强效细胞毒性化合物的靶向递送
1.相关申请
2.本技术请求依据35u.s.c.
§
119(e)于2019年1月28日提交的美国临时申请第62/797,881号的权益,其整体内容通过引用并入本文中。
3.联邦资助研究的声明
4.本发明在美国国立卫生研究院的国立普通医学科学研究所授予r01 gm073857的政府资助下完成。美国政府享有本发明的特定权利。
5.作为引用并入的序列表
6.名称为“40984-514001wo_sl.txt”的序列表的文字文件的内容为2020年1月27日建立的,且其大小为96,467字节,其整体通过引用并入本文中。
技术领域
7.本发明关于化学治疗。
背景技术:
8.例如非靶向化学治疗的癌症治疗的传统方法具有毒性且无法很好地辨别正常及肿瘤组织,且因其对病患有害的副作用而使其使用受限。相较于传统化学治疗,靶向治疗更加有效及安全,且近年来得到越来越多的关注。强效细胞毒性药物至病变细胞的靶向递送保证于减少副作用的同时最大化其等治疗效果。靶向治疗的主要目标为递送细胞毒性分子至病变组织中的细胞,同时避免向正常组织的显著递送。
技术实现要素:
9.本发明提供强效细胞毒性化合物的特定靶向及细胞内递送至酸性病变组织中的细胞的问题的解决方案,以主要于病变组织中诱导细胞死亡,同时避开健康细胞。
10.由于肿瘤的快速代谢作用,细胞外酸中毒普遍存在于肿瘤中,包括原发性肿瘤和转移性肿瘤。肿瘤细胞通过将酸度输出至细胞外环境以稳定其等的细胞质ph值。由于酸性通量及膜电位,癌细胞表面的细胞外ph值最低,显着低于正常生理组织的ph值或肿瘤中的整体细胞外ph值。即使于灌注良好的肿瘤区域,低ph值区域持续存在于癌细胞表面。癌细胞表面的酸度为不受克隆选择影响的可靶向特征,且酸度水平为肿瘤入侵和侵略的预测因子,因为越快速生长的肿瘤细胞越酸。低ph插入肽为一种水溶性膜肽,其于中性ph下与细胞膜微弱地相互作用,不会插入脂双层;然而,于微酸ph下,插入细胞膜中且形成稳定的穿膜螺旋。通过将或等效物结合至细胞毒性化合物,可特异性靶向细胞且直接递送所述细胞毒性化合物至癌细胞以及其等的胶质溶胶,因其等的酸性细胞表面。
11.使用肽递送强效细胞毒性化合物因此可选择性靶向病变组织(例如,肿
瘤),以提高治疗效果。此方法的显著优点为本文所述的构建物所介导的细胞毒性化合物的靶向递送对于病患的副作用很少,其为使用有校历的细胞毒性化合物的主要问题。因此,本文所述构建体的使用与治疗指数(therapeutic index)的增加以及治疗窗的增加相关。
12.因此,本发明的特征为一种包括强效细胞毒性化合物及肽的组合物,例如,其中,所述细胞毒性化合物包括诱导细胞死亡的小分子。所述细胞毒性化合物典型为质量2,000道尔顿或更少。所述强效细胞毒性化合物无法单独使用,因其会于正常组织中诱导显著的毒性,可能导致危及生命的副作用,例如,根据common terminology criteria for adverse events(ctcae)v5.0,2017年11月27日的nci指南所定义的等级3至5的不良事件。所述有效的的细胞毒性化合物仅可用作靶向治疗的一部分。强效细胞毒性化合物的实例可选自微管蛋白抑制剂、rna聚合酶抑制剂及dna损伤剂的类别。细胞毒性微管蛋白结合化合物的实例包括美登素(maytansines)、美登素及美登素类(maytansinoids)的化学衍生物及美登素的类似物。这些化合物结合至微管蛋白美登素结合位点且使微管组成脱稳。
13.细胞毒性化合物为接近或经细胞所吸收时,抑制细胞生长或促进细胞死亡的化合物,此外,于递送至细胞时(至靶向细胞的内部或所述细胞的表面),能够杀伤细胞或者抑制细胞增殖。强效细胞毒性化合物太过致命以致于无法单独施用。举例而言,于作为癌症治疗的化学治疗中,通常依赖细胞毒性剂杀伤或破坏正在繁殖的细胞的能力;其优先靶向快速分裂的癌细胞。例如,强效细胞毒性化合物以临床上不接受的水平杀伤正常细胞,且因此必须特异性递送至癌细胞(或其它不想要的细胞),例如,经由如本文所述的肽的高度特异肿瘤靶向工具。因此,本文所述的化合物及方法的显著优点为肽的使用使得先前临床上不可接受的药物现在可安全且有效的临床使用。
14.细胞毒性rna聚合酶结合化合物的实例包括鹅膏毒素(amatoxins),包括α-鹅膏蕈碱(amanitin)及α-鹅膏蕈碱的化学衍生物及类似物,其结合rna聚合酶且停止蛋白质合成。
15.例示性细胞毒性dna损伤化合物包括:i)烯二炔(enediyne)抗肿瘤抗生素,包括加利车霉素(calicheamicin)化合物及加利车霉素的化学衍生物及类似物,其结合于dna小沟中且导致链断裂;以及ii)拓扑异构酶(topoisomerase)i抑制剂化合物,包括喜树碱(camptothecin)及其结构类似物、依沙替康(exatecan)及依沙替康的其它化学衍生物及类似物,其结合且稳定拓扑异构酶i及dna的复合物,导致dna损伤。
16.本发明提供问题的解决方案,因为肽序列介导肿瘤细胞酸度的靶向以及所述强效细胞毒性化合物至病变组织中的细胞中的随后特异性递送。所述递送基于化合物跨质膜(绕过内吞摄取)直接转移至靶向细胞的细胞质中,同时很少或不会转移至正常细胞中。因此,开发所述肽以允许使用强效细胞毒性化合物作为靶向高度特异性的化疗剂。
17.如本文中所用,术语“药物”包括细胞毒性化合物,例如,强效细胞毒性化合物。举例而言,所述细胞毒性化合物无法单独使用,因其可能于正常组织中诱导显著的毒性,可能导致危及生命的副作用,例如,根据common terminology criteria for adverse events
(ctcae)v5.0,2017年11月27日的nci指南所定义的等级3至5的不良事件,通过引用并入本文中。关于细胞毒性化合物的效力及评估效力的方法的其它参考文献包括kummar,s.等人,br j clin pharmacol(2006)62(1);第15至26页以及florento l.等人,int j biomed sci.(2012)8(1);第76至80页,其等整体通过引用并入本文中。所述强效细胞毒性化合物仅可用作靶向治疗的一部分。
18.于一些实施例中,所述组合物还包括所述细胞毒性化合物(药物)及肽之间的连接子。例示性连接子包括二硫键或酸不稳定键。于一些实施例中,所述连接子为可裂解的。例示性可裂解连接子包括其为自我牺牲者。自我牺牲的清除通过熵的增加结合热力学稳定产物(例如,co2)的不可逆形成所驱动。所述连接子所具有的优点在于如果货物/治疗剂(药物)具有适当的nh2或-oh基团,其可以未经修饰的形式释放,如下列示意图中所绘示:
[0019][0020]
所述连接子所具有的优点在于强效货物/治疗剂以未经修饰的形式释放。实例亦包括具有于递送后裂解的二硫键的连接子。
[0021]
极性调节剂任选地包括于所述组合物中。所述调节剂改变构建体的整体极性以优化向肿瘤细胞或肿瘤肿块的递送。举例而言,如果所述货物使得所述组合物过于极性,添加调节剂以降低整体组合物极性,或者如果所述货物极性不足,添加调节剂以增加所述组合物极性。例如,包括所述调节剂的连接子具有增强药物递送至胞质溶胶的效率或改善肿瘤相对于正常组织的靶向的优点。于一些实施例中,所述构建体可包括极性调节剂;于其它实施例中(例如于极性药物的情况中),所述构建体可包括较疏水的调节剂,以促进向细胞中的递送。举例而言,当货物为极性(logp《-0.4),疏水性调节剂将增加[货物-调节剂]的logp(logp》-0.4)。假如货物为疏水性logp》2.5,极性调节剂将减少[货物-调节剂]的logp(logp《2.5)。调节剂的非限制性实例为脂肪酸、peg聚合物、疏水性荧光染料或环肽。
[0022]
于一些实施例中,所述组合物包括2种或更多肽。例示性构建体包括下列结构:肽
–
连接
–
b,
[0023]
其中,“肽”为包括序列addqnpwrayldllfptdtllldllwxa(seq id no:1)或adqdnpwrayldllfptdtllldllwxa(seq id no:2)的第一肽,“b”为包括序列addqnpwrayldllfptdtllldllwxa(seq id no:1)或adqdnpwrayldllfptdtllldllwxa(seq id no:2)的第二肽,其中,大写“x”表示任何氨基酸残基且可包括赖氨酸(lys)、半胱氨酸(cys)或含叠氮氨基酸;以及“连接”为聚乙二醇连接子,以及每个
“–”
为共价键。
[0024]
亦于本发明中的为一种肿瘤治疗的方法,包括向受试者施用包括如上所述的强效
细胞毒性化合物及肽的组合物。于一些实施例中,所述强效细胞毒性化合物为破坏微管功能且诱导细胞死亡的化合物。于一些实施例中,所述强效细胞毒性化合物为导致dna链断裂且诱导细胞死亡的化合物。于一些实施例中,所述强效细胞毒性化合物为结合rna聚合酶,停止蛋白质合成以及诱导细胞死亡的化合物。举例而言,所述受试者包括肿瘤。
[0025]
使用本领域已知的方法施用所述组合物,例如,所述组合物直接注射至肿瘤肿块中,或者通过膀胱内灌注局部施用,或局部应用,或者所述组合物为全身施用。因为所述构建体的靶向特性,所述细胞毒性化合物特异性靶向肿瘤细胞且递送至其等细胞质中。
[0026]
特定应用包括肠胃外、局部或全身施用的制剂,包括-连接子-药物,如本文所揭露。
[0027]
亦提供静脉内、动脉内、腹膜内、脑内、脑室内、鞘内、心内、海绵体内、骨内、眼内、皮下或玻璃体内施用的包括-连接子-药物的制剂。
[0028]
于一方面,本文提供一种肌内、皮内、经皮、经粘膜、病灶内、皮下、局部、表皮、羊膜外、阴道内、膀胱内、鼻或口服施用的包括-连接子-药物的制剂。
[0029]
本技术的主题亦包括一种膀胱内灌注的制剂,包括本文公开的-连接子-药物。于一些实施例中,所述制剂用于癌症(例如,实体肿瘤)或其它酸性病变组织的治疗。
[0030]
本文亦提供一种全身施用的包括-连接子-药物的制剂,其包含多个肽。于特定实施例中,所述制剂用于癌症或发炎或动脉粥样硬化或靶向衰老细胞的治疗。
[0031]
本文提供一种于受试者中治疗癌症或发炎或动脉粥样硬化或靶向衰老细胞的方法,包括向所述受试者施用有效量的ph触发化合物,其中,所述化合物包括细胞毒性化合物。癌症的非限制性实例包括膀胱癌、结肠癌、前列腺癌、乳癌、肺癌、皮肤癌、肝癌、骨癌、卵巢癌、胃癌、胰脏癌、睾丸癌及脑癌。于一些实施例中,所述癌症为膀胱癌。
[0032]
本文亦包括于受试者中检测及/或成像病变组织(如癌症组织)的方法,包括向所述受试者施用接合成像剂(i.a.)的-连接子-药物,例如,-连接子-药物。
[0033]
由于所述组合物中的存在,所述细胞毒性化合物主要递送至酸性病变组织,以主要于所述靶向组织中诱导生物效应及细胞死亡。举例而言,于存在下递送的所述细胞毒性化合物,例如于包含两种组分(如所述肽及细胞毒性化合物),的治疗指数至少相较所述组合物中不存在下所递送的细胞毒性化合物的治疗指数高10%、20%、50%、2倍、5倍或更高。
[0034]
相较于健康组织,所述组合物优先将所述强效细胞毒性化合物靶向病变组织,从而最小化所述健康组织的损伤。于不存在下,所述强效细胞毒性化合物诱导不想
要的且危及生命的副作用,包括下列的一些或全部:肺毒性、神经毒性、耳毒性、肾毒性、肝毒性、心动过速、骨髓抑制(myelosuppression)、深静脉血栓形成、口腔粘膜炎、味觉障碍、厌食症、恶心、呕吐、腹泻或便秘、腹痛、认知障碍、焦虑、抑郁症、肌肉疲劳及其它。相对地,于存在下,不会观察到这些副作用。
[0035]
本文包括的药物组合物包含ph触发化合物及其药学上可接受的载体。
[0036]
如本文中所用,当“有效”指化合物的含量时是指,当以本揭露的方法使用时,足以产生所需反应的化合物的量,而不具有与合理的益处/风险比相称的过度不良副作用。
[0037]
于一些实施例中,受试者为哺乳动物。于特定实施例中,所述哺乳动物为啮齿动物(例如,小鼠或大鼠)、灵长类动物(例如,黑猩猩、大猩猩、猴子、长臂猿、狒狒)、牛、骆驼、狗、猫、马、羊驼(llama)、绵羊、山羊或猪。于优选实施例中,所述受试者为人类。
[0038]
本文揭示的每个实施例预期适用于每个其它公开的实施例,故本文描述的各种元素的所有组合均于本发明的范围内。
[0039]
由以下优选实施例的描述以及自权利要求书,本发明的其它特征及优点为显而易见。除非另外定义,本文使用的所有技术及科学用语具有与属于本发明的领域技术人员所通常理解的相同意义,虽然本发明的实施或测试中可以使用相近或等于本文所述的那些方法和材料,但下文描述合适的方法和材料。
附图说明
[0040]
图1为具有药物的构建体的图,所述药物为细胞毒性化合物。
[0041]
图2a至c为具有药物及调节剂(m)分子的构建体的图。所述调节剂可附接至肽(图2a)、连接子(图2b)或药物(图2c)。
[0042]
图3为具有2种(或更多)药物的构建体的图。所述药物可为相同或不同。
[0043]
图4a至c为具有2种(或更多)药物及m分子的构建体的图。所述药物可为相同或不同。所述调节剂可附接至肽(图4a)、连接子(图4b)或药物(图4c)。
[0044]
图5为具有位于膜非插入端的药物及成像剂(i)的构建体的图。
[0045]
图6a至c为具有位于膜非插入端的药物、m分子及成像剂的构建体的图。所述调节剂可附接至肽(图6a)、连接子(图6b)或药物(图6c)。
[0046]
图7为具有多个位于膜非插入端的药物及成像剂的构建体的图。所述药物可为相同或不同。
[0047]
图8a至c为具有多个位于膜非插入端的药物、m分子及成像剂的构建体的图。所述药物可为相同或不同。所述调节剂可附接至肽(图8a)、连接子(图8b)或药物(图8c)。
[0048]
图9为通过peg聚合物(或者以紫色显示的任何其它聚合物)彼此连接的两种或更
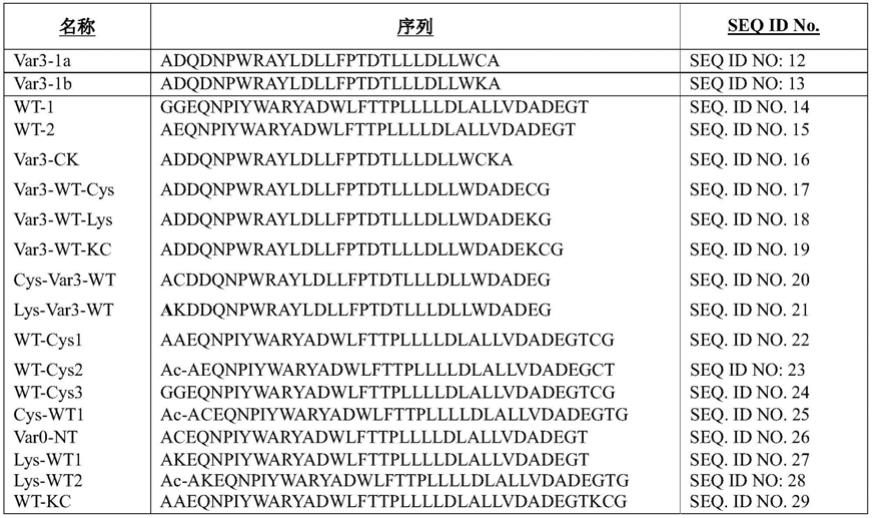
多肽的图,其具有经由连接子分子连接在一起的药物。所述药物可为相同或不同。
[0049]
图10a至c为具有通过peg聚合物(或任何其它聚合物,以紫色显示)彼此连接的两种或更多肽,以及经由连接子分子连接在一起的药物的例示性构建体的图。所述药物可为相同或不同。所述调节剂可附接至肽(图10a)、连接子(图10b)或药物(图10c)。
[0050]
图11为具有通过peg聚合物彼此连接的两种或更多肽(或任何其它聚合物,以紫色显示),以及经由连接子分子连接在一起的药物的例示性构建体的图。所述药物可为相同或不同。
[0051]
图12a至c为具有两种或更多肽及经由连接子分子连接在一起的药物的例示性构建体的图。所述药物可为相同或不同。所述调节剂可附接至肽(图12a)、连接子(图12b)或药物(图12c)。
[0052]
图13为例示性异双功能自我牺牲连接子的结构,其可用于制备含有药物(治疗性货物)的构建体。
[0053]
图14a为具有与肽的cys残基s-s键交换的药物的例示性自我牺牲连接子的结构。
[0054]
图14b为具有于肽与lys残基接合的药物的例示性自我牺牲连接子的结构。
[0055]
图15为美登素类,mertansine(dm1;cas编号:139504-50-0)的化学结构,其中,r=ch2ch2sh。
[0056]
图16为α-鹅膏蕈碱的化学结构。
[0057]
图17为烯二炔抗生素,加利车霉素的化学结构。
[0058]
图18为喜树碱化学类似物,依沙替康(dx-8951f)的化学结构。
[0059]
图19a为phlip-s-s-mertansine构建体的化学结构。
[0060]
图19b绘示相较于未接受任何治疗的对照组,以phlip-s-s-mertansine(参见图21a)进行肿瘤内(it)及腹膜内(ip)治疗后所收集的乳腺mda-mb-231肿瘤的图像。于it治疗组中由
“‑”
指示的位置表示肿瘤于治疗的过程中消失而无法收集。
[0061]
图19c绘示呈现相较于对照(未经治疗)组,以phlip-s-s-mertansine进行ip及it治疗后所收集的肿瘤重量的平均值(实心方块)、中位数、25及75的百分位数(箱形本身)及标准偏差值的箱形图。p水平使用双侧检验进行计算。
[0062]
图20a为绘示肿瘤质量减少的条状图,其于phlip-s-s-鹅膏蕈碱的多次ip施用后,于携带鼠类jc和4t1乳癌的小鼠身上进行测试。p水平使用双侧检验进行计算。
[0063]
图20b为绘示于ph 7.4及ph 6.0下以增加的浓度的phlip-s-s-鹅膏蕈碱(以对数标度)治疗后的细胞活力的线形图。
[0064]
图21a绘示经用于与含有单个cys残基的phlip直接接合的3-(2-吡啶基二硫代)丙
酸琥珀酰亚胺酯(spdp)所修饰的加利车霉素的化学结构的图像。
[0065]
图21b绘示相较于未接受任何治疗的对照组,以phlip-s-s-加利车霉素进行ip治疗后所收集的宫颈hela肿瘤的图像。
[0066]
图21c绘示呈现相较于对照(未经治疗)组,以phlip-s-s-加利车霉素进行ip及it治疗后的肿瘤重量的平均值(实心方块)、中位数、25及75的百分位数(箱形本身)及标准偏差值的箱形图。p水平使用双侧检验进行计算。
[0067]
图22绘示经用于与含有单个cys残基的直接接合的spdp修饰的伊沙替康的化学结构的图像。
具体实施方式
[0068]
本发明提供包含于低ph下相较于正常ph下对膜脂双层具有较高亲和力的肽的接合至细胞毒性药物的组合物,能够靶向酸性病变组织且通过跨膜插入递送药物至细胞中。现有的药物无法很好的辨别病变及正常组织,从而影响两者,导致危及生命的副作用及受限的治疗效果。目前使用的一些化合物,例如阿霉素(doxorubicin),实际上相较于肿瘤组织更加靶向正常组织。特别而言,强效细胞毒性化合物因其等危及生命的副作用无法单独使用。然而,所述细胞毒性化合物可用作靶向治疗的一部分。
[0069]
所述组合物及方法用于使用靶向肿瘤,以特异性递送细胞毒性化合物至病变组织(癌症)的细胞中,且因此主要于所述靶向组织内促进细胞死亡。本发明提供3个主要特征:i)细胞毒性化合物向肿瘤的靶向,以主要于肿瘤内诱导细胞杀伤;ii)于健康器官及组织中的正常细胞的间隔,以减少与所述细胞毒性化合物单独使用相关的危及生命的副作用;以及iii)细胞毒性化合物穿过病变组织中的细胞膜的直接递送,从而避免内体摄取,其需要细胞毒性化合物的内体逃逸才能发挥作用。
[0070]
强效细胞毒性化合物的靶向递送的优点
[0071]
可靶向于病变组织中的细胞表面过度表达的各种蛋白质生物标志物,从而提高治疗效果。虽然存在许多可用于改善肿瘤靶向和治疗结果的生物标志物,例如于一些癌细胞表面过度表达的各种受体,但并非所有肿瘤中均存在有用的标志物。此外,单独的肿瘤中及不同病患的肿瘤之间的癌细胞群的异质性限制生物标志物靶向技术的有效使用。又,快速的突变增加不表达高水平靶向生物标志物的癌细胞表型的选择的可能性。生物标志物靶向可作为一种导致耐药性发展及病患预后不佳的选择方法。
[0072]
因此,目前靶向化疗领域的一个挑战为寻找替代的、更可靠的肿瘤细胞靶向生物标志物。同样重要的为强效细胞毒性药物的细胞内递送的方法。如果靶向可结合递送,非常重要的进展就在手边。本文所述的组合物及方法实现这些目标,因此表现出优于先前方法的显着优势。
[0073]
微管蛋白抑制剂
[0074]
微管蛋白抑制剂为直接干扰微管蛋白系统的药物。微管蛋白为微管的主要构件,几乎存在于所有真核细胞中,由α-微管蛋白和β-微管蛋白亚单元所组成。微管的动态组装和拆卸涉及许多细胞过程,如细胞结构维持、细胞分裂和细胞内运输。微管的破坏诱导细胞周期停滞于g2/m期,使得微管成为药物发现的具有吸引力的靶标。临床使用的大部份微管
蛋白抑制剂为天然产物及其等合成性衍生物。微管/微管蛋白抑制剂包括促进微管蛋白聚合及稳定微管结构以阻挡功能所需的动态组装/拆卸的剂(例如,紫杉醇(paclitaxel)、埃博霉素(epothilones)、圆皮海绵内酯(discodermolide)及根薯酮内酯(taccalonolides)),以及抑制微管蛋白的聚合及使微管结构脱稳,使所述微管无法形成的其它剂(例如,美登素类、澳瑞他汀(auristatin)、长春碱(vinblastine)及长春新碱(vincristine))。这些抑制剂进一步分为基于其等微管蛋白结合位点分为六种不同类别:美登素、长春花(vince)、紫衫烷(taxane)、秋水仙碱(colchicine)、pironetin及laulimalide/peloruside结合位点。结合紫衫烷及laulimalide/peloruside位点的分子使微管稳定,而靶向美登素、秋水仙碱、长春碱或pironetin位点的化合物使微管脱稳。
[0075]
rna聚合酶抑制剂
[0076]
rna聚合酶的抑制剂预防遗传信息自dna至rna的转录,其停止蛋白质合成、核酶产生及rna的调节功能,导致细胞死亡。一些化合物(例如,阿糖胞苷(cytarabine)盐酸阿糖胞苷(cytosine arabinoside))具有包括与dna和rna聚合酶及dna的结合的多种作用机制。一些其它化合物包括对rna聚合酶具有高度选择性的利福霉素(rifamycins)(例如利福平(rifampicin或rifampicin)、利福布汀(rifabutin)、利福喷汀(rifapentine)、利福拉齐(rifalazil)及利福昔明(rifaximin))及鹅膏菌毒素(amanita toxins)(例如,α-鹅膏蕈碱、β-鹅膏蕈碱、γ-鹅膏蕈碱、ε-鹅膏蕈碱、amanullin、amanullic acid、amaniamide、amanin和proamanullin)。
[0077]
dma损伤剂
[0078]
已知的dna损伤剂包括直接修饰dna碱基、插入碱基之间或于dna中形成交联的化合物。dna烷化剂包括用于临床的氮芥(例如,环磷酰胺、苯丁酸氮芥及美法仑(melphalan))、亚硝基脲类(例如,卡莫司汀(carmustine)、洛莫司汀(lomustine)及司莫司汀(semustine))和三氮烯类(triazenes)(例如,达卡巴嗪(dacarbazine)和替莫唑胺(temozolomide))、烷基化样铂剂(例如,顺铂(cisplatin))及铂基类似物(例如,卡铂(carboplatin)和奥沙利铂(oxaliplatin))。抗代谢物包括模拟正常细胞分子及干扰dna复制的嘧啶类似物(例如,5-氟尿嘧啶、卡培他滨(capecitabine)、脱氧氟尿苷(floxuridine)和吉西他滨(gemcitabine))和嘌呤类似物(例如,6-巯基嘌呤、8-氮鸟嘌呤、氟达拉滨(fludarabine)和克拉屈滨(cladribine))。细胞毒性dna损伤剂包括蒽环类药物(例如,阿霉素、柔红霉素(daunorubicin)、表阿霉素(epirubicin)、伊达比星(idarubicin)、吡柔比星(pirarubicin)、阿克拉霉素(aclarubicin)及米托蒽醌(mitoxantrone))、博来霉素(bleomycins)、丝裂霉素(mitomycins)、放线菌素(actinomycin)及烯二炔抗生素的(例如,加利车霉素及类似物),其于dna的两条链上的碱基对之间插入(dna插入),或/及导致链断裂,或/及产生高反应性自由基,或/及诱导dna烷基化。dna损伤剂的另一类别为拓扑异构酶i抑制剂(例如,喜树碱及类似物),其结合拓扑异构酶i且稳定拓扑异构酶i及dna的复合物。因此,预防导致dna损伤的dna再连接。
[0079]
强效细胞毒性化合物的靶向治疗及靶向递送
[0080]
显著比例的癌症病患于长期化疗后出现获得性耐药性或复发。此外,非靶向药物具有损害正常组织的非特异性毒性,导致严重的副作用且限制其等的疗效。相较于传统化疗,靶向治疗设计为主要影响病变组织,但避开健康细胞。目前靶向癌症治疗的主要类别是
小分子(包括丝氨酸/苏氨酸激酶抑制剂及酪氨酸激酶抑制剂)、单克隆抗体(mab)及抗体药物接合物(adc)。此外,用于靶向治疗的各种纳米技术方法正在开发中。
[0081]
单独的小分子或mab靶向治疗通常由于其细胞毒性低和对实体肿瘤的渗透性差,因此显示不充分的治疗活性。一种改进的方法为使用adc,其中,mab通过适当的连接子与高度强效的细胞毒性药物(或药物)接合。adc设计为仅靶向且杀伤癌细胞同时避开健康细胞的靶向治疗。adc的开发面临许多挑战:i)adc中使用的抗体可能引起免疫原性反应并导致从循环中快速清除;ii)细胞毒性药物可能不够有效,且进一步的结合可能导致效力降低;iii)病变细胞表面靶向抗原的呈递不足会导致adc效力降低或失效;iv)抗体的肿瘤渗透性差;v)抗体的内吞摄取需要药物的内体逃逸以找到其细胞靶点;vi)对于许多肿瘤,尚未发现可用的表面表位,以及vii)肿瘤异质性可能导致耐药细胞的克隆选择,导致肿瘤生长恢复。
[0082]
于强效细胞毒性化合物中,美登素类及其等衍生物及类似物为微管蛋白抑制剂的示例,鹅膏蕈碱及其衍生物及类似物为rna聚合酶抑制剂的示例,而加利车霉素及喜树碱、其等衍生物及类似物为dna损伤剂的示例。每个这些及其它强效细胞毒性化合物需要靶向肿瘤且递送至细胞中,此为使用技术所达成的任务。
[0083]
构建体
[0084]
本发明提供使用靶向肿瘤的组合物及方法,以特异性递送强效细胞毒性化合物至酸性病变组织中(例如,肿瘤),绕过内吞摄取且将细胞毒性化合物递送至靶向病变组织中的细胞的细胞质中,从而特异性仅(或主要)于靶向组织中促进细胞杀伤。如上所述,强效细胞毒性化合物必须靶向所述病变组织,否则副作用将妨碍其临床使用。
[0085]
包括肽及细胞毒性化合物的化合物的一般代表显示于图1至16中,且描述如下。
[0086]
图1显示连接至肽的细胞毒性化合物(药物)分子:
[0087]
图2至12中显示的组合为图1显示的方案的变体。
[0088]
一种或更多调节剂分子(m)任选地附接至肽膜插入端或连接子或药物,通过减少药物至健康组织的非特异性递送,可能增强治疗效果(图2)。调节剂分子可为极性分子,以防止疏水性及中等疏水性的细胞内递送。药物分子以正常细胞外ph穿过健康细胞的膜。
[0089]
图3显示连接至单个肽的多个药物分子。
[0090]
图4a至4c显示连接至具有一种或更多调节剂分子的单个肽的多个药物分子。
[0091]
图5、图6a至6c、图7及图8a至8c绘示可于肽膜非插入端携带一种或更多成像剂(i)(或其它分子)的化合物。
[0092]
图9显示经由连接子分子连接在一起的具有药物货物的两种或更多肽。
[0093]
例示性构建体包括var3序列addqnpwrayldllfptdtllldllwca(seq id no:3)或addqnpwrayldllfptdtllldllwka(seq id no:4)adqdnpwrayldllfptdtllldllwca(seq id no:5)或adqdnpwrayldllfptdtllldllwka(seq id no:6)或其变体,例如,下列表格及本文引用的参考文献中(通过引用并入本文)所提供的序列。
[0094]
于一实施例中,所述药物为微管蛋白抑制剂分子。微管蛋白抑制剂分子可靶向微管蛋白系统以促进微管蛋白聚合从而稳定微管结构,或者抑制微管蛋白聚合从而使微管结构脱稳。举例而言,微管蛋白抑制剂分子结合微管蛋白美登素位点,导致微管动态的压抑并导致细胞周期停滞于g2/m期。非限制性实例为美登素、安丝菌素(ansamitocin)、美登醇(maytansinol)、d-丙氨酰基美登素(d-alanyl maytansine)及具有二硫或硫醇基团的美登素衍生物及类似物(dm1、dm3及dm4)及其等衍生物。美登素类的示例为mertansine(或s-甲基-dm1或dm1)。
[0095]
或者,所述药物为rna聚合酶抑制剂。rna聚合酶抑制剂结合rna聚合酶且停止功能性rna的生产。举例而言,包括用于蛋白质合成的核酶、trna、调控rna及mrna。例如,rna聚合酶抑制剂结合rna聚合酶ii及iii。非限制性实例包括鹅膏菌毒素及其等衍生物及类似物。鹅膏菌毒素的示例为α-鹅膏蕈碱。
[0096]
于另一实施例中,所述药物为dna损伤剂。dna损伤剂导致链断裂或结合拓扑异构酶i-dna复合物。举例而言,dna损伤剂结合dna或拓扑异构酶i。非限制性实例为烯二炔抗生素及拓扑异构酶i抑制剂。烯二炔抗生素及拓扑异构酶i的示例分别为加利车霉素及喜树碱、其等衍生物及类似物。
[0097]
药物经由可裂解连接与肽连接。举例而言,所述可裂解连接可为二硫键或酸不稳定键。于其它实施例中,所述可裂解连接为自我牺牲连接。
[0098]
美登素类
[0099]
强效美登素类及澳瑞他汀细胞毒性微管蛋白抑制剂由于肿瘤靶向的缺乏以及高毒性,治疗窗差(低治疗指数)。所述低治疗指数导致这些强效微管蛋白抑制剂作为化疗抗癌剂的失败。举例而言,美登素作为抗癌药物具有显著的剂量限制毒性,包括神经毒性、胃肠道毒性、虚弱、恶心、呕吐及腹泻。仅有靶向治疗可解决这些限制。目前,美登素-及澳瑞他汀-衍生物为adc中验证的药物且已被广泛地研究。
[0100]
美登素类为衍生自美登素的抗有丝分裂微管蛋白抑制剂,结合美登素位点,导致微管动态的压抑且细胞周期停滞于g2/m期。所述美登素位点为β-微管蛋白的独特的位点,其位于微管中的纵向微管蛋白
–
微管蛋白接口。结合此位点的抑制剂易于使微管组装脱稳。已经引入靶向所述美登素位点的三种明显不同的配体:美登素、pm060184及根瘤菌素(rhizoxin)。美登素为具有皮摩尔ic
50
值以及相较于用作抗癌化学治疗剂的紫杉烷-及长春碱-位点抑制剂较高的效力的第一强效细胞毒性化合物之一。美登素生产自安丝菌素,其得自发酵微生物珍贵束丝放线菌(actinosynnema pretiosum)。例如美登素类似物(dm1、dm3及dm4)的美登素类通过半合成策略的应用所引入。mertansine(dm1以及,于其一些形式,emtansine)为美登素类似物的一者。
[0101]
澳瑞他汀衍生自由海兔截尾海兔(dolabella auricularia)分离的尾海兔素-10(dolastatin-10)的天然产物。尾海兔素-10及其类似物抑制微管蛋白依赖性gtp的结合及
阻挡長春花生物鹼(vinca alkaloid)已非竞争性方式与微管蛋白的结合。其等广泛用作adc药物。肽的化合物的wt家族与单甲基澳瑞他汀f(mmaf)及单甲基澳瑞他汀e(mmae)接合,于小鼠模型中表现出显着的治疗效果且不具有明显毒性[美国专利申请第20170267727号;burns ke,hensley h,robinson mk,th
é
venin d.therapeutic efficacy of a family of phlip-mmaf conjugates in cancer cells and mouse models.mol pharm.2017 14(2),415-422;burns ke,robinson mk,th
é
venin d.inhibition of cancer cell proliferation and breast tumor targeting of phlip-monomethyl auristatin e conjugates.mol pharm.2015 12(4),1250-1258]。
[0102]
鹅膏毒素
[0103]
α-鹅膏毒素属于数种蘑菇属鹅膏菌属(amanita)(条纹毒鹅膏菌(amanita phalloides)及鱗柄白鵝膏菌(a.virosa)及双孢鹅膏(a.bisporigera))中发现的所有鹅膏毒素的最致命种类。α-鹅膏蕈碱为rna聚合酶ii的强效抑制剂、rna聚合酶iii的中度抑制剂以及rna聚合酶iv的弱抑制剂。其阻挡rna聚合酶且抑制包括trna、核酶、mirna及mrna的功能性rna的转录,且因此蛋白质的随后合成,导致细胞死亡48小时。其为一种无法有效地分散在血浆膜上的极性环肽,除了于肝细胞中以外,其具有用于摄取小循环分子的特殊运输系统,如鬼笔毒素(phallo toxin)及鵝膏菌毒素(amanita toxin)。于人类癌细胞系上测试接合抗epcam抗体的α-鹅膏蕈碱的抗增殖效应,并在携带皮下人类胰脏癌异种移植肿瘤的免疫受损小鼠中进行体内评估。与α-鹅膏蕈碱接合的肽的化合物于癌细胞系上测试浓度依赖及ph依赖的细胞毒性[wyatt lc,moshnikova a,crawford t,engelman dm,andreev oa,reshetnyak yk.peptides of phlip family for targeted intracellular and extracellular delivery of cargo molecules to tumors.proc natl acad sci u s a.2018,115(12),e2811-e2818;moshnikova a,moshnikova v,andreev oa,reshetnyak yk.antiproliferative effect of phlip-amanitin.biochemistry.2013,52(7),1171-1178]。最近的研究表明一些肿瘤将特别容易受到α-鹅膏蕈碱治疗的影响,因为一些癌症中tp53基因的基因组缺失(编码p53蛋白者)与相邻基因的抑制有关,例如polr2a基因,其编码rna聚合酶ii的催化亚基。举例而言,polr2a表达水平与人类结肠直肠癌中的基因拷贝数密切相关。因此,预期使用α-鹅膏蕈碱用于结直肠癌的潜在治疗(或具有半合子tp53和polr2a缺失的类似癌症)特别有效。
[0104]
加利车霉素
[0105]
加利车霉素化合物属于通过双链裂解损伤dna的天然烯二炔抗生素的化学群组。加利车霉素起初自细菌棘孢小单胞菌(micromonospora echinospora)的分离,且被认为是尚未鉴定的最有效的抗肿瘤剂。加利车霉素γ为adc中使用最活跃的化合物之一。加利车霉素于细胞中通过谷胱甘肽进行还原键切割,接着为自发的环化和双自由基的产生,随后形成来自dna的抽象氢原子,导致双链双自由基。于氧的存在下,dna双链经裂解,之后为细胞死亡。数种应用加利车霉素作为其等药物的adc目前已于临床试验中进行测试。
[0106]
喜树碱类似物
[0107]
喜树碱为自中国观赏树喜树(camptothecaacuminata)分离的dna拓扑异构酶i抑制剂。伊立替康(irinotecan)(喜树碱-11)为喜树碱的半合成类似物。已知为甲磺酸伊沙替
康(sn38及dx-8951f)的伊立替康活性代谢物用作adc的药物。dx-8951f为更加水溶性的喜树碱类似物,其非mdr1底物。数种应用喜树碱类似物及其衍生物作为其等药物的adc目前已于临床使用中测试,且获得突破治疗的称号。
[0108]
肽
[0109]
野生型肽(wt)的实例为aeqnpiywaryadwlfttplllldlallvdadegt(seq id no:7),其中,aeqnpiy(seq id no:8)代表侧翼序列,waryadwlfttplllldlallv(seq id no:9)代表膜插入序列,以及dadegt(seq id no:10)代表侧翼序列。
[0110]
其它例示性肽显示于下列表格中。
[0111]
表1:例示性肽
[0112]
[0113][0114]
表2:例示性肽
[0115][0116]“ac”意指乙酰化n末端
[0117]“am”意指酰胺化n末端
[0118]
表3:包括l-异构体、d-异构体、α-异构体、β-异构体、乙二醇-及甲基-修饰的编码及例示性非编码的氨基酸。
[0119]
[0120]
[0121][0122]
表4:包含l-异构体、d-异构体、α-异构体和β-异构体的可质子化残基的非限制性实例及其取代。
[0123][0124]
表5:编码氨基酸的实例
[0125][0126]
表6:属于不同肽群组的膜插入序列的非限制性实例。每个可质子化残基(以粗体表示)可经其自表4的取代所替换。每个非极性残基可经其自表5的编码氨基酸取代及/或自表3的非编码氨基酸取代所替换。
[0127][0128]
表7:序列的非限制性实例。可将半胱氨酸、赖氨酸、经叠氮修饰的氨基酸或经炔基修饰的氨基酸并入肽的n-末端(首6个残基)或c-末端(末6个残基)部分,以接合货物及连接子。
[0129]
[0130][0131]
治疗癌症、发炎、动脉粥样硬化或靶向受试者中衰老细胞的方法
[0132]
本文包括一种于有此需要的受试者中预防或治疗癌症、发炎、动脉粥样硬化或靶向受试者中衰老细胞(例如,于酸性病变组织中杀伤细胞)的方法。于另一具体实施例中,所述方法包括向所述受试者施用有效量的组合物,包括肽及细胞毒性化合物(例如,细胞毒性微管蛋白抑制剂、细胞毒性rna聚合酶抑制剂、细胞毒性dna损伤化合物或拓扑异构酶i抑制剂)。
[0133]
举例而言,预防或治疗癌症、发炎、动脉粥样硬化或靶向受试者中的衰老细胞的方法(例如,于酸性病变组织中杀伤细胞)包括施用包括有效量的组合物的组合物,包括肽及细胞毒性化合物。
[0134]
于其它实施例中,所述治疗癌症、发炎、动脉粥样硬化或靶向受试者中衰老细胞的方法包括向受试者施用根据本文所述的方法生产的包括肽及细胞毒性化合物的
14a-b显示例示性-连接子-药物构建体。如上所述,单独使用美登素类、α-鹅膏蕈碱、加利车霉素、喜树碱及其等衍生物及类似物作为抗癌剂不可能达到可接受的治疗窗口,因此需要靶向递送。本文所述的构建体,例如-连接子-药物构建体,介导美登素类、α-鹅膏蕈碱、加利车霉素、喜树碱及其等衍生物及类似物向肿瘤细胞(由于其等低ph表面)的靶向,且减少这些强效细胞毒性化合物向正常细胞(具有正常细胞外ph)的递送,从而避免或最小化副作用,增加治疗窗且增强治疗指数。靶向强效细胞毒性化合物,于此情况中为美登素类或α-鹅膏蕈碱、加利车霉素或喜树碱及这些化合物的衍生物及类似物,的问题通过本发明/药物组合物所解决。
[0146]
连接子可为相对小,例如仅几个原子,或者相对大(4至5kda)。图13a显示例示性异双功能连接子,一端与游离硫醇反应以自发形成二硫键,硫吡啶作为离去基团,另一端于dipea及一些情况下的dmap或其它活化剂碱的存在下与活化的胺或羟基基团反应,以分别形成氨基甲酸酯或碳酸盐。
[0147]
图14a为于具有半胱氨酸残基的肽与游离硫醇反应以自发形成二硫键的连接子-药物接合物,硫吡啶作为离去基团。图14b为于具有赖氨酸残基的与游离氨基反应的连接子-药物接合物,如果于其氨基末端受到例如n-乙酰化的保护。
[0148]
于一些实施例中,可使用下列交联连接子:3-(2-吡啶二硫代)丙酸琥珀酰亚胺酯(spdp);lc-spdp(6-(3(2-吡啶二硫代)丙酰氨基)己酸琥珀酰亚胺酯);sulfo-lc-spdp(6-(3’(2-吡啶二硫代)丙酰氨基)己酸磺酸基琥珀酰亚胺酯);peg4-spdp(经peg修饰,长链spdp交联连接子);peg12-spdp(经peg修饰,长链spdp交联连接子);smcc(4-(n-马来酰亚胺基甲基)环己烷-1-羧酸琥珀酰亚胺酯);sulfo-smcc(4-(n-马来酰亚胺基甲基)环己烷-1-羧酸磺酸基琥珀酰亚胺酯);smpt(4-马来酰亚胺基氧基羰基-α-甲基-α(2-吡啶二硫代)甲苯);dtme(二硫代双马来酰亚胺乙烷)。
[0149]
化合物具有下式:
[0150]
肽
–
mod
–
连接子
–
药物(1),
[0151]
其中:
[0152]
肽为肽,
[0153]
mod为调节剂且为任选的。mod包括调节连接子-药物的整体极性的化学实体,以通过最佳化细胞内递送。为达到最佳化靶向,可通过logp引导mod-连接子-药物的整体极性,其中,p为所测得的辛醇-水分配系数,优选为-1《logp《1的范围内。任选地,所述mod可附接至的插入端、连接子或药物。
[0154]
于一些情况中,强效细胞毒性化合物或药物为极性或中等疏水性。药物所测得logp的平均值约为2至3。例示性药物为如下列特征所定义的极性、中等疏水性或疏水性。极性:logp《-0.4;中等疏水性:2.5《logp《-0.4;以及疏水性:logp》2.5。所递送的药物或化合物的极性及/或疏水性可使用本领域已知的方法测量,例如,通过测定logp,其中,p为辛醇-水分配系数。将物质溶解于辛醇-水混合物中,混合并达到平衡。接着测量物质于每个(或一
个)相的数量。测量本身可为本领域中已知的多种方法,例如,通过测量吸光度或使用nmr、hplc或其它已知方法测定数量。假如货物为极性(logp《-0.4),疏水性调节剂将增加(货物-调节剂)的logp(logp》-0.4)。假如货物为logp《2.5的疏水性,极性调节剂将降低(货物-调节剂)的logp(logp《2.5)。
[0155]
连接子包括共价键或化学连接子,使(1)选自下列:
[0156][0157]
每个y的出现可为存在或不存在,且独立地为1至4的整数;
[0158]
每个x的出现独立地选自ch2、ch(烷基)及c(烷基)2所组成的群组;
[0159]
每个b的出现可为存在或不存在,且独立地选自由烷基、芳基及peg所组成的群组;
[0160]
键a形成于a中的半胱氨酸残基的硫及硫醇取代之间;
[0161]
键b形成于所述药物上的取代及碳之间,其中,所述取代选自由羟基、羰基、胺、氨基、硫酸、磺胺、磷酸盐及磷酰胺所组成的群组;
[0162]
键c形成于药物上的取代及羰基之间,其中,所述氨基酸选自由伯胺、仲胺和羟基所组成群组;
[0163]
键d形成于b及a中的氨基酸残基之间,其中,所述氨基酸选自由丝氨酸、苏氨酸、酪氨酸、色氨酸、组氨酸、赖氨酸及半胱氨酸所组成群组,且包括胺、酯、氨基甲酸酯、碳酸盐或马来酰亚胺键;
[0164]
药物包括或由具有抗癌活性的强效细胞毒性药物或化合物所组成。所述强效细胞毒性化合物(或高效活性药物成分,hpapi)定义为以8小时时间加权平均值具有等于或低于10μg/m3的空气的职业接触限值(oel)及具有高选择性及/或于低剂量可能导致癌症、突变、发育影响或生殖毒性的药理活性成分或中间体的剂。
[0165]
例示性药物描述如下。所述药物为微管蛋白抑制剂,结合下列微管蛋白结合位点的一者:美登素、长春花、紫衫烷、秋水仙碱、pironetin及laulimalide/peloruside。式(1)
的组合物可包括为微管蛋白抑制剂的药物。或者,所述药物选自由微管蛋白抑制剂及其等衍生物,包括微管脱稳剂:美登素及其衍生物、海兔素-10及其衍生物、念珠藻素-1、念珠藻素-52、tubulysins d、hemiasterlin及其衍生物(hti-286)、秋水仙碱及ca4,以及微管稳定剂:紫杉醇、圆皮海绵内酯、根薯酮内酯a及b及其等衍生物(根薯酮内酯af及根薯酮内酯aj)及根薯酮内酯ai环氧化物、laulimalide及埃博霉素a及b所组成的群组:
[0166][0167]
式(1)的化合物,其中,药物为美登素类、微管蛋白抑制剂,其结合美登素微管蛋白结合位点。
[0168]
式(1)的化合物,其中,药物选自由美登素类所组成的群组:
[0169]
安丝菌素
[0170][0171]
美登醇
[0172][0173]
dm1-sme,其中,r=ch2ch2ssme
[0174][0175]
dm3-sme,其中,r=ch2ch2ch(ch3)ssme
[0176][0177]
dm4-sme,其中,r=ch2ch2c(ch3)2ssme
[0178][0179]
mertansine(dm1),其中,r=ch2ch2sh
[0180][0181]
dm3,其中,r=ch2ch2ch(ch3)sh
[0182][0183]
dm4,其中,r=ch2ch2c(ch3)2sh
[0184][0185]
及其等类似物及衍生物。
[0186]
药物为结合rna聚合酶ii的鹅膏毒素。
[0187]
式(1)的化合物,其中,药物为鹅膏毒素。鹅膏毒素的序列为ile-trp-gly-ile-gly-cys-asn-pro(seq id no:11),于trp及cys之间具有经由亚砜(s=o)部分的交联。
[0188]
式(1)的化合物,其中,药物选自由α-鹅膏蕈碱、β-鹅膏蕈碱、γ-鹅膏蕈碱、ε-鹅膏蕈碱、amanullin、amanullic acid、amaniamide、amanin及proamanullin所组成的群组:
[0189][0190]
其中,α-鹅膏蕈碱(r1=oh;r2=oh;r3=nh2;r4=oh;r5=oh)、β-鹅膏蕈碱(r1=oh;r2=oh;r3=oh;r4=oh;r5=oh)、γ-鹅膏蕈碱(r1=oh;r2=h;r3=nh2;r4=oh;r5=oh)、ε-鹅膏蕈碱(r1=oh;r2=h;r3=oh;r4=oh;r5=oh)、amanullin(r1=h;r2=h;r3=nh2;r4=oh;r5=oh)、amanullinicacid(r1=h;r2=h;r3=oh;r4=oh;r5=oh)、amaninamide(r1=oh;r2=oh;r3=nh2;r4=h;r5=oh)、amanin(r1=oh;r2=oh;r3=oh;r4=h;r5=oh)及proamanullin(r1=h;r2=h;r3=nh2;r4=oh;r5=h)。
[0191]
式(1)的化合物,其中,药物为α-鹅膏蕈碱:
[0192][0193]
及其类似物及衍生物。
[0194]
药物为结合dna的小沟且导致链断裂的dna损伤剂。
[0195]
式(1)的化合物,其中,药物为烯二炔抗生素。
[0196]
式(1)的化合物,其中,药物为加利车霉素:
[0197][0198]
及其类似物及衍生物。
[0199]
药物为结合拓扑异构酶i的dna损伤剂。
[0200]
式(1)的化合物,其中,药物选自由喜树碱化合物所组成的群组:
[0201][0202]
式(1)的化合物,其中,药物为伊沙替康(dx-8951f):
[0203]
[0204]
及其类似物或衍生物。
[0205]
实施例2:介导的肿瘤靶向及细胞毒性微管蛋白抑制剂、mertansine、微管蛋白的美登素位点的抑制剂的群组的细胞质递送
[0206]
将mertansine(dm1;cas编号:139504-50-0)经由可裂解s-s连接接合至肽膜插入端(图19a)。研究中使用的肽:(seq id no:3)通过固相合成进行制备。首先,phlip-cys与aldrithiol
tm-2(2,2'-二吡啶二硫,cas编号2127-03-9)接合,以得到phlip-s-s-吡啶。phlip-s-s-吡啶使用反相hplc进行纯化(梯度:水及具有0.05%三氟乙酸(tfa)的乙腈)。接着,将mertansine与phlip-s-s-吡啶接合以交换s-s键并得到phlip-s-s-mertansine,之后通过rp-hplc纯化(梯度:水及具有0.05%三氟乙酸(tfa)的乙腈)及冷冻干燥。所述构建体的纯度及特性分别通过分析rp-hplc及表面增强激光解吸电离飞行时间质谱(seldi-tof)质谱所建立。构建体浓度以肽于280nm的吸光度,自mertansine的吸光度归一化进行计算(参见图19a)。
[0207]
将溶解于pbs中的phlip-s-s-mertansine以多次腹膜内(ip)或肿瘤内(it)注射(3至4天中一次)给予雌性无胸腺裸鼠的侧腹携带mda-mb-231人类三阴性乳癌或hela宫颈肿瘤的小鼠。phlip-s-s-mertansine的多次ip注射的总合并剂量为~10mg/kg,且phlip-s-s-mertansine的多次it注射的总合并剂量为~2.5mg/kg。当对照(未经治疗)组中的肿瘤达到约1cm3(约1g)的尺寸时,牺牲动物;收集(图19b)并将肿瘤秤重(图19c)。
[0208]
于phlip-s-s-mertansine的ip施用后观察到约60%的肿瘤重量减少,且于phlip-s-s-mertansine的it施用后观察后大于90%的肿瘤重量减少。于一些情况中,肿瘤于所述构建体的it施用后消失。
[0209]
于个别的实验中进行it注射,肿瘤消失且小鼠留置数周。于这些例子的60%中,没有观察到肿瘤再生长,且于ip或it治疗的过程中没有观察到毒性的征兆。
[0210]
实施例3:介导的肿瘤靶向及细胞毒性rna聚合酶抑制剂、α-鹅膏蕈碱、鹅膏毒素的群组的细胞质递送
[0211]
将α-鹅膏蕈碱经由可裂解s-s连接接合至肽膜插入端。研究中使用的所述肽:(seq id no:3)于固相合成中制备。为制备phlip-s-s-鹅膏蕈碱,将α-鹅膏蕈碱接合至3-(2-吡啶基二硫代)丙酸琥珀酰亚胺酯(spdp)交联剂,接着使用rp-hplc(梯度:水及具有0.05%三氟乙酸(tfa)的乙腈)进行纯化,且spdp-鹅膏蕈碱接合至肽的c末端半胱氨酸(上述序列中的粗体及底线)残基。phlip-s-s-鹅膏蕈碱的反应及纯化过程使用rp-hplc(梯度:水及具有0.05%三氟乙酸(tfa)的乙腈),接着冷冻干燥。所述构建体的纯度及特性分别通过分析rp-hplc及表面增强激光解吸电离飞行时间质谱(seldi-tof)质谱所建立。所述构建体的浓度以肽于3100nm的吸光度,其中,α-鹅膏蕈碱,ε
310
=13,000m-1
cm-1
。
[0212]
将溶解于pbs中的phlip-s-s-鹅膏蕈碱以多次腹膜内(ip)注射(3至4天中一次)给
予侧腹携带jc或4t1鼠乳腺肿瘤的雌性balb/c小鼠。phlip-s-s-鹅膏蕈碱的多次注射的总合并剂量为~0.3mg/kg。每次注射伴随于phlip-s-s-鹅膏蕈碱施用的30分钟前ip施用的葡萄糖溶液(200μl)的6.3mg/kg。当对照(未经治疗)组中的肿瘤达到约1cm3(约1g)的尺寸时,牺牲动物;收集并将肿瘤秤重。观察到约40至50%的肿瘤重量减少(图20a)。
[0213]
如鹅膏蕈碱的细胞毒性及高极性剂可能于局部应用中特别有效。例如,当所述构建体通过膀胱内滴注施用时,可使用phlip-s-s-鹅膏蕈碱治疗表面膀胱癌。于ph 7.4及ph 6.0的一组膀胱癌细胞系上进行测试phlip-s-s-鹅膏蕈碱以建立比率,此证明构建体性能于不同ph下的差异,且其可以解释为治疗指数。
[0214]
将癌细胞装载于96孔板的孔中(5,000细胞/孔)并培育过夜。以不含fbs的培养基替换标准生长培养基,ph 6.0或7.4,含有增加含量的phlip-s-s-鹅膏蕈碱(自0至2.0μm)。ph 6.0的培养基通过于1l的去离子水中混合13.3g的干dmem进行制备。与phlip-s-s-鹅膏蕈碱培育两小时后,移除构建体且替换为标准生长培养基。单独使用鹅膏蕈碱以高达2μm的浓度治疗两小时并未诱导细胞死亡。于48小时后使用通过490nm的吸收测量的比色法celltiter 96aq
ueous one solution cell proliferation assay以评估细胞活力(图20b呈现于scaber鳞状细胞癌上所得数据的实施例)。
[0215]
于没有构建体的情况下,使用培养基培养的细胞的活力于ph 7.4及ph 6.0没有差异;因此,ph的作用排除于考良之外。治疗指数(ti)根据公式进行计算:
[0216][0217]
下列表8含有ph 7.4及ph 6.0的治疗所得ec
50
值以及所计算的治疗指数,其清楚表示phlip-s-s-鹅膏蕈碱的ph依赖性细胞作用。不同细胞系的比率(ti)自3.5至8不等。phlip-s-s-鹅膏蕈碱辨别具有正常细胞表面ph(ph 7.4)的健康细胞,结合且插入具有低细胞表面ph(ph 5.5至6.5)的癌细胞的细胞膜。
[0218]
表8:ph 7.4及ph 6.0的膀胱癌细胞系使用phlip-s-s-鹅膏蕈碱的治疗所计算以nm为单位的ec
50
值
[0219] ec
50ph6.0
ec
50ph7.4
tiht-1197细胞:膀胱癌193.8804.84.25637细胞:第ii级上皮细胞癌89.9378.24.2ht-1376细胞:第iii级上皮细胞癌766.73002.33.9scaber细胞:鱗狀細胞癌127.81017.08.0j82细胞:移行细胞癌83.7549.16.6um-uc-3细胞:移行细胞癌446.22143.04.8sw780细胞:移行细胞癌144.9887.86.1t-24细胞:移行细胞癌163.0575.43.5tccsur细胞:iv移行细胞癌72.1391.35.4rt4细胞:移行细胞乳头状瘤107.3473.34.4
[0220]
实施例4:介导的肿瘤靶向及细胞毒性dna损伤化合物、加利车霉素、烯二
炔抗生素的群组的细胞质递送
[0221]
以spdp修饰的加利车霉素由cfm,gmbh所合成且纯化(图21a)。使用加利车霉素-spdp与肽的膜插入端的cys残基接合,以得到phlip-s-s-加利车霉素。研究中使用的所述肽:addqnpwrayldllfptdtllldllwca(seq id no:3)于固相合成中制备。使用rp-hplc(梯度:水及具有0.05%三氟乙酸(tfa)的乙腈)进行phlip-s-s-加利车霉素的纯化,接着冷冻干燥。所述构建体的纯度及特性分别通过分析rp-hplc及表面增强激光解吸电离飞行时间质谱(seldi-tof)质谱所建立。所述构建体的浓度通过肽于280nm的吸光度及加利车霉素的吸光度校正进行计算。
[0222]
将溶解于pbs中的phlip-s-s-加利车霉素以多次腹膜内(ip)或肿瘤内(it)注射(3至4天中一次)给予雌性无胸腺裸鼠的侧腹携带hela人类宫颈肿瘤的小鼠。
[0223]
phlip-s-s-加利车霉素的多次注射的总合并剂量为~1.5mg/kg。当对照(未经治疗)组中的肿瘤达到约0.5g的尺寸时,牺牲动物;收集(图21b)并将肿瘤秤重(图21c)。
[0224]
phlip-s-s-加利车霉素的ip施用后观察到约88%的肿瘤重量减少。
[0225]
实施例5:介导的肿瘤靶向及细胞毒性dna损伤化合物、拓扑异构酶i抑制剂、依沙替康、喜树碱化合物的群组的细胞质递送
[0226]
以spdp修饰的依沙替康由cfm,gmbh所合成且纯化(图22)。使用依沙替康-spdp与肽的膜插入端的cys残基接合,以得到phlip-s-s-依沙替康。研究中使用的所述肽:addqnpwrayldllfptdtllldllwca(seq id no:3)于固相合成中制备。使用rp-hplc(梯度:水及具有0.05%三氟乙酸(tfa)的乙腈)进行phlip-s-s-依沙替康的纯化,接着冷冻干燥。所述构建体的纯度及特性分别通过分析rp-hplc及表面增强激光解吸电离飞行时间质谱(seldi-tof)质谱所建立。所述构建体的浓度通过肽于280nm的吸光度及依沙替康的吸光度校正进行计算。
[0227]
将溶解于pbs中的phlip-s-s-依沙替康以多次腹膜内(ip)注射(3至4天中一次)给予雌性无胸腺裸鼠的侧腹携带hela人类宫颈肿瘤的小鼠。当对照(未经治疗)组中的肿瘤达到约1cm3(约1g)的尺寸时,牺牲动物;收集并将肿瘤秤重。
[0228]
phlip-s-s-依沙替康的ip施用后观察到约50至60%的肿瘤重量减少,且于phlip-s-s-依沙替康的it施用后观察到大于70%的肿瘤重量减少。
[0229]
实施例6
[0230]
于方面中,本文提供一种包括强效细胞毒性化合物及肽的组合物。举例而言,所述细胞毒性化合物为细胞毒性微管蛋白抑制剂化合物,其抑制微管蛋白的聚合及微管结构的脱稳。于其它实施例中,所述细胞毒性微管蛋白抑制剂化合物结合美登素位点。
[0231]
于实施例中,所述细胞毒性微管蛋白抑制剂化合物包括美登素、安丝菌素、美登醇、d-丙氨酰基美登素或具有二硫或硫醇基团的美登素类似物及其等衍生物。举例而言,所述细胞毒性微管蛋白抑制剂化合物包括mertansine或其衍生物。
[0232]
于实施例中,所述细胞毒性化合物为细胞毒性rna聚合酶抑制剂。举例而言,所述
细胞毒性rna聚合酶抑制剂化合物为鹅膏毒素。于其它实施例中,所述细胞毒性rna聚合酶抑制剂化合物为α-鹅膏蕈碱或其衍生物。
[0233]
于实施例中,所述细胞毒性化合物为细胞毒性dna损伤化合物。举例而言,所述细胞毒性dna损伤化合物为烯二炔抗生素。于其它实施例中,所述细胞毒性dna损伤化合物为加利车霉素或其衍生物。于实施例中,所述细胞毒性dna损伤化合物为拓扑异构酶i抑制剂。举例而言,所述细胞毒性拓扑异构酶i抑制剂化合物为喜树碱化合物。于实施例中,所述细胞毒性拓扑异构酶i抑制剂化合物为依沙替康或其衍生物。
[0234]
于实施例中,所述细胞毒性化合物包括肿瘤的受限靶向。
[0235]
于实施例中,本文所述的组合物还包括位于所述细胞毒性化合物及肽之间的连接子。例如,所述连接子包括二硫键或酸不稳定键。于实施例中,所述连接子为可裂解的。于其它实施例中,所述连接子为不可裂解的。于实施例中,所述连接子为自我牺牲的。
[0236]
于实施例中,本文所述的组合物还包括极性调节剂。
[0237]
于实施例中,本文亦提供一种包括肽的组合物,其中,所述肽具有序列addqnpwrayldllfptdtllldllwxa(seq id no:1)或adqdnpwrayldllfptdtllldllwxa(seq id no:2),其中,大写“x”表示任何氨基酸且可包括赖氨酸(lys)、半胱氨酸(cys)或含叠氮氨基酸。
[0238]
于实施例中,所述组合物具有下列结构:肽
–
连接
–
b,
[0239]
其中,“肽”为包括序列addqnpwrayldllfptdtllldllwxa(seq id no:1)或adqdnpwrayldllfptdtllldllwxa(seq id no:2)的第一肽,“b”为包括序列addqnpwrayldllfptdtllldllwxa(seq id no:1)或adqdnpwrayldllfptdtllldllwxa(seq id no:2)的第二肽,其中,大写“x”表示任何氨基酸残基且可包括赖氨酸(lys)、半胱氨酸(cys)或含叠氮氨基酸;“连接”为聚乙二醇连接子,以及每个
“–”
为共价键。
[0240]
于方面中,本文提供一种于酸性病变组织中杀伤细胞的方法,包括向受试者施用包括细胞毒性化合物及肽的组合物。
[0241]
于其它实施例中,所述方法包括所述受试者具有实体肿瘤。举例而言,于实施例中,所述受试者具有膀胱肿瘤。
[0242]
于实施例中,所述组合物直接施用至肿瘤肿块中。于实施例中,所述组合物灌注至膀胱。
[0243]
于实施例中,所述组合物为局部应用。于其它实施例中,所述组合物为全身施用。于其它实施例中,所述微管蛋白抑制剂化合物递送至癌细胞的胞质溶胶中。于实施例中,所述细胞毒性化合物递送至衰老细胞的胞质溶胶中。
[0244]
于实施例中,所述细胞毒性化合物靶向酸性组织,以主要于靶向组织内诱导生物效应。
[0245]
于实施例中,所述细胞毒性化合物细胞内递送,以诱导生物效应。于其它实施例中,所述细胞毒性化合物于所述不存在下具有受限的肿瘤靶向能力。
[0246]
于实施例中,相较于健康组织,所述组合物优先将所述细胞毒性化合物靶向病变
组织,从而最小化所述健康组织的损伤。
[0247]
一般定义
[0248]
除非另外明确定义,本文中所用的所有技术及科学术语应视为与本领域技术人员(例如,细胞培养、分子遗传学和生物化学)所一般理解的意义相同。
[0249]
如本文中所用,术语“约”于数值或范围的内容中意指所记载或请求的数值或范围的
±
10%,除非内容中要求更限定的范围。
[0250]
于上述说明及权利要求书中,例如“至少一者”或“一或更多”的短语出现后有元素或特征的连接列表。术语“及/或”自可出现于两种或更多元素或特征的列表中。除非与其所使用的内容另外隐含或明确矛盾,所述短语意指任何单独列出的元素或特征,或与任何所述元素或特征与任何其它所述元素或特征的组合。举例来说,短语“a及b的至少一者”、“a及b的一者或更多”及“a及/或b”各自意指“a单独、b单独或a及b一起”。类似的解释亦适用于包含三个或更多项目的列表。例如,短语“a、b及c的至少一者”、“a、b及c的一者或更多”及“a、b及/或c”各自意指“a单独、b单独、c单独、a及b一起、a及c一起、b及c一起或a和b及c一起”。此外,于上述及权利要求书所用术语“基于”意指“至少部分基于”,使未记载的特征或元素亦为允许的。
[0251]
应理解,提供参数范围下,本发明亦提供所述范围内的所有整数及其十分之一。举例而言,“0.2至5mg”为0.2mg、0.3mg、0.4mg、0.5mg、0.6mg等,直到且包括5.0mg。
[0252]
小分子为质量小于2000道尔顿的化合物。所述小分子的分子量优选小于1000道尔顿,更优选小于600道尔顿,例如,所述化合物小于500道尔顿、400道尔顿、300道尔顿、200道尔顿或100道尔顿。
[0253]
如本文中所用,“分离的”或“纯化的”核酸分子、聚核苷酸、聚肽或蛋白质实质上不具有其它细胞物质,或通过重组技术生产的培养基,或化学合成的化学前驱物或其他化学物。纯化的化合物为目标化合物的重量(干燥重量)的至少60%。优选地,制剂为目标化合物的重量的至少75%,更优选至少90%,最优选至少99%。举例而言,纯化的化合物为所需化合物的重量的至少90%、91%、92%、93%、94%、95%、98%、99%或100%(w/w)。通过任何合适的标准方法测量纯度,例如,通过柱色谱、薄层色谱或高效液相色谱(hplc)分析。纯化或分离的多核苷酸(核糖核酸(rna)或脱氧核糖核酸(dna))或不含氨基酸序列的多肽,或于其天然存在状态下位于其侧翼的核酸序列。纯化亦定义施用至人类受试者为安全的无菌程度,例如,缺乏传染性或毒性剂。纯化或分离的多核苷酸(核糖核酸(rna)或脱氧核糖核酸(dna))于其天然存在的状态下不含位于其侧翼的基因或序列。纯化或分离的多肽于其天然存在的状态下不含位于其侧翼的氨基酸或序列。
[0254]
类似地,“实质上纯的”是指已经与天然伴随的组分分离的核苷酸或多肽。通常,当核苷酸和多肽以重量计为至少60%、70%、80%、90%、95%或甚至99%时,其等实质上为纯的,不含蛋白质以及与其等自然结合的天然存在的有机分子。
[0255]
与“包含”、“含有”或“特征在于”同义的过渡性术语“包括”是包括性或开放式,且不排除附加的、未记载的元素或方法步骤。相较于此,过渡性短语“由
……
组成”排除权利要求的范围中未指定的任何元素、步骤或成分。过渡短语“基本上由
……
组成”将权利要求的范围限制为特定的材料或步骤“以及不会实质性影响所请求保护的发明的基本及新颖特征者”。
[0256]
本文所用术语“受试者”、“患者”、“受试者”等的目的并非限制性,且通常可以互换使用。换言之,描述为“患者”的受试者不一定具有特定疾病,而可能仅为寻求医疗建议。
[0257]
如本文所用,除非上下文另有明确规定,否则单数形式“一”及“所述”包括复数的提及。因此,例如,对“疾病”、“疾病状态”或“核酸”的提及是对一个或多个所述实施例的提及,且包括本领域技术人员已知的其等同物等等。
[0258]
如本文中所用,“治疗”包括例如疾病进展的抑制、复原或停滞。治疗亦包括预防或改善所述疾病的任何症状。如本文所用,“抑制”受试者中的疾病进展或疾病并发症是指预防或减少受试者中的疾病进展及/或疾病并发症。
[0259]
如本文中所用,与疾病相关的“症状”包括与疾病相关的任何临床或实验室表现,且不限于受试者所能感觉到或观察到者。
[0260]
如本文中所用,当指治疗化合物的量时,“有效”是指当以本公开的方式使用时,化合物的量足以产生所需的治疗反应而不具有与合理的效益/风险比相称的过度不良副作用(例如毒性、刺激和过敏反应)。
[0261]
如本文中所用,“药学上可接受的”载体或赋形剂是指适用于人类及/或动物而不具有与合理的效益/风险比相称的过度不良副作用(例如毒性、刺激和过敏反应)。其可为例如药学上可接受的溶剂、悬浮剂或媒介物,用于将本发明化合物递送至所述受试者。
[0262]
以下提供实例以加速更完整的理解本发明。以下实施例说明制备和实施本发明的例示性方式。然而,本发明的范围不限于这些实施例中公开的具体实施例,这些实施例仅为说明的目的,因为可以使用替代方法得到相似结果。
[0263]
通过于比较窗中内比较两个最佳比对的序列确定“序列同一性百分比”,其中,与参考序列(不包括添加或删除)相比,于比较窗口中的多核苷酸或多肽序列部分可包含添加或删除(例如,空位),以实现两个序列的最佳比对。通过确定两个序列中出现相同核酸碱基或氨基酸残基的位置的数量,得到匹配位置的数量,将匹配位置的数量除以比较窗口中的位置总数,并将结果乘以100以得到序列同一性的百分比。
[0264]
在两个或更多核酸或多肽序列的内容中,术语“相同”或“同一性”百分比是指当使用序列比较算法或通过手动比对和目视检查,于比较窗口或指定区域上进行比较和比对以获得最大对应时,两个或更多相同或具有特定百分比的氨基酸残基或核苷酸的序列或子序列为相同(例如,于指定区域,例如,整个多肽序列或其单结构域的50%、55%、60%、65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或更多的同一性)。至少约80%相同的序列称为“实质上相同”。在一些实施例中,两个序列是100%相同的。在特定实施例中,两个序列于序列的一者的整个长度上为100%相同的(例如,于序列具有不同长度的情况下,两个序列中较短的一者)。在各种实施例中,同一性可指测试序列的互补。于一些实施例中,同一性存在于至少约10至约100、约20至约75、约30至约50个氨基酸或核苷酸长度的区域。在特定实施例中,同一性存在于至少约50个氨基酸长度的区域,或更优选地为100至500、100至200、150至200、175至200、175至225、175至250、200至225、200至250个或更多个氨基酸的长度的区域。
[0265]
对于序列比较,通常一个序列做为参考序列,与测试序列进行比较。于各种实施例中,当使用序列比较算法时,将测试和参考序列输入电脑,如果需要,指定子序列坐标,并且指定序列算法程式参数。优选地,可以使用预设程式参数,或者可以指定替代参数。接着,序
列比较算法根据程式参数计算测试序列相对于参考序列的序列同一性百分比。
[0266]“比较窗口”是指多个连续位置中的任何一个的片段(例如,至少约10至约100、约20至约75、约30至约50、100至500、100至200、150至200、175至200、175至225、175至250、200至225、200至250),其中,于最佳比对两个序列后,可以将序列与具有相同数量连续位置的参考序列进行比较。于各种实施例中,比较窗口为两个比对序列的一者或两者的全长。于一些实施例中,所比较的两个序列包含不同的长度,且比较窗口是两个序列中较长或较短序列的全长。用于比较的序列比对方法为本领域中已知的。用于比较的序列的最佳比对可通过,例如,通过smith&waterman,adv.appl.math.2:482(1981)的局部同源性算法;通过needleman&wunsch,j.mol.biol.48:443(1970)的同源性比对算法;通过pearson&lipman,proc.nat'l.acad.sci.usa85:2444(1988)的相似方法的寻找;通过这些算法的计算机化实现(gap,bestfit,fasta及tfasta in the wisconsin genetics software package,genetics computer group,575science dr.,madison,wis.),或通过手动比对和目视检查(参见,例如,current protocols in molecular biology(ausubel等人编辑,1995增刊))进行。
[0267]
于各种实施例中,适合确定序列同一性百分比及序列相似度的算法为blast及blast 2.0算法,其分别描述于altschul等人,nuc.acids res.25:3389-3402(1977)及altschul等人,j.mol.biol.215:403-410(1990)。可以使用blast及blast 2.0与本文所述的参数以确定核酸和蛋白质的序列同一性百分比。如本领域已知,执行blast分析的软件可通过国家生物技术信息中心(national center for biotechnology information)公开取得。此算法首先通过识别查询序列中长度为w的短词以识别高得分序列对(hsp),当与数据库序列中相同长度的词对齐时,这些词匹配或满足一些正值阈值分数t。t被称为邻域词得分阈值(altschul等人,supra)。这些最初的邻域词命中作为启动搜索以找到包含其等的更长hsp的种子。单词命中沿着每个序列在两个方向上扩展,只要累积对齐分数可以增加。对于核苷酸序列,使用参数m(一对匹配残基的奖励分数;总是》0)及n(错配残基的惩罚分数;总是《0)计算累积分数。对于氨基酸序列,使用分数矩阵计算累积分数。在以下情况下,单词命中于每个方向的扩展将停止:累积对齐分数从其最大实现值下降数量x;由于一个或多个负分数残基比对的累积,累积分数变为零或更低;或到达任一序列的末端。blast算法参数w、t及x确定比对的敏感度及速度。blastn程式(针对核苷酸序列)使用预设字长(w)为11,期望值(e)为10,m=5,n=-4以及两条链的比较。对于氨基酸序列,blastp程式使用预设字长为3、期望值(e)为10以及blosum62评分矩阵(参见henikoff&henikoff,proc.natl.acad.sci.usa 89:10915(1989))、比对(b)为50,期望值(e)为10,m=5,n=-4以及两条链的比较。
[0268]
其它实施例
[0269]
虽然本发明已经结合其详细说明进行描述,但前述说明的目的在于说明而非限制本发明的范围,本发明的范围由所附权利要求书的范围所限定。其它方面、优点及修饰落于以下权利要求的范围内。
[0270]
于本文提及的专利及科学文献确立本领域技术人员可取得的知识。本文中引用的所有美国专利及公开或未公开的美国专利申请均通过引用并入本文中。本文中引用的所有已公开的外国专利和专利申请均通过引用并入本文中。由本文引用的登录号所表示的
genbank及ncbi提交物通过引用并入本文中。本文中引用的所有其它已公开的参考文献、文件、原稿及科学文献均通过引用并入本文中。
[0271]
虽然本发明已经具体展示及描述其优选实施例,但本领域技术人员将理解,于不脱离所附的权利要求书所涵盖的本发明的范围下,可以于形式及细节进行各种改变。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
