明通过对黑骨藤煎煮提取方法研究,采用水作为提取溶剂,不会破坏黑骨藤中的天然成分;并对煎煮方法、浓缩以及干燥方法进行优化,减少了制备过过程中天然成分的损失,最大限度地保留了黑骨藤中的天然成分。本发明提供的黑骨藤标准汤剂的制备方法,能有效地提取黑骨藤中的天然成分,且提率高,破坏率低。
69.2、绿原酸是黑骨藤的主要成分之一,有抗氧化、抗肿瘤、抗炎、抗菌、抗病毒等多种药理作用。本发明通过实验研究发现,黑骨藤标准汤剂中绿原酸含量较高,对黑骨藤标准汤剂的质量具有较高的参考价值。本发明提供黑骨藤标准汤剂的绿原酸含量色谱检测方法,对色谱系统、供试品制备方法、色谱条件等均进行了选择和优化,具有分离效果好、灵敏度高、重现性好、回收率高等优点。
70.3、本发明提供的黑骨藤标准汤剂特征图谱的表征方法,明确了6个共有峰及其相对保留时间、相对峰面积,并指认了其中5个特征峰:峰1为原儿茶酸,峰2为新绿原酸,峰4为绿原酸,峰为5为隐绿原酸,峰6为咖啡酸;特征图谱表征较全面,客观真实地反映了黑骨藤表准汤剂活性成分的种类与数量,能有效地表征黑骨藤标准汤剂产品的质量。
71.4、本发明从出膏率、绿原酸含量、特征图谱对黑骨藤标准汤剂进行了质量检测,绿原酸含量采用优化的液相色谱件进行分析,具有分离效果好、灵敏度高、重现性好等优点;特征图谱采用优化的高效液相色谱法,具有分离效果好、专属性好、整体性好、精密度高、稳定、重现性好等优点。本发明提的黑骨藤标准汤剂质量检测控制方法,从有效成分提取率、绿原酸含量、特征图谱等方面对黑骨藤标准汤剂的质量进行综合性控制,具有测量准、稳定性强、全面、参考价值高等特点,提高了黑骨藤标准汤剂的质量控制标准,可有效地控制黑骨藤水煎剂产品质量,从而确保其临床疗效。
72.为能更好的阐述本发明的有益效果,本发明还列举了如下研究过程,旨在说明本发明的有益效果,但决不仅限于本发明的保护范围。
73.一、黑骨藤标准汤剂制备方法研究
74.本发明根据《中药配方颗粒质量控制与标准制定技术要求》、《医疗机构中药煎药室管理规范》、《中药饮片生产过程质量标准通则(试行)》对黑骨藤标准汤剂的提取工艺、浓缩干燥工艺进行了研究。
75.1.1制备工艺考察
76.1.1.1提取工艺考察
77.(1)溶剂用量:本发明通过实验发现黑骨藤饮片吸水率约为0.7倍,黑骨藤药材吸水率考察情况如表1所示,其中,吸水率%=(煎煮完成后药材重量-药材取样量)/药材取样量*100%。结合浸过药面2-5厘米的原则,确定黑骨藤提取一煎加水量为10-14倍,二煎加水量为8-12倍。
78.表1黑骨藤药材吸水率考察
[0079][0080]
(2)浸泡时间:黑骨藤药材浸泡到30分钟以上,水分能渗透到组织内部,药材变软,确定浸泡时间为30分钟以上,浸泡时间考察如表2所示。
[0081]
表2黑骨藤标准汤剂浸泡时间考察
[0082][0083]
(3)煎煮时间:固定一煎加水量为12倍,二煎加水量为10倍;研究不同煎煮时间下黑骨藤藤标准汤剂的出膏率;研究结果显示,一煎时间在30-60min,二煎时间在20-40min,黑骨藤标准汤剂的出膏率的的较高,可达到8.04%以上,说明该提取时间下黑骨藤药材中的成分提取率高;一煎时间为30min,二煎时间为20min时的提取效果最优,黑骨藤标准汤剂的出膏率可达到8.76%;
[0084]
(4)过滤条件:本发明考察了不同目数滤布对黑骨藤煎煮汤的过滤效果,采用200目及以下滤布过滤,易过滤,但滤液出现浑浊;采用400目及以上滤布过滤,难过滤;综合考虑过滤难易度、过滤效果,选择250-350目数滤布过滤,优选300目滤布,过滤顺利,药液澄明,无明显固形沉淀物。
[0085]
1.1.2浓缩方法考察
[0086]
为了最大限度地保留了黑骨藤提取液中的有效成分,本发明采用低温减压浓缩的方法进行浓缩,并对浓缩条件进行了优化研究;研究发现在65
±
5℃、-0.085~-0.090mpa下浓缩不仅能保持较好的浓缩效率,同时能避免有效成分的损失;浓缩后相对密度1.02~1.03g/ml,浸膏粘稠度适中,流动性好,方便转移,含固量在10%-20%之间,适合冻干。
[0087]
1.1.3干燥工艺考察
[0088]
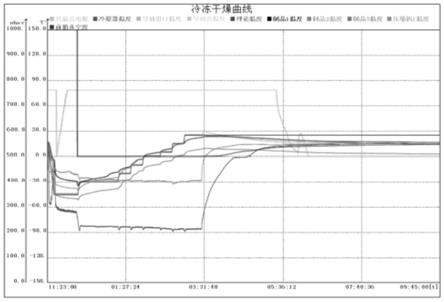
为了最大限度地保留了黑骨藤浓缩液中的有效成分,本发明采用冷冻干燥的方法进行干燥,并对冷冻干燥条件进行了优化研究。黑骨藤浓缩液真空冷冻干燥机共晶点测试结果为-28.0℃,保持干燥过程中的真空度为0.2mbar,先-45℃下预冻4-5h,然后在-30℃~0℃下升化干燥12-14,再在0℃~25℃下解析干燥7-9h,可获得的质地疏松的棕黄色固体,冷冻干燥工艺参数如表3所示,冻干曲线图如图1所示。
[0089]
表3黑骨藤标准汤剂冷冻干燥参数设置
[0090][0091]
1.2制备工艺验证
[0092]
黑骨藤样品信息:2019030601-01(产地:毕节市织金县三塘乡)、2019030801-01(产地:贵阳市修文县六厂乡)2019030701-01(安顺市普定县补郎乡)
[0093]
1.2.1提取浓缩工艺验证
[0094]
分别称取3份黑骨藤药材,每份约200g,第一煎加12倍水,第二煎加10倍水,浸泡30分钟后开始煎煮。第一煎煎煮30分钟。趁热分别300目滤过。第二煎煎煮20分钟。趁热分别300目滤过。合并相同目数下的煎液,将煎液在65℃下浓缩后取样测定出膏率和(指标成分)。结果如表4和表5所示。
[0095]
表4提取工艺验证
[0096][0097]
表5浓缩工艺验证
[0098]
[0099][0100]
1.2.2冷冻干燥工艺验证
[0101]
将1.2.1制备得到的的黑骨藤标准汤浓缩液,调节固含量至10.0%~20.0%,置于烧杯中,磁力搅拌条件下分装至10ml西林瓶中,每瓶分装约2.0ml,按冻干参数进行冻干制得标准汤剂,标准汤剂的收率见表6。
[0102]
表6冻干工艺验证
[0103][0104]
经3批分别平行3次的验证试验可知,上述黑骨藤标汤的制备工艺稳定可行。
[0105]
二、黑骨藤汤剂出膏率研究
[0106]
2.1将表7中的15批黑骨藤样品按照“黑骨藤标准汤剂制备方法研究”确定的方法制成15批黑骨藤标准汤剂。
[0107]
表7 15批黑骨藤样品的信息
[0108][0109]
2.2出膏率计算:
[0110]
出膏率以标准汤剂冻干粉量计。冷冻干燥分装前称定包材(西林瓶 胶塞)重量,黑骨藤标准汤剂浓缩液进行分装,记录分装的浓缩液重量,冷冻干燥后称定冻干粉和包材总重量,计算出膏率。
[0111]
计算公式:
[0112][0113]
15批黑骨藤标标准汤剂的测定结果,如表8所示。
[0114]
表8 15批黑骨藤标准汤剂出膏率结果
plus、welch xtimate、kromasil、agientzorbax extend、ageladurashell、waters symmetry、phenomenex gemini-nx、kshiseido、capcell pak、acchromtature等c18色谱柱对黑骨藤供试品溶液的分离效果。结果显示:菲罗门superlu c18色谱柱,分离效果差;agilent zorbax eclipse plus c18(分离度20.09%)和welch xtimatec18的分离效果均较好,但welch xtimate c18色谱柱的分离度、对称因子、理论塔板数均较agilent zorbax eclipse plus c18色谱柱优,故选择welch xtimate c18(4.6mm
×
250mm,5μm)作为本次实验的优选色谱柱,岛津色谱柱测定结果如图2所示。
[0124]
(2)柱温流速考察
[0125]
考察了柱温25℃、30℃、35℃,不同柱温下的绿原酸含量的rsd为0.99%,表明柱温30℃
±
5℃对绿原酸含量测定几乎无影响;考察了0.9ml/min、1.0ml/min、1.1ml/min的柱流速,不同流速下的绿原酸含量的rsd为0.99%,结果表明流速0.9~1.1ml/min,各色谱峰分离基本无影响。
[0126]
综上,色谱条件为:流动相为乙腈:0.5%甲酸水溶液=8-10:90-92;十八烷基硅烷键合硅胶柱为welch xtimate c18色谱柱,规格为4.6mm
×
250mm,粒径5μm;0.9~1.1ml/min流速进行洗脱;柱温25℃~35℃;检测波长为327nm;理论板数按绿原酸计算应不低于5000。
[0127]
3.2供试品溶液制备方法确定
[0128]
原料:黑骨藤样品:2019030601-01,毕节市织金县三塘乡;按“黑骨藤标准汤剂制备方法研究”优化后的方法制成黑骨藤标准汤剂。
[0129]
(1)提取溶剂考察:取黑骨藤标准汤剂(20190916-1)粉碎精密称定0.1g,平行7组,每组2份,置具塞锥形瓶中,分别精密加入稀乙醇、75%乙醇、无水乙醇、50%甲醇、75%甲醇、甲醇、水各25ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用相应的溶剂补足减失重量,摇匀,滤过,取续滤液。精密吸取10μl,注入液相色谱仪。测定结果显示:采用无水乙醇作为提取黑骨藤标准汤剂,不能提取其中的绿原酸,色谱图中未显示出绿原酸的特征峰;采用其他溶剂处理均能提取黑骨藤标准汤剂(冻干粉)中的绿原酸,50%甲醇的提取效果更佳,绿原酸含量最高,可达到2.12%,色谱峰对称性也好。
[0130]
(2)提取方式考察:考察了超声提取、回流提取、振荡提取对供试品溶液绿原酸含量测定的影响,色谱测定结果显示:三种提取方式下,对黑骨藤标准汤剂中绿原酸的提取率无明显差异,选择超声作为提取方式。
[0131]
(3)提取时考察:采用超声提取,考察了超声处理20min、30min、60min、90min对供试品溶液绿原酸含量的印象。结果显示:超声处理20min绿原酸的提取率偏低;超声处理90min,供试品溶液绿原酸含量最高(2.17%),但相较于30min并无较明显差异,从时间和能耗方面考虑,选取30min超声条件。
[0132]
3.3方法学验证
[0133]
(1)专属性考察:准备黑骨藤标准汤剂(20190916-1)供试品溶液、绿原酸对照品溶液、空白溶剂,精密吸取10μl,注入液相色谱仪,按照“2.1色谱条件研究”确定的色谱条件测定。实验结果表明空白溶剂色谱中在与绿原酸相应的保留时间没有色谱峰,说明溶剂对绿原酸的测定无干扰,本发明提供的方法测定黑骨藤标准汤剂中绿原酸的含量具有专属性。
[0134]
(2)峰纯度考察:准备黑骨藤标准汤剂(20190916-1)供试品溶液、绿原酸对照品溶液,精密吸取10μl,注入液相色谱仪,按照“2.1色谱条件研究”确定的色谱条件;以dad检测
器进行190nm~400nm扫描检测,计算峰纯度。结果显示:match匹配度为1000,峰纯度符合要求。
[0135]
(3)线性考察:分别精密吸取绿原酸对照品溶液(79.76μg/ml)1、4、7、10、13、16、20μl,注入液相色谱仪,按照“2.1色谱条件研究”确定的色谱条件,以峰面积积分值为纵坐标,绿原酸质量(μg)为横坐标,绘制标准曲线。结果显示:线性回归方程为:y=1867.5x-14.99(y为峰面积、x为绿原酸质量),相关系数r2=1,表明在含量为0.0798μg~1.5952μg的范围内绿原酸浓度与峰面积线性关系良好。
[0136]
(4)精密度、稳定性考察:黑骨藤标准汤剂(20190916-1)供试品溶液,色谱条件:“2.1色谱条件研究”确定的色谱条件。骨藤标准汤剂供试品溶液,连续进样6次,峰面积在1477.2~1490.3,峰面积rsd为0.33%,精密度良好。骨藤标准汤剂供试品溶液分别在0、4、8、12、16、20、24小时进样,峰面积rsd为0.50%,表明供试品溶液在24小时内稳定。
[0137]
(5)中间精密度考察:黑骨藤标准汤剂(20190916-1)供试品溶液,色谱条件:“2.1色谱条件研究”确定的色谱条件。不同分析人员在员在不同日期和不同色谱仪下进行操作分析。实验结果表明,不同分析人员在不同日期和不同色谱仪下操作,绿原酸含量结果rsd《2%,该分析方法中间精密度良好。
[0138]
3.4黑骨藤标准汤剂中绿原酸含量的确定
[0139]
(1)黑骨藤标准汤剂中绿原酸含量确定
[0140]
原料:表7中的15批黑骨藤样品
[0141]
采用前述而黑骨藤标准汤剂的制备方法、供试品制备方法,制备黑骨藤标准汤剂供试品溶液;对15批黑骨藤标准汤剂供试品溶液分别进样10μl,采用前述色谱条件检测,记录色谱图及峰面积,计算含量。结果显示:黑骨藤标准汤剂中绿原酸的转移率均值为8.94%,sd为4.68%,均值70%~130%的范围为6.26%~11.63%;绿原酸含量的均值为1.41%,sd为0.93%,含量均值
±
3倍sd为-1.38%~4.20%%,均值的70%~130%为0.98%~1.83%,绿原酸含量及转移率如表9所示,绿原酸含量和转移率数据分析如表10所示,黑骨藤标准汤剂特征图谱如图3所示。其中有一批黑骨藤标汤(bt20190924-2)的含量很低,与其它批次差异大,因此作为异常数据没有纳入对照图谱的生成。
[0142]
表9 15批黑骨藤标准汤剂绿原酸含量及转移率结果
[0143][0144]
表10 15批黑骨藤标准汤剂中绿原酸含量和转移率数据分析
[0145][0146]
(2)范围实验
[0147]
15批黑骨藤标准汤剂供试品溶液经检测,黑骨藤(2019040301)的绿原酸含量最高;黑骨藤(2019051406)的绿原酸含量最低,以这两个数值作为绿原酸范围值,结合重复性和准确度进行实验。
[0148]
重复性实验:取批号为2019040301的黑骨藤标准汤剂约0.2g,精密称定,平行称定6份,制备供试品溶液6份。采用前述而黑骨藤标准汤剂的检测条件,吸取10μl测定。结果显示,黑骨藤(2019040301)标准汤供试品溶液的平均绿原酸含量为2.74%,rsd值为1.6%;表明本分析方法的重复性良好。
[0149]
准确度实验:分别取批号为2019040301、2019051406的黑骨藤标准汤剂约0.1g,精密称定,各平行称定6份,每份加入相近量的绿原酸对照品,采用前述供试品的制备方法制成供试品溶液,采用前述色谱条件吸取10μl测定。结果显示,黑骨藤(2019040301)标准汤供试品溶液的绿原酸回收率为1.02%;黑骨藤(2019051406)标准汤供试品溶液的回收率为98.03%。根据据中国药典2015版四部“药品质量标准分析方法验证指导原则”规定样品中待测成分含量为0.1%-1%时,回收率限度为92%-105%;待测成分含量为1%-10%时,回
收率限度为95%~102%。高低含量的回收率均在药典规定的限度范围内,表明该方法准确度良好。
[0150]
四、黑骨藤标准汤剂特征图谱研究
[0151]
中药色谱图能够全面客观表征中药成分,对中药材的质量控制具有重要意义。现有技术中有关于黑骨藤指纹图谱的基础性研究,但还不够完善。
[0152]
因此,本发明力图寻找更加黑骨藤标准汤剂的指纹图谱表征方法,使得图谱反映信息丰富、共有峰多、且成分确认、共有峰的来源清晰。主要进行了供试品制备的研究、色谱条件研究、方法学考察研究、黑骨藤标准汤剂样品检测研究等内容。
[0153]
4.1色谱条件研究
[0154]
(1)检测波长
[0155]
准备新绿原酸参照物溶液进行了全波长检测;记录紫外吸收图谱,结果显示,绿原酸在327nm附近有最大吸收波长,故选择327nm作为检测波长。绿原酸全波长扫描图如图4所示,新绿原酸对照品的紫外吸收图如图5所示。
[0156]
准备黑骨藤标准汤剂供试品溶液,进行了全波长检测;记录紫外吸收的dad-3d图谱,如图6所示;结果显示,在280nm左右,黑骨藤标准汤剂样品溶液可以检测到的色谱峰最多,且各色谱峰的峰面积较大,综合考虑,选择280nm为检测波长。
[0157]
(2)色谱柱考察:本发明在试验过程中,考察了waters、塞默飞、岛津、kromasil、agientzorbax extend、agienteclipseplus、ageladurashell等品牌共不同型号的色谱柱对黑骨藤标准汤剂供试品溶液分离效果的影响。结果显示岛津(inertsil ods-3 2.1
×
100mm2μm)的分离度最高,说明岛津(inertsil ods-3 2.1
×
100mm 2μm)的分离效果优于其他色谱柱,确定色谱柱为岛津(inertsil ods-3 2.1
×
100mm 2μm)。
[0158]
(3)柱温考察:采用岛津(inertsil ods-3 2.1
×
100mm 2μm),考察了不同柱温对黑骨藤标准汤剂供试品溶液分离效果的影响。结果表明柱温对黑骨藤标汤的出峰情况有一定影响,柱温在30℃
±
5℃时的分离效果较好,柱温为30℃时的分离效果最好。
[0159]
(4)流速考察:采用岛津(inertsil ods-3 2.1
×
100mm 2μm)色谱柱,考察了0.25ml/min、0.30ml/min、0.35ml/min等流速对对黑骨藤标准汤剂供试品溶液分离效果的影响。结果表明柱温对出峰时间影响较大,对分离效果影响较小,综合考虑,确定流速为0.30ml/min。
[0160]
(5)采用岛津(inertsil ods-3 2.1
×
100mm 2μm)色谱柱;以乙腈为流动相a,以0.1%磷酸溶液为流动相b;在不同梯度条件下对黑骨藤标准汤剂供试品溶液进行梯度洗脱;在大梯度条件(0~30min,
[0161]5→
75%a,95%
→
25%b;31~31min,75
→
5%a,25%
→
95%b;31~36min,5%a,95%b)下记录的色谱图结果显示黑骨藤标准汤剂中主要存在大极性成分。在大梯度条件的基础上再不断细化调整梯度条件下,记录其色谱图,结果显示在如表11所示的梯度的洗脱条件下色谱峰分离效果好。
[0162]
表11梯度洗脱条件
[0163][0164]
(6)流动相考察
[0165]
在前述确定的梯度洗脱条件下,对水相(b相)中的有机酸种类、有机相(a相)进行了考察。
[0166]
水相中有机酸考察:在试验过程中考察了乙腈-0.1%甲酸、乙腈-0.1%醋酸、乙腈-0.1%磷酸作为流动相获得的色谱图。结果显示:采用乙腈-0.1%醋酸,色谱峰不能显示出来;采用乙腈-0.1%甲酸,色谱峰的分离度、对称性差,出现连峰;采用乙腈-0.1%磷酸,色谱峰分离度、对称性好。在采用乙腈-0.1%磷酸作为流动相的基础上,本发明还考察了磷酸浓度对供试品溶液色谱图的影响,磷酸浓度为0.05-0.15%范围内可获得分离、对称性良好的色谱图,优选为0.1%。
[0167]
有机相考察:在试验过程中考察了甲醇-0.1%磷酸、乙腈-0.1%磷酸作为流动相对黑骨藤标准汤剂供试品溶液色谱图的影响;结果显示采用乙腈-0.1%磷酸作为流动相,色谱峰的分离度、对称性都要优于甲醇-0.1%磷酸;确定采用乙腈作为流动相a。
[0168]
4.2供试品制备方法研究
[0169]
经hplc初步检测后,发现黑骨藤标准汤剂中含有大量极性的化合物,极性相近且色谱峰众多,因此样品需进行预处理,以得到分离度较好的更多的色谱峰信号。
[0170]
(1)提取溶剂考察:取0.1g黑骨藤标准汤剂(冻干粉),加入20ml不同溶剂(水、50%甲醇、75%甲醇、无水甲醇、稀乙醇、75%乙醇、无水乙醇),超声处理30分钟,放冷,再称定重量,用对应的溶剂补足减失的重量,滤过,取续滤液,即制成供试品溶液。按照前述确定的色谱条件对供试品溶液进行测定。测定结果显示无水甲醇、无水乙醇对样品中有效成分的提取率较低,其他溶剂相差不大,综合考虑样品保存、过滤情况,选择50-75%甲醇作为提取溶剂,优选为75%甲醇。
[0171]
(2)提方式考察:考察了超声提取、震荡提取、加热回流提取所制得的黑骨藤标准汤剂供试品溶液测定结果的影响。结果显示,振振荡提取的提取效率较差,回流和超声的提取效率差异不大,综合考虑方便性,选择超声提取。
[0172]
(3)提取时间考察:考察了超声处理10、15、30、45分钟对黑骨藤标准汤剂供试品溶液测定结果的影响;结果显示,10min黑骨藤标准汤剂的提取效率较差,在15min至45min之间对黑骨藤标准汤剂的提取效率没有显著性差异。
[0173]
4.3方法学研究
[0174]
按照指纹图谱相关技术要求,对指纹图谱检测方法进行精密度、稳定性和重复性等考察,并采用指纹图谱相似度评价软件计算相关结果,其结果均符合要求。
[0175]
黑骨藤标准汤剂供试品溶液:“3.2供试品制备方法研究”确定的方法制备成的;色
谱条件:“3.1色谱条件研究”确定的色谱条件。
[0176]
(1)专属性:对黑骨藤标准汤剂供试品溶液以及所用的空白溶剂进行检测,黑骨藤标准汤剂专属性考察结果如图7所示;检测结果显示对溶剂对黑骨藤标准汤剂图谱中的特征峰没有干扰。
[0177]
(2)整体性:考察了对黑骨藤标准汤剂供试品溶液在较长时间内的出峰情况。如图8所示,黑骨藤标准汤剂在45min后无明显的色谱峰,整体性较好。
[0178]
(3)精密度:将黑骨藤标准汤剂供试品溶液进样6次,每次1μl,考察特征峰相对保留时间和相对峰面积的一致性。实验结果如表12-1、12-2所示,结果显示各特征峰相对保留时间及相对峰面积值rsd均小于3%,仪器精密度良好。
[0179]
表12-1精密度实验结果(相对保留时间)
[0180][0181]
表12-2精密度实验结果(相对峰面积)
[0182][0183][0184]
(4)稳定性:取黑骨藤标准汤剂供试品溶液,每隔2小时进样一次,共测定12小时,分别进样1μl,考察特征峰相对保留时间和相对峰面积的一致性。实验结果如表13-1、13-2所示,结果显示,各特征峰的相对保留时间及相对峰面积的rsd均小于3%,供试液在12小时内稳定性良好。
[0185]
表13-1稳定性实验结果(相对保留时间)
[0186][0187]
表13-2稳定性实验结果(相对峰面积)
[0188][0189]
(5)重复性实验:制备6份黑骨藤标准汤剂供试品溶液,进样1μl分析,考察特征峰相对保留时间和相对峰面积的一致性。实验结果如表14-1、14-2所示,结果显示,各特征峰的相对保留时间及相对峰面积值的rsd均小于3%,该方法的重复性良好。
[0190]
表14-1重复性实验结果(相对保留时间)
[0191][0192]
表14-2重复性实验结果(相对峰面积)
[0193][0194]
(6)中间精密度:制备6份黑骨藤标准汤剂供试品溶液,进样1μl分析,考察6位不同操作人员在不同日期和不同色谱仪下重复2次操作的特征峰相对保留时间和相对峰面积的一致性。实验结果如表15-1、15-2所示,结果显示:特征峰相对保留时间rsd在0.000~0.836%,各特征峰相对峰面积值rsd在0.000~7.661%,说明本发明提供的方法中间精密度良好。
[0195]
表15-1黑骨藤标准汤剂特征图谱中间精密度结果(相对保留时间)
[0196][0197]
表15-2黑骨藤标准汤剂特征图谱中间精密度结果(相对峰面积)
[0198][0199]
4.4样品检测研究
[0200]
对表7中15批黑骨藤样品制成的黑骨藤标准汤剂进行指纹图谱检测,并采用指纹图谱相似度评价软件对检测结果进行分析,呈现6个分离度较好的共有峰。其中新绿原酸分离度较好,周围没有其它色谱峰干扰,故以新绿原酸为参照峰(即s峰),采用对照品进行指认,峰2为新绿原酸。特征图谱中有6个特征峰,与新绿原酸参照物质对应的峰2为s峰,计算各特征峰与s峰的相对保留时间,其相对保留时间应该在规定值的
±
5%之内,规定值为:0.776(峰1)、1.000(峰2)、1.712(峰3)、1.814(峰4)、3.222(峰5)、3.391(峰6);计算各特征峰与s峰的相对峰面积,其相对保留峰面积应在规定范围内,规定值为:0.038~0.714(峰1)、0.641~1.022(峰3)、0.831~1.041(峰4)、0.201~0.404(峰5)、0.061~0.233(峰6)。如表16所示。
[0201]
表16各特征峰的相对保留时间和相对峰面积
[0202][0203]
4.5共有峰的指认
[0204]
利用lc-ms方法对共有峰进行分析指认,通过测定不同批次黑骨藤标准汤剂样品的uplc图谱,采用国家药典委员会推荐的“中药色谱指纹图谱相似度评价系统(2012版)”进
行结果分析,选择其共有峰。黑骨藤标准汤剂的特征图谱如图9所示,积分参数:平滑峰宽0.1,最小峰面积0.1。
[0205]
4.5.1、实验设备及条件
[0206]
uplc条件:waters acquity uplc色谱仪;色谱柱:waters t3色谱柱(2.1*150mm,1.8μm);流动相系统:乙腈(a):0.1%甲酸水(b);按照表17梯度程序洗脱;流速:0.36ml/min;检测波长280,254,210,330nm;柱温:30℃;进样量:0.8μl;0.5μl(对照品)。
[0207]
质谱条件:waters xevo g2-xs qtof质谱仪,esi离子源正负离子检测;源电压:2.5kv,n2流速:800l/h,碰撞气体为氮气;毛细管温度400℃;锥孔气体流速:100l/h;气源温度:120℃;采用全扫描方式,分子量扫描范围50~1500;碰撞诱导解离电压:6v(低能量)、30~60v(高能量);供试品处理:取黑骨藤标准汤冻干粉1支,0.08g,精密称定,置具塞锥形瓶中,精密加入甲醇20ml,称定重量,超声处理(功率500w,频率40khz)30min,放冷,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
[0208]
表17共有峰指认的梯度提取程序
[0209][0210]
4.5.2lc-ms分析
[0211]
对黑骨藤标准汤剂供试品溶液进行lc-ms分析,检测波长280nm;对黑骨藤标准汤剂供试品溶液的lc-ms图显示的色谱峰进行标注,共标注了11个色谱峰;其保留时间为峰1(3.17min),峰2(3.17min),峰3(4.53min),峰4(5.56min),峰5(6.16min),峰6(6.57min),峰7(12.16min),峰8(13.19min),峰9(18.83min),峰10(19.66min),峰11(22.69min)。lc-ms质谱轮廓图如图10所示,自上而下分别为bpi模式质谱图和280nm下紫外色谱图。
[0212]
对标注的11个色谱峰进行lc-ms鉴定:
[0213]
(1)峰1:dad图谱显示,最大吸收在220,259和292nm处;质谱图显示,该成分的准分子离子峰[m-h]为153.03841;[m-coo]:109.04851,提示分子中含有一分子甲酸,因此推测该成分为原儿茶酸。峰1的dad图谱如图11所示,化学结构如图12所示,质谱图如图13所示。
[0214]
(2)峰2:dad图谱显示,最大吸收在324nm处;质谱图显示,该成分的准分子离子峰[m-h]为353.10025;[m-coo]:191.07466为分子中脱去一分子咖啡酰基形成的奎尼酸离子碎片,135.04630为咖啡酸脱去一分子甲酸,提示该化合物为咖啡酰基奎尼酸,考虑到5.57min和6.16min的色谱峰与其一致,表明他们为同分异构体,结合在c18色谱柱中的出峰时间先后,确定3.72min,5.57min和6.16min分别为新绿原酸、绿原酸和隐绿原酸。峰2的dad图谱如图14所示,质谱图如图15所示。
[0215]
(3)峰3:dad图谱显示,最大吸收在277,308nm处;质谱图显示,该成分的准分子离子峰[m-h]为137.04369,93.05349为脱去一分子甲酸形成的离子碎片,因此该化合物鉴定为邻羟基苯甲酸。峰3的dad图谱如图16所示,质谱图如图17所示。
[0216]
(4)峰4:dad图谱和质谱图显示与峰2一致,结合峰2结果,确定峰4为绿原酸。峰4的dad图谱如图18所示,质谱图如图19所示。
[0217]
(5)峰5:dad图谱和质谱图显示与峰2、峰4一致,结合峰2和峰4结果,确定峰4为隐绿原酸。峰5的dad图谱如图20所示,质谱图如图21所示。
[0218]
(6)峰6:dad图谱所示,最大吸收在322nm处,提示为咖啡酸类成分;质谱图显示,179.05367为其准分子离子峰,135.06416为脱去一分子甲酸的离子碎片,因此,峰6鉴定为咖啡酸。峰6的dad图谱如图22所示,质谱图如图23所示。
[0219]
(7)峰7:dad图谱显示,最大吸收波长在321nm处,提示为咖啡酸类衍生物。质谱显示,该成分的准分子离子峰[m-h]为559.14489;[m-90]:353.09978和191.07461碎片提示分子中有一分子奎尼酸和一分子咖啡酸结构,结构待定。峰7的dad图谱如图24所示,质谱图如图25所示。
[0220]
(8)峰8:dad图谱显示知,最大吸收波长在321nm处,提示该化合物为咖啡酸衍生物;质谱显示,该成分的准分子离子峰[m-h]为529.13643;[m-90]:353.09925和191.07443碎片提示分子中有一分子奎尼酸和一分子咖啡酸结构,结构待定。峰8的dad图谱如图26所示,质谱图如图27所示。
[0221]
(9)峰9:dad图谱所示,最大吸收波长在324nm,提示,该化合物为咖啡酸类成分;质谱图所示,该成分的准分子离子峰[m-h]为515.12179;353.09866为脱去一分子咖啡酰基形成的碎片离子,191.07447为脱去另一分子咖啡酰基的形成的奎尼酸碎片离子,提示分子中有2分子咖啡酰基和1分子奎尼酸,结合19.66min和22.69min的图谱信息,表明三者为结构中含2分子咖啡酰基和1分子奎尼酸的同分异构体,结合在c18色谱柱的出峰先后,18.83,19.66和22.69min分别为3,4-o-二咖啡酰基奎尼酸,3,5-o-二咖啡酰基奎尼酸和4,5-o-二咖啡酰基奎尼酸。峰9的dad图谱如图28所示,质谱图如图29所示。
[0222]
(10)峰10:dad和质谱图显示,与峰9基本一致,结合峰9信息,峰10为3,5-o-二咖啡酰基奎尼酸。峰10的dad图谱如图30所示,质谱图如图31所示。
[0223]
(11)峰11:其dad图谱和质谱与峰9、峰10基本一致,峰11鉴定为4,5-o-二咖啡酰基奎尼酸。峰11的dad图谱如图32所示,质谱图如图33所示。
[0224]
11个色谱峰的鉴定结果如表18所示
[0225]
表18指纹图谱色谱峰鉴定结果
[0226]
[0227][0228]
4.6特征峰的指认
[0229]
在“4.1色谱条件研究”已确定的色谱条件下,分别进样原儿茶酸、绿原酸、隐绿原酸、新绿原酸、咖啡酸对照品溶液和黑骨藤标准汤剂供试品溶液,对特征图谱的色谱峰进行指认。
[0230]
结果表明,经指认后,确定特征图谱中在黑骨藤准汤剂特征图谱中能找到与原儿茶酸、绿原酸、隐绿原酸、新绿原酸、咖啡酸保留时间一致的特征峰,峰1为原儿茶酸,峰2为新绿原酸,峰4为绿原酸,峰为5为隐绿原酸,峰6为咖啡酸。黑骨藤标汤特征图谱峰指认如图34所示,其中,图a为标汤样品,图b为原儿茶酸,图c为绿原酸,图d为隐绿原酸,图e为新绿原酸,图f为咖啡酸。
附图说明
[0231]
图1为黑骨藤标准汤剂冻干曲线图。
[0232]
图2为岛津色谱柱测定结果。
[0233]
图3为黑骨藤标准汤剂特征图谱。
[0234]
图4为绿原酸全波长扫描图。
[0235]
图5为新绿原酸对照品的紫外吸收图。
[0236]
图6为黑骨藤标准汤剂样品的dad-3d图。
[0237]
图7为黑骨藤标准汤剂专属性考察结果。
[0238]
图8为黑骨藤标准汤剂整体性考察结果。
[0239]
图9为黑骨藤标准汤剂的特征图谱。
[0240]
图10为黑骨藤标准汤lc-ms图(自上而下分别为bpi模式质谱图和280nm下紫外色谱图)。
[0241]
图11为峰1的dad图谱。
[0242]
图12为该成分的化学结构。
[0243]
图13为峰1的质谱图。
[0244]
图14为峰2的dad图谱。
[0245]
图15为峰2的质谱图。
[0246]
图16为峰3的dad图谱。
[0247]
图17为峰3的质谱图。
[0248]
图18为峰4的dad图谱。
[0249]
图19为峰4的质谱图。
[0250]
图20为峰5的dad图谱。
[0251]
图21为峰5的质谱图。
[0252]
图22为峰6的dad图谱。
[0253]
图23为峰6的质谱图。
[0254]
图24为峰7的dad图谱。
[0255]
图25为峰7的质谱图。
[0256]
图26为峰8的dad图谱。
[0257]
图27为峰8的质谱图。
[0258]
图28为峰9的dad图谱。
[0259]
图29为峰9的质谱图。
[0260]
图30为峰10的dad图谱。
[0261]
图31为峰10的质谱图。
[0262]
图32为峰11的dad图谱。
[0263]
图33为峰11的质谱图。
[0264]
图34为黑骨藤标汤特征图谱峰指认图,其中,a为标汤样品,b为原儿茶酸,c为绿原酸,d为隐绿原酸,e为新绿原酸,f为咖啡酸。
具体实施方式
[0265]
下面结核具体的实施方式来对本发明的技术方案做进一步的限定,但要求保护的范围不仅局限于所作的描述。
[0266]
实施例1
[0267]
1、黑骨藤标准汤剂样品:
[0268]
取黑骨藤(批号:2019040306)药材饮片,加10倍量水浸泡30min后,武火煮沸后,文火煎煮30min,300目滤布过滤;药渣再加入10倍量水,武火煮沸后,文火煎煮20min,300目滤布过滤;然后将两次获得的滤液合并,在65
±
5℃下浓缩至相对密度为1.02~1.03g/ml,得黑骨藤标浓缩液;将浓缩液冷冻干燥,得到黑骨藤标准汤剂。其中,冷冻干燥分为三个阶段:先在-45℃下预冻4h;然后在-30℃~0℃下升化干燥12h;再在0℃~25℃下解析干燥7h。
[0269]
2、质量检测:
[0270]
出膏率:冷冻干燥分装前称定包材(西林瓶 胶塞)重量,黑古藤标准汤剂浓缩液进行分装,记录分装的浓缩液重量,冷冻干燥后称定冻干粉和包材总重量,按以下公式计算出膏率:
[0271][0272]
测得黑骨藤出膏率为7.85%。
[0273]
含量测定:
[0274]
色谱条件与系统适用性试验:welch xtimate c18色谱柱,规格为4.6mm
×
250mm,粒径5μm,以乙腈:0.5%甲酸水溶液=8:90为流动相;0.9ml/min流速进行洗脱;柱温30℃;检测波长为327nm;理论板数按绿原酸计算应不低于5000;
[0275]
对照品液的制备:取绿原酸标准品,精密称定,加流动相制成每1ml含80ug的溶液;
[0276]
供试品溶液的制备:取黑骨藤标准汤剂粉碎,称取0.1g,加入75%甲醇25ml,称定重量;超声提取25min,放冷,再称定重量,用75%甲醇补足减失重量,摇匀,滤过,取续滤液过0.22μm微孔滤膜,即得。
[0277]
测定法:分别精密吸取对照品溶液与供试品溶液10μl,注入液相色谱仪,测定,即得。
[0278]
测得供试品的绿原酸含量为1.43%,转移率为8.94%。
[0279]
特征图谱:
[0280]
色谱条件与系统适用性试验:inertsil ods-3 2.1
×
100mm 2μm色谱柱;以乙腈为流动相a,以0.05%磷酸溶液为流动相b,进行梯度洗脱;柱温为25℃;流速为0.25ml/min;检测波长为280nm;理论板数按新绿原酸计算应不低于5000;
[0281]
所述梯度洗脱条件为:
[0282]
0-5min,5%
→
8%a,95%
→
92%b;
[0283]
5-12min,8%
→
10%a,92%
→
90%b;
[0284]
12-18min,10%
→
15%a 90%
→
85%b;
[0285]
18-38min,15%
→
20%a 85%
→
80%b;
[0286]
38-39min,20%
→
5%a,80%
→
95%b;
[0287]
39-45min 5%
→
5%a 95%
→
95%b。
[0288]
参照物溶液的制备:称取新绿原酸标准品,加50%甲醇制成每1ml含新绿原酸0.5mg,即得新绿原酸标准品溶液;
[0289]
供试品溶液的制备:精密称取0.1g黑骨藤标准汤剂,按20ml 50%甲醇,称定重量;超声提取45min,放冷,再称定重量,用50%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即可。
[0290]
测定法:分别精密吸取对照品溶液与供试品溶液各1μl,注入液相色谱仪,测定,记录色谱图,即得。
[0291]
供试品的特征图谱为:特征图谱包含6个峰,与新绿原酸参照物质对应的峰2为s峰,计算各特征峰与s峰的相对保留时间;相对保留时间为:0.776(峰1)、1.000(峰2)、1.712(峰3)、1.814(峰4)、3.222(峰5)、3.391(峰6);相对峰面积为:0.056(峰1)、1.000(峰2)、0.825(峰3)、0.917(峰4)、0.301(峰5)、0.119(峰6)。
[0292]
实施例2
[0293]
1、黑骨藤标准汤剂样品:
[0294]
取黑骨藤(批号:2019030801-01)药材饮片,加12倍量水浸泡30min后,武火煮沸后,文火煎煮30min,过滤;药渣再加入10倍量水,武火煮沸后,文火煎煮20-40min;然后将两次获得的滤液合并,在65
±
5℃下浓缩至相对密度为1.02~1.03g/ml,得黑骨藤标准汤剂浓缩液;将浓缩液冷冻干燥,得到黑骨藤标准汤剂。其中,冷冻干燥分为三个阶段:先在-45℃下预冻4h;然后在-30℃~0℃下升化干燥14h;再在0℃~25℃下解析干燥7h。
[0295]
2、质量检测:
[0296]
出膏率:冷冻干燥分装前称定包材(西林瓶 胶塞)重量,黑古藤标准汤剂浓缩液进行分装,记录分装的浓缩液重量,冷冻干燥后称定冻干粉和包材总重量,按以下公式计算出膏率:
[0297][0298]
测得出膏率为7.79%。
[0299]
含量测定:
[0300]
色谱条件与系统适用性试验:welch xtimate c18色谱柱,规格为4.6mm
×
250mm,粒径5μm,以乙腈:0.5%甲酸水溶液=9:91为流动相;1ml/min流速进行洗脱;柱温30℃;检测波长为327nm;理论板数按绿原酸计算应不低于5000;
[0301]
对照品液的制备:取绿原酸标准品,精密称定,加流动相制成每1ml含80ug的溶液;
[0302]
供试品溶液的制备:取黑骨藤标准汤剂粉碎,称取0.1g,加入50%甲醇25ml,称定重量;超声提取30min,放冷,再称定重量,用50%甲醇补足减失重量,摇匀,过0.22μm微孔滤膜,取续滤液,即得。
[0303]
测定法:分别精密吸取对照品溶液与供试品溶液10μl,注入液相色谱仪,测定,即得。
[0304]
测得供试品的绿原酸含量为1.40%;转移率为8.91%。
[0305]
特征图谱:
[0306]
色谱条件与系统适用性试验:inertsil ods-3 2.1
×
100mm 2μm色谱柱;以乙腈为流动相a,以0.05%磷酸溶液为流动相b,进行梯度洗脱;柱温为30℃;流速为0.30ml/min;检测波长为280nm;理论板数按新绿原酸计算应不低于5000;
[0307]
所述梯度洗脱条件为:
[0308]
0-5min,5%
→
8%a,95%
→
92%b;
[0309]
5-12min,8%
→
10%a,92%
→
90%b;
[0310]
12-18min,10%
→
15%a 90%
→
85%b;
[0311]
18-38min,15%
→
20%a 85%
→
80%b;
[0312]
38-39min,20%
→
5%a,80%
→
95%b;
[0313]
39-45min 5%
→
5%a 95%
→
95%b。
[0314]
参照物溶液的制备:称取新绿原酸标准品,加50%甲醇制成每1ml含新绿原酸0.5mg,即得新绿原酸标准品溶液;
[0315]
供试品溶液的制备:精密称取0.1g黑骨藤标准汤剂,按20ml 50%甲醇,称定重量;超声提取30min,放冷,再称定重量,用50%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即可。
[0316]
测定法:分别精密吸取对照品溶液与供试品溶液各1μl,注入液相色谱仪,测定,记录色谱图,即得。
[0317]
供试品的特征图谱为:特征图谱包含6个峰,与新绿原酸参照物质对应的峰2为s峰,计算各特征峰与s峰的相对保留时间;相对保留时间为:0.775(峰1)、1.000(峰2)、1.712(峰3)、1.814(峰4)、3.217(峰5)、3.387(峰6);相对峰面积为:0.039(峰1)、1.000(峰2)、
0.965(峰3)、0.980(峰4)、0.246(峰5)、0.089(峰6)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
