1.本发明属于生物医用高分子材料技术领域,具体涉及一种相转变可调控的聚合物/锂藻土纳米粒子复合物热致水凝胶及其制备方法与应用。
背景技术:
2.水凝胶具有类似软组织的机械性能、高度的亲水性以及优异的生物相容性等特点,使其在生物医学领域具有良好的应用前景。其中,随温度的升高,发生溶胶-溶胶转变的聚合物热致水凝胶在药物递送、可注射组织工程等应用方面显示了巨大的潜力。
3.具有热致凝胶化性质的聚合物通常是两亲性的,亲水嵌段常采用生物相容的聚乙二醇(peg),疏水嵌段可选择可生物降解的聚氨基酸等。亲水嵌段和疏水嵌段合适的比例是形成热致水凝胶的关键。虽然聚乙二醇-聚氨基酸嵌段聚合物具有良好的生物相容性且具有明确的热致凝胶化性能,但,令人遗憾的是,很多组成条件下的聚乙二醇-聚氨基酸嵌段聚合物材料的溶液-凝胶相转变点温度远高于人体温度,无法满足体内注射并原位形成凝胶的需求。
4.故而,考虑到实际应用方面的具体需求,寻求一种热致凝胶化温度在室温和人体温度之间以满足材料使用时的形态需求,并兼具良好的生物相容性及载药性能的复合物水凝胶体系就成为了本领域技术人员亟需解决的技术问题。
技术实现要素:
5.有鉴于此,本发明的第一个目的是针对现有技术中存在的问题,提供一种热致凝胶化温度在室温和人体温度之间,且具有良好的生物相容性的水凝胶材料。
6.为了实现上述目的,本发明采用如下技术方案:
7.一种相转变可调控的聚合物/锂藻土纳米粒子复合物热致水凝胶,包括0.1-8wt%的溶胶型xls型号的锂藻土纳米粒子、0.5-20wt%的两亲性嵌段共聚物和余量的溶媒。
8.值得说明的是,锂藻土是一种硅酸镁锂(na
0.7
[mg
5.5
li
0.3
si8o
20
(oh)4]-0.7
)纳米粒子,其表面荷有较高的负电荷,边缘荷有的电荷呈现ph依赖性(在ph小于9时呈正电荷),其电荷可与药物如阿霉素或者蛋白质药物等通过静电作用相互复合,实现缓释的目的。另外,研究发现,锂藻土纳米粒子的共混不仅可调节水凝胶的机械性能,而且可降低材料的毒性并促进成纤维细胞在材料表面增殖。
[0009]
本发明通过锂藻土纳米粒子共混的方式调控peg/聚氨基酸嵌段聚合物水体系的相转变温度,使其可实现体内注射并原位形成凝胶的应用需求。同时,通过锂藻土纳米粒子与负载药物静电相互作用,实现复合水凝胶体系中药物的缓慢释放,并可促进细胞的粘附和增殖以满足组织工程的应用。
[0010]
优选的,所述两亲性嵌段共聚物的含量为2-25wt%。
[0011]
进一步优选的,所述两亲性嵌段共聚物由平均分子量为400-8000的亲水性聚乙二
醇嵌段a和平均分子量为500-40000的疏水性聚氨基酸嵌段b组成,其中嵌段a的含量为10-90wt%,嵌段b的含量为90-10wt%。
[0012]
更进一步优选的,所述疏水性聚氨基酸嵌段为聚l-丙氨酸、聚d,l-丙氨酸、聚d-丙氨酸、聚苯丙氨酸、聚亮氨酸、聚赖氨酸、聚谷氨酸、聚天冬氨酸,聚酪氨酸、聚缬氨酸中的一种或多种的组合。
[0013]
更进一步优选的,所述嵌段共聚物为aba或bab型的三嵌段共聚物、ab型的两嵌段共聚物、(a-b)n或(b-a)n的星形嵌段共聚物以及a(ba)n或b(ab)n构型的多嵌段共聚物,其中n为2至10的整数。
[0014]
优选的,所述水凝胶的相转变温度为4-37℃,降解周期为3-90天。
[0015]
值得说明的是,本发明由锂藻土纳米粒子与两亲性嵌段共聚物混合得到的复合体系,与以水为主体的分散介质为溶媒,两者构成的水体系;该锂藻土纳米粒子与两亲性嵌段聚合物共混得到的复合水体系具有可调的相转变温度和可控的降解周期,且该复合体系的相转变温度介于室温与人体之间。当体系温度低于溶胶-凝胶转变温度时,复合水体系处于可流动的溶液状态,具有良好的可注射性;当体系温度温度高于溶胶-凝胶转变温度时,则能够自发转变为半固体水凝胶,具有良好的机械性能和较长的降解周期。
[0016]
优选的,所述溶媒包括纯水、注射用水、生理盐水、缓冲溶液、动植物/人体体液、组织培养液、细胞培养液及不以有机溶剂为主体的介质。
[0017]
优选的,所述水凝胶还包括0.01-15wt%的调节剂,所述调节剂包括糖、盐、羧甲基纤维素钠、(碘)甘油、二甲硅油、丙二醇、卡波姆、甘露醇、山梨醇、吐温20、吐温40、吐温80、木糖醇、低聚糖、软骨素、甲壳素、壳聚糖、明胶、蛋白胶、透明质酸、聚乙二醇中的一种或多种的组合。
[0018]
本发明的第二个目的在于提供一种如上所述相转变可调控的聚合物/锂藻土纳米粒子复合物热致水凝胶的制备方法。
[0019]
为了实现上述目的,本发明采用如下技术方案:
[0020]
一种如上所述相转变可调控的聚合物/锂藻土纳米粒子复合物热致水凝胶的制备方法:
[0021]
首先配制嵌段共聚物水溶液,然后加入锂藻土纳米粒子,溶解均匀后成为锂藻土纳米粒子共混聚合物溶液,在-20℃或以下储存备用,使用前复溶;或,
[0022]
首先分别配制嵌段共聚物水溶液和锂藻土纳米粒子注射液,在-20℃或以下储存备用,使用前充分混匀,然后制成锂藻土纳米粒子共混聚合物溶液;或,
[0023]
首先配制得到锂藻土纳米粒子水溶液,然后与嵌段共聚物混合并溶解均匀后成为锂藻土纳米粒子共混聚合物溶液,在-20℃或以下储存备用,使用前复溶;或,
[0024]
首先将嵌段共聚物与锂藻土纳米粒子混合,然后加入溶媒,溶解均匀后成为锂藻土纳米粒子共混聚合物溶液,在-20℃或以下储存备用,使用前复溶。
[0025]
值得说明的是,本发明制备的相转变可调控的聚合物/锂藻土纳米粒子复合物热致水凝胶只通过简单的物理共混的方式即可调控复合溶液的相转变温度,不涉及复杂的且周期较长的化学合成来改变聚合物的单体比例或分子量来调控溶液的相转变温度。
[0026]
本发明的第三个目的在于提供一种如上所述相转变可调控的聚合物/锂藻土纳米粒子复合物热致水凝胶的应用。
[0027]
所述相转变可调控的聚合物/锂藻土纳米粒子复合物热致水凝胶在药物递送、组织工程修复中的应用。
[0028]
与现有技术相比,本发明所制备的复合物热致水凝胶,通过锂藻土纳米粒子的浓度调控,即可实现满足体内注射热致水凝胶的制备,且锂藻土纳米粒子特有的表面电荷可通过与荷正电或荷负电的药物的相互作用,实现药物的长效缓释。同时,锂藻土复合后,还可调控热致水凝胶体内的降解周期,从而可实现药物缓释周期和载体降解周期匹配的缓释制剂的制备,在慢性病如:糖尿病、关节炎、椎间盘退变等疾病的治疗中,解决局部长期多次注射时材料的清除问题。peg/聚氨基酸材料具有良好的生物相容性,通过锂藻土纳米粒子的复合可增加热致水凝胶在组织工程的应用时细胞的粘附,并且可控的降解周期在组织工程领域也具有极强的应用潜力。
附图说明
[0029]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
[0030]
图1为本发明实施例20中copolymer-4(5wt%)溶液的变温动态流变曲线。
[0031]
图2为本发明实施例21中copolymer-4(8wt%)溶液的变温动态流变曲线。
[0032]
图3为本发明实施例22中共混聚合物lp3p8溶液的变温动态流变曲线。
[0033]
图4为本发明实施例23中共混聚合物lp3p11溶液的变温动态流变曲线。
[0034]
图5为本发明实施例24中共混聚合物lp1.5p8溶液的变温动态流变曲线。
具体实施方式
[0035]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0036]
实施例1
[0037]
首先将0.500g(0.25mmol)mpeg
2000-nh2与50ml的甲苯共沸除水,待剩余溶剂体积为5ml,密封体系并冷却至室温。将0.240g(2.09mmol)l-丙氨酸-n-羧基环内酸酐(l-ala-nca)和0.065g(0.34mmol)l-苯丙氨酸-n-羧基环内酸酐(l-phe-nca)单体加入到20ml经无水硫酸镁干燥的chcl3/dmf(v/v 3:1)混合溶剂中,并室温下磁力搅拌30min。在氩气的氛围下,将混合单体的溶液转移到干燥的mpeg
2000-nh2中,随后将体系温度升至37℃,在氩气的氛围下,磁力搅拌反应3天。随后将体系冷却至室温,加入20ml的chcl3,待反应产物完全溶解后,逐滴加入到冰乙醚中进行沉降,并重复上述操作三次。最后,将产物置于25℃的真空烘箱中干燥48h,得到两嵌段聚合物mpeg
2000-paf,产率约67%,通过凝胶渗透色谱(gpc,聚苯乙烯为标样)测得上述两嵌段聚合物(mpeg
2000-paf,copolymer-1)的重均与数均分子量(mw,mn)分别为3300和2570,分子量分布系数(mw/mn,)为1.28,其聚合物水体系具有热致凝胶化性能。
[0038]
实施例2
[0039]
首先将0.500g(0.10mmol)mpeg
5000-nh2与50ml的甲苯共沸除水,待剩余溶剂体积为5ml,密封体系并冷却至室温。将0.012g(0.10mmol)l-丙氨酸-n-羧基环内酸酐(l-ala-nca)单体加入到20ml经无水硫酸镁干燥的chcl3/dmf(v/v 3:1)混合溶剂中,并室温下磁力搅拌30min。在氩气的氛围下,将混合单体的溶液转移到干燥的mpeg
5000-nh2中,随后将体系温度升至37℃,在氩气的氛围下,磁力搅拌反应3天。随后将体系冷却至室温,加入20ml的chcl3,待反应产物完全溶解后,逐滴加入到冰乙醚中进行沉降,并重复上述操作三次。最后,将产物置于25℃的真空烘箱中干燥48h,得到两嵌段聚合物mpeg
5000-pa,产率约67%,通过凝胶渗透色谱(gpc,聚苯乙烯为标样)测得上述两嵌段聚合物(mpeg
5000-pa,copolymer-2)的重均与数均分子量(mw,mn)分别为8500和7230,分子量分布系数(mw/mn,)为1.18,其聚合物水体系具有热致凝胶化性能。
[0040]
实施例3
[0041]
首先将0.500g(0.25mmol)双氨基的peg
2000
与50ml的甲苯共沸除水,待剩余溶剂体积为5ml,密封体系并冷却至室温。将0.321g(2.80mmol)l-丙氨酸-n-羧基环内酸酐(l-ala-nca)和0.178g(0.93mmol)l-苯丙氨酸-n-羧基环内酸酐(l-phe-nca)单体加入到20ml经无水硫酸镁干燥的chcl3/dmf(v/v 3:1)混合溶剂中,并室温下磁力搅拌30min。在氩气的氛围下,将混合单体的溶液转移到干燥的nh
2-peg
2000-nh2中,随后将体系温度升至37℃,在氩气的氛围下,磁力搅拌反应3天。随后将体系冷却至室温,加入20ml的chcl3,待反应产物完全溶解后,逐滴加入到冰乙醚中进行沉降,并重复上述操作三次。最后,将产物置于25℃的真空烘箱中干燥48h,得到三嵌段聚合物paf-peg
2000-paf,产率约70%,通过凝胶渗透色谱(gpc,聚苯乙烯为标样)测得上述两嵌段聚合物(paf-peg
2000-paf,copolymer-3)的重均与数均分子量(mw,mn)分别为5200和4330,分子量分布系数(mw/mn,)为1.20,其聚合物水体系具有热致凝胶化性能。
[0042]
实施例4
[0043]
首先将0.500g(0.10mmol)mpeg
5000-nh2与50ml的甲苯共沸除水,待剩余溶剂体积为5ml,密封体系并冷却至室温。将0.313g(2.72mmol)l-丙氨酸-n-羧基环内酸酐(l-ala-nca)和0.074g(0.39mmol)l-苯丙氨酸-n-羧基环内酸酐(l-phe-nca)单体加入到20ml经无水硫酸镁干燥的chcl3/dmf(v/v3:1)混合溶剂中,并室温下磁力搅拌30min。在氩气的氛围下,将混合单体的溶液转移到干燥的mpeg
5000-nh2中,随后将体系温度升至37℃,在氩气的氛围下,磁力搅拌反应3天。随后将体系冷却至室温,加入20ml的chcl3,待反应产物完全溶解后,逐滴加入到冰乙醚中进行沉降,并重复上述操作三次。最后,将产物置于25℃的真空烘箱中干燥48h,得到三嵌段聚合物mpeg
5000-paf,产率约70%,通过凝胶渗透色谱(gpc,聚苯乙烯为标样)测得上述两嵌段聚合物(mpeg
5000-paf,copolymer-4)的重均与数均分子量(mw,mn)分别为7500和5560,分子量分布系数(mw/mn,)为1.35,其聚合物水体系具有热致凝胶化性能。
[0044]
实施例5
[0045]
首先将0.500g(0.25mmol)双氨基的peg
2000
与50ml的甲苯共沸除水,待剩余溶剂体积为5ml,密封体系并冷却至室温。将0.375g(2.48mmol)l-丙氨酸-n-羧基环内酸酐(l-ala-nca)单体加入到20ml经无水硫酸镁干燥的chcl3/dmf(v/v 3:1)混合溶剂中,并室温下磁力
搅拌30min。在氩气的氛围下,将混合单体的溶液转移到干燥的nh
2-peg
2000-nh2中,随后将体系温度升至37℃,在氩气的氛围下,磁力搅拌反应3天。随后将体系冷却至室温,加入20ml的chcl3,待反应产物完全溶解后,逐滴加入到冰乙醚中进行沉降,并重复上述操作三次。最后,将产物置于25℃的真空烘箱中干燥48h,得到三嵌段聚合物pa-peg
2000-pa,产率约70%,通过凝胶渗透色谱(gpc,聚苯乙烯为标样)测得上述两嵌段聚合物(pa-peg
2000-pa,copolymer-5)的重均与数均分子量(mw,mn)分别为5000和4270,分子量分布系数(mw/mn,)为1.17,其聚合物水体系具有热致凝胶化性能。
[0046]
实施例6
[0047]
首先将0.500g(0.67mmol)mpeg
750-nh2与50ml的甲苯共沸除水,待剩余溶剂体积为5ml,密封体系并冷却至室温。将1.76g(6.70mmol)γ-苄基-l-谷氨酸-n-羧基环内酸酐(bgl-nca)单体加入到20ml经无水硫酸镁干燥的chcl3/dmf(v/v 3:1)混合溶剂中,并室温下磁力搅拌30min。在氩气的氛围下,将混合单体的溶液转移到干燥的mpeg
750-nh2中,随后将体系温度升至37℃,在氩气的氛围下,磁力搅拌反应3天。随后将体系冷却至室温,加入20ml的chcl3,待反应产物完全溶解后,逐滴加入到醋酸和甲醇混合物(v/v,1:3)中进行沉降,得到白色固体产物。最后,将产物置于25℃的真空烘箱中干燥48h,得到两嵌段聚合物mpeg
750-pbgl。随后通过催化氢化的方式去除苄基保护基团。具体为将0.500g的mpeg-pbgl溶解于15ml的thf中,少量的pd/c(10%)悬浮于5ml的甲醇中。然后将两种液体添加到氢化器中,用氮气吹扫三次后,在1mpa氢气压力下,在50℃下搅拌反应混合物3天。除去pd/c粉末后,将溶液滴入过量的石油醚中,沉淀物在40℃下真空干燥24h,得到最终产物peg-pgl。通过凝胶渗透色谱(gpc,聚苯乙烯为标样)测得上述两嵌段聚合物(mpeg
750-pgl,copolymer-6)的重均与数均分子量(mw,mn)分别为2750和2200,分子量分布系数(mw/mn,)为1.25,其聚合物水体系具有热致凝胶化性能。
[0048]
实施例7
[0049]
首先将0.500g(0.67mmol)双氨基peg
2000
与50ml的甲苯共沸除水,待剩余溶剂体积为5ml,密封体系并冷却至室温。将3.94g(15mmol)γ-苄基-l-谷氨酸-n-羧基环内酸酐(bgl-nca)单体加入到20ml经无水硫酸镁干燥的chcl3/dmf(v/v 3:1)混合溶剂中,并室温下磁力搅拌30min。在氩气的氛围下,将混合单体的溶液转移到干燥的nh
2-peg
2000-nh2中,随后将体系温度升至37℃,在氩气的氛围下,磁力搅拌反应3天。随后将体系冷却至室温,加入20ml的chcl3,待反应产物完全溶解后,逐滴加入到醋酸和甲醇混合物(v/v,1:3)中进行沉降,得到白色固体产物。最后,将产物置于25℃的真空烘箱中干燥48h,得到三嵌段聚合物pbgl-peg
2000-pbgl。随后通过催化氢化的方式去除苄基保护基团。具体为将0.500g的pbgl-peg-pbgl溶解于15ml的thf中,少量的pd/c(10%)悬浮于5ml的甲醇中。然后将两种液体添加到氢化器中,用氮气吹扫三次后,在1mpa氢气压力下,在50℃下搅拌反应混合物3天。除去pd/c粉末后,将溶液滴入过量的石油醚中,沉淀物在40℃下真空干燥24h,得到最终产物pgl-peg-pgl。通过凝胶渗透色谱(gpc,聚苯乙烯为标样)测得上述三嵌段聚合物(pgl-peg
2000-pgl,copolymer-7)的重均与数均分子量(mw,mn)分别为5750和5000,分子量分布系数(mw/mn,)为1.15,其聚合物水体系具有热致凝胶化性能。
[0050]
实施例8
[0051]
首先将10g(10mmol)的mpeg
1000
与100ml甲苯共沸除水,待剩余体积约为10ml,密闭
体系并冷却至室温。将15.3g(50mmol)的ε-羧苄氧基-l-赖氨酸-n-羧基环内酸酐(z-l-lys-nca)单体加入到60ml经无水硫酸镁干燥的chcl3/dmf(v/v 3/1)混合溶剂中,并在室温下磁力搅拌30min。在氩气的氛围下,将单体的溶液转移到干燥的mpeg
1000-nh2中,随后将体系温度升至37℃,磁力搅拌反应2天。然后,向反应混合物中添加10.35g(90mmol)l-丙氨酸-n-羧基环内酸酐(l-ala-nca),继续在37℃下反应2天。随后将体系冷却至室温,加入20ml的chcl3,待反应产物完全溶解后,逐滴加入到冰乙醚中进行沉降,并重复上述操作三次。最后,将产物置于25℃的真空烘箱中干燥48h。最后,在室温下,将聚合物的ε-羧苄氧基-l-赖氨酸基团在三氟乙酸/乙酸中的溴酸(15ml/15ml)中脱保护3h。再用氢氧化钠(1.0m)中和至ph约7后,使用截留分子量为1000da的透析膜透析聚合物并冷冻干燥。产率约为60%。通过凝胶渗透色谱(gpc,聚苯乙烯为标样)测得上述两嵌段聚合物(mpeg
1000-pk-pa,copolymer-8)的重均与数均分子量(mw,mn)分别为3900和3080,分子量分布系数(mw/mn,dm)为1.27,其聚合物水体系具有热致凝胶化性能。
[0052]
实施例9
[0053]
称量1g实施例1中的copolymer-1(mpeg
2000-paf)至三口烧瓶中,colymer-1的末端具有氨基端基。随后在体系中加入含0.10g(0.33mmol)偶联剂偶氮苯-4,4-二羧基二氯的二氯甲烷溶液,0.11ml(0.79mmol)的三乙胺催化剂。随后,将体系置于15℃的条件下反应6h,然后,将反应体系在正己烷中进行纯化,得到的产物在25℃的真空烘箱中干燥48h。产率约为60%。通过凝胶渗透色谱(gpc,聚苯乙烯为标样)测得上述三嵌段聚合物(mpeg
2000-paf-z-paf-mpeg
2000
,copolymer-9)的重均与数均分子量(mw,mn)分别为12000和10400,分子量分布系数(mw/mn,dm)为1.15,其聚合物水体系具有热致凝胶化性能。
[0054]
实施例10
[0055]
首先将0.500g(0.10mmol)4-arm-peg
5000-nh2与50ml的甲苯共沸除水,待剩余溶剂体积为5ml,密封体系并冷却至室温。将0.313g(2.72mmol)l-丙氨酸-n-羧基环内酸酐(l-ala-nca)和0.074g(0.39mmol)l-苯丙氨酸-n-羧基环内酸酐(l-phe-nca)单体加入到20ml经无水硫酸镁干燥的chcl3/dmf(v/v3:1)混合溶剂中,并室温下磁力搅拌30min。在氩气的氛围下,将混合单体的溶液转移到干燥的4-arm-peg
5000-nh2中,随后将体系温度升至37℃,在氩气的氛围下,磁力搅拌反应3天。随后将体系冷却至室温,加入20ml的chcl3,待反应产物完全溶解后,逐滴加入到冰乙醚中进行沉降,并重复上述操作三次。最后,将产物置于25℃的真空烘箱中干燥48h,得到星型嵌段聚合物4-arm-peg
5000-paf,产率约50%,通过凝胶渗透色谱(gpc,聚苯乙烯为标样)测得上述星型嵌段聚合物(4-arm-peg
5000-paf,copolymer-10)的重均与数均分子量(mw,mn)分别为7800和5780,分子量分布系数(mw/mn,dm)为1.35,其聚合物水体系具有热致凝胶化性能。
[0056]
实施例11
[0057]
按照实施例1-10给出的基本步骤,用不同分子量的peg或mpeg与不同的单体合成其他嵌段共聚物,其性能列于表1:
[0058]
表1
[0059][0060]
上述表中的嵌段聚合物均具有热致凝胶化的性能。将聚合物配成一定浓度的水溶液,其在温度低于凝胶转变温度时为溶液状态,随温度的上升转变为半固体的凝胶。上述的聚合物中peg/聚氨基酸嵌段聚合物水体系的溶胶-凝胶的转变是不可逆的。
[0061]
实施例12
[0062]
将锂藻土纳米粒子溶于去离子水中,使得最终的浓度为1.5wt%。
[0063]
实施例13
[0064]
将锂藻土纳米粒子溶于生理盐水中,使得最终的浓度为3wt%。
[0065]
实施例14
[0066]
将锂藻土纳米粒子溶于磷酸盐缓冲溶液中,使得最终的浓度为5wt%。
[0067]
实施例15
[0068]
将实施例4中所得聚合物copolymer-4与锂藻土纳米粒子共混,然后加入去离子水,使得最终的colymper-4的浓度为8wt%,锂藻土纳米粒子的浓度为3wt%,即为lp3p8。
[0069]
实施例16
[0070]
将实施例4中所得聚合物copolymer-4与锂藻土纳米粒子共混,然后加入去离子
水,使得最终的colymper-4的浓度为11wt%,锂藻土纳米粒子的浓度为3wt%,即为lp3p11。
[0071]
实施例17
[0072]
将实施例4中所得聚合物copolymer-4与锂藻土纳米粒子共混,然后加入去离子水,使得最终的colymper-4的浓度为8wt%,锂藻土纳米粒子的浓度为1.5wt%,即为lp1.5p8。
[0073]
实施例18
[0074]
将实施例5中所得聚合物colymper-5配制成10wt%的生理盐水溶液,与实施例14的锂藻土纳米粒子水体系等体积混合,得到锂藻土纳米粒子共混的聚合物水体系。
[0075]
实施例19
[0076]
将实施例6所得聚合物colymper-6配制成20wt%的生理盐水溶液,锂藻土纳米粒子配制成5wt%的生理盐水溶液,然后将两组分以体积比2:1混合,得到锂藻土纳米粒子共混的聚合物水体系。
[0077]
实施例20
[0078]
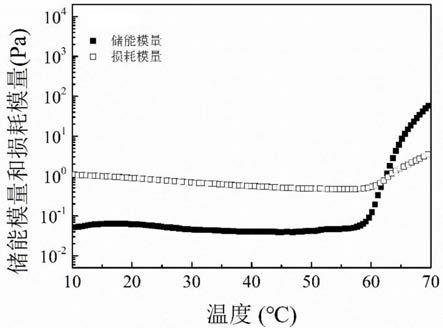
将实施例4中所得聚合物copolymer-4配制成5wt%的去离子水体系,采用旋转流变仪测量聚合物水体系的模量、粘度等流变学性质随温度的变化。在固定剪切频率(w=10rad/s)下,以0.5℃/min的升温速率进行温度扫描。结果记录于图1。如图1所示,5wt%的colymer-4聚合物溶液在室温下储能模量较小,体系具有很好的流动性,而在60℃附近储能模量急剧上升,溶液-凝胶的相转变点为62℃。
[0079]
实施例21
[0080]
将实施例4中所得聚合物copolymer-4配制成8wt%的去离子水体系,采用旋转流变仪测量聚合物水体系的模量、粘度等流变学性质随温度的变化。在固定剪切频率(w=10rad/s)下,以0.5℃/min的升温速率进行温度扫描。结果记录于图2。如图2所示,8wt%的colymer-4聚合物溶液在室温下储能模量较小,体系具有很好的流动性,而在50℃附近储能模量急剧上升,溶液-凝胶的相转变点为59℃。
[0081]
实施例22
[0082]
将实施例15中所得锂藻土共混的聚合物水体系lp3p8,采用旋转流变仪测量聚合物水体系的模量、粘度等流变学性质随温度的变化。在固定剪切频率(w=10rad/s)下,以0.5℃/min的升温速率进行温度扫描。结果记录于图3。如图3所示,lp3p8共混聚合物溶液在室温下储能模量较小,体系具有很好的流动性,而在25℃附近储能模量急剧上升,溶液-凝胶的相转变点为33℃。
[0083]
实施例23
[0084]
将实施例16中所得锂藻土共混的聚合物水体系lp3p11,采用旋转流变仪测量聚合物水体系的模量、粘度等流变学性质随温度的变化。在固定剪切频率(w=10rad/s)下,以0.5℃/min的升温速率进行温度扫描。结果记录于图4。如图4所示,lp3p11共混聚合物体系在测试温度范围内其储能模量均已高于损耗模量,表明其在测试温度范围内一直处于凝胶的状态。
[0085]
实施例24
[0086]
将实施例17中所得锂藻土共混的聚合物水体系lp1.5p8,采用旋转流变仪测量聚合物水体系的模量、粘度等流变学性质随温度的变化。在固定剪切频率(w=10rad/s)下,以
0.5℃/min的升温速率进行温度扫描。结果记录于图5。如图5所示,lp1.5p8共混聚合物溶液在室温下储能模量较小,体系具有很好的流动性,而在30℃附近储能模量急剧上升,溶液-凝胶的相转变点为48℃。
[0087]
实施例25
[0088]
将实施例5中的copolymer-5用生理盐水配制成5wt%的水体系,采用流变仪测得该聚合物水体系的溶胶-凝胶相转变温度为65℃,随后在该体系中加入锂藻土纳米粒子,使得最终纳米粒子的浓度为2wt%,流变实验测得该复合体系的相转变温度降低至45℃,然后,将锂藻土纳米粒子的浓度提高至5wt%,流变实现表明该复合体系的相转变温度降低至35℃,可以满足体内注射的应用需求。
[0089]
实施例26
[0090]
将实施例6中的copolymer-6用磷酸盐缓冲溶液配制成2wt%的水体系,采用流变仪测得该聚合物水体系的溶胶-凝胶相转变温度为57℃,随后在该体系中加入锂藻土纳米粒子,使得最终纳米粒子的浓度为1wt%,流变实验测得该复合体系的相转变温度降低至46℃,然后,将锂藻土纳米粒子的浓度提高至2.5wt%,流变实现表明该复合体系的相转变温度降低至34℃,可以满足体内注射的应用需求。
[0091]
实施例27
[0092]
将实施例7中的copolymer-7用去离子水配制成15wt%的水体系,采用流变仪测得该聚合物水体系的溶胶-凝胶相转变温度为46℃,随后在该体系中加入锂藻土纳米粒子,使得最终纳米粒子的浓度为0.5wt%,流变实验测得该复合体系的相转变温度降低至40℃,然后,将锂藻土纳米粒子的浓度提高至1.5wt%,流变实现表明该复合体系的相转变温度降低至32℃,可以满足体内注射的应用需求。
[0093]
实施例28
[0094]
将实施例8中的copolymer-8用细胞培养基溶液配制成8wt%的水体系,采用流变仪测得该聚合物水体系的溶胶-凝胶相转变温度为62℃,随后在该体系中加入锂藻土纳米粒子,使得最终纳米粒子的浓度为5wt%,流变实验测得该复合体系的相转变温度降低至40℃,然后,将锂藻土纳米粒子的浓度提高至7wt%,流变实现表明该复合体系的相转变温度降低至32℃,可以满足体内注射的应用需求。
[0095]
实施例29
[0096]
将实施例9中的copolymer-9用去离子水配制成10wt%的水体系,采用流变仪测得该聚合物水体系的溶胶-凝胶相转变温度为70℃,随后在该体系中加入锂藻土纳米粒子,使得最终纳米粒子的浓度为3wt%,流变实验测得该复合体系的相转变温度降低至48℃,然后,将锂藻土纳米粒子的浓度提高至8wt%,流变实现表明该复合体系的相转变温度降低至33℃,可以满足体内注射的应用需求。
[0097]
实施例30
[0098]
取4mg模型蛋白bsa加入到实施例16所制备的共混聚合物lp3p8水体系中,混合均匀。取0.5g上述载药聚合物溶液加入试管中,于37℃水浴摇床放置15min,待其成胶后,加5ml磷酸盐缓冲溶液(ph7.4),定期取点测吸光度,采用uv-vis分光光度计在测药物的释放,药物可实现至少二周的释放周期。
[0099]
实施例31
[0100]
将实施例15所制备的共混聚合物lp3p8水体系通过0.22μm滤膜过滤后,在37℃形成凝胶,随后将mc3t3-e1细胞种植于凝胶表面,随后激光共聚焦显微镜观察发现lp3p8水凝胶可促进细胞的粘附和增殖。
[0101]
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。