一种
α-酮戊二酸钠盐的合成工艺
技术领域
1.本发明涉及有机合成领域,特别是涉及一种α-酮戊二酸钠盐的合成工艺。
背景技术:
2.α-酮戊二酸现阶段只能通过发酵法生产,由于发酵法生产过程中的控制水平不高,导致不同批次的α-酮戊二酸产品的质量差异较大。对于诊断试剂行业而言,α-酮戊二酸的产品质量不稳定严重影响体外检测产品的质量均一性和稳定性,因此,体外诊断行业通常要求以钠盐等形式提供高纯度(纯度通常要求大于99%)优质原料。然而,α-酮戊二酸作为α-酮戊二酸钠盐的直接原料,其规模化来源是发酵法,其产物纯度往往低于95%,受到发酵产业的规模限制,不同批次的α-酮戊二酸产品之间的含量不同,且批间差异很大,特别是产品外观,包括色泽、结晶形态、水溶液浊度等指标均存在明显的批间差,纯度低的甚至不到80%,难以满足体外诊断等下游试剂行业对该类产品严苛的质量要求,这直接导致现有技术的产品合格率低,不合格品需要经过多次返工精制才能勉强合格。
技术实现要素:
3.为了弥补上述现有技术的不足,本发明提出一种α-酮戊二酸钠盐的合成工艺。
4.本发明的技术问题通过以下的技术方案予以解决:
5.一种α-酮戊二酸钠盐的连续流合成工艺,包括如下步骤:
6.(1)将naoh用h2o溶解配制为质量分数为5-30%的naoh溶液,作为物料a;
7.(2)将α-酮戊二酸用溶剂配制为质量分数为10-30%的α-酮戊二酸溶液,作为物料b;
8.(3)将物料a和物料b分别通过进料泵注入混合器内,然后进入连续流微反应器,在预设的反应温度下,在所述连续流微反应器内反应停留预定时间后,收集反应液;其中,在反应过程中,naoh和α-酮戊二酸的摩尔比为1.0~2.2:1;所述物料a和所述物料b在所述连续流微反应器内的反应停留时间为1-10min;
9.(4)将所收集的反应液经过后处理后,得到固体产物。
10.优选地,当naoh和α-酮戊二酸的摩尔比为1.0~1.1:1时,所述步骤(4)中的固体产物为α-酮戊二酸单钠,当naoh和α-酮戊二酸的摩尔比为2.0~2.2:1时,所述步骤(4)中的固体产物为α-酮戊二酸二钠。
11.优选地,当naoh和α-酮戊二酸的摩尔比为1.03~1.05:1时,所述步骤(4)中的固体产物为α-酮戊二酸单钠,当naoh和α-酮戊二酸的摩尔比为2.05~2.1:1时,所述步骤(4)中的固体产物为α-酮戊二酸二钠。
12.优选地,步骤(3)中在所述连续流微反应器内的反应温度不超过15℃,优选地,反应温度为0-5℃,更优选地,反应温度为0-0.5℃。
13.优选地,所述步骤(2)中的溶剂为水、甲醇和乙醇中的至少一种。
14.优选地,所述步骤(2)中的α-酮戊二酸的纯度》98%,还包括α-酮戊二酸的精制步
骤:将α-酮戊二酸用无水乙醇和/或无水甲醇在30-60℃溶解后,过滤除杂,再降温进行重结晶提纯,并通过真空干燥脱除溶剂,得到纯度》98%的α-酮戊二酸结晶粉末。
15.一种α-酮戊二酸单钠的合成工艺,包括如下步骤:
16.s1、将na2co3和/或nahco3完全溶解于水中,并冷却到0-10℃,得到溶液c;
17.s2、将α-酮戊二酸粉末缓慢加入所述溶液c中,或者将所述溶液c缓慢加入α-酮戊二酸粉末中,控制反应温度在5-15℃下反应,其中,溶液c中na
与α-酮戊二酸的摩尔比为:n(na
):n(α-酮戊二酸)=1-1.16:1;
18.s3、将经过步骤s2反应后的反应液进行后处理,得到α-酮戊二酸单钠固体。
19.优选地,步骤s2中的α-酮戊二酸粉末的纯度》98%,还包括α-酮戊二酸粉末的精制步骤:将α-酮戊二酸用无水乙醇和/或无水甲醇在30-60℃溶解后,过滤除杂,再降温进行重结晶提纯,并通过真空干燥脱除溶剂,得到纯度》98%的α-酮戊二酸结晶粉末。
20.优选地,在所述步骤s1中,当采用na2co3时,所述溶液c中的na2co3的质量分数为30-32%;当采用nahco3时,所述溶液c中的nahco3的质量分数为18-20%;当采用na2co3和nahco3两者的混合物时,所述溶液c中的混合物的质量分数为20-32%。
21.一种α-酮戊二酸二钠的连续流合成工艺,包括如下步骤:
22.(1)将naoh用水溶解配制为质量分数为25-30%的naoh溶液,作为物料d;
23.(2)将α-酮戊二酸单钠与水配置成质量分数为30-50%的α-酮戊二酸单钠水溶液,作为物料e;
24.(3)将物料d和物料e分别通过进料泵注入混合器内,然后进入连续流微反应器,在预设的反应温度下,在所述连续流微反应器内反应停留预定时间后,收集反应液;其中,在反应过程中,naoh和α-酮戊二酸单钠的摩尔比为1.0~1.05:1;所述物料d和所述物料e在所述连续流微反应器内的反应停留时间为5-6min;所述预设的反应温度是0-5℃;
25.(4)将所收集的反应液经过后处理后,得到固体产物。
26.优选地,所述α-酮戊二酸单钠由所述的合成工艺制备得到。
27.本发明与现有技术对比的有益效果包括:
28.本发明通过改进合成工艺,提供了一种操作简便、收率高、易于放大、产品质量稳定、纯度高,并能满足各项产品质量要求的α-酮戊二酸钠盐合成工艺,实现低成本连续化生产,产品色泽为白色、类白色或米黄色,产品配制的30wt%水溶液的浊度小于0.7,且经透光判断无明显肉眼可见的悬浮物和漂浮物。同时,本发明也提高了产品质量的一致性,降低了批间差。
附图说明
29.图1是本发明第一具体实施方式中的α-酮戊二酸钠盐的连续流合成工艺流程图。
30.图2是本发明实施例1制得的α-酮戊二酸二钠的核磁氢谱图。
具体实施方式
31.下面对照附图并结合优选的实施方式对本发明作进一步说明。需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。在下文中,如无特别说明,“纯度”均指的“核磁纯度”。
32.第一具体实施方式
33.如图1所示,在该具体实施方式中,一种α-酮戊二酸钠盐的连续流合成工艺,包括如下步骤:
34.(1)将naoh用h2o溶解配制为质量分数为5-30%的naoh溶液,作为物料a;
35.(2)将α-酮戊二酸用溶剂配制为质量分数为10-30%的α-酮戊二酸溶液,作为物料b;
36.(3)将物料a和物料b分别通过进料泵1、2注入混合器3内,然后进入连续流微反应器4,在预设的反应温度下,在所述连续流微反应器4内反应停留预定时间后,收集反应液;其中,在反应过程中,naoh和酮戊二酸的摩尔比为1.0~2.2:1,所述物料a和所述物料b在所述连续流微反应器内的反应停留时间为1-10min;
37.根据氢氧化钠的投料量不同,可以得到α-酮戊二酸单钠盐、α-酮戊二酸二钠盐,或这两种盐的混合物,反应方程式为:naoh c5h6o5=nac5h5o5 h2o;2naoh c5h6o5=na2c5h4o5 h2o。
38.(4)将所收集的反应液经过后处理后,得到固体产物。
39.该具体实施方式采用连续流微反应的优势在于:1、各原料的投料量可精准控制,易于实现最佳物料配比;2、设备换热效果优异,参与反应的物料管径细微,换热面积大且换热效率极高,可以精准控制反应温度,不会发生局部温度过热现象,能实现反应温度的精准控制;3、可以通过调节泵压和阀门开度控制进料液体流速,控制物料在反应器内的摩尔比,通过控制物料在反应器内的停留时间从而精确控制反应时长;4、几乎没有混料返流现象,可减少副反应的发生;5、氢氧化钠在水中的溶解度高,在整个反应过程中不会产生气体,其作为连续流进料,可以避免反应器管道发生堵塞,也可以实现较高浓度的配料,减少水分的带入量,降低后处理过程的脱水成本。因此,该实施方式可以得到质量均一稳定、纯度较高的α-酮戊二酸钠盐的高纯度结晶粉末状。
40.在优选的实施方式中,当naoh和α-酮戊二酸的摩尔比为1.0~1.1:1时,所述步骤(4)中的固体产物为α-酮戊二酸单钠,当naoh和α-酮戊二酸的摩尔比为2.0~2.2:1时,所述步骤(4)中的固体产物为α-酮戊二酸二钠;当naoh和α-酮戊二酸的摩尔比在1.1~2.0:1时,得到固体产物为α-酮戊二酸二钠和α-酮戊二酸单钠的混合物,后续可以通过进一步的处理进行两者的分离。
41.在优选的实施方式中,当naoh和α-酮戊二酸的摩尔比为1.03~1.05:1时,所述步骤(4)中的固体产物为α-酮戊二酸单钠,当naoh和α-酮戊二酸的摩尔比为2.05~2.1:1时,所述步骤(4)中的固体产物为α-酮戊二酸二钠;当naoh和α-酮戊二酸的摩尔比在1.05~2.05:1时,得到固体产物为α-酮戊二酸二钠和α-酮戊二酸单钠的混合物,后续可以通过进一步的处理进行两者的分离。
42.在优选的实施方式中,步骤(2)中的α-酮戊二酸的纯度》98%,还包括α-酮戊二酸的精制步骤:将α-酮戊二酸用无水乙醇和/或无水甲醇在30-60℃溶解后,过滤除杂,再降温进行重结晶提纯,并通过真空干燥脱除溶剂,得到纯度》98%的α-酮戊二酸结晶粉末。
43.在优选的实施方式中,步骤(3)中在所述连续流微反应器内的反应温度不超过10℃,优选地,反应温度为0-5℃,更优选地,反应温度为0-0.5℃。
44.在优选的实施方式中,所述步骤(2)中的溶剂为水、甲醇和乙醇中的至少一种。
45.在优选的实施方式中,物料a的进料速度为0.1-2ml/min,物料b的进料速度为0.5-2ml/min。
46.以下通过实施例1对第一具体实施方式的α-酮戊二酸钠盐的连续流合成工艺进行阐述。
47.实施例1
48.α-酮戊二酸二钠盐的连续流合成工艺包括如下步骤:
49.(1)将24.96g氢氧化钠溶于240ml去离子水,得到245ml氢氧化钠溶液,作为物料a;
50.(2)将α-酮戊二酸先用无水乙醇在30-60℃溶解后,过滤除杂,再降温至0℃左右,重结晶提纯,并通过真空干燥脱除溶剂,得到纯度》98%的精制α-酮戊二酸结晶粉末;将43.83g的经过精制的α-酮戊二酸溶解于115ml乙醇中,抽滤,向得到的α-酮戊二酸乙醇溶液中加入115ml去离子水,得到251ml的α-酮戊二酸溶液,作为物料b;
51.(3)将物料a和物料b分别通过进料泵1、2注入混合器3内,然后进入连续流微反应器4,在所述连续流微反应器4内反应停留预定时间后,收集反应液;其中,在反应过程中,naoh和酮戊二酸的摩尔比为2.08:1;其中,在本例中,连续流微反应器采用ezone mf300连续流微通道固定床反应器(型号mrhc-200,购于深圳市一正科技有限公司),进料泵采用注射泵(型号hp-10-2107-0204,购于欧世盛(北京)科技有限公司);物料a的进料速度是1ml/min,物料b的进料速度是1.024ml/min,停留时间为5.6min,反应温度为0℃,收集得到的反应液的ph为9.3,α-酮戊二酸二钠的质量分数约为20wt%。
52.(4)将所收集的反应液经过后处理后,得到固体产物,经图2所示的核磁氢谱确认,该固体产物为α-酮戊二酸二钠。其中,后处理过程为:将反应液旋蒸浓缩体积,到其成为黄色油状液体,向反应液中加入50ml无水乙醇,不断搅拌除水,待溶液分层后,下层呈现黄色油状液体,分液将上层清液倒掉,继续加入50ml无水乙醇,静置分液后移除上层清液,对下层液体过滤后收集滤液,重复此步骤2-3次,待有大量固体析出后,抽滤,得到固体产品,采用真空或负压干燥,控制温度不超过70℃脱除全部残留溶剂,得到固体产物。
53.本实施例所得的结果:产品的质量收率为94%,产品的纯度为92%,收率86.5%(收率=质量收率*纯度)。后续,可以再经过重结晶精制,可以得到产品纯度大于99%的产物。用上述得到的固体产物或者重结晶精制后的产物配制质量分数为30%的水溶液,均无明显悬浮物,符合工艺要求。
54.第二具体实施方式
55.该具体实施方式提供一种α-酮戊二酸单钠的合成工艺,包括如下步骤:
56.s1、将na2co3和/或nahco3完全溶解于水中,并冷却到0-10℃,得到溶液c;
57.s2、将α-酮戊二酸粉末缓慢加入所述溶液c中,或者将所述溶液c缓慢加入α-酮戊二酸粉末中,控制反应温度在5-15℃下反应,其中,溶液c中na
与α-酮戊二酸的摩尔比为:n(na
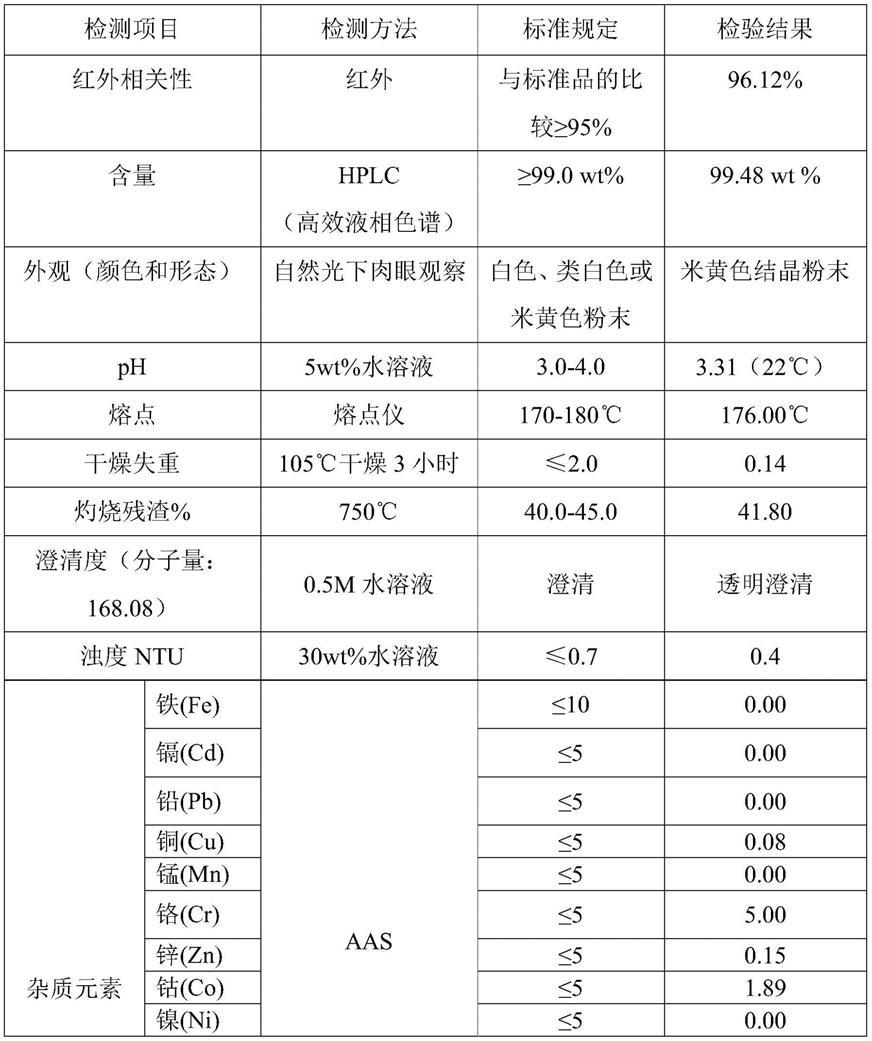
):n(α-酮戊二酸)=1-1.16:1;反应方程式为:na2co3 2c5h6o5=2nac5h5o5 h2o co2;nahco3 c5h6o5=nac5h5o5 h2o co2。
58.s3、将经过步骤s2反应后的反应液进行后处理,得到α-酮戊二酸单钠固体。
59.该具体实施方式是釜式反应合成工艺,由于合成α-酮戊二酸钠盐的成盐反应是酸碱中和反应,具有强放热特征,釜式反应在加料搅拌过程中很难避免局部温度偏高,使得产物颜色偏黄而合格率低,因此,为了控制反应料液温度不会整体偏高而导致副反应增多,本
具体实施方式中在配置溶液c时,将其冷却到5-10℃备用,然后在步骤s2中通过缓慢加料的形式(缓慢加料的目的是为了控制反应温度始终维持在5-15℃下,避免局部温度超过15℃,以免反应过于激烈,导致副反应失控,甚至无法得到目标产物的问题)
60.在优选的实施方式中,步骤s2中的α-酮戊二酸粉末的纯度》98%,还包括α-酮戊二酸粉末的精制步骤:将α-酮戊二酸用无水乙醇和/或无水甲醇在30-60℃溶解后,过滤除杂,再降温进行重结晶提纯,并通过真空干燥脱除溶剂,得到纯度》98%的α-酮戊二酸结晶粉末。
61.在优选的实施方式中,在所述步骤s1中,当采用na2co3时,所述溶液c中的na2co3的质量分数为30-32%;当采用nahco3时,所述溶液c中的nahco3的质量分数为18-20%;当采用na2co3和nahco3两者的混合物时,所述溶液c中的混合物的质量分数为20-32%。
62.以下通过实施例2对第二具体实施方式的α-酮戊二酸单钠的合成工艺进行阐述。
63.实施例2
64.α-酮戊二酸单钠的合成工艺包括如下步骤:
65.(1)将500g无水na2co3充分溶解在35-36℃的1000ml纯净水中,快速精密过滤去除不溶物,得到的滤液再通过冰浴等方式强制冷却到5-10℃,得到溶液c;
66.(2)将α-酮戊二酸先用无水乙醇在30-60℃溶解后,过滤除杂,再降温至0℃左右,重结晶提纯,并通过真空干燥脱除溶剂,得到纯度》98%的精制α-酮戊二酸结晶粉末,本步骤中,共精制得到约1500g的α-酮戊二酸备用;
67.(3)在搅拌下,按投料摩尔比为n(na2co3):n(酮戊二酸)=0.5-0.58:1,将步骤(2)的α-酮戊二酸粉末缓慢加入步骤(1)的溶液c中,本步骤中约投入1250-1380g的α-酮戊二酸粉末,控制反应温度在5-15℃,且局部温度不超过15℃,搅拌反应4小时,直至大部分悬浮物溶解,溶液变得清澈后,快速过滤除杂,将滤液放入结晶釜内,将温度降低到0-4℃区间,维持8-12小时,可自然析晶;
68.(4)过滤收集步骤(3)的晶体,用少量无水乙醇洗涤2-3次后,沥干,常温下真空干燥脱除溶剂,得到α-酮戊二酸单钠盐固体产品,本实施例所得的结果:产品的质量收率为95.6%,产品的纯度为99%,收率94.6%(收率=质量收率*纯度。将所得产品送检,质量合格(如下表1所示,为本例得到的α-酮戊二酸单钠的检测结果),本例中,得到约1440~1585g产品。
69.表1
[0070][0071][0072]
第三具体实施方式
[0073]
发明人在实验中发现,如果用第一具体实施方式的连续流合成方法,将氢氧化钠与α-酮戊二酸合成α-酮戊二酸二钠,因为要保证氢氧化钠水溶液(物料a)和α-酮戊二酸的乙醇溶液(物料b),以及产品α-酮戊二酸二钠在乙醇-水溶剂体系中均始终保持溶液状态,不得有固体析出的情况发生,因此,受制于上述三者中溶解度最低的那个质量分数,得到的产物浓度(质量分数)较低(通常低于20wt%),这样,通过微通道连续流合成得到的产物α-酮戊二酸二钠在溶液中的浓度就很难提高到一个较高的水平(比如超过30wt%),那么反应完毕,在后处理的析晶过程中,尤其是放大实验时,就需要使用大量的乙醇,导致生产成本较高。为此,本发明提供第三具体实施方式,改进了α-酮戊二酸二钠的合成工艺,包括如下步骤:
[0074]
(1)将naoh用水溶解配制为质量分数为25-30%的naoh溶液,作为物料d;
[0075]
(2)将α-酮戊二酸单钠与水配置成质量分数为30-50%的α-酮戊二酸单钠水溶液,作为物料e;
[0076]
(3)将物料d和物料e分别通过进料泵注入混合器内,然后进入连续流微反应器,在预设的反应温度下,在所述连续流微反应器内反应停留预定时间后,收集反应液;其中在反应过程中,naoh和α-酮戊二酸单钠的摩尔比为1.0~1.05:1;所述物料d和所述物料e在所述连续流微反应器内的反应停留时间为5-6min;所述预设的反应温度是0-5℃;反应方程式为:naoh nac5h5o5=na2c5h4o5 h2o;
[0077]
(4)将所收集的反应液经过后处理后,得到固体产物。
[0078]
在优选的实施方式中,所述α-酮戊二酸单钠可以由第二具体实施方式所述的合成工艺制备得到。
[0079]
在优选的实施方式中,物料d与物料e的进料流量比为1:1.68。
[0080]
以下通过实施例3对第三具体实施方式的α-酮戊二酸二钠的连续流合成工艺进行阐述。
[0081]
实施例3
[0082]
α-酮戊二酸二钠的连续流合成工艺,包括如下步骤:
[0083]
(1)将400g~450g氢氧化钠投入1000ml纯净水中,充分溶解后,自然冷却到常温,再通过冰浴等强制冷却手段,将其温度下降到0-10℃,静置保存备用,即可得到浓度约为25wt%~30wt%的naoh水溶液,即物料d,本例中,naoh水溶液的浓度为25wt%;
[0084]
(2)将1000-1500g实施例2制得的α-酮戊二酸单钠在常温下溶于1000ml纯净水中,充分溶解后,自然冷却至常温,再通过冰浴等强制冷却手段,将其温度降低到5-10℃,静置备用,即可得到30wt%~50wt%的α-酮戊二酸单钠水溶液,即物料e,本例中,α-酮戊二酸单钠水溶液的浓度为50wt%;
[0085]
(3)将物料d和物料e分别通过进料泵注入混合器内,然后进入连续流微反应器,在所述连续流微反应器内反应停留预定时间后,收集反应液;其中,在反应过程中,naoh和α-酮戊二酸单钠的摩尔比为1-1.05:1;其中,在本例中,连续流微反应器采用ezone mf300连续流微通道固定床反应器(型号mrhc-200,购于深圳市一正科技有限公司),进料泵采用注射泵(型号hp-10-2107-0204,购于欧世盛(北京)科技有限公司);物料d与物料e的进料流量比为1:1.68,停留时间为330-340秒,反应温度为0.5~1℃,收集得到的反应液中,α-酮戊二酸二钠的质量分数为40-42wt%。
[0086]
4)将所收集的反应液经过后处理后,得到固体产物。其中,后处理过程为:在反应液中加入2l无水乙醇,搅拌均匀后静置10分钟,待溶液分层后,移除上层清液,再次加入1l无水乙醇后,重复上述步骤,即可析出α-酮戊二酸二钠固体,经过抽滤脱除溶剂后,采用真空干燥脱除残留溶剂,得到结晶粉末,将结晶粉末再经过重结晶精制,可以得到产品纯度大于99%的产物。本实施例重结晶后所得的产物的结果:产品的质量收率为87.4%,产品的纯度为99.5%,收率87%(收率=质量收率*纯度)。
[0087]
用上述重结晶精制后的产物配制质量分数为30%的水溶液,未见悬浮物,其它各项检测结果均满足产品合格指标,检测结果如下表2所示。
[0088]
表2
[0089]
[0090][0091]
以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的技术人员来说,在不脱离本发明构思的前提下,还可以做出若干等同替代或明显变型,而且性能或用途相同,都应当视为属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。