一种lncrna tug1靶向调控hsa-mir-4638-3p的方法
技术领域
1.本发明涉及生物技术领域,具体涉及一种lncrna tug1靶向调控hsa-mir-4638-3p的方法。
背景技术:
2.目前人类平均寿命逐年增加,但癌症对人类的威胁日益突出,成为危害人类生命安全的重要疾病之一。随着医疗技术的提高、抗肿瘤药物研究的不断发展及肿瘤综合诊治水平的提高,多种肿瘤以慢性病模式长期存在,肿瘤患者生存期延长的同时,抗肿瘤治疗所引起的心血管毒性也与日俱增,尤以蒽环类药物为代表。从长期来看,心血管疾病导致的死亡风险超过了多种肿瘤复发的风险;高龄、合并多种疾病以及需要长期或强化治疗的患者,心血管的毒性风险增加。
3.lncrna是一类长度大于200bp且不编码蛋白质的rna分子,由于功能和种类的不明确,长期以来被认为是不具蛋白编码能力的无功能序列而未受到重视。然而,近年来的研究发现,lncrna能够在转录、转录后和翻译等水平调控基因表达从而参与细胞的发育、分化、生长及凋亡等生物学过程。并且,越来越多的研究表明lncrna在心脏发育和功能维持中具有重要作用。
4.牛磺酸调节基因1(taurine up-regμlated gene 1,tug1)是一种在小鼠视网膜中发现的lncrna,定位在22q12.2染色体上,参与血管生成、细胞耐药等过程,还与人体肿瘤发生有关在人体正常细胞功能维持以及疾病发生过程中发挥关键作用。tug1可调节细胞凋亡,抑制某些细胞系统的凋亡,如β细胞、膀胱癌细胞和肝细胞癌,但在其他细胞如晶状体上皮细胞、神经元和胶质瘤中促进凋亡。但在受到炎症刺激的h9c2心肌细胞中,tug1抑制细胞凋亡和炎症反应,而在缺氧条件下,tug1促进细胞凋亡,加重缺氧所致的损伤,提示tug 1的双重作用与细胞类型和应激刺激有关。那么tug1是否也可以通过作用于心肌细胞中或蒽环类药物心肌损伤中的特定mirnas来调节细胞死亡,tug1参与蒽环类药物心血管毒性的具体分子生物学机制仍有待进一步深入研究。
技术实现要素:
5.本发明的目的是提供一种lncrna tug1靶向调控hsa-mir-4638-3p的方法,该方法确定了lncrna tug1和hsa-mir-4638-3p的靶向调控关系,为进一步研究它们在蒽环类药物心脏毒性作用及分子机制奠定了实验基础。
6.为了实现上述目的,本发明的技术方案具体如下:
7.本发明提供一种lncrna tug1靶向调控hsa-mir-4638-3p的方法,包括以下步骤:
8.步骤1、野生型tug1双报告载体构建;
9.或者突变预测hsa-mir-4638-3p第一结合位点载体构建;
10.或者突变预测hsa-mir-4638-3p第二结合位点载体构建;
11.或者双突变预测hsa-mir-4638-3p结合位点载体构建;
12.第一结合位点的序列为:tgtccagg;
13.第一结合位点突变后的序列为:gagttcaa;
14.第二结合位点的序列为:tgtccag;
15.第二结合位点突变后的序列为:gagttca;
16.步骤2、microrna与质粒共转染细胞
17.采用阳离子脂质体法进行细胞转染;
18.步骤3、lucifersae检测
19.对步骤2获得的样品进行luciferase活性检测。
20.在上述技术方案中,步骤1中野生型tug1双报告载体构建的步骤如下:
21.1、tug1序列;
22.第一结合位点的序列为:tgtccagg;
23.第二结合位点的序列为:tgtccag;
24.2、扩增引物序列:
25.tug1-xhoif:
26.5'ccgctcgagtggtgctcaccaagtggtacagccctaagc 3';
27.tug1-notir:
28.5'ataagaatgcggccgcgaagaaatgaaatgcacactcatggaac 3';
29.3、pcr钓取基因:
30.1》模板cdna制备
31.1、总rna抽提
32.1)离心收集人的293t细胞,加入1mltrizol溶液,吹打混匀,使的细胞充分裂解,静置5min;
33.2)加入200μl氯仿,剧烈振荡混匀30s,使水相和有机相充分接触,室温静置15min;
34.3)4℃下,10000g离心15min,将上层水相中的rna移至另一个新的rnase free ep管;
35.4)沉淀rna:加入0.5ml异丙醇,混匀,室温静置10min;
36.5)4℃下,10000g离心10min,收集rna沉淀,去上清;
37.6)用75%乙醇洗涤两次,超净台风干;
38.7)加入15~50μldepc水溶解沉淀;
39.2、总rna纯度和完整性检测
40.1)纯度检测:取1μlrna样品50倍稀释,测定od值,od260/od280的比值大于1.8,说明制备的rna较纯,无蛋白质污染;
41.2)总rna完整性检测:取rna样品1μl,1%琼脂糖凝胶电泳80v
×
20min,eb染色10min后用凝胶成像系统观察总rna的5s rrna,18s rrna和28s rrna条带,三条条带完整的话即可证明总rna抽提比较完整;
42.3、逆转录
43.1)在rnase free的pcr管中配置下列溶液:total rna0.75μg,加水至总体积为12μl;
44.2)将上述溶液吹打均匀,置65℃保温5min,使rna变性;随后立即冰上致冷,以防止
rna复性;
45.3)在该pcr管中加入下列试剂:oligo 0.5μl,random primer 0.5μl,10mm dntp 2.0μl,rnase inhibitor 0.5μl,5x buffer 4.0μl,m-mlv 0.5μl;
46.4)将上述20μl反应溶液30℃保温10min;
47.5)42℃保温60min;
48.6)72℃保温10min;
49.7)放置零下20
°
保存;
50.2》pcr反应体系
51.在0.2ml ep管中配制以下体系,模板原液稀释20倍后取0.5μl扩增tug1:
52.2mm dntp mixture 2.5μl,10
×
kod buffer 2.5μl,25mm mgso41.5μl,模板0.5μl,引物tug1 f 0.3μl,引物tug1 r 0.3μl,kod plus neo 0.3μl,ddh2o 17.1μl;
53.3》扩增条件
54.混匀后扩增,tug1基因的扩增条件:94℃、1min,98℃、15sec,58℃、15sec,68℃、1min,68℃、5min,30个循环;16℃保存;
55.4》pcr产物回收
56.8)pcr产物经1%凝胶电泳后,在紫外灯下,用手术刀切取含目的基因片段的凝胶条带至干净1.5ml ep管中,称重后,按100mg凝胶对应100μl溶液bd的比例向离心管中加入溶液bd;
57.9)60℃水浴10min至凝胶完全溶化,水浴期间振荡混合3次;
58.10)将溶液转移至dna纯化柱中,静置2min,室温12000rpm离心1min,弃滤液;
59.11)向柱上加入500μl溶液pe,室温12000rpm离心1min,弃滤液;
60.12)重复上一操作一次;
61.13)室温空柱12000rpm离心1min以彻底去除纯化柱中残留的液体;
62.14)将柱子置于新的1.5ml ep管上,向柱中央加入30μl 60℃预热的无菌水,13400g离心1min以洗脱出dna;
63.4、pcr回收产物及载体双酶切
64.在2个无菌的0.2ml ep反应管,分别取tug1 pcr回收产物和psicheck-2载体各15μl,分别用xhoi/noti双酶切,酶切体系如下:xhoi 1.5μl,noti 1.5μl,10
×
buffer 5μl,ddh2o 27μl;混匀后,37℃反应2.5-3.5h;
65.5、酶切产物回收
66.1)酶切产物经1%凝胶电泳后,在紫外灯下,用手术刀分别切取含目的片段和载体的凝胶条带至干净的1.5ml ep管中,按100mg凝胶对应100μl溶液bd的比例向离心管中加入溶液bd;
67.2)60℃水浴10min至凝胶完全溶化,水浴期间振荡混合3次;
68.3)将溶液转移至dna纯化柱中,静置2min,室温12000rpm离心1min,弃滤液;
69.4)向柱上加入500μl溶液pe,室温12000rpm离心1min,弃滤液;
70.5)重复上一操作一次;
71.6)空柱12000rpm离心1min以彻底去除纯化柱中残留的液体;
72.7)将柱子置于新的1.5ml ep管上,向柱中央加入30μl 60℃预热的无菌水,13400g
离心1min以洗脱出dna;
73.6、目段片段与载体连接:
74.向0.2ml ep管中加入以下试剂:酶切回收的tug1 pcr产物3μl,酶切回收的psicheck-2载体2μl,10
×
ligase buffer 1μl,t4 dna ligase 1μl,ddh2o 3μl;16℃连接1h;
75.7、连接产物的转化
76.1)冰浴中将5μl连接产物分别加入至50μl dh5α感受态细胞中,旋转混匀,冰浴30min;
77.2)42℃水浴热休克90s;
78.3)将管转移至冰浴中,冰浴2min;
79.4)分别加入200μl lb培养基,混匀,37℃、200rpm振荡培养1h;
80.5)于超净工作台中,将菌液均匀涂布于含氨苄青霉素的lb平板上,室温下放置,至液体吸收;
81.6)倒置平板,转移入37℃生化培养箱过夜培养;
82.8、质粒酶切鉴定阳性克隆
83.1》从平板上挑取若干单克隆于3ml lb管中摇床过夜培养;
84.2》质粒提取
85.a)用1.5ml ep管收集3μl菌液,12000rpm离心1min,去上清;
86.b)加入250μl溶液i/rnasea混合液,重悬菌体;
87.c)加入250μl溶液ii,温和反复颠倒混匀6次,室温放置2min;
88.d)加入350μl溶液iii,温和反复颠倒混匀6次;
89.e)12000rpm离心10min,吸去上清至dna纯化柱中,静置2min;
90.f)12000rpm离心1min,弃滤液;
91.g)加入500μl溶液pb至柱中,12000rpm离心1min,弃滤液;
92.h)加入500μl溶液w至柱中,12000rpm离心1min,弃滤液,重复一次;
93.i)空柱12000rpm离心3min;
94.j)将柱取出置于新的1.5ml ep管中,加入50μl无菌水,静置2min,13400rpm离心1min以洗脱出质粒;
95.3》酶切鉴定提取质粒
96.酶切反应体系如下:提取的质粒tug13μl,xhoi0.4μl,noti 0.4μl,10
×
buffer 1μl,ddh2o 5.2μl;37℃酶切2h;酶切产物以含溴化乙锭的1%琼脂糖凝胶电泳分离,uvp凝胶成像系统成像;
97.溶液ⅰ:50mml葡萄糖/10mml edta/25mml tris-hcl,ph=8.0,即重悬液;
98.溶液ⅱ:0.2ml naoh/1%sds,即裂解液;
99.溶液ⅲ:3ml醋酸钾/2ml醋酸,即中和沉淀液。
100.在上述技术方案中,步骤1中突变预测hsa-mir-4638-3p第一结合位点载体构建的步骤如下:
101.1、tug1序列
102.第一结合位点的序列为:tgtccagg;
103.第一结合位点突变后的序列为:gagttcaa;
104.2、扩增引物序列:
105.muttug1-1f:
106.5'ggcaaaacatgagagagttcaaattggaggttgaaaagatttcactacagtgttctg 3';
107.muttug1-1r:
108.5'ttttcaacctccaatttgaactctctcatgttttgccacagaatttcaagctttgag 3';
109.3、基因突变:
110.1》pcr反应体系
111.在0.2ml pcr反应管中配置以下体系,模板为构建好的tug1质粒,质粒稀释50倍后取1μl做模板:
112.2mm dntp mixture 2.5μl,10
×
kod buffer 2.5μl,25mm mgso41.5μl,引物muttug1-1f 0.3μl,引物muttug1-1r 0.3μl,kod plus neo 0.3μl,ddh2o16.6μl;
113.2》扩增条件
114.混匀后扩增,muttug1-1基因的扩增条件:94℃、1min,98℃、15sec,58℃、15sec,68℃、4min30 sec,68℃、5min,20个循环;16℃保存;
115.3》pcr产物用dpni酶切
116.在pcr管中加入1μldpni酶,在37℃消化4h;
117.4》dpni处理的pcr产物转化,步骤同野生型tug1双报告载体构建的步骤7;
118.5》挑1个菌落提取质粒,步骤同野生型tug1双报告载体构建的步骤8;
119.6》送样测序。
120.在上述技术方案中,步骤1中突变预测hsa-mir-4638-3p第二结合位点载体构建的步骤如下:
121.1、tug1序列
122.第二结合位点的序列为:tgtccag;
123.第二结合位点突变后的序列为:gagttca;
124.2、扩增引物序列:
125.muttug1-2f:
126.5'gaagacctgagtttcgagttcaaccctcagtgcaaactgaggatgctccatccaaag 3';
127.muttug1-2r:
128.5'gcactgagggttgaactcgaaactcaggtcttctaattcccagtaacagtgctggtg 3';
129.3、基因突变:
130.1》pcr反应体系
131.在0.2ml pcr反应管中配置以下体系,模板为构建好的tug1质粒,质粒稀释50倍后取1μl做模板;
132.2mm dntp mixture 2.5μl,10
×
kod buffer 2.5μl,25mm mgso41.5μl,引物muttug1-2f 0.3μl,引物muttug1-2r 0.3μl,kod plus neo 0.3μl,ddh2o 16.6μl;
133.2》扩增条件
134.混匀后扩增,muttug1-2基因的扩增条件:94℃、1min,98℃、15sec,58℃、15sec,68℃、4min30 sec,68℃、5min,20个循环;16℃保存;
135.3》pcr产物用dpni酶切
136.在pcr管中加入1μldpni酶,在37℃消化4h;
137.4》dpni处理的pcr产物转化,步骤同野生型tug1双报告载体构建的步骤7;
138.5》挑1个菌落提取质粒,步骤同野生型tug1双报告载体构建的步骤8;
139.6》送样测序。
140.在上述技术方案中,双突变预测hsa-mir-4638-3p结合位点载体构建的步骤如下:
141.1、tug1序列;
142.第一结合位点的序列为:tgtccagg;
143.第一结合位点突变后的序列为:gagttcaa;
144.第二结合位点的序列为:tgtccag;
145.第二结合位点突变后的序列为:gagttca;
146.2、扩增引物序列:
147.muttug1-2f:
148.5'gaagacctgagtttcgagttcaaccctcagtgcaaactgaggatgctccatccaaag 3'
149.muttug1-2r:
150.5'gcactgagggttgaactcgaaactcaggtcttctaattcccagtaacagtgctggtg 3'
151.3、基因突变:
152.1》pcr反应体系
153.在0.2mlpcr反应管中配置以下体系,模板为突变好的muttug1-1质粒,质粒稀释50倍后取1μl做模板:
154.2mm dntp mixture 2.5μl,10
×
kod buffer 2.5μl,25mm mgso41.5μl,引物muttug1-2f 0.3μl,引物muttug1-2r 0.3μl,kod plus neo 0.3μl,ddh2o 16.6μl;
155.2》扩增条件
156.混匀后扩增,muttug1-3基因的扩增条件:94℃、1min,98℃、15sec,58℃、15sec,68℃、4min30 sec,68℃、5min,20个循环;16℃保存;
157.3》pcr产物用dpni酶切
158.在pcr管中加入1μldpni酶,在37℃消化4h;
159.4》dpni处理的pcr产物转化,步骤同野生型tug1双报告载体构建的步骤7;
160.5》挑1个菌落提取质粒,步骤同野生型tug1双报告载体构建的步骤8;
161.6》送样测序。
162.在上述技术方案中,步骤2具体包括以下步骤:
163.(1)转染前一天,细胞按2
×
104个/孔接种于24孔板上,培养基为含10%fbs的dmem高糖;
164.(2)转染当天,细胞汇合度约为50-60%,吸去旧的培养基,用pbs洗涤两次,然后每孔加入300μl opti-mem培养基,置于5%co2、37℃培养箱中;
165.(3)每个孔用opti-mem培养基稀释cellfectin ii reagent1μl,终体积为50μl,室温下静置5min;
166.(4)每个孔用加入20μm浓度的hsa-mir-4638-3p1μl或hsa-mir-4638-3p抑制剂和0.5μg质粒,再加入opti-mem至总体积50μl,室温下静置5min;
tug1和hsa-mir-4638-3p的靶向调控关系,为进一步研究它们在蒽环类药物心脏毒性作用及分子机制奠定了实验基础。本发明采用萤火虫/海肾双荧光素酶报告系统进行靶标验证,以确定lncrna tug1与hsa-mir-4638-3p基因存在的确切的靶标关系。
188.第一部分:野生型tug1双报告载体构建
189.1、tug1序列:
190.第一结合位点的序列为:tgtccagg;
191.第二结合位点的序列为:tgtccag;
192.预测结果参见图1。
193.2、扩增引物序列:(苏州上海生工合成page纯化2od):
194.tug1-xhoif:5'ccgctcgagtggtgctcaccaagtggtacagccctaagc 3';
195.tug1-notir:5'ataagaatgcggccgcgaagaaatgaaatgcacactcatggaac 3';
196.3、pcr钓取基因:
197.1》模板cdna制备
198.1、总rna抽提
199.1)离心收集人的293t细胞,加入1ml trizol(invitrogen)溶液,吹打混匀,使的细胞充分裂解,静置5min;
200.2)加入200μl氯仿,剧烈振荡混匀30s,使水相和有机相充分接触,室温静置15min;
201.3)4℃下,10,000g离心15min,可见分为三层,rna在上层水相,移至另一个新的rnase free ep管;
202.4)沉淀rna:加入0.5ml异丙醇,轻柔地充分混匀,室温静置10min;
203.5)4℃下,10,000g离心10min,收集rna沉淀,去上清;
204.6)用75%乙醇洗涤两次,超净台风干;
205.7)加入15~50μldepc水溶解沉淀。
206.2、总rna纯度和完整性检测
207.1)纯度检测:取1μl rna样品50倍稀释,在beckman coμlter520uv/vis spectrophotometer上测定od值,od260/od280的比值大于1.8,说明制备的rna较纯,无蛋白质污染。
208.2)总rna完整性检测:取rna样品1μl,1%琼脂糖凝胶电泳80v
×
20min,eb染色10min后用凝胶成像系统观察总rna的5s rrna,18s rrna和28s rrna条带,三条条带完整的话即可证明总rna抽提比较完整。
209.3、逆转录
210.1)在rnase free的pcr管中配置下列溶液:
[0211][0212]
2)将上述溶液吹打均匀,置65℃保温5min,使rna变性。随后立即冰上致冷,以防止rna复性;
[0213]
3)在该pcr管中加入下列试剂(promega):
[0214][0215]
4)将上述20μl反应溶液30℃保温10min;
[0216]
5)42℃保温60min;
[0217]
6)72℃保温10mn。
[0218]
7)放置零下20
°
保存。
[0219]
2》pcr反应体系
[0220]
在0.2ml ep管中配制以下体系,模板原液稀释20倍后取0.5μl扩增tug1:
[0221][0222]
注:kod plus neo dnapolymerase购于东洋纺公司,货号:kod 401;
[0223]
3》扩增条件
[0224]
混匀后,置于geneamp pcr system 2400型pcr扩增仪扩增。
[0225]
tug1基因的扩增条件:
[0226][0227]
4》pcr产物回收(dna凝胶回收试剂盒,dongsheng biotech)
[0228]
8)pcr产物经1%凝胶电泳后,在紫外灯下,用手术刀切取含目的基因片段的凝胶
条带至干净1.5ml ep管中,称重后,按100mg凝胶对应100μl溶液bd的比例向离心管中加入溶液bd。
[0229]
9)60℃水浴10min至凝胶完全溶化,水浴期间振荡混合3次。
[0230]
10)将溶液转移至dna纯化柱中,静置2min,室温12000rpm离心1min,弃滤液。
[0231]
11)向柱上加入500μl溶液pe,室温12000rpm离心1min,弃滤液。
[0232]
12)重复上一操作一次。
[0233]
13)室温空柱12000rpm离心1min以彻底去除纯化柱中残留的液体。
[0234]
14)将柱子置于新的1.5ml ep管上,向柱中央加入30μl 60℃预热的无菌水,13400g离心1min以洗脱出dna。
[0235]
5》pcr产物切胶回收时照片见图2;
[0236]
由图2可知,tug1用模板已扩增出来,大小与预测一致(扩增条带大概在marker 1000bp的位置);证明tug1目的基因已扩增出来;
[0237]
4、pcr回收产物及载体双酶切
[0238]
在2个无菌的0.2ml ep反应管,分别取tug1 pcr回收产物和psicheck-2载体各15μl,分别用xhoi/noti双酶切,酶切体系如下:
[0239][0240]
混匀后,37℃反应3h左右。
[0241]
5、酶切产物回收(dna凝胶回收试剂盒,dongsheng biotech)
[0242]
1)酶切产物经1%凝胶电泳后,在紫外灯下,用手术刀分别切取含目的片段和载体的凝胶条带至干净的1.5ml ep管中,按100mg凝胶对应100μl溶液bd的比例向离心管中加入溶液bd。
[0243]
2)60℃水浴10min至凝胶完全溶化,水浴期间振荡混合3次。
[0244]
3)将溶液转移至dna纯化柱中,静置2min,室温12000rpm离心1min,弃滤液。
[0245]
4)向柱上加入500μl溶液pe,室温12000rpm离心1min,弃滤液。
[0246]
5)重复上一操作一次。
[0247]
6)空柱12000rpm离心1min以彻底去除纯化柱中残留的液体。
[0248]
7)将柱子置于新的1.5ml ep管上,向柱中央加入30μl 60℃预热的无菌水,13400g离心1min以洗脱出dna。
[0249]
6、目段片段与载体连接:
[0250]
向0.2ml ep管中加入以下试剂(t4 dnaligase酶购于takara公司,货号:d2011a;)
[0251][0252]
16℃连接1h。
[0253]
7、连接产物的转化
[0254]
1)冰浴中将5μl连接产物分别加入至50μl dh5α感受态细胞中。轻轻旋转混匀,冰浴30min。
[0255]
2)42℃水浴热休克90s。
[0256]
3)快速将管转移至冰浴中,冰浴2min。
[0257]
4)分别加入200μl lb培养基,混匀,37℃、200rpm振荡培养1h。
[0258]
5)于超净工作台中,将菌液均匀涂布于含氨苄青霉素(amp)(100μg/ml)的lb平板上,室温下放置,至液体吸收。
[0259]
6)倒置平板,转移入37℃生化培养箱过夜培养。
[0260]
8、质粒酶切鉴定阳性克隆
[0261]
1》从平板上挑取若干单克隆于3ml lb管中摇床过夜培养;
[0262]
2》质粒提取,(高纯质粒小量提取试剂盒,g-shun)
[0263]
a)用1.5ml ep管收集3μl菌液,12000rpm离心1min,去上清。
[0264]
b)加入250μl溶液i/rnasea混合液,重悬菌体。
[0265]
c)加入250μl溶液ii,温和反复颠倒混匀6次,室温放置2min。
[0266]
d)加入350μl溶液iii,温和反复颠倒混匀6次。
[0267]
e)12000rpm离心10min,小心吸去上清至dna纯化柱中,静置2min。
[0268]
f)12000rpm离心1min,弃滤液。
[0269]
g)加入500μl溶液pb至柱中,12000rpm离心1min,弃滤液。
[0270]
h)加入500μl溶液w至柱中,12000rpm离心1min,弃滤液。重复一次。
[0271]
i)空柱12000rpm离心3min。
[0272]
j)将柱取出置于新的1.5ml ep管中,加入50μl无菌水(60℃预热),静置2min,13400rpm离心1min以洗脱出质粒。
[0273]
所述溶液ⅰ为50mml葡萄糖/10mml edta/25mml tris-hcl,ph=8.0,即重悬液。
[0274]
所述溶液ⅱ为0.2ml naoh/1%sds,即裂解液。
[0275]
所述溶液ⅲ为3ml醋酸钾/2ml醋酸,即中和沉淀液。
[0276]
溶液pb为质粒提取试剂,购买于东盛生物(dongsheng biotech),溶液pb中含有碱及蛋白变性剂,不要直接接触皮肤。溶液w为质粒提取试剂,购买于东盛生物(dongsheng biotech),溶液w初次使用前用无水乙醇按1:1.5稀释,即含60%乙醇。溶液bd为dna凝胶回收试剂,购买于东盛生物(dongsheng biotech),溶液bd中含有变性剂,不要直接接触皮肤。
[0277]
3》酶切鉴定所提取质粒
[0278]
酶切反应体系如下:
[0279][0280]
37℃酶切2h。
[0281]
酶切产物以含溴化乙锭(eb)的1%琼脂糖凝胶电泳分离,uvp凝胶成像系统成像。结果参见图3。由图3可知:tug1(1000bp)在相应的位置切出一条目的条带(下方箭头所指),说明筛到有阳性克隆,送tug1阳性质粒去测序。
[0282]
9、测序结果报告单
[0283]
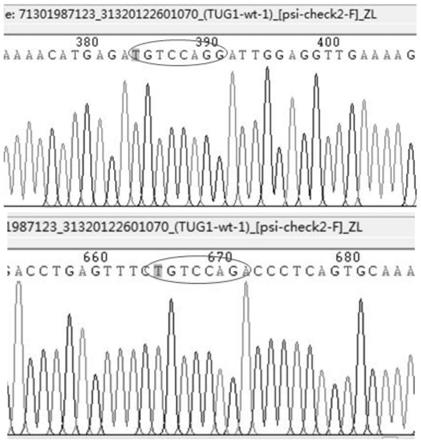
送tug1到上海生工基因公司测序,测序结果如序列表中的seq id no:3所示,tug1序列如序列表中的seq id no:4所示。测序结果blast分析:tug1已成功克隆至psicheck-2载体中,与ncbi上已知序列进行blast 100%一致,因此,可用于后续双报告实验。核心结合位点测序峰图参见图4,图4可以说明野生型tug1双报告载体构建成功。
[0284]
第二部分:突变预测hsa-mir-4638-3p第一结合位点载体构建
[0285]
1、tug1序列
[0286]
第一结合位点的序列为:tgtccagg;
[0287]
第一结合位点突变后的序列为:gagttcaa;
[0288]
2、扩增引物序列:(苏州上海生工合成page纯化2od):
[0289]
muttug1-1f:5'ggcaaaacatgagagagttcaaattggaggttgaaaagatttcactacagtgttctg 3';
[0290]
muttug1-1r:5'ttttcaacctccaatttgaactctctcatgttttgccacagaatttcaagctttgag 3';
[0291]
3、基因突变:
[0292]
1》pcr反应体系
[0293]
在0.2mlpcr反应管中配置以下体系,模板为构建好的tug1质粒,质粒稀释50倍后取1μl做模板:
[0294][0295]
注:kod plus neo dnapolymerase购于toyobo公司,货号:kod 401;
[0296]
2》扩增条件
[0297]
混匀后,置于geneamp pcr system 2400型pcr扩增仪扩增。
[0298]
muttug1-1基因的扩增条件:
[0299][0300][0301]
3》pcr产物用dpni酶切(以消化掉含甲基化的模板dna);
[0302]
在pcr管中加入1μldpni酶(产自promega公司,货号:r6231),在37℃消化4h。
[0303]
4》dpni处理的pcr产物转化,步骤同第一部分步骤7。
[0304]
5》挑1个菌落提取质粒,步骤同第一部分步骤8。
[0305]
6》送样测序
[0306]
4、测序结果报告单
[0307]
送到上海生工基因公司测序,测序结果如序列表中的seq id no:7所示,测序结果blast分析:tug1序列已成功在目的突变区域实现突变,因此,可用于后续双报告实验。核心结合位点突变测序峰图参见图5,由图5可知突变预测hsa-mir-4638-3p第一结合位点载体构建成功。
[0308]
第三部分:突变预测hsa-mir-4638-3p结合位点载体构建
[0309]
1、tug1序列
[0310]
第二结合位点的序列为:tgtccag;
[0311]
第二结合位点突变后的序列为:gagttca;
[0312]
2、扩增引物序列:(苏州上海生工合成page纯化2od):
[0313]
muttug1-2f:5'gaagacctgagtttcgagttcaaccctcagtgcaaactgaggatgctccatccaa
ag 3';
[0314]
muttug1-2r:5'gcactgagggttgaactcgaaactcaggtcttctaattcccagtaacagtgctggtg 3';
[0315]
3、基因突变:
[0316]
1》pcr反应体系
[0317]
在0.2mlpcr反应管中配置以下体系,模板为构建好的tug1质粒,质粒稀释50倍后取1μl做模板:
[0318][0319]
注:kod plus neo dnapolymerase购于toyobo公司,货号:kod 401;
[0320]
2》扩增条件
[0321]
混匀后,置于geneamp pcr system 2400型pcr扩增仪扩增。
[0322]
muttug1-2基因的扩增条件:
[0323][0324]
3》pcr产物用dpni酶切(以消化掉含甲基化的模板dna);
[0325]
在pcr管中加入1μldpni酶(产自promega公司,货号:r6231),在37℃消化4h。
[0326]
4》dpni处理的pcr产物转化,步骤同第一部分步骤7
[0327]
5》挑1个菌落提取质粒,步骤同第一部分步骤8
[0328]
6》送样测序
[0329]
4、测序结果报告单
[0330]
送到上海生工基因公司测序,测序结果如序列表中的seq id no:10所示,测序结果blast分析:tug1序列已成功在目的突变区域实现突变,因此,可用于后续双报告实验。核心结合位点突变测序峰图参见图6,由图6可知突变预测hsa-mir-4638-3p第二结合位点载
体构建成功。
[0331]
第四部分:双突变预测hsa-mir-4638-3p结合位点载体构建
[0332]
1、tug1序列
[0333]
第一结合位点的序列为:tgtccagg;
[0334]
第一结合位点突变后的序列为:gagttcaa;
[0335]
第二结合位点的序列为:tgtccag;
[0336]
第二结合位点突变后的序列为:gagttca;
[0337]
2、扩增引物序列:(苏州上海生工合成page纯化2od):
[0338]
muttug1-2f:5'gaagacctgagtttcgagttcaaccctcagtgcaaactgaggatgctccatccaaag 3';
[0339]
muttug1-2r:5'gcactgagggttgaactcgaaactcaggtcttctaattcccagtaacagtgctggtg 3';
[0340]
3、基因突变:
[0341]
1》pcr反应体系
[0342]
在0.2mlpcr反应管中配置以下体系,模板为突变好的muttug1-1质粒,质粒稀释50倍后取1μl做模板:
[0343][0344]
注:kod plus neo dnapolymerase购于toyobo公司,货号:kod 401;
[0345]
2》扩增条件
[0346]
混匀后,置于geneamp pcr system 2400型pcr扩增仪扩增。
[0347]
muttug1-3基因的扩增条件:
[0348]
[0349]
3》pcr产物用dpni酶切(以消化掉含甲基化的模板dna);
[0350]
在pcr管中加入1μldpni酶(产自promega公司,货号:r6231),在37℃消化4h。
[0351]
4》dpni处理的pcr产物转化,步骤同第一部分步骤7
[0352]
5》挑1个菌落提取质粒,步骤同第一部分步骤8
[0353]
6》送样测序
[0354]
4、测序结果报告单
[0355]
送到上海生工基因公司测序,测序结果如序列表中的seq id no:13所示,测序结果blast分析:tug1序列已成功在目的突变区域实现突变因此,可用于后续双报告实验。核心结合位点突变测序峰图参见图7,由图7可知:双突变预测hsa-mir-4638-3p结合位点载体构建成功。
[0356]
二、microrna与质粒共转染细胞
[0357]
阳离子脂质体法进行细胞转染:
[0358]
(1)转染前一天,细胞按2
×
104个/孔接种于24孔板上,培养基为含10%fbs的dmem高糖;
[0359]
(2)转染当天,细胞汇合度约为50-60%,吸去旧的培养基,用pbs洗涤两次,然后每孔加入300μl opti-mem(invitrogen公司)培养基,置于5%co2、37℃培养箱中;
[0360]
(3)每个孔用opti-mem培养基稀释cellfectin ii reagent(invitrogen公司)1μl,终体积为50μl,室温下静置5min;
[0361]
(4)每个孔用加入20μm浓度的hsa-mir-4638-3p1μl或hsa-mir-4638-3p抑制剂和0.5ug质粒,再加入opti-mem至总体积50μl,室温下静置5min;(最终孵育液中的hsa-mir-4638-3p浓度为50nm,hsa-mir-4638-3p抑制剂浓度为100nm)
[0362]
(5)复合(3)和(4)中的稀释液,室温下静置20min;
[0363]
(6)每孔加入100μl(5)中的转染复合液,晃动24孔板稍加混匀;
[0364]
(7)在5%co2、37℃培养箱中孵育5h,用新鲜的完全培养基(含血清)替换含有转染复合物的培养基。
[0365]
注:所用tip头已做去rnase处理。
[0366]
三、lucifersae检测
[0367]
用promega公司的dual-luciferase reporterassay system(e1910)进行样品luciferase活性检测。
[0368]
转染48h后,吸去旧的培养基,用pbs清洗洗两次,每孔细胞加入100μl的plb(passive lysis buffer),室温轻微振摇15分钟,收集细胞裂解液。
[0369]
1、使用手动的双萤光检测仪(promega,glomax生物发光检测仪)
[0370]
1)将20μl细胞裂解液加入发光板后,用glomax生物发光检测仪读取背景值2s。
[0371]
2)每样品加入100μl lar ii工作液,快速混匀,读值2s。
[0372]
3)读值完毕后,每样品再加入100μl stop&reagent,快速混匀后,放入发光检测仪中,读值2s。
[0373]
4)保存数据。
[0374]
三、检测结果分析
[0375]
萤火虫萤光素酶 f
[0376]
海肾萤光素酶 r
[0377]
△
活性倍数=(r/f)样品/(r/f)对照
[0378]
2》细胞转染试验组设置(每组数据设3个复孔)
[0379][0380]
图8为tug1与hsa-mir-4638-3p共转染萤光素酶表达活性图,从图8分析tug1与hsa-mir-4638-3p共转染萤光素酶表达活性倍数为45%;突变预测mir-4638-3p第一个结合位点萤光素酶活性倍数为81%;突变预测mir-4638-3p第二个结合位点萤光素酶活性倍数为60%;说明预测mir-4638-3p两个结合位点均可以通过结合tug13’utr进行调控,从结果来看,第一个结合位点抑制性比第二个结合位点稍强。同时突变第一/二结合位点萤光素酶活性倍数为100%,说明已经完全突变。
[0381]
图9为转染克隆有tug1基因3’utr质粒各组活性比较图,从图9可知在转染克隆有tug1基因3’utr质粒的实验中mir组与空白组相比较,**p《0.01差异极显著(p=0.0009),
mir组与nc组相比,**p《0.01差异极显著(p=0.0006),说明hsa-mir-4638-3p能通过结合tug1基因3’utr区域影响其荧光活性改变。hsa-mir-4638-3p inhibitor组与空白组相比较,**p《0.01差异极显著(p=0.0052),hsa-mir-4638-3p inhibitor组和nc inhibitor组相比较,**p《0.01差异极显著(p=0.0022),说明293t细胞中内源性mir-4638-3p表达抑制了tug1活性,在加入hsa-mir-4638-3p inhbitor后导致与blank/nc inhibitor相比,活性显著升高。
[0382]
图10为转染克隆有muttug1-1基因3’utr质粒各组活性比较图,由图10可知在转染克隆有muttug1-1基因3’utr质粒的实验中mir组与空白组相比较,**p《0.01差异极显著(p=0.0009),mir组与nc组相比,*p《0.05差异显著(p=0.0123),说明hsa-mir-4638-3p能通过结合muttug1-1基因3’utr区域影响其荧光活性改变。hsa-mir-4638-3p inhibitor组与空白组相比较,*p《0.05差异显著(p=0.0197),hsa-mir-4638-3p inhibitor组和nc inhibitor组相比较,**p《0.01差异极显著(p=0.0021),说明293t细胞中内源性mir-4638-3p表达抑制了muttug1-1活性,在加入hsa-mir-4638-3p inhbitor后导致与blank/nc inhibitor相比,活性显著升高。
[0383]
图11为转染克隆有muttug1-2基因3’utr质粒各组活性比较图,由图11可知在转染克隆有muttug1-2基因3’utr质粒的实验中mir组与空白组相比较,**p《0.01差异极显著(p=0.0019),mir组与nc组相比,**p《0.01差异极显著(p=0.0027),说明hsa-mir-4638-3p能通过结合muttug1-2基因3’utr区域影响其荧光活性改变。hsa-mir-4638-3p inhibitor组与空白组相比较,*p《0.05差异显著(p=0.0158),hsa-mir-4638-3p inhibitor组和nc inhibitor组相比较,*p《0.05差异显著(p=0.0244),说明293t细胞中内源性mir-4638-3p表达抑制了muttug1-2活性,在加入hsa-mir-4638-3p inhbitor后导致与blank/nc inhibitor相比,活性显著升高。
[0384]
图12为转染克隆有muttug1-3质粒各组活性比较图,由图12可知在转染克隆有muttug1-3质粒的实验中mir组与空白组相比较,p》0.05无统计学差异(p=0.9635),mir组与nc组相比,p》0.05无统计学差异(p=0.0925),说明hsa-mir-4638-3p不能通过结合muttug1-3影响其荧光活性改变。hsa-mir-4638-3p inhibitor组与空白组相比较,p》0.05无统计学差异(p=0.3673),hsa-mir-4638-3p inhibitor组和nc inhibitor组相比较,p》0.05无统计学差异(p=0.6597);muttug1-3质粒已实现完全定点突变。
[0385]
图9-12的结果分析中:*为显著差异,**为极显著差异。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
