具有潜在铁螯合活性的单胺氧化酶b抑制剂及其应用
技术领域
1.本发明涉及有机合成和药物化学领域,具体涉及一类具有铁离子螯合活性的单胺氧化酶b抑制剂及其制备方法,以及其在制备治疗抑郁症和神经退行性综合征(如帕金森和阿尔兹海默症)等疾病的药物中的用途。
背景技术:
2.单胺氧化酶(mao,ec 1.4.3.4)是一类含有共价结合的氧化还原辅因子黄素腺嘌呤二核苷酸(fad)的酶。这种酶广泛存在于哺乳动物的线粒体外膜组织中,分布在中枢和外周神经系统的不同细胞,该酶的作用包括催化内源性和外源性胺氧化脱氨生成相应的醛。在人体中,它主要以两种亚型存在:mao-a和mao-b。这种分类基于它们催化的底物:mao-a氧化5-羟色胺、肾上腺素和去甲肾上腺素,而mao-b氧化β-苯乙胺和苄胺。此外,多巴胺是其两者共同的底物,但优先被mao-b代谢。
3.鉴于单胺氧化酶在神经递质的代谢中起到了重要的作用,单胺氧化酶抑制剂(maois)被广泛应用于抑郁症和神经退行性综合征(如帕金森和阿尔兹海默症)的治疗。目前上市的mao-b抑制剂有4个,分别是司来吉兰(selegiline)、雷沙吉兰(rasagiline)、沙芬酰胺(safinamide)和优降宁(pargyline)。但这些被大量引入临床的maos抑制剂由于选择性差以及肝毒性、直立性低血压、高血压危象等不良反应而被限制应用。基于这些原因,新一代maois的开发旨在发现选择性和可逆的治疗药物。
4.色酮是一类重要的含氧杂环化合物,具有苯并-γ-吡喃酮环,它们是类黄酮家族的一部分。色酮衍生物在自然界中含量丰富,与许多其他天然化合物一样有助于植物色素沉着,为授粉昆虫提供有吸引力的颜色。它们还提供了对真菌病原体或紫外线辐射的保护,从而确保了植物的生存和繁殖。此外,色酮母核已被公认为天然或人工来源的大量生物活性分子的药效团,是自然界中普遍存在的天然化合物的核心组成部分。这些化合物作为抗氧化剂、抗炎剂、ache抑制剂、抗aβ纤维形成、抗β分泌酶、mao抑制剂、神经保护剂、自由基清除剂和金属螯合剂的有效性已经得到了证实。其中大量文献指出3位取代的色酮对mao-b具有很好的抑制活性和选择性。因此,色酮母核为新型单胺氧化酶抑制剂的设计提供了最佳骨架。
5.另外,由于铁在ad(阿尔茨海默病)发病机制中的重要作用,铁螯合剂治疗已成为ad等神经退行性疾病的重要治疗手段。设计和开发无毒、临床可用、具口服活性和可透过血脑屏障的铁螯合剂需要兼顾许多不同的性质。在众多的铁螯合剂中,以3,4-hpos为母核的铁螯合剂去铁酮作为一种治疗铁超载相关疾病的口服药物脱颖而出。随后,越来越多的研究采用结构修饰、官能团拼接、药效团融合等方式来实现多靶点铁螯合剂的设计和开发。
6.因此,选择合适的化学链接臂将具有铁螯合活性的吡啶酮衍生物和具有mao-b抑制作用的色酮母核融合到一个分子中,开发出一类具有铁离子螯合活性的单胺氧化酶b抑制剂,在多靶点单胺氧化酶抑制剂研发中极具研究前景。
技术实现要素:
7.本发明基于多靶点配体策略、计算机辅助药物设计、药效团的拼接和融合原理、合理药物设计、类药性等原则设计合成了一类具有铁离子螯合活性的单胺氧化酶抑制剂,提供色酮杂合吡啶酮衍生物或其可药用盐的制备方法,并通过抑制单胺氧化酶、螯合金属铁离子来预防或治疗相关疾病尤其是阿尔兹海默症和帕金森病的药物中的应用。
8.为实现上述目的,本发明提供如下技术方案:
9.第一方面,本发明提供了一种如式(i)所示的色酮/吡啶酮杂合衍生物及其药学上可接受的盐:
[0010][0011]
式(i)中:
[0012]
r1为h、c
1-c6直链或支链的烷基、oh、och3或cooh;
[0013]
r2、r3各自独立为h或c
1-c6直链或支链的烷基;
[0014]
r4为h、c
1-c6直链或支链的烷烃、cl、f、br、ch3、cf3、oh、och3、cooh、cooch3或苄基;
[0015]
r5、r6、r7各自独立为h、c
1-c
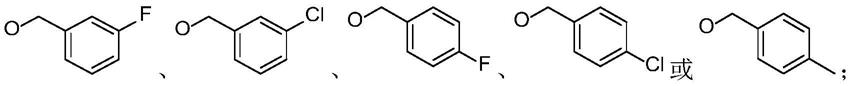
16
直链或支链的烷烃、cl、f、br、ch3、cf3、oh、och3、cooh、cooch3或其中,y1、z1各自独立为h、ch3、cl、f、cf3、oh、och3、ch(ch3)2或c(ch3)3;
[0016]
m为
[0017]
n为ch2的个数,n为1-6。
[0018]
优选地,r1、r2或r3各自独立为h或c
1-c4直链或支链的烷基;r4各自独立为h、ch3或och3;r5或r7各自独立为h、ch3、och3、oh或obn;r6为h、ch3、och3或其中,y1、z1各自独立为h、ch3、cl、f或och3,n为1-3。
[0019]
优选地,r1为ch3;r2、r3、r4和r7为h;r5为h、ch3或och3;r6为h、obn、为h、obn、n为2。
[0020]
具体地,式(i)所示的色酮/吡啶酮杂合衍生物为下列化合物之一:
[0021]
[0022][0023]
具体地,所述药学上可接受的盐为式(i)所示的色酮/吡啶酮杂合衍生物的盐酸盐。
[0024]
第二方面,本发明还提供一种上述式(i)所示的色酮/吡啶酮杂合衍生物或其药学上可接受的盐在制备通过抑制单胺氧化酶(尤其是单胺氧化酶b)、螯合金属铁离子、抗aβ或抗氧化来预防或治疗相关疾病的药物中的应用。更优选式(i)所示的色酮/吡啶酮杂合衍生物或其药学上可接受的盐在制备通过抑制单胺氧化酶或螯合金属铁离子来预防或治疗相关疾病的药物中的应用。
[0025]
优选地,所述疾病为阿尔兹海默症或帕金森病。
[0026]
进一步优选所述色酮/吡啶酮杂合衍生物为a1-a12、b1-b12所示化合物之一。
[0027]
本发明特别优选所述色酮/吡啶酮杂合衍生物为化合物b1、b5、b6、b7或b12,最优选化合物b5。
[0028]
第三方面,本发明还提供一种式(i)所示的色酮/吡啶酮杂合衍生物的制备方法,合成思路为:式1所示的氨基取代的羟基保护的吡啶酮的制备方法请参考专利cn 112552232 a;以式2所示的不同位置取代的邻羟基苯乙酮为原料合成得到式3所示的色酮醛,以式3所示的色酮醛合成式4所示的色酮酸;再通过缩合反应将吡啶酮与式4色酮酸连接得到式5所示的化合物(m为),通过还原胺化反应将吡啶酮与式3色酮醛连接得到式5所示的化合物(m为),最后脱保护得到式(i)所示的目标化合物。
[0029]
具体的,本发明所述的式(i)所示的色酮/吡啶酮杂合衍生物具体按如下方法制备:
[0030]
(1)将式2所示的邻羟基苯乙酮类化合物溶于无水n,n-二甲基甲酰胺(dmf)a中,于-15℃下搅拌1h,将三氯氧磷置于恒压滴液漏斗中,于-15℃下滴加,滴加完毕后转移到室温继续搅拌反应14h,得到反应液a,所得反应液a经后处理a得到式3所示化合物;所述的邻羟基苯乙酮类化合物和三氯氧磷的物质的量之比为1:2~3;
[0031]
进一步,步骤(1)中所述的n,n-二甲基甲酰胺a的体积以所述的式2所示的邻羟基苯乙酮类化合物的物质的量计为2~3ml/mmol;所述后处理a为:将所述反应液a倒入冰水中析出固体,过滤,用水洗涤再用乙醚洗涤得到式3所示化合物。
[0032]
(2)将步骤(1)中所述的式3所示化合物溶于二氯甲烷a中,再加入氨基磺酸的水溶液,于-2℃下搅拌0.5-2h,将亚氯酸钠的水溶液置于恒压滴液漏斗中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,得到反应液b,反应结束后,所得反应液b经后处理b得到式4所示的化合物;所述的式3所示化合物、所述氨基磺酸的水溶液中的氨基磺酸与所述亚氯酸钠的水溶液中的亚氯酸钠的物质的量比为1:5~6:5~6;
[0033]
进一步,步骤(2)中所述的二氯甲烷a的体积以所述的式3所示化合物的物质的量
计为5~20ml/mmol。所述后处理b为:将所述反应液b减压蒸馏除去溶剂,二氯甲烷萃取,水洗,干燥,减压蒸馏除去溶剂后用甲醇重结晶得到式4所示的化合物。
[0034]
(3)将步骤(2)中所述式4所示的化合物溶于二氯甲烷b中,滴加n,n-二甲基甲酰胺b、氯化亚砜,于30℃下反应6-12h(生成酰氯中间体),反应结束后,减压蒸馏除去溶剂,加入二氯甲烷c,得到为混合液;将式1所示的化合物溶于二氯甲烷d中,加入三乙胺,滴加所述混合液,于室温下反应6-24h,反应结束后,得到反应液c经后处理c得到式5所示的化合物;式5中m为所述的式4所示的化合物、氯化亚砜、式1所示的化合物与三乙胺的物质的量之比为1:2~3:2~3:4~6;所述n,n-二甲基甲酰胺b的体积以所述式4所示的化合物的物质的量计为0.1ml/mmol;
[0035]
进一步,步骤(3)中所述二氯甲烷b、二氯甲烷c或二氯甲烷d的体积以所述式4化合物的物质的量计为5~20ml/mmol。所述后处理c为:将所述反应液c减压浓缩,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,收集含目标产物(通过tlc点板检测)的洗脱液,减压浓缩除去溶剂得到式5所示的化合物。
[0036]
(4)将步骤(1)中式3所示的化合物溶于二氯甲烷e中,加入式1化合物、三乙酰氧基硼氢化钠,氮气保护下,于25℃下反应2-12h,反应结束后,所得反应液d经后处理d得到式5化合物;所述的式3所示的化合物、式1化合物、三乙酰氧基硼氢化钠的物质的量之比为2:1~3:1~3;式5中m为
[0037]
进一步,步骤(4)中所述的二氯甲烷e的体积以所述的式3化合物的物质的量计为5~20ml/mmol。所述后处理d为:将所述反应液d减压浓缩,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,收集含目标产物(通过tlc点板检测)的洗脱液,减压浓缩除去溶剂得到式5所示的化合物。
[0038]
(5)将步骤(4)所得式5化合物溶于无水二氯甲烷中,在氮气保护下,-78℃~-48℃下缓慢滴加0.02~0.1mmol/ml三氯化硼(bcl3)的无水二氯甲烷溶液,滴加完毕后转移到室温继续搅拌反应12-24h,得到反应液g,加入甲醇淬灭,经后处理g得到式(i)所示的化合物;所述的式5化合物与三氯化硼的无水二氯甲烷溶液中三氯化硼的物质的量之比为1:3~6。
[0039]
进一步,步骤(5)中所述无水二氯甲烷的体积以所述式5化合物的物质的量计为30-40ml/mmol;所述甲醇的的体积以式5化合物的物质的量计为30-40ml/mmol;所述后处理g为:所得反应液g减压蒸馏除去溶剂,用体积比为1:7的甲醇和乙醚的混合溶剂重结晶,得到式(i)所示的化合物。
[0040][0041]
以上各步骤中的a、b、c、d等字母只是为了区分不同阶段的物质,无其它特殊含义。
[0042]
与现有技术相比,本发明的有益效果在于:本发明合成了一类具有铁离子螯合活性的单胺氧化酶抑制剂,创新性地将具有铁离子螯合活性的吡啶酮衍生物与具有mao-b抑制活性的色酮母核有机结合在一起,对于发病机制复杂的阿尔茨海默症以及帕金森等神经退行性疾病具有显著优势。
具体实施方式
[0043]
下面结合具体实例对本发明作进一步阐述,但本发明不局限于这些实施例。
[0044]
实施例1
[0045]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-4h-苯并吡喃-3-甲酰胺(a1)的制备方法
[0046]
于100ml单口瓶中加入2-羟基苯乙酮(1.36g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析出固体,过滤,用水洗涤,再用乙醚洗涤,得淡黄色固体(1.55g),收率89%。
[0047]
在250ml单口瓶中加入上述淡黄色固体(0.522g,3mmol),溶解于50ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(40ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.393g),收率69.0%。
[0048]
于50ml单口瓶中加入3-羧基色酮(0.19g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,加入二氯甲烷(10ml)。于50ml单口瓶中加入1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),三乙胺(0.606g,6mmol),二氯甲烷(10ml),滴加上述反应液,室温反应12h,反应结束后,浓缩反应液,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析经硅胶柱层析纯化,减压浓缩后得油状液体(0.139g),收率
32.3%。
[0049]
于50ml单口瓶中加入上述油状液体(0.129g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a1(0.097g)收率95.1%,纯度为97.92%。
[0050]
m.p.268-270℃;esi-hrms:m/z calcd for c
18h16
n2o5[m h]
:341.1132;found:341.1125;1h nmr(400mhz,dmso-d6)δ9.30(t,j=6.0hz,1h),8.90(s,1h),8.16(d,j=8.0hz,1h),8.09(d,j=6.8hz,1h),7.91(t,j=7.6hz,1h),7.76(d,j=8.4hz,1h),7.60(t,j=7.6hz,1h),7.30(d,j=6.8hz,1h),4.5 3(t,j=5.6hz,2h),4.01(q,j=6.0hz,2h),2.59(s,3h);
13
c nmr(100mhz,dmso-d6)δ176.0,162.9,162.7,158.8,155.6,142.8,141.8,138.6,135.3,126.6,125.4,123.5,118.69,115.1,110.5,55.2,38.3,12.6.
[0051]
实施例2
[0052]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-6-甲基-4h-苯并吡喃-3-甲酰胺(a2)的制备方法
[0053]
于100ml单口瓶中加入2-羟基-5-甲基苯乙酮(1.50g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析出固体,过滤,用水洗涤,再用乙醚洗涤,得淡黄色固体(1.64g),收率87.2%。
[0054]
在250ml单口瓶中加入上述淡黄色固体(0.564g,3mmol),溶解于51ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(42ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.379g),收率61.9%。
[0055]
于50ml单口瓶中加入6-甲基-3-色酮酸(0.204g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,加入二氯甲烷(12ml)。于50ml单口瓶中加入1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),三乙胺(0.606g,6mmol),二氯甲烷(10ml),滴加上述反应液,室温反应12h,反应结束后,浓缩反应液,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.137g),收率30.9%。
[0056]
于50ml单口瓶中加入上述油状液体(0.133g,0.3mmol),无水二氯甲烷(12ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a2(0.093g)收率87.6%,纯度为99.08%。
[0057]
m.p.265-267℃;esi-hrms:m/z calcd for c
19h18
n2o5[m h]
:355.1288;found:355.1273;1h nmr(400mhz,dmso-d6)δ9.32(t,j=6.1hz,1h),8.97(s,1h),8.09(d,j=7.0hz,1h),7.95(s,1h),7.74(dd,j=8.6,1.9hz,1h),7.68(d,j=8.6hz,1h),7.29
–
7.23
(m,1h),4.54(t,j=5.8hz,2h),3.78(q,j=5.5,5.1hz,2h),2.60(s,3h),2.47(s,3h);
13
c nmr(100mhz,dmso-d6)δ176.40,163.30,163.22,159.27,154.35,143.30,142.25,139.01,136.87,136.77,125.03,123.72,118.96,115.36,111.00,55.63,38.73,20.98,13.09.
[0058]
实施例3
[0059]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-7-甲基-4h-苯并吡喃-3-甲酰胺(a3)的制备方法
[0060]
于100ml单口瓶中加入2-羟基-4-甲基苯乙酮(1.50g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析出固体,过滤,用水洗涤,再用乙醚洗涤,得淡黄色固体(1.59g),收率84.6%。
[0061]
在250ml单口瓶中加入上述淡黄色固体(0.564g,3mmol),溶解于51ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(42ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.360g),收率58.8%。
[0062]
于50ml单口瓶中加入7-甲基-3-色酮酸(0.204g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,加入二氯甲烷(12ml)。于50ml单口瓶中加入1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),三乙胺(0.606g,6mmol),二氯甲烷(10ml),滴加上述反应液,室温反应12h,反应结束后,浓缩反应液,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.145g),收率32.7%。
[0063]
于50ml单口瓶中加入上述油状液体(0.133g,0.3mmol),无水二氯甲烷(12ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a3(0.099g)收率93.2%,纯度为98.88%。
[0064]
m.p.270-272℃;esi-hrms:m/z calcd for c
19h18
n2o5[m h]
:355.1288;found:355.1276;1h nmr(400mhz,dmso-d6)δ9.33(t,j=6.1hz,1h),8.95(s,1h),8.07(dd,j=10.1,7.6hz,2h),7.60(s,1h),7.44(d,j=8.2hz,1h),7.24(d,j=6.8hz,1h),4.53(t,j=5.8hz,2h),3.78(q,j=5.9hz,2h),2.59(s,3h),2.50(s,3h);
13
c nmr(100mhz,dmso-d6)δ176.28,163.29,163.16,159.29,156.15,147.01,143.31,142.22,139.02,128.44,125.66,121.75,118.66,115.42,111.00,55.64,38.72,21.74,13.09.
[0065]
实施例4
[0066]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-6-甲氧基-4h-苯并吡喃-3-甲酰胺(a4)的制备方法
[0067]
于100ml单口瓶中加入2-羟基-5-甲氧基苯乙酮(1.66g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析
出固体,过滤,用水洗涤,再用乙醚洗涤,得淡黄色固体(1.73g),收率84.8%。
[0068]
在250ml单口瓶中加入上述淡黄色固体(0.612g,3mmol),溶解于50ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(40ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.415g),收率62.9%。
[0069]
于50ml单口瓶中加入6-甲氧基-3-色酮酸(0.220g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,加入二氯甲烷(10ml)。于50ml单口瓶中加入1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),三乙胺(0.606g,6mmol),二氯甲烷(10ml),滴加上述反应液,室温反应12h,反应结束后,浓缩反应液,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.151g),收率32.8%。
[0070]
于50ml单口瓶中加入上述油状液体(0.138g,0.3mmol),无水二氯甲烷(12ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a4(0.090g)收率81.1%,纯度为98.42%。
[0071]
m.p.260-262℃;esi-hrms:m/z calcd for c
19h18
n2o6[m na]
:393.1057;found:393.1057;1h nmr(400mhz,dmso-d6)δ9.32(t,j=6.1hz,1h),8.97(s,1h),8.08(d,j=7.0hz,1h),7.77
–
7.73(m,1h),7.51(dd,j=7.3,2.9hz,2h),7.24(d,j=7.0hz,1h),4.53(t,j=5.8hz,2h),3.89(s,3h),3.78(q,j=6.0hz,2h),2.59(s,3h);
13
c nmr(100mhz,dmso-d6)δ176.11,163.36,163.02,159.24,157.77,150.81,143.29,142.29,139.02,124.84,124.58,120.83,114.76,111.01,105.77,56.37,55.66,38.74,13.10.
[0072]
实施例5
[0073]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-7-甲氧基-4h-苯并吡喃-3-甲酰胺(a5)的制备方法
[0074]
于100ml单口瓶中加入2-羟基-4-甲氧基苯乙酮(1.66g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析出固体,过滤,用水洗涤,再用乙醚洗涤,得淡黄色固体(1.62g),收率79.4%。
[0075]
在250ml单口瓶中加入上述淡黄色固体(0.612g,3mmol),溶解于51ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(42ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.386g),收率58.5%。
[0076]
于50ml单口瓶中加入7-甲氧基-3-色酮酸(0.220g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,
保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a6(0.105g)收率78.5%,纯度为100.00%。
[0086]
m.p.225-227℃;esi-hrms:m/z calcd for c
25h22
n2o6[m h]
:447.1551;found:447.1539;1h nmr(400mhz,dmso-d6)δ9.37(t,j=6.1hz,1h),8.92(s,1h),8.07(t,j=7.8hz,2h),7.50(d,j=7.0hz,2h),7.46
–
7.36(m,4h),7.26(dd,j=8.9,2.3hz,1h),7.17(d,j=6.9hz,1h),5.31(s,2h),4.52(t,j=5.7hz,2h),3.77(q,j=7.4,6.5hz,2h),2.59(s,3h);
13
c nmr(100mhz,dmso-d6)δ175.69,163.97,163.33,163.01,159.11,157.86,143.27,142.33,139.04,136.31,129.06,128.74,128.49,127.40,117.70,116.87,115.29,110.93,102.42,70.75,55.68,38.67,13.08.
[0087]
实施例7
[0088]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-7-(3-甲基苄氧基)-4h-苯并吡喃-3-甲酰胺(a7)的制备方法
[0089]
于100ml单口瓶中加入2,4-二羟基苯乙酮(2.28g,15mmol),碳酸钾(3.105g,22.5mmol),丙酮(50ml),滴加3-甲基苄溴(3.33g,18mmol),57℃回流8小时。反应结束后,过滤,将滤液减压浓缩,用甲醇重结晶,烘干得白色固体(2.7g),收率70.3%。
[0090]
于100ml单口瓶中加入2-羟基-4-(3-甲基苄氧基)苯乙酮(2.56g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析出固体,过滤,用水洗涤,再用乙醚洗涤,得淡棕色固体(2.44g),收率83%。
[0091]
在250ml单口瓶中加入上述淡棕色固体(0.882g,3mmol),溶解于50ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(42ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.558g),收率60%。
[0092]
于50ml单口瓶中加入7-(3-甲基苄氧基)-3-色酮酸(0.310g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,加入二氯甲烷(12ml)。于50ml单口瓶中加入1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),三乙胺(0.606g,6mmol),二氯甲烷(10ml),滴加上述反应液,室温反应12h,反应结束后,浓缩反应液,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.176g),收率32.0%。
[0093]
于50ml单口瓶中加入上述油状液体(0.165g,0.3mmol),无水二氯甲烷(12ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a7(0.109g)收率79.0%,纯度为100.00%。
[0094]
m.p.238-240℃;esi-hrms:m/z calcd for c
26h24
n2o6[m h]
:461.1707;found:461.1708.;1h nmr(400mhz,dmso-d6)δ1h nmr(400mhz,dmso-d6)δ9.37(t,j=6.1hz,1h),
8.92(s,1h),8.07(dd,j=7.9,4.0hz,2h),7.36
–
7.34(m,1h),7.33
–
7.21(m,5h),7.17(d,j=6.9hz,1h),5.24(s,2h),4.52(t,j=5.6hz,2h),3.76(q,j=5.6hz,2h),2.58(s,3h),2.32(s,3h).
13
c nmr(100mhz,dmso-d6)δ175.29,163.60,162.91,162.62,158.85,157.46,142.88,141.71,138.61,137.85,135.82,128.95,128.61,128.55,126.98,125.18,117.28,116.46,114.88,110.53,101.97,70.38,55.22,38.27,21.05,12.66.
[0095]
实施例8
[0096]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-7-(4-甲基苄氧基)-4h-苯并吡喃-3-甲酰胺(a8)的制备方法
[0097]
于100ml单口瓶中加入2,4-二羟基苯乙酮(2.28g,15mmol),碳酸钾(3.105g,22.5mmol),丙酮(50ml),滴加4-甲基苄溴(3.33g,18mmol),57℃回流8小时。反应结束后,过滤,将滤液减压浓缩,用甲醇重结晶,烘干得白色固体(2.66g),收率69.2%。
[0098]
于100ml单口瓶中加入2-羟基-4-(4-甲基苄氧基)苯乙酮(2.56g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析出固体,过滤,用水洗涤,再用乙醚洗涤,得淡棕色固体(2.54g),收率86.4%。
[0099]
在250ml单口瓶中加入上述淡棕色固体(0.882g,3mmol),溶解于50ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(42ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.562g),收率60.4%。
[0100]
于50ml单口瓶中加入7-(4-甲基苄氧基)-3-色酮酸(0.310g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,加入二氯甲烷(12ml)。于50ml单口瓶中加入1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),三乙胺(0.606g,6mmol),二氯甲烷(10ml),滴加上述反应液,室温反应12h,反应结束后,浓缩反应液,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.179g),收率32.5%。
[0101]
于50ml单口瓶中加入上述油状液体(0.165g,0.3mmol),无水二氯甲烷(12ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a8(0.112g)收率81.2%,纯度为98.05%。
[0102]
m.p.234-236℃;esi-hrms:m/z calcd for c
26h24
n2o6[m h]
:461.1707;found:461.1724;1h nmr(400mhz,dmso-d6)δ9.37(t,j=6.1hz,1h),8.92(s,1h),8.05(dd,j=7.8,5.3hz,2h),7.40
–
7.33(m,3h),7.22(d,j=8.0hz,3h),7.15(d,j=6.9hz,1h),5.24(s,2h),4.50(t,j=5.5hz,2h),3.77
–
3.73(m,2h),2.57(s,3h),2.31(s,3h).
13
c nmr(100mhz,dmso-d6)δ175.29,163.60,162.91,162.62,159.08,157.46,142.95,141.42,138.60,137.65,132.86,129.19,128.21,126.95,117.24,116.50,114.88,110.54,101.98,70.27,55.15,38.28,20.87,12.63.
[0103]
实施例9
[0104]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-7-(3-氟苄氧基)-4h-苯并吡喃-3-甲酰胺(a9)的制备方法
[0105]
于100ml单口瓶中加入2,4-二羟基苯乙酮(2.28g,15mmol),碳酸钾(3.105g,22.5mmol),丙酮(50ml),滴加3-氟苄溴(3.402g,18mmol),57℃回流8小时。反应结束后,过滤,将滤液减压浓缩,用甲醇重结晶,烘干得白色固体(2.82g),收率72.3%。
[0106]
于100ml单口瓶中加入2-羟基-4-(3-氟苄氧基)苯乙酮(2.60g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析出固体,过滤,用水洗涤,再用乙醚洗涤,得淡棕色固体(2.53g),收率84.9%。
[0107]
在250ml单口瓶中加入上述淡棕色固体(0.894g,3mmol),溶解于50ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(42ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.584g),收率62%。
[0108]
于50ml单口瓶中加入7-(3-氟苄氧基)-3-色酮酸(0.314g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,加入二氯甲烷(12ml)。于50ml单口瓶中加入1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),三乙胺(0.606g,6mmol),二氯甲烷(10ml),滴加上述反应液,室温反应12h,反应结束后,浓缩反应液,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.182g),收率32.8%。
[0109]
于50ml单口瓶中加入上述油状液体(0.166g,0.3mmol),无水二氯甲烷(12ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a9(0.105g)收率75.4%,纯度为98.00%。
[0110]
m.p.237-239℃;esi-hrms:m/z calcd for c
25h21
fn2o6[m h]
:465.1456;found:465.1475;1h nmr(400mhz,dmso-d6)δ9.35(t,j=6.2hz,1h),8.91(s,1h),8.07(dd,j=9.5,8.0hz,2h),7.47(td,j=7.8,6.3hz,1h),7.36(d,j=2.3hz,1h),7.34(d,j=7.6hz,2h),7.26(dd,j=8.9,2.3hz,1h),7.21
–
7.18(m,2h),5.33(s,2h),4.52(t,j=5.9hz,2h),3.76(q,j=6.0hz,2h),2.58(s,3h).
13
c nmr(100mhz,dmso-d6)δ175.28,163.32,162.90,162.64,162.23(d,1j=242hz),158.72,157.42,142.85,141.88,138.82(d,3j=8hz),138.61,130.76,130.68,127.04,123.92(d,4j=2hz),117.43,116.40,115.085(d,2j=21hz),114.63(d,2j=22hz),110.54,102.08,69.41,55.26,38.27,12.68.
[0111]
实施例10
[0112]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-7-(4-氟苄氧基)-4h-苯并吡喃-3-甲酰胺(a10)的制备方法
[0113]
于100ml单口瓶中加入2,4-二羟基苯乙酮(2.28g,15mmol),碳酸钾(3.105g,
22.5mmol),丙酮(50ml),滴加4-氟苄溴(3.402g,18mmol),57℃回流8小时。反应结束后,过滤,将滤液减压浓缩,用甲醇重结晶,烘干得白色固体(2.78g),收率71.3%。
[0114]
于100ml单口瓶中加入2-羟基-4-(4-氟苄氧基)苯乙酮(2.60g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析出固体,过滤,用水洗涤,再用乙醚洗涤,得淡棕色固体(2.54g),收率85.2%。
[0115]
在250ml单口瓶中加入上述淡棕色固体(0.894g,3mmol),溶解于50ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(42ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.574g),收率60.9%。
[0116]
于50ml单口瓶中加入7-(4-氟苄氧基)-3-色酮酸(0.314g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,加入二氯甲烷(12ml)。于50ml单口瓶中加入1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),三乙胺(0.606g,6mmol),二氯甲烷(10ml),滴加上述反应液,室温反应12h,反应结束后,浓缩反应液,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.182g),收率32.8%。
[0117]
于50ml单口瓶中加入上述油状液体(0.166g,0.3mmol),无水二氯甲烷(12ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a10(0.102g)收率73.3%,纯度为100.00%。
[0118]
m.p.246-248℃;esi-hrms:m/z calcd for c
25h21
fn2o6[m h]
:465.1456;found:465.1470;1h nmr(400mhz,dmso-d6)δ9.36(t,j=6.2hz,1h),8.91(s,1h),8.07(dd,j=8.0,3.9hz,2h),7.57
–
7.54(m,2h),7.36(d,j=2.3hz,1h),7.27
–
7.23(m,3h),7.19(d,j=6.9hz,1h),5.28(s,2h),4.52(t,j=5.9hz,2h),3.76(q,j=6.0hz,2h),2.58(s,3h);
13
c nmr(100mhz,dmso-d6)δ175.69,163.87,163.65,163.33,163.04,161.22,159.05,157.86,143.26,142.37,139.05,132.58(d,4j=3hz),130.89(d,3j=9hz),127.41,117.73(d,1j=244hz),116.85(d,2j=22hz),115.91,110.92,102.42,70.01,55.70,38.66,13.08.
[0119]
实施例11
[0120]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-7-(3-氯苄氧基)-4h-苯并吡喃-3-甲酰胺(a11)的制备方法
[0121]
于100ml单口瓶中加入2,4-二羟基苯乙酮(2.28g,15mmol),碳酸钾(3.105g,22.5mmol),丙酮(50ml),滴加3-氯苄溴(3.69g,18mmol),57℃回流8小时。反应结束后,过滤,将滤液减压浓缩,用甲醇重结晶,烘干得白色固体(2.92g),收率70.4%。
[0122]
于100ml单口瓶中加入2-羟基-4-(3-氯苄氧基)苯乙酮(2.77g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,
25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析出固体,过滤,用水洗涤,再用乙醚洗涤,得淡棕色固体(2.47g),收率78.5%。
[0123]
在250ml单口瓶中加入上述淡棕色固体(0.944g,3mmol),溶解于50ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(42ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.588g),收率59.3%。
[0124]
于50ml单口瓶中加入7-(3-氯苄氧基)-3-色酮酸(0.331g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,加入二氯甲烷(12ml)。于50ml单口瓶中加入1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),三乙胺(0.606g,6mmol),二氯甲烷(10ml),滴加上述反应液,室温反应12h,反应结束后,浓缩反应液,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.188g),收率32.9%。
[0125]
于50ml单口瓶中加入上述油状液体(0.171g,0.3mmol),无水二氯甲烷(12ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a11(0.111g)收率77.1%,纯度为98.16%。
[0126]
m.p.230-232℃;esi-hrms:m/z calcd for c
25h21
cln2o6[m h]
:481.1161;found:481.1167;1h nmr(400mhz,dmso-d6)δ9.36(t,j=6.1hz,1h),8.91(s,1h),8.07(d,j=8.7hz,2h),7.57(s,1h),7.45(t,j=4.4hz,3h),7.36(d,j=2.1hz,1h),7.28
–
7.21(m,2h),5.31(s,2h),4.52(t,j=5.5hz,2h),3.76(q,j=6.8,6.1hz,2h),2.58(s,3h).
13
c nmr(100mhz,dmso-d6)δ175.69,163.70,163.31,163.05,159.21,157.82,143.29,142.19,139.02,138.90,133.66,131.01,128.64,128.09,127.46,126.97,117.85,116.80,115.31,110.94,102.49,69.73,55.64,38.68,13.07.
[0127]
实施例12
[0128]
n-(2-(2-甲基-3-羟基-1(4h)-4-氧代吡啶基)乙基)-4-氧代-7-(4-氯苄氧基)-4h-苯并吡喃-3-甲酰胺(a12)的制备方法
[0129]
于100ml单口瓶中加入2,4-二羟基苯乙酮(2.28g,15mmol),碳酸钾(3.105g,22.5mmol),丙酮(50ml),滴加4-氯苄溴(3.69g,18mmol),57℃回流8小时。反应结束后,过滤,将滤液减压浓缩,用甲醇重结晶,烘干得白色固体(2.89g),收率69.6%。
[0130]
于100ml单口瓶中加入2-羟基-4-(4-氯苄氧基)苯乙酮(2.77g,10mmol),和无水n,n-二甲基甲酰胺(20ml),在-15℃下搅拌1h,再通过恒压滴液漏斗将三氯氧磷(3.825g,25mmol)缓慢滴加到反应液中,滴加完毕后转移到室温继续搅拌反应14h,反应结束后,倒入冰水中,析出固体,过滤,用水洗涤,再用乙醚洗涤,得淡棕色固体(2.62g),收率83.3%。
[0131]
在250ml单口瓶中加入上述淡棕色固体(0.944g,3mmol),溶解于50ml二氯甲烷中,再将氨基磺酸(1.748g,18mmol)用水(42ml)溶解后加入,于-2℃下搅拌0.5h,再通过恒压滴液漏斗将亚氯酸钠(1.628g,18mmol)的水溶液(25ml)缓慢滴加到反应液中,于-2℃下滴加,
滴加完毕后转移到室温继续搅拌反应12h,反应结束后,加入二氯甲烷(10ml)萃取,用20ml
×
3水洗,合并有机层,用无水硫酸钠干燥,减压浓缩,用甲醇重结晶,得淡黄色固体(0.578g),收率58.3%。
[0132]
于50ml单口瓶中加入7-(4-氯苄氧基)-3-色酮酸(0.331g,1mmol)、dmf(0.1ml)、二氯甲烷(10ml),和氯化亚砜(0.357g,3mmol),在30℃下搅拌12h,反应结束后,减压蒸馏除去溶剂,加入二氯甲烷(12ml)。于50ml单口瓶中加入1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),三乙胺(0.606g,6mmol),二氯甲烷(10ml),滴加上述反应液,室温反应12h,反应结束后,浓缩反应液,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.179g),收率31.3%。
[0133]
于50ml单口瓶中加入上述油状液体(0.171g,0.3mmol),无水二氯甲烷(12ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体a12(0.108g)收率74.8%,纯度为98.33%。
[0134]
m.p.241-243℃;esi-hrms:m/z calcd for c
25h21
cln2o6[m h]
:481.1161;found:481.1170;1h nmr(400mhz,dmso-d6)δ9.36(t,j=6.1hz,1h),8.91(s,1h),8.07(dd,j=7.9,3.9hz,2h),7.54
–
7.46(m,4h),7.36(d,j=2.2hz,1h),7.26
–
7.18(m,2h),5.30(s,2h),4.52(t,j=5.7hz,2h),3.76(q,j=5.7hz,2h),2.58(s,3h).
13
c nmr(100mhz,dmso-d6)δ175.70,163.79,163.32,163.04,159.29,157.85,143.33,142.11,139.06,135.39,133.32,130.31,129.07,127.45,117.81,116.85,115.32,110.94,102.50,69.86,55.62,38.68,13.05.
[0135]
实施例13
[0136]
2-甲基-3-羟基-1-(2-(((4氧代-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b1)的制备方法
[0137]
于100ml单口瓶中加入4-氧代-4h-苯并呋喃-3-甲醛(0.348g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.333g),收率40.0%。
[0138]
于50ml单口瓶中加入上述油状液体(0.125g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b1(0.089g)收率92.0%,纯度为99.69%。
[0139]
m.p.248-250℃;esi-hrms:m/z calcd for c
18h18
n2o4[m h]
:327.1339;found:327.1332;1h nmr(400mhz,dmso-d6)δ9.81(s,2h),8.72(s,1h),8.28(d,j=7.0hz,1h),8.12(dd,j=8.0,1.5hz,1h),7.88(ddd,j=8.7,7.2,1.7hz,1h),7.73(d,j=8.4hz,1h),7.59
–
7.55(m,1h),7.31(d,j=6.9hz,1h),4.74(t,j=6.8hz,2h),4.05(s,2h),3.45(s,2h),2.56(s,3h).
13
c nmr(100mhz,dmso-d6)δ175.94,159.26,158.71,155.89,143.08,142.07,138.62,134.88,126.13,125.10,123.04,118.69,115.05,111.00,51.53,45.04,41.40,
13.00.
[0140]
实施例14
[0141]
2-甲基-3-羟基-1-(2-(((4氧代-6-甲基-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b2)的制备方法
[0142]
于100ml单口瓶中加入6-甲基-4-氧代-4h-苯并呋喃-3-甲醛(0.376g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.370g),收率43.0%。
[0143]
于50ml单口瓶中加入上述油状液体(0.129g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b2(0.092g)收率90.2%,纯度为99.08%。
[0144]
m.p.259-261℃;esi-hrms:m/z calcd for c
19h20
n2o4[m h]
:341.1496;found:341.1490;1h nmr(400mhz,dmso-d6)δ9.87(s,2h),8.69(s,1h),8.29(d,j=7.0hz,1h),7.89(s,1h),7.69(dd,j=8.6,2.0hz,1h),7.62(d,j=8.6hz,1h),7.32(d,j=7.0hz,1h),4.74(t,j=6.7hz,2h),4.04(s,2h),3.44(t,j=5.9hz,2h),2.56(s,3h),2.45(s,3h);
13
c nmr(100mhz,dmso-d6)δ176.37,159.82,158.95,154.66,143.56,142.32,139.06,136.37,136.25,124.67,123.19,118.93,115.29,111.43,51.94,45.53,42.01,20.94,13.38.
[0145]
实施例15
[0146]
2-甲基-3-羟基-1-(2-(((4氧代-7-甲基-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b3)的制备方法
[0147]
于100ml单口瓶中加入7-甲基-4-氧代-4h-苯并呋喃-3-甲醛(0.376g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.382g),收率44.4%。
[0148]
于50ml单口瓶中加入上述油状液体(0.129g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b3(0.093g)收率91.2%,纯度为99.17%。
[0149]
m.p.251-253℃;esi-hrms:m/z calcd for c
19h20
n2o4[m h]
:341.1496;found:341.1490;1h nmr(400mhz,dmso-d6)δ9.80(s,2h),8.69
–
8.64(m,1h),8.32
–
8.24(m,1h),7.99(d,j=8.1hz,1h),7.55(s,1h),7.36(dd,j=24.6,8.2hz,2h),4.78
–
4.68(m,2h),4.03(s,2h),3.44(s,2h),2.56(s,3h),2.48(s,3h).
13
c nmr(100mhz,dmso-d6)δ176.19,159.74,158.85,156.47,146.46,143.54,142.39,139.07,127.93,125.31,121.25,118.62,115.37,111.43,51.96,45.52,41.96,21.74,13.39.
[0150]
实施例16
[0151]
2-甲基-3-羟基-1-(2-(((4氧代-6-甲氧基-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b4)的制备方法
[0152]
于100ml单口瓶中加入6-甲氧基-4-氧代-4h-苯并呋喃-3-甲醛(0.408g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.388g),收率43.4%。
[0153]
于50ml单口瓶中加入上述油状液体(0.134g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b4(0.099g)收率92.07%,纯度为99.32%。
[0154]
m.p.267-269℃;esi-hrms:m/z calcd for c
19h20
n2o5[m h]
:357.1445;found:357.1448;1h nmr(400mhz,dmso-d6)δ9.89(s,2h),8.71(s,1h),8.31(d,j=7.0hz,1h),7.70(d,j=9.1hz,1h),7.53
–
7.41(m,2h),7.36(d,j=7.0hz,1h),4.75(t,j=6.6hz,2h),4.05(s,2h),3.87(s,3h),3.45(s,2h),2.56(s,3h).
13
c nmr(100mhz,dmso-d6)δ175.70,159.31,158.47,156.96,150.70,143.12,142.00,138.66,123.89,123.80,120.41,114.23,111.01,104.82,55.91,51.57,45.06,41.51,12.99.
[0155]
实施例17
[0156]
2-甲基-3-羟基-1-(2-(((4氧代-7-甲氧基-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b5)的制备方法
[0157]
于100ml单口瓶中加入7-甲氧基-4-氧代-4h-苯并呋喃-3-甲醛(0.408g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.392g),收率43.9%。
[0158]
于50ml单口瓶中加入上述油状液体(0.134g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b5(0.097g)收率90.8%,纯度为97.35%。
[0159]
m.p.252-254℃;esi-hrms:m/z calcd for c
19h20
n2o5[m h]
:357.1445;found:357.1460;1h nmr(400mhz,dmso-d6)δ9.74(s,2h),8.63(s,1h),8.28(d,j=7.0hz,1h),8.01(d,j=8.9hz,1h),7.31(d,j=7.0hz,1h),7.22(d,j=2.3hz,1h),7.13(dd,j=8.9,2.3hz,1h),4.73(t,j=6.7hz,2h),4.02(s,2h),3.91(s,3h),3.44(s,2h),2.56(s,3h);
13
c nmr(100mhz,dmso-d6)δ175.60,164.70,159.78,158.59,158.29,143.55,142.37,139.07,126.97,117.22,115.82,115.31,111.44,101.44,56.76,51.95,45.54,41.95,13.39.
[0160]
实施例18
[0161]
2-甲基-3-羟基-1-(2-(((4氧代-7-苄氧基-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b6)的制备方法
[0162]
于100ml单口瓶中加入7-苄氧基-4-氧代-4h-苯并呋喃-3-甲醛(0.56g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.420g),收率40.2%。
[0163]
于50ml单口瓶中加入上述油状液体(0.157g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b6(0.109g)收率84.1%,纯度为98.34%。
[0164]
m.p.169-171℃;esi-hrms:m/z calcd for c
25h24
n2o5[m h]
:433.1758;found:433.1769;1h nmr(400mhz,dmso-d6)δ9.73(s,2h),8.62(s,1h),8.27(d,j=7.0hz,1h),8.02(d,j=8.9hz,1h),7.49(d,j=7.0hz,2h),7.44
–
7.27(m,5h),7.21(dd,j=8.9,2.3hz,1h),5.29(s,2h),4.72(t,j=6.6hz,2h),4.02(s,2h),3.43(s,2h),2.55(s,3h);
13
c nmr(100mhz,dmso-d6)δ175.60,163.65,159.77,158.61,158.16,143.55,142.38,139.08,136.40,129.04,128.69,128.44,127.04,117.35,116.32,115.29,111.44,102.43,70.65,51.96,45.52,41.99,13.38.
[0165]
实施例19
[0166]
2-甲基-3-羟基-1-(2-(((4氧代-7-(3-甲基苄氧基)-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b7)的制备方法
[0167]
于100ml单口瓶中加入7-(3-甲基苄氧基)-4-氧代-4h-苯并呋喃-3-甲醛(0.588g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.461g),收率43.0%。
[0168]
于50ml单口瓶中加入上述油状液体(0.161g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b7(0.116g)收率,纯度为100.00%。。
[0169]
m.p.211-213℃;esi-hrms:m/z calcd for c
26h26
n2o5[m h]
:447.1914;found:447.1934;1h nmr(400mhz,dmso-d6)δ9.72(s,2h),8.62(s,1h),8.25(d,j=7.0hz,1h),8.02(d,j=8.9hz,1h),7.32
–
7.25(m,5h),7.22
–
7.16(m,2h),5.24(s,2h),4.71(t,j=6.5hz,2h),4.02(s,2h),3.43(s,2h),2.55(s,3h),2.33(s,3h);
13
c nmr(100mhz,dmso-d6)δ175.59,163.69,160.05,158.61,158.16,143.62,142.02,139.05,138.24,136.32,129.31,128.98,128.94,127.03,125.53,117.34,116.29,115.30,111.45,102.39,70.70,51.87,45.53,41.95,21.46,13.35.
[0170]
实施例20
[0171]
2-甲基-3-羟基-1-(2-(((4氧代-7-(4-甲基苄氧基)-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b8)的制备方法
[0172]
于100ml单口瓶中加入7-(4-甲基苄氧基)-4-氧代-4h-苯并呋喃-3-甲醛(0.588g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.465g),收率43.3%。
[0173]
于50ml单口瓶中加入上述油状液体(0.161g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b8(0.112g)收率83.6%,纯度为98.00%。
[0174]
m.p.134-136℃;esi-hrms:m/z calcd for c
26h26
n2o5[m h]
:447.1914;found:447.1908;1h nmr(400mhz,dmso-d6)δ9.64(s,2h),8.60(s,1h),8.22(d,j=5.6hz,1h),8.01(d,j=8.9hz,1h),7.37(d,j=8.0hz,2h),7.30(d,j=2.3hz,1h),7.24
–
7.17(m,4h),5.23(s,2h),4.69(t,j=5.7hz,2h),4.02(s,2h),3.43(s,2h),2.54(s,3h),2.31(s,3h);
13
c nmr(100mhz,dmso-d6)δ175.58,163.68,159.99,158.61,158.16,143.60,142.10,139.05,138.00,133.37,129.57,128.56,127.00,117.31,116.34,115.29,111.45,102.40,70.58,51.88,45.51,41.91,21.27,13.36.
[0175]
实施例21
[0176]
2-甲基-3-羟基-1-(2-(((4氧代-7-(3-氟苄氧基)-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b9)的制备方法
[0177]
于100ml单口瓶中加入7-(3-氟苄氧基)-4-氧代-4h-苯并呋喃-3-甲醛(0.596g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.472g),收率43.7%。
[0178]
于50ml单口瓶中加入上述油状液体(0.162g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b9(0.112g)收率83.0%,纯度为97.86%。
[0179]
m.p.215-217℃;esi-hrms:m/z calcd for c
25h23
fn2o5[m h]
:451.1664;found:451.1676;1h nmr(400mhz,dmso-d6)δ9.78(s,2h),8.63(s,1h),8.27(d,j=7.0hz,1h),8.03(d,j=8.9hz,1h),7.47(q,j=7.6hz,1h),7.35
–
7.28(m,4h),7.20(ddd,j=14.5,8.2,2.0hz,2h),5.32(s,2h),4.73(t,j=6.7hz,2h),4.02(s,2h),3.43(s,2h),2.55(s,3h);
13
c nmr(100mhz,dmso-d6)δ175.19,163.00,162.24(d,1j=243hz),159.53,158.24,157.72,143.19,141.76,138.92(d,3j=7hz),138.65,130.71(d,3j=9hz),126.69,123.86(d,4j=
3hz),117.07,115.85,115.04(d,1j=210hz),114.90,114.56(d,1j=220hz),111.03,102.09,69.30,51.49,45.12,41.55,12.95.
[0180]
实施例22
[0181]
2-甲基-3-羟基-1-(2-(((4氧代-7-(4-氟苄氧基)-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b10)的制备方法
[0182]
于100ml单口瓶中加入7-(4-氟苄氧基)-4-氧代-4h-苯并呋喃-3-甲醛(0.596g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.468g),收率43.3%。
[0183]
于50ml单口瓶中加入上述油状液体(0.162g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b10(0.110g)收率81.5%,纯度为98.00%。
[0184]
m.p.218-220℃;esi-hrms:m/z calcd for c
25h23
fn2o5[m h]
:451.1664;found:451.1683;1h nmr(400mhz,dmso-d6)δ9.74(s,2h),8.62(s,1h),8.26(d,j=7.0hz,1h),8.02(d,j=8.9hz,1h),7.55(dd,j=8.4,5.6hz,2h),7.32(d,j=2.2hz,1h),7.24(ddd,j=21.6,7.9,6.0hz,4h),5.27(s,2h),4.72(t,j=6.6hz,2h),4.02(s,2h),3.44(s,2h),2.55(s,3h);
13
c nmr(100mhz,dmso-d6)δ175.59,163.53,162.41(d,1j=242hz),159.63,158.63,158.14,143.52,142.55,139.07,132.68,132.65,130.81(d,1j=8hz),127.05,117.38,116.28,115.98,115.77,115.29,111.43,102.42,69.92,51.98,45.52,41.95,13.40.
[0185]
实施例23
[0186]
2-甲基-3-羟基-1-(2-(((4氧代-7-(3-氯苄氧基)-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b11)的制备方法
[0187]
于100ml单口瓶中加入7-(3-氯苄氧基)-4-氧代-4h-苯并呋喃-3-甲醛(0.629g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.473g),收率42.5%。
[0188]
于50ml单口瓶中加入上述油状液体(0.167g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b11(0.113g)收率80.7%,纯度为100.00%。
[0189]
m.p.226-228℃;esi-hrms:m/z calcd for c
25h23
cln2o5[m h]
:467.1368;found:467.1388.;1h nmr(400mhz,dmso-d6)δ9.63(s,2h),8.61(s,1h),8.22(d,j=7.0hz,1h),
8.03(d,j=8.9hz,1h),7.57(s,1h),7.47
–
7.42(m,3h),7.32(d,j=2.3hz,1h),7.24
–
7.20(m,2h),5.31(s,2h),4.69(t,j=6.8hz,2h),4.02(s,2h),3.43(s,2h),2.54(s,3h);
13
c nmr(100mhz,dmso-d6)δ175.60,163.40,160.18,158.64,158.14,143.65,141.85,139.06,139.00,133.66,130.99,128.60,128.06,127.11,126.93,117.51,116.25,115.33,111.45,102.52,69.64,51.85,45.55,42.00,13.32.
[0190]
实施例24
[0191]
2-甲基-3-羟基-1-(2-(((4氧代-7-(4-氯苄氧基)-4h-苯并吡喃-3-基)甲基)氨基)乙基)吡啶-4(1h)-酮(b12)的制备方法
[0192]
于100ml单口瓶中加入7-(4-氯苄氧基)-4-氧代-4h-苯并呋喃-3-甲醛(0.629g,2mmol)、二氯甲烷(20ml)、1-(2-氨乙基)-2-甲基-3-苄氧基吡啶-4-酮(0.774g,3mmol),和三乙酰氧基硼氢化钠(0.636g,3mmol),氮气保护下,于25℃下反应6h,反应结束后,减压蒸馏除去溶剂,以体积比为100:1、80:1、60:1、40:1、20:1的二氯甲烷和甲醇的混合溶液为洗脱剂进行硅胶柱层析,减压浓缩后得油状液体(0.480g),收率43.1%。
[0193]
于50ml单口瓶中加入上述油状液体(0.167g,0.3mmol),无水二氯甲烷(10ml),将1.0mol/l三氯化硼(0.90ml)用无水二氯甲烷溶液(15ml)稀释后置于恒压滴液漏斗中,在n2保护,-48℃下缓慢滴加稀释后的三氯化硼的无水二氯甲烷溶液,滴毕,转移到室温继续搅拌反应12h,待原料转化完全后,加入10ml甲醇淬灭,反应0.5h后,将反应液减压浓缩,用甲醇/乙醚(v:v=1:7)混合溶液重结晶得白色固体b12(0.112g)收率80.0%,纯度为100.00%。
[0194]
m.p.189-191℃;esi-hrms:m/z calcd for c
25h23
cln2o5[m h]
:467.1368;found:467.1363;1h nmr(400mhz,dmso-d6)δ9.68(s,2h),8.61(s,1h),8.25(d,j=7.0hz,1h),8.02(d,j=8.9hz,1h),7.52(d,j=8.5hz,2h),7.48(d,j=8.5hz,2h),7.31(d,j=2.3hz,1h),7.25(d,j=6.8hz,1h),7.21(dd,j=8.9,2.4hz,1h),5.29(s,2h),4.71(t,j=6.7hz,2h),4.02(s,2h),3.43(s,2h),2.55(s,3h);
13
c nmr(100mhz,dmso-d6)δ175.59,163.46,159.98,158.63,158.14,143.60,142.11,139.07,135.49,133.27,130.27,129.05,127.08,117.45,116.28,115.32,111.45,102.50,69.76,51.91,45.53,41.99,13.35.
[0195]
实施例25
[0196]
下面是本发明部分化合物的药理实验数据:
[0197]
1、化合物对mao-b的抑制活性的测定
[0198]
实验方法:
[0199]
从sigma购买mao-b试剂盒并储存在-20℃备用,按试剂盒操作预先准备好酶溶液和底物溶液。取5μl不同浓度的待测化合物(1nm,10nm,50nm,100nm,200nm,500nm,1μm)和25μl酶溶液混合加入到cornin g底读96孔微量测试板中于37℃振摇孵育10分钟,再加入20μl底物溶液,在多检测微孔板荧光读取器中基于所产生的荧光(激发,535nm;发射,587nm)对结果进行定量。
[0200]
结果表明本发明实施例1~24所制备的化合物对mao-b的抑制作用较好,当药物浓度为100nm时最高可达61.43
±
2.42%,显著优于阳性对照药pargyline的48.93
±
1.17%,最有效化合物的ic
50
值达到67.02nm,显著优于阳性对照药pargyline的ic
50
值111.30nm。是一个比较有潜力的先导化合物。
[0201]
表1.实施例1~24对mao-b的抑制率
[0202][0203][0204]
表2.部分实施例抑制活性的ic
50
值
[0205][0206]
表3.相关专利实施例抑制活性的ic
50
值
[0207]
[0208][0209]
通过ic
50
值的测定我们发现,实施例b1(ic
50
=67.27nm)、实施例b5(ic
50
=67.02nm)在本案的24个化合物中表现最为出色。另外,我们将本发明实施例与单胺氧化酶抑制剂的相关专利的实施例进行比较:专利cn110804045a与专利cn 110218207 b的主要区
别在于单胺氧化酶抑制剂活性基团与铁螯合剂活性基团的桥连基团不同,专利cn110218207b的实施例2e(ic
50
=424.6nm)、实施例1d(ic
50
=335.7nm)等化合物对mao-b的抑制活性较差,因此专利cn110804045a的实施例a1b1(ic
50
=173.3nm)、a1b3(ic
50
=194.6nm)、a5b1(ic
50
=187.8nm)将桥连基团替换成酰胺键后提高了化合物对mao-b的抑制活性,然而ic
50
依然在200nm左右,专利cn 111995567 a保持桥连基团不变,将香豆素母核替换为苯环后活性并没有得到较好的改善。因此本发明将上述专利实施例的香豆素母核替换为色酮母核,半数以上的化合物的ic
50
达到在100nm以下,显著提高了对mao-b的抑制活性;专利cn109678848 b的实施例ⅱg(ic
50
=1.457μm)、实施例ⅱh(ic
50
=0.856μm)与本发明实施例b1(ic
50
=67.27nm)主要区别在于桥连基团,本发明实施例显著提高了对mao-b的抑制活性,使ic
50
上升了一个数量级,达到了纳摩尔级别。因此,实施例b1(ic
50
=67.27nm)、实施例b5(ic
50
=67.02nm)是非常有潜力的先导化合物。
[0210]
2、化合物对mao-a的抑制活性的测定
[0211]
实验方法:
[0212]
从sigma购买mao-a试剂盒并储存在-20℃备用,按试剂盒操作预先准备好酶溶液和底物溶液。取5μl不同浓度的待测化合物(50nm,500nm,1μm,2μm,4μm,10μm,20μm)和25μl酶溶液混合加入到cornin g底读96孔微量测试板中于25℃振摇孵育10分钟,再加入20μl底物溶液,在多检测微孔板荧光读取器中基于所产生的荧光(激发,535nm;发射,587nm)对结果进行定量。
[0213]
结果表明,本发明以下实施例所制备的化合物均表现出对mao-b良好的选择性,选择性最好的化合物b1的si值达到30.67,接近于阳性对照药pargyline的si值37.59。是一个比较有潜力的先导化合物。
[0214]
表2.部分实施例抑制活性的ic
50
值和si值
[0215][0216]
3、化合物对铁离子螯合能力的测定
[0217]
根据分光光度法原理,采用一套自动滴定系统(automatic titration system)对pka进行测定。该系统包括:自动滴定器(metrohm dosimat 765liter ml syringe)、ph计[mettler toledo mp230 with metrohm ph electrode(6.0133.100)and a reference electrode(6.0733.100)]、紫外-可见分光光度计(hp 8453)和设定好vb程序的计算机。
[0218]
pka的滴定:于光程为50mm的比色皿中加入45ml(0.1m)氯化钾溶液,校正基线,加入40μl饱和na2edta溶液,加入1.5m盐酸,酸化至ph约为2。加入20μl浓度为50mm待测化合物dmso溶液,恒速搅拌。待吸收光谱稳定后开始自动滴定。即自动滴定管滴加0.1m koh溶液,滴加量控制在刚好能使溶液的ph增加0.1单位,加毕,待体系ph平衡之后再过30s,系统自动采集一次全波长光谱。(待测溶液ph平衡的判定标准为:在3秒内ph值的变化不超过0.001,可认为读数稳定)。系统自动重复自动滴定的操作直至ph达到规定的终点ph值。整个过程所
测得的所有数据均通过内部的visual basic程序记录并备份。采集的光谱结果采用hypspec2014程序进行分析并做pka计算。
[0219]
logβ1的测定:向光路为50mm的石英比色皿中加入45ml浓度为0.1m的氯化钾溶液,校正基线,用1.5m的盐酸酸化至ph约为2.1。加入60μl浓度为50mm的工作溶液,稳定2min。按照配体/铁离子=1.1/1加入fecl3的酸性溶液,恒速搅拌。od值稳定后,自动滴定管添加一定量的(4m)hcl溶液,使得溶液的ph下降0.1单位。待ph稳定后开始监测光谱,若光谱吸收曲线的最大值也达到了稳定就采集保存一次全部光谱信号。(ph稳定的判定标准为:当ph值在3秒内变化不超过0.001;光谱吸收稳定标准为:2min内变化率不超1%)。重复循环直至达到规定的ph值。整个过程所测得的所有数据都通过内部的visual basic程序记录并备份。
[0220]
logβ2和logβ3的测定:向光程为50mm的石英比色皿中加25ml浓度为0.1m的kcl溶液,20ml dmso。待光谱稳定再校正基线。采用1.5m的盐酸酸化至ph约为2.5。加入浓度为50mm的待测化合物溶液,稳定2min。按配体/铁离子=5/1的比例加入fecl3的酸性溶液,恒速搅拌。od值稳定后,自动滴定管添加一定量的浓度0.1m的hcl溶液,使溶液的ph上升0.1单位。ph值稳定1min后,开始采集并保存一次全谱信号。(ph稳定的判定标准为:当ph值在3秒内变化不超过0.001时,可认为ph读数稳定。重复该循环直至达到规定的ph值。整个过程所测得的所有数据都通过内部的visual basic程序记录并备份。
[0221]
根据以上方法测定的pk
a1
,pk
a2
,logβ1,logβ2和logβ3的数值,再用hyss软件拟合计算可得pfe
3
[0222]
结果表明实施例a1~12、b1~12中半数以上的pfe
3
值基本均大于18以上,说明该系列化合物在铁离子螯合活性上表现得很出众。特别是实施例a4,其pfe
3
值为19.08,十分接近本实验中所采用的对照药物deferiprone。说明该系列化合物也是十分具有进一步研究的潜力。
[0223]
表4.实施例1~24的pfe
3
值
[0224]
[0225][0226]
a:pka及logβ1测量体系:0.1m kcl溶液;logβ2及logβ3测量体系:dmso:kcl(0.1m)=2:3(v/v)
[0227]
b:在0.1m kcl溶液中测量
[0228]
c:文献xie yy,lu zd,kong xl,zhou t,bansal s,hider r.systematic comparison of the mono-,dimethyl-and trimethyl 3-hydroxy-4(1h)-pyridones-attempted optimization of the orally active iron chelator,deferiprone.eur j med chem 2016;115:132-140)报道的deferiprone参考数值(0.1kcl)
[0229]
[0230][0231]
通过铁离子螯合能力的测定我们发现,实施例a4(pfe
3
=19.08)在本案的24个化合物中表现最为出色。另外,我们将本发明实施例与单胺氧化酶抑制剂的相关专利的实施例进行比较:专利cn110804045a与专利cn 110218207 b的主要区别在于单胺氧化酶抑制剂活性基团与铁螯合剂活性基团的桥连基团不同,专利cn110218207b的实施例1e、1h、3d等化合物的pfe
3
值均在17以下,专利cn110804045a的实施例a12b1、a22b1、a5b1等化合物将桥连基团替换成酰胺键后铁离子螯合活性并没有得到改善,pfe
3
值依然在17以下。因此本发明将上述专利实施例的香豆素母核替换为色酮母核,半数以上的pfe
3
值基本均大于18以上,显著改善了铁离子螯合活性;专利cn109678848 b的实施例ⅱg(pfe
3
=17.1)与本发明实施例b1(pfe
3
=18.62)主要区别在于桥连基团,可以看出实施例b1的铁离子螯合活性得到了提高。
[0232]
表5.相关专利实施例mao-b抑制活性的ic
50
值以及pfe
3
值
[0233][0234]
同时结合mao-b抑制活性和铁离子螯合能力数据,我们可以发现,以上专利的实施
例在mao-b抑制活性和铁离子螯合能力方面的活性都不是很理想。而色酮母核已被公认为天然或人工来源的大量生物活性分子的药效团,是自然界中普遍存在的天然化合物的核心组成部分。本发明将上述专利实施例的香豆素母核替换为色酮母核,通过酰胺键将单胺氧化酶活性基团与铁螯合剂活性基团连接起来,显著改善了上述专利实施例的mao-b抑制活性和铁离子螯合能力。说明该系列化合物十分具有进一步研究的潜力。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。