1.本发明涉及一种2-乙基-1-丁醇的制备方法,属于有机合成技术领域。
背景技术:
2.2-乙基-1-丁醇,cas:97-95-0,无色透明液体,有特臭气味,用作硝基喷漆、合成树脂清漆的助溶剂或稀释剂、印刷油墨溶剂,还用于香料、表面活性剂、增塑剂制造和润滑油添加剂合成,同时也是新冠药物瑞德西韦重要中间体l-丙氨酸2-乙基丁醇酯的基础原料。
3.2-乙基-1-丁醇的合成工艺主要为:以2-乙基已醇(如用乙醇和丁醇为原料,通过特殊催化剂、高温高压下经过氧化还原反应生成多种成份)合成的副产物,通过特定精馏技术得到[applied catalysis a:general,2019,588,117265],反应方程式如下:
[0004][0005]
该方法并不属于精细化工范畴,其对设备的要求更高。通过该方法得到2-乙基-1-丁醇主要为副产物,产量、质量与需求不能更好的满足日益增长的市场需求。
[0006]
文献[journal of organic chemistry,1986,51,4000]和[bulletin of the chemical society of japan,1984,57,1948]采用2-乙基丁酸酯还原制得,反应方程式如下:
[0007][0008]
该反应收率高,收率分别是87%和84%,纯度相对较高,分离容易。但原料价格更贵,不具备经济效益。
[0009]
文献[green chemistry,2017,19,169]和[synthetic communication s,1995,25,3089]采用2-乙基丁醛还原制得,收率分别是87%和80%。由于反应醛还原成醇过程中原料会有剩余,后续分离过程存在少量原料情况,不具备经济效益。
[0010]
文献[journal of the american chemical society,1932,54,4680]采用2,2-二乙基二丙酸250℃脱羧,随后氢化制得,该方法反应温度过高,难以实现。反应方程式如下:
[0011][0012]
针对上述方法的不足,本发明采用流程简便,对设备要求低,不需要高温高压、氧化还原等一系列复杂反应制备,并且通过巧妙的后处理方式,得到品质更高的2-乙基-1-丁醇,以满足日益增长的市场需求。
技术实现要素:
[0013]
为了克服上述技术缺陷,本发明以3-卤代戊烷为原料与金属镁制备格氏试剂,格
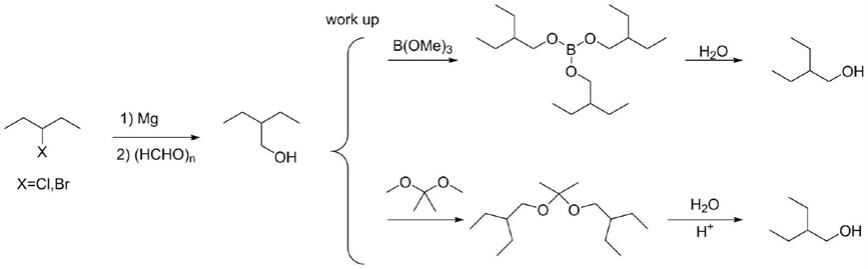
氏试剂与多聚甲醛反应,水解得到2-乙基-1-丁醇粗品。粗品与硼酸三甲酯或丙酮缩二甲醇酯交换反应,处理后得到2-乙基-1-丁醇。本发明工艺流程简便,对设备相对要求低,无需精馏即可得到高纯度、高含量的2-乙基-1-丁醇,且有利于新冠药物瑞德西韦中间体l-丙氨酸-2-乙基丁醇酯的工业需求。
[0014]
本发明所述一种2-乙基-1-丁醇的制备方法,反应方程式为:
[0015]
包括如下步骤:
[0016]
反应阶段:将3-卤代戊烷、金属镁与有机溶剂混合,升温引发后,滴加剩余3-卤代戊烷反应,制备格氏试剂后,降温加入多聚甲醛,水解后得到2-乙基-1-丁醇粗品;
[0017]
纯化处理a:将2-乙基-1-丁醇粗品与硼酸三甲酯混合,升温下减压蒸馏交换掉的甲醇,接着150-180℃减压蒸馏得到硼酸三(2-乙基-丁基酯),随后将硼酸三(2-乙基-丁基酯)加入水中,处理后得到2-乙基-1-丁醇。
[0018]
纯化处理b:将2-乙基-1-丁醇粗品与丙酮缩二甲醇混合,升温下减压蒸馏交换掉的甲醇,接着120-140℃减压蒸馏得到丙酮缩二(2-乙基-丁基酯),随后将丙酮缩二(2-乙基-丁基酯)滴加到酸水中,处理后得到2-乙基-1-丁醇。
[0019]
进一步地,在上述技术方案中,反应阶段,所述3-卤代戊烷选自3-氯代戊烷或3-溴代戊烷。
[0020]
进一步地,在上述技术方案中,反应阶段,有机溶剂选自2-甲基四氢呋喃或四氢呋喃。
[0021]
进一步地,在上述技术方案中,反应阶段,3-卤代戊烷、镁屑与多聚甲醛摩尔比为1:1.10-1.15:2.0-4.0。
[0022]
进一步地,在上述技术方案中,纯化处理a,所述2-乙基-1-丁醇粗品与硼酸三甲酯摩尔比为1:0.30-0.35。
[0023]
进一步地,在上述技术方案中,纯化处理b,所述2-乙基-1-丁醇粗品与丙酮缩二甲醇摩尔比为1:0.45-0.50。
[0024]
发明有益效果
[0025]
1、通过格氏试剂与多聚甲醛反应水解直接得到目标产物,避免高温高压或氧化还原反应,对设备要求相对较低,操作简单。
[0026]
2、后处理通过酯交换反应,水解后得到的产品质量和纯度更高,对精馏技术(如回流比控制、再沸器温度控制等)几乎无要求。
具体实施例
[0027]
下面通过具体实例对本发明进行进一步说明。这些实施例应理解为仅用于说明本发明而不用于限制本发明的保护范围。在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等效变化和修改同样落入本发明权利要求所限定的范围。
[0028]
实施例1
[0029][0030]
氮气保护下,室温下将3-氯戊烷5.3g(0.05mol)、2粒碘、镁屑14g(0.575mol)和300ml2-甲基四氢呋喃,装上回流装置,缓慢升温至50℃,搅拌下引发,随后缓慢滴加3-氯戊烷48g(0.45mol)溶于100ml2-甲基四氢呋喃混合溶液,并控制温度在55-60℃,滴加结束反应4小时。降温至-15℃,加入45g(1.5mol)多聚甲醛的2-甲基四氢呋喃溶液。在-15℃反应3小时,缓慢升温至10℃,加入1mol/l盐酸淬灭,调节ph=2-3,分层,水相mtbe萃取,合成有机相,加入无水硫酸镁干燥,过滤,滤液减压浓缩蒸除溶剂得到2-乙基-1-丁醇粗品44.4g,gc:93.6%,gc外标含量89.3%。
[0031]
后处理:
[0032][0033]
氮气保护下,室温下将2-乙基-1-丁醇粗品44.4g(含量89.3%,0.388mol)和硼酸三甲酯12.9g(0.124mol)混合,改为常压蒸馏装置,升温至50℃反应1小时,随后升温至66℃,常压蒸馏交换的甲醇,逐渐升温至70℃,待不流液后,改为减压蒸馏装置,随后150-180℃减压蒸馏得到硼酸三(2-乙基-丁基酯),将硼酸三(2-乙基-丁基酯)滴加到水中,升温至30℃反应2小时,静置分层,收集有机层,水相mtbe萃取,合并有机相,无水硫酸镁干燥,有机相减压蒸馏得到2-乙基-1-丁醇35.6g,总收率69.6%,gc:99.8%,外标含量99.7%。
[0034]
实施例2
[0035][0036]
氮气保护下,室温下将3-溴戊烷7.6g(0.05mol)、2粒碘、镁屑14g(0.575mol)和300ml四氢呋喃,装上回流装置,缓慢升温至50℃,搅拌下引发,随后缓慢滴加3-溴戊烷68g(0.45mol)溶于150ml四氢呋喃混合溶液,并控制温度在55-60℃,滴加结束反应2小时。降温至-15℃,加入45g(1.5mol)多聚甲醛的2-甲基四氢呋喃溶液。在-15℃反应3小时,缓慢升温至10℃,加入1mol/l盐酸淬灭调ph=2-3,分层,水相mtbe萃取,合成有机相,加入无水硫酸镁干燥,过滤,滤液减压浓缩蒸除溶剂得到2-乙基-1-丁醇粗品45.1g,gc:94.1%,gc外标含量85.7%。
[0037]
后处理:
[0038][0039]
氮气保护下,室温下将2-乙基-1-丁醇粗品45.1g(含量85.7%,0.3785mol)和丙酮缩二甲醇18.2g(0.175mol)混合,改为常压蒸馏装置,升温至50℃反应1小时,随后升温至66℃,常压蒸馏交换的甲醇,逐渐升温至85℃,待不流液后,改为减压蒸馏装置,随后120-140℃减压蒸馏得到丙酮缩二(2-乙基-丁基酯),随后将丙酮缩二(2-乙基-丁基酯)滴加到1mol/l盐酸水溶液中,室温反应2小时,静置分层,收集有机层,水相mtbe萃取,合并有机相,无水硫酸镁干燥,有机相减压蒸馏得到2-乙基-1-丁醇34.3g。总收率67.1%,gc:99.7%,外标含量99.6%。
[0040]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
