一种瑞德西韦d-核糖酸内脂的合成方法
技术领域
1.本发明属于化学药物技术领域,具体涉及一种瑞德西韦d-核糖酸内脂的合成方法。
背景技术:
2.瑞德西韦(remdesivir),是一种核苷类似物,cas登记号:1809249-37-3,结构式ⅱ所示,由美国吉利德科学公司研制,美国fda对瑞德西韦(remdesivir)开绿灯,批准该药物用于住院的新冠肺炎患者。瑞德西韦同时也是治疗埃博拉病毒感染的孤儿药,也成为美国首个获准用于新冠患者的药物。用于2019-ncov感染者的急症治疗。具有抗病毒活性,在hae细胞中,对 sars-cov 和 mers-cov 的 ec50 值为74 nm,在延迟脑肿瘤细胞中,对鼠肝炎病毒的ec50 值为30 nm,瑞德西韦在抗新型冠状病毒时展现出较好的疗效,是治疗新型冠状病毒的潜在药品,国内外报道瑞德西韦关键化合物2,3,5-三苄氧基-1,4-内脂(结构式ⅲ所示,cas登记号:55094-52-5)化合物合成制备方法。
3.现阶段制备2,3,5-三苄氧基-1,4-内脂方法(合成路线如ⅳ所示)大多对反应温度较高,易产生杂质,收率偏低,使用高毒物质,环保压力较大等因素;
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀⅱꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀⅲ
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀⅳ
。
4.如专利文献wo2013/158746和wo2014/78778报道用溴素和水氧化,但溴素是易制毒品,具有刺激性和强腐蚀性,不易运输和操作安全系数低,另外wo2018/053706和wo2013/084165报道以d-核糖为原料,利用浓硫酸甲氧基化,苄基化,水解,采用dmp(戴斯-马丁高碘烷)氧化剂氧化,每一步收率偏低,并且dmp原料价格昂贵等问题不适宜工业化。
5.而国内专利关于2,3,5-三苄氧基-1,4-内脂,现有技术中的合成路线如下:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀⅴ
如上述结构式
ⅴ
所示,通过以d-核糖为起始原料,在甲醇中,用草酰氯等做催化剂,合成甲氧基化合物2,之后加入氢化钠和氯化苄或溴化苄,高温反应合成出化合物3,取化合物3用稀硫酸或稀盐酸催化,回流搅拌过夜处理后,得到化合物4,溶于二氯甲烷和水,加入碳酸氢钠和四甲基哌啶氮氧化物,用10%次氯酸钠氧化,室温搅拌过夜处理,得到化合物5,该方法路线和反应时间偏长,产率低只有40%左右,并使用较危险化合物氢化钠同时高温次数多易产生杂质和不易固化等现象,同时,合成瑞德西韦后期工序脱去苄基保护,与瑞德西韦侧链(2-乙基丁基 ((s)-(五氟苯氧基)(苯氧基)磷酰基)-l-丙氨酸酯)对接易衍生杂质,如文献us2016122374报道为了避免产生杂质,脱去苄基后,为了避免杂质再用异亚丙
基保护邻位羟基,然后与侧链对接,而且乙酰基保护极易脱除,影响了异丙叉保护。为此,需要一种新的技术方案来解决上述技术方案。
技术实现要素:
6.本发明的目的在于提供一种瑞德西韦d-核糖酸内脂的合成方法,以解决上述背景技术中提出的现阶段瑞德西韦d-核糖酸内脂的合成方法存在工序繁多,成本高,极易产生杂质,收率低,环保压力大的问题。
7.为实现上述目的,本发明提供如下技术方案:一种瑞德西韦d-核糖酸内脂的合成方法,其特征在于,具体的合成路线如下:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀⅰ
。
8.所述瑞德西韦d-核糖酸内脂的具体合成步骤为:s1、化合物rd-1的制备:以d-核糖作为起始原料,与摩尔比1:0.01~1.0的稳定剂,和质量体积比1:5的溶剂进行搅拌溶解,在0~5℃下加入摩尔比1:0.1~3.0的氧化剂,在0~5℃条件下进行氧化反应,反应结束后加入亚硫酸氢钠在0~5℃条件下进行20-40分钟的淬灭,再在45~50℃真空条件下浓缩形成反应液,然后再用无水乙醇进行热过滤,浓缩得到化合物rd-1,其中,所述的稳定剂为二氮杂二环,所述的溶剂为四氢呋喃、50%四氢呋喃水溶液的其中一种,所述的氧化剂为氯铬酸吡啶盐;s2、化合物rd-2的制备:将s1中得到的化合物rd-1与质量比1:5的反应溶剂和摩尔比1:0.01的4-二甲氨基吡啶进行搅拌溶解后,再加入三乙胺,降温至0℃,在氮气的保护条件下,滴加与摩尔比1:0.1~2.0的保护剂,搅拌1-2h,再加入2m盐酸在-5~5℃的条件下进行0.5-1.5小时的淬灭,饱和碳酸氢钠洗涤,水洗,浓缩得到化合物rd-2,其中,所述的反应溶剂为二氯甲烷,所述的保护剂为乙酸酐;s3、化合物rd-3的制备:将s2中得到的化合物rd-2与质量比1:3的混合保护剂进行搅拌溶解后,在室温条件下滴加摩尔比1:0.01的酸性催化剂,再在0~5℃滴加摩尔比1:1.5的三乙胺进行淬灭反应,而后浓缩至干加入乙酸乙酯和水洗涤,浓缩后用异丙醚精制得到目标产物化合物 rd-3,其中,所述的混合保护剂为丙酮和甲醇的混合物,所述丙酮与甲醇的摩尔比为1:1,所述的酸性催化剂为氯化亚砜、盐酸、硫酸的其中一种。
9.与现有技术相比,本发明的有益效果是:1.本发明的瑞德西韦d-核糖酸内脂合成方法利用氯铬酸吡啶盐氧化反应、乙酰基
保护和丙酮叉保护羟基等反应,使得合成工艺路线短,有效地简化了操作难度,且无高温反应,使得合成过程更加安全可靠,有效地节约了能源,提高了收率,无需使用剧毒物质就能完成合成工艺,有效地避免有毒有害废水的产生,使得环境得以改善,从而能够很好地实现工业化生产。
10.2.本发明的瑞德西韦d-核糖酸内脂合成方法采用价格低廉易得的d-核糖作为起始原料,使得生产成本得以降低,有效地简化了合成工序,同时也使得环保压力得到了有效地改善。
附图说明
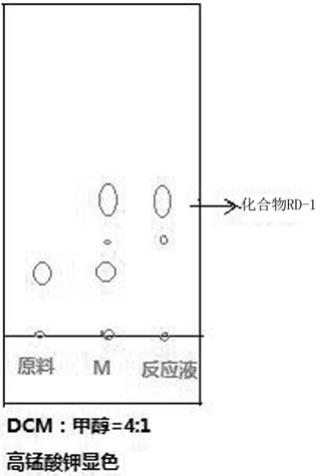
11.图1为本发明的s1的tlc图;图2为本发明的s2原料反应情况的tlc图;图3为本发明的s3产物的tlc图。
具体实施方式
12.以下实施例用来进一步说明本发明的内容,并不限制本发明的应用。
13.实施例1:s1、化合物 rd-1的制备称取100g,0.67mol的d-核糖,25.5g,0.165mol,0.25eq的二氮杂二环,500ml的四氢呋喃,158g,0.73mol,1.1eq的氯铬酸吡啶盐;首先按照上述称取量将d-核糖与二氮杂二环和四氢呋喃进行充分搅拌溶解,在氮气的保护条件下,降温至0~5℃,再分批缓慢加入氯铬酸吡啶盐,在tlc监控下进行2~3小时的反应(如图1所示),反应结束后,加入6.5g,62.5mmol的亚硫酸氢钠在0~5℃的条件下进行淬灭,搅拌30分钟后,在45~50℃真空条件下进行浓缩形成反应液,然后再用无水乙醇进行热过滤,浓缩得到85g白色固体的化合物rd-1,其收率为86%。
14.s2、化合物 rd-2的制备称取70g,0.5mol的化合物rd-1,350g的二氯甲烷,0.6g,0.005mol,0.01eq的4-二甲氨基吡啶,48.5g,0.475mol,0.95eq的乙酸酐;先按照上述称取量将化合物rd-1与二氯甲烷和4-二甲氨基吡啶进行充分搅拌溶解,再加入2ml的三乙胺,降温至0℃,在氮气保护的条件下,滴加乙酸酐,搅拌1-2小时,在tlc监控下进行4~6小时的保温反应(如图2所示),而后加入2m盐酸在-5~5℃的条件下进行1小时的淬灭,用饱和碳酸氢钠水溶液洗涤,水洗,浓缩得到77g油状物的化合物rd-2,其收率为80%。
15.s3、化合物 rd-3的制备称取77g ,0.4mol的化合物rd-2,231g的丙酮,231g的甲醇,0.5g,0.004mol,0.01eq的氯化亚砜,60.7g,0.6mol,1.5eq的三乙胺;在氮气保护的条件下,按照上述称取量将化合物rd-2与丙酮和甲醇进行充分搅拌溶解,在室温条件下缓慢滴加氯化亚砜,在tlc监控下进行2~3小时的反应(如图3所示),再在0~5℃的条件下滴加三乙胺进行淬灭反应,而后浓缩至干加入乙酸乙酯和水洗涤,浓缩后用异丙醚精制得到75g白色固体的化合物rd-3,即目标产物2,3-异亚丙基-5-乙酰基-1,
4-内脂,其收率为82%。
16.实施例2:s1、化合物 rd-1的制备称取100g,0.67mol的d-核糖,20.5g,0.135mol,0.20eq的二氮杂二环,500ml的四氢呋喃,288g, 1.34mol, 2.0eq的氯铬酸吡啶盐;首先按照上述称取量将d-核糖与二氮杂二环和四氢呋喃搅拌,在氮气的保护条件下,降温至0~5℃,再分批缓慢加入氯铬酸吡啶盐,在tlc 监控下进行2~3小时的反应(如图1所示),反应结束后,加入6.5g,62.5mmol的亚硫酸氢钠在0~5℃条件下进行淬灭,搅拌30分钟后,在 45~50℃真空条件下浓缩形成反应液,然后再用无水乙醇热过滤,浓缩得66g白色固体的化合物rd-1,其收率为65%。
17.s2、化合物 rd-2的制备称取60g,0.40mol的化合物rd-1,330g的二氯甲烷,0.5g,0.004mol,0.01eq的4-二甲氨基吡啶,41.4g,0.4mol,0.95eq的乙酸酐;先按照上述称取量将化合物 rd-1与二氯甲烷和4-二甲氨基吡啶进行充分搅拌溶解,再加入1.9ml三乙胺,降温至0℃,在氮气保护的条件下,滴加乙酸酐,搅拌1-2小时,在tlc监控下进行4~6小时的保温反应(如图2所示),而后加入2m盐酸在-5~5℃的条件下进行1小时的淬灭,用饱和碳酸氢钠水溶液洗涤,水洗,浓缩得到66g油状物的化合物rd-2,其收率为80%。
18.s3、化合物 rd-3的制备称取60g ,0.35mol的化合物rd-2,180g的丙酮,180g的甲醇,0.13g,0.0035mol,0.01eq的盐酸,53.6g,0.53mol,1.5eq的三乙胺;在氮气保护的条件下,按照上述称取量将化合物rd-2与丙酮和甲醇进行充分搅拌溶解,在室温条件下缓慢滴加盐酸,在tlc监控下进行2~3小时的反应(如图3所示),再在0~5℃条件下滴加三乙胺进行淬灭反应,而后浓缩至干加入乙酸乙酯和水洗涤,浓缩后用异丙醚精制得到56g白色固体的化合物 rd-3,即目标产物2,3-异亚丙基-5-乙酰基-1,4-内脂,其收率为71%。
19.实施例3:s1、化合物 rd-1的制备称取100g,0.67mol的d-核糖,25.5g,0.165mol,0.25eq的二氮杂二环,500ml的50%四氢呋喃水溶液,100g, 0.5mol, 0.6eq的氯铬酸吡啶盐;首先按照上述称取量将d-核糖与二氮杂二环和50%四氢呋喃水溶液进行充分搅拌溶解,在氮气的保护条件下,降温至0~5℃,再分批缓慢加入氯铬酸吡啶盐,在tlc监控下进行2~3小时的反应(如图1所示),反应结束后,加入6.5g,62.5mmol的亚硫酸氢钠在0~5℃条件下进行淬灭,搅拌30分钟后,在45~50℃真空条件下进行浓缩形成反应液,然后再用无水乙醇进行热过滤,浓缩得到60g白色固体的化合物rd-1,其收率为59%。
20.s2、化合物 rd-2的制备称取60g,0.40mol的化合物rd-1,300g的二氯甲烷,0.5g,0.004mol,0.01eq的4-二甲氨基吡啶,41.4g,0.41mol,0.95eq的乙酸酐;先按照上述称取量将化合物rd-1与二氯甲烷和4-二甲氨基吡啶进行充分搅拌溶
解,再加入1.9ml三乙胺,降温至0℃,在氮气保护的条件下,滴加乙酸酐,搅拌1-2小时,在tlc监控下进行4~6小时的保温反应(如图2所示),而后加入2m盐酸在0~5℃的条件下进行1小时的淬灭,用饱和碳酸氢钠水溶液洗涤,水洗,浓缩得到64g油状物的化合物rd-2,其收率为75%。
21.s3、化合物 rd-3的制备称取64g ,0.35mol的化合物rd-2,180g的丙酮,180g的甲醇,0.35g,0.0035mol,0.01eq的硫酸,53.6g,0.53mol,1.5eq的三乙胺;在氮气保护的条件下,按照上述称取量将化合物rd-2与丙酮和甲醇进行充分搅拌溶解,在室温条件下缓慢滴加硫酸,在tlc监控下进行2~3小时的反应(如图3所示),在0~5℃条件下滴加三乙胺进行淬灭反应,而后浓缩至干加入乙酸乙酯和水洗涤,浓缩后用异丙醚精制得到52g白色固体的化合物rd-3,即目标产物2,3-异亚丙基-5-乙酰基-1,4-内脂,其收率为64%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。