1.本发明涉及止血粉末,其包含至少10wt.%的颗粒附聚物,所述颗粒附聚物包含:
2.·
亲电聚噁唑啉颗粒,其含有携带至少3个反应性亲电基团的亲电聚噁唑啉,所述反应性亲电基团能够在形成共价键的情况下与血液中的胺基团反应;以及
3.·
亲核聚合物颗粒,其含有携带至少3个反应性亲核基团的亲核聚合物,所述反应性亲核基团能够在水的存在下在所述亲电聚噁唑啉与所述亲核聚合物之间形成共价键的情况下与所述亲电聚噁唑啉的所述反应性亲电基团反应。
4.当将本发明的止血粉末应用至出血部位时,颗粒附聚物迅速形成凝胶,同时与血液和周围组织中存在的蛋白质结合,从而加速止血。
5.发明背景
6.止血是严格调控的过程,其在发生对组织损伤的血栓反应的同时维持通过脉管系统的血流。维持止血需要血管壁、血小板以及凝血和纤维蛋白溶解系统的复杂相互作用。止血的两个主要阶段是:初级(即,细胞阶段)和次级(即,体液阶段)。
7.初级止血在内皮破坏后立即开始,其特征在于血管收缩,血小板粘附和形成软附聚栓塞。在损伤发生后,存在血管平滑肌的暂时局部收缩和血流减慢,促进血小板粘附和活化。在受伤的20秒内,循环的von willebrand因子附着于损伤部位的内皮下,并粘附至血小板表面上的糖蛋白。当血小板粘附至损伤表面时,它们通过与结合循环纤维蛋白原的暴露胶原的受体接触而被活化。形成附聚的血小板和纤维蛋白原的软栓塞。该止血阶段是短暂的,并且软栓塞可以容易地从损伤表面断裂。
8.软血小板栓塞在次级止血期间被稳定以形成凝块。血管收缩和由此产生的血流减少通过血小板分泌的5-羟色胺、前列腺素和血栓素维持,同时启动凝血级联。凝血级联是一系列涉及几种血浆蛋白、钙离子和血小板的依赖性反应,其导致纤维蛋白原转化成纤维蛋白。凝血因子由肝脏产生,并以非活性形式循环,直到启动凝血级联。然后通过一系列连续和依赖的凝血因子活化反应启动和完成级联的每个步骤。在最后的步骤中,凝血酶将可溶性血浆蛋白纤维蛋白原转化为不溶性蛋白纤维蛋白,同时将xiii因子转化为xiiia因子。这种因子转化使纤维蛋白稳定并导致纤维蛋白单体的交联,产生稳定的凝块。
9.在手术期间,保持出血与凝血之间的微妙平衡是重要的,使得血液继续流向手术部位的组织而不会过度失血,以优化手术的成功和患者的预后。在手术期间,从弥漫性小毛细血管或小静脉中持续出血可能使手术视野模糊,延长操作时间,增加生理并发症的风险,并使患者暴露于与输血相关的风险。
10.外科医生具有一系列控制出血的选择,包括机械和热技术和设备以及药物疗法和局部药剂。
11.最早的局部止血剂之一是纱布海绵形式的棉制品。尽管这种材料通过物理吸附来聚集血液和凝血产物,但它们不被身体吸收,并且在移除时,凝块可能被清除,导致进一步的出血。自此开发了可吸收的局部止血剂,并且当常规止血方法无效或不实用时提供了有用的辅助治疗。局部止血剂可直接应用于出血部位,并且可以防止持续不能缓解的出血。使
用局部药剂的止血也可以避免全身止血药物的副作用,例如“不希望的”血凝块。此外,在失血量不可预见的手术程序中,局部止血剂可以在失血较少时少量使用,而在严重出血时大量使用。
12.许多局部止血剂目前可用于手术。它们可以分成两类:以生物活性方式提供其对凝血级联的作用机制的那些和通过接触活化和促进血小板附聚而被动作用的那些。被动局部止血剂包括胶原、纤维素和明胶,而活性剂包括凝血酶和其中凝血酶与被动剂组合以提供活性整体产品的产品。
13.us 2003/0064109描述了通过包括以下步骤的方法制备的干止血粉末:提供包含与至少一种再水合助剂组合的明胶的水溶液;干燥所述溶液以产生固体;研磨所述固体以产生粉末;使所述粉末交联;除去至少50%(w/w)的再水合助剂;以及将交联后的明胶干燥以产生粉末。再水合助剂可以包含选自聚乙二醇(peg)、聚乙烯吡咯烷酮(pvp)和葡聚糖的至少一种材料。
14.wo 2010/059280描述了无水纤维片,其包含第一组分的纤维聚合物和第二组分,所述聚合物含有亲电基团或亲核基团,当所述片暴露于与生物组织接触的水性介质时,所述第二组分能够交联所述第一组分以形成交联水凝胶,所述交联水凝胶粘附至所述生物组织;其中:
15.a)其中第二组分是具有与第一组分的纤维聚合物相同或不同的主链结构的纤维聚合物,并且如果第一组分含有亲核基团,则第二组分含有亲电基团,或者如果第一组分含有亲电基团,则第二组分含有亲核基团;
16.b)第二组分是第一组分的纤维聚合物上的涂层,并且其中如果第一组分含有亲核基团,则所述涂层含有亲电基团,或者如果第一组分含有亲电基团,则所述涂层含有亲核基团;或者
17.c)第二组分是分散和截留在第一组分的纤维聚合物的间隙内的干燥粉末,其中如果第一组分含有亲核基团,则所述粉末含有亲电基团,或者如果第一组分含有亲电基团,则所述粉末含有亲核基团。
18.us 2012/0021058描述了制备止血组合物的方法,所述方法包括:a)提供生物相容性聚合物的干颗粒制剂;以及b)用凝血诱导剂(例如凝血酶溶液)的制剂涂覆所述干颗粒制剂中的颗粒。生物相容性聚合物可以选自明胶、可溶性胶原、白蛋白、血红蛋白、纤维蛋白原、纤维蛋白、酪蛋白、纤连蛋白、弹性蛋白、角蛋白、层粘连蛋白、及其衍生物或组合。
19.us 2013/0316974描述了止血材料,其包含压实的orc粉末,该粉末包含平均纵横比为约1至约18的颗粒。止血材料可以进一步包含选自多糖、钙盐、抗感染剂、止血促进剂、明胶、胶原的添加剂。
20.us 2016/0271228描述了止血组合物,其包含:
21.·
颗粒形式的止血生物相容性聚合物,其选自蛋白质、多糖、生物聚合物、非生物聚合物、及其衍生物和组合,其中所述颗粒形式的止血生物相容性聚合物以中值直径为50μm至700μm的颗粒状颗粒存在;
22.·
一种包含亲电反应性基团的亲水性交联剂,其中所述亲电反应性基团保持其反应性直到所述组合物暴露于患者的血液,其中所述亲电反应性基团被配置成与所述患者的血液蛋白交联以形成具有密封和止血性质的凝胶;以及
23.·
粘合剂,其不与所述一种亲水性交联剂的亲电反应性基团反应;
24.其中所述止血组合物呈糊剂形式。
25.wo 2012/057628描述了用于生产生物相容性交联聚合物的试剂盒,所述试剂盒包含亲电活化的聚噁唑啉(el-pox),所述el-pox包含m个亲电基团;并且所述亲核交联剂包含n个亲核基团,其中所述m个亲电基团能够与所述n个亲核基团反应以形成共价键;其中m》2,n》2且m n》5;其中所述m个亲电基团中的至少一个是侧基亲电基团。
26.wo 2016/056901描述了选自经涂覆的网、经涂覆的泡沫或经涂覆的粉末的粘附性止血产品,所述止血产品包含:
27.·
多孔固体基材,其具有至少5vol.%的孔隙率并且包含外表面,所述外表面包含含有反应性亲核基团的亲核聚合物;
28.·
覆盖所述固体基材的至少一部分的粘附性涂层,所述涂层包含亲电活化的聚噁唑啉(el-pox),所述el-pox平均包含至少1个反应性亲电基团。
技术实现要素:
29.发明人已经开发了止血粉末,其可以方便地用于控制手术期间的出血,甚至用于抗凝血。
30.本发明的止血粉末包含至少10wt.%的颗粒附聚物,所述颗粒附聚物具有1μm至500μm的直径并且包含:
31.·
亲电聚噁唑啉颗粒,其含有携带至少3个反应性亲电基团的亲电聚噁唑啉,所述反应性亲电基团能够在形成共价键的情况下与血液中的胺基团反应;以及
32.·
亲核聚合物颗粒,其含有携带至少3个反应性亲核基团的水溶性亲核聚合物,所述反应性亲核基团能够在水的存在下在所述亲电聚噁唑啉与所述亲核聚合物之间形成共价键的情况下与所述亲电聚噁唑啉的所述反应性亲电基团反应。
33.当应用于出血部位时,本发明的止血粉末变成凝胶,同时与存在于血液中和周围组织上的蛋白质结合。止血粉末的突出止血能力是由于高活性诱导的血液凝结和粘附至组织的强胶凝血凝块的形成。止血粉末可以容易地分布在出血部位。如果需要,可以添加额外的粉末,因为这将形成新的胶凝血凝块层,该胶凝血凝块层将粘附至胶凝血凝块的底层。
34.尽管发明人不希望受理论束缚,但据信当止血粉末与血液接触时,含有亲电聚噁唑啉的亲电聚噁唑啉颗粒快速溶解并同时与血液中的蛋白质和亲核聚合物颗粒中的水溶性亲核聚合物的反应性亲核基团反应。因此,形成了胶凝血凝块层。溶解的亲电聚噁唑啉也将与出血部位的周围组织中的蛋白质反应,从而将胶凝血凝块层固定至组织并密封出血区域。
35.与包含亲电聚噁唑啉和水溶性亲核聚合物的分子混合物的颗粒相比,本发明的颗粒附聚物提供的优点在于它们提供了更好的密封性能。据信这是由于以下事实:当颗粒附聚物与血液接触时,亲电聚噁唑啉与水溶性亲核聚合物以相对慢的速率反应,从而允许亲电聚噁唑啉不仅与水溶性亲核聚合物反应,还与出血部位的血液和组织中的蛋白质反应。
36.与由亲电聚噁唑啉颗粒和水溶性亲核聚合物颗粒组成的粉末混合物相比,本发明的颗粒附聚物提供的优点在于形成了更均匀的强凝胶。据信,当颗粒附聚物与血液接触时,形成亲电聚噁唑啉和水溶性亲核聚合物的非常均匀的分散体,并且当聚合物组分溶解在其
中并开始反应时,形成均匀的强凝胶。
37.本发明还提供制备止血颗粒附聚物的方法,所述止血颗粒附聚物具有:
38.(a)亲电聚噁唑啉颗粒,其含有携带至少3个反应性亲电基团的亲电聚噁唑啉,所述反应性亲电基团能够在形成共价键的情况下与血液中的胺基团反应;以及
39.(b)亲核聚合物颗粒,其含有携带至少3个反应性亲核基团的水溶性亲核聚合物,所述反应性亲核基团能够在所述亲电聚噁唑啉与所述亲核聚合物之间形成共价键的情况下与所述亲电聚噁唑啉的所述反应性亲电基团反应;
40.所述方法包括在非水性制粒液的存在下将100重量份的所述亲电聚噁唑啉颗粒与10重量份至1000重量份的所述亲核聚合物颗粒组合的步骤。
41.发明人出乎意料地发现,通过使用亲电聚噁唑啉不溶于其中并且亲核聚合物在某种程度上可溶于其中的非水性制粒液,可以将亲电聚噁唑啉和水溶性亲核聚合物组合成单个颗粒,同时使亲电聚噁唑啉之间的交联反应最小,并且使亲电聚噁唑啉的降解最小。尽管发明人不希望受理论束缚,但据信使用亲电聚噁唑啉不溶于其中的非水性制粒液确保在制粒期间,亲电聚噁唑啉与亲核聚合物之间不会发生交联反应。同样,以这种方式使亲电聚噁唑啉的降解(水解)最小化。与亲电聚噁唑啉相反,一些亲核聚合物将溶解在非水性制粒液中,从而形成能够将亲电聚噁唑啉颗粒和亲核聚合物颗粒粘合在一起的粘性物质。
42.进一步提供了生物相容性柔性止血片,其包括:
43.·
包含三维互连的间隙空间的粘性纤维载体结构;以及
44.·
分布在所述间隙空间内的本发明的所述止血粉末。
具体实施方式
45.因此,本发明的第一方面涉及止血粉末,其包含至少10wt.%的颗粒附聚物,所述颗粒附聚物具有1μm至500μm的直径并且包含:
46.·
亲电聚噁唑啉颗粒,其含有携带至少3个反应性亲电基团的亲电聚噁唑啉,所述反应性亲电基团能够在形成共价键的情况下与血液中的胺基团反应;以及
47.·
亲核聚合物颗粒,其含有携带至少3个反应性亲核基团的水溶性亲核聚合物,所述反应性亲核基团能够在水的存在下在亲电聚噁唑啉与亲核聚合物之间形成共价键的情况下与亲电聚噁唑啉的反应性亲电基团反应。
48.如本文使用的术语“颗粒附聚物”是指包含全部结合在一起的两个或更多个颗粒的颗粒。
49.如本文使用的术语“聚噁唑啉”是指聚(n-酰基亚烷基亚胺)或聚(芳酰基亚烷基亚胺),并且还被称为pox。pox的实例是聚(2-乙基-2-噁唑啉)。术语“聚噁唑啉”也包括pox共聚物。
50.如本文使用的术语“水溶性亲核聚合物”是指在3至7的ph下,在20℃的脱盐水中具有至少50g/l的水溶性的亲核聚合物。壳聚糖是水溶性亲核聚合物的实例。在ph《6下,壳聚糖在水中是水溶性的。为了测定亲核聚合物在不同ph下的水溶性,使用盐酸调节脱盐水的ph。
51.如本文使用的术语“胶原”是指动物体内各种结缔组织的细胞外间隙中的主要结构蛋白。胶原形成三条多肽链的特征性三螺旋。根据矿化程度,胶原组织可以是刚性的(骨
骼)或顺应性的(肌腱)或具有从刚性到顺应性的梯度(软骨)。除非另外说明,术语“胶原”还包括明胶以外的改性胶原(例如,交联胶原)。
52.如本文使用的术语“明胶”是指通过从动物(例如家养的牛、鸡、猪和鱼)的皮肤、骨骼和结缔组织提取的胶原的部分水解产生的肽和蛋白质的混合物。在水解期间,各胶原链之间的天然分子键被分解成更容易重排的形式。如本文使用的术语“明胶”还包括改性明胶,例如交联明胶和减少交联的明胶。
53.如本文使用的术语“减少交联的明胶”是指已经部分水解的交联明胶。交联明胶中的肽键的部分水解可以通过例如碱性处理来实现。交联明胶的水解导致游离羧基和游离胺基团的密度增加和水溶性增加。
54.除非另外说明,如本文使用的术语“止血片”是指具有阻止来自受损组织出血的能力的片。本发明的止血片可以通过将血液转变成凝胶和/或通过形成封闭伤口部位的密封来实现止血。
55.如本文使用的关于纤维载体结构的术语“耐水性”是指该结构不是水溶性的,并且在中性ph条件(ph 7)和37℃的温度下不会在水中崩解而形成胶态分散体。
56.如本文使用的术语“间隙空间”是指纤维载体结构内的空隙(“空的”)空间。纤维载体结构内的间隙空间允许将止血粉末引入结构中。而且,血液和其它体液可以进入间隙空间,从而允许止血粉末发挥其止血效果和/或为止血片提供组织粘附性。
57.除非另外说明,如本文使用的术语“蛋白质”也包括交联和水解的蛋白质。同样,除非另外说明,每当提及特定的蛋白质种类,例如明胶或胶原时,也包括该蛋白质种类的水解和交联版本。
58.止血粉末、颗粒附聚物、亲电聚噁唑啉颗粒和亲核聚合物颗粒的直径分布可以适当地通过使用malvern mastersizer 2000结合不锈钢样品分散单元的激光衍射来测定。样品分散单元填充有约120ml的环己烷,其在1800rpm的搅拌速度下稳定5至10分钟,随后进行背景测量(空白测量)。将样品管摇动并且水平转动20次。然后,将约50mg分散在含有环己烷的样品分散单元中。在将样品引入分散单元之后,在进行测量之前,将样品在1800rpm下搅拌1.5分钟以确保所有颗粒被适当地分散。对分散的颗粒不进行超声。平均粒度表示为d[4,3],体积加权平均直径表示为(σnidi4)/(σnidi3)。
[0059]
除了包含亲电聚噁唑啉颗粒和亲核聚合物颗粒的颗粒附聚物之外,本发明的止血粉末还可以包含其它颗粒组分,例如生物相容性(生物)聚合物,如明胶海绵(gelfoam)或淀粉。优选地,止血粉末含有至少30wt.%,更优选至少60wt.%和最优选至少80wt.%的颗粒附聚物。
[0060]
止血粉末中的颗粒附聚物优选含有至少10wt.%,更优选至少25wt.%和最优选至少40wt.%的亲电聚噁唑啉。
[0061]
亲核聚合物优选以至少10wt.%,更优选至少12wt.%和最优选至少30wt.%的浓度包含在颗粒附聚物中。
[0062]
亲电聚噁唑啉和亲核聚合物的组合通常占颗粒附聚物的至少10wt.%。更优选地,这两种聚合物的组合占颗粒附聚物的至少50wt.%,最优选至少75wt.%。
[0063]
除了亲电聚噁唑啉和亲核聚合物之外,颗粒附聚物还可以包含一种或多种其它组分,例如多糖。可用于其中的多糖的实例包括直链淀粉、麦芽糖糊精、支链淀粉、淀粉、葡聚
糖、透明质酸、肝素、软骨素硫酸盐、皮肤素硫酸盐、乙酰肝素硫酸盐、角质素硫酸盐、葡聚糖硫酸盐、戊聚糖多硫酸盐、海藻酸盐及其组合。通常,颗粒附聚物中的多糖的量不超过50wt.%。如果多糖包含在颗粒附聚物中,亲电聚噁唑啉、亲核聚合物和多糖的组合优选占颗粒附聚物的至少60wt.%,更优选至少80wt.%和最优选至少90wt.%。
[0064]
根据另一个有利的实施方案,颗粒附聚物包含干燥缓冲体系。优选地,缓冲体系的缓冲ph为7至11,更优选8至10。优选地,缓冲体系具有至少10mmol.l-1
.ph-1
的缓冲能力。更优选缓冲容量为至少25mmol.l-1
.ph-1
,最优选缓冲容量为至少50mmol.l-1
.ph-1
。可用于颗粒附聚物的生物相容性缓冲体系的实例包括磷酸钠/碳酸钠缓冲液;硼酸钠十水合物缓冲液;tris缓冲蛋白质;hepes缓冲盐水和碳酸氢钠/碳酸盐缓冲液。
[0065]
如将在下面解释的,本发明的颗粒附聚物可以在不使用制粒粘合剂的情况下制备,所述制粒粘合剂即在制粒期间使用以提供亲电聚噁唑啉颗粒与亲核聚合物颗粒之间的粘附的组分。因此,在优选实施方案中,颗粒附聚物不包含制粒粘合剂。
[0066]
本发明的止血粉末优选含有至少10wt.%的直径为1μm至200μm的颗粒附聚物,更优选至少10wt.%的直径为1μm至100μm的颗粒附聚物,最优选至少10wt.%的直径为1μm至75μm的颗粒附聚物。
[0067]
止血粉末的体积加权平均直径(d[4,3],(∑nid4)/(∑nid3))优选为10μm至1000μm,更优选15μm至500μm,最优选30μm至300μm。
[0068]
止血粉末中的颗粒附聚物的d[4,3]优选为10μm至200μm,更优选15μm至100μm,最优选30μm至60μm。
[0069]
颗粒附聚物中的亲电聚噁唑啉颗粒的体积加权平均直径(d[4,3])优选为1μm至100μm,更优选50μm至50μm,最优选10μm至40μm。
[0070]
颗粒附聚物中的亲核聚合物颗粒的体积加权平均直径(d[4,3])优选为10μm至300μm,更优选15μm至200μm,最优选20μm至100μm。
[0071]
亲电聚噁唑啉颗粒优选含有至少30wt.%,更优选至少50wt.%和最优选至少80wt.%的亲电聚噁唑啉。
[0072]
亲电聚噁唑啉在20℃的蒸馏水中的溶解度优选为至少100g/l,更优选为至少300g/l。
[0073]
亲电聚噁唑啉在20℃下在异丙醇中的溶解度优选小于10mg/l,更优选小于5mg/l,最优选小于1mg/l。
[0074]
亲电聚噁唑啉优选具有至少2kda的分子量。更优选地,亲电聚噁唑啉具有5kda至200kda,最优选10kda至100kda的分子量。
[0075]
如本文前面所解释的,发明人已经发现了将亲电聚噁唑啉和水溶性亲核聚合物组合成单个颗粒的方式,其中亲电聚噁唑啉之间的交联反应最小,并且亲电聚噁唑啉的降解最小。因此,在本发明非常优选的实施方案中,颗粒附聚物中的亲电聚噁唑啉的多分散性指数(pdi)小于2.0,更优选小于1.8,最优选小于1.5。
[0076]
亲电聚噁唑啉优选含有至少4个反应性亲电基团,更优选至少8个反应性亲电基团,甚至更优选至少16个反应性亲电基团,最优选至少32个反应性亲电基团。
[0077]
亲电聚噁唑啉通常平均携带至少10个,更优选至少20个反应性亲电基团。
[0078]
亲电聚噁唑啉优选衍生自其重复单元由下式(i)表示的聚噁唑啉:
[0079]
(chr1)mncor2[0080]
其中r2和每个r1独立地选自h、任选取代的c
1-22
烷基、任选取代的环烷基、任选取代的芳烷基、任选取代的芳基;并且m是2或3。
[0081]
优选地,式(i)中的r1和r2选自h和c
1-8
烷基,甚至更优选选自h和c
1-4
烷基。r1最优选为h。式(i)中的整数m优选等于2。
[0082]
根据优选实施方案,聚噁唑啉是聚合物,甚至更优选是2-烷基-2-噁唑啉的均聚物,所述2-烷基-2-噁唑啉选自2-甲基-2-噁唑啉、2-乙基-2-噁唑啉、2-丙基-2-噁唑啉、2-丁基-2-噁唑啉及其组合。优选地,聚噁唑啉是2-丙基-2-噁唑啉或2-乙基-噁唑啉的均聚物。最优选地,聚噁唑啉是2-乙基-噁唑啉的均聚物。
[0083]
根据特别优选的实施方案,亲电聚噁唑啉包含至少20个噁唑啉单元,更优选至少30个噁唑啉单元,最优选至少80个噁唑啉单元。亲电聚噁唑啉优选每个噁唑啉残基平均包含至少0.05个反应性亲电基团。甚至更优选地,亲电聚噁唑啉每个噁唑啉残基平均包含至少0.1个反应性亲电基团。最优选地,亲电聚噁唑啉每个噁唑啉残基平均包含0.12个至0.5个反应性亲电基团。
[0084]
聚噁唑啉可以在其侧链携带反应性亲电基团(侧基反应性亲电基团)、在其末端处携带反应性亲电基团,或者在其侧链和末端处携带反应性亲电基团。根据本发明使用的亲电聚噁唑啉有利地包含一个或多个侧基反应性亲电基团。通常,亲电聚噁唑啉每个单体含有0.03至0.5个侧基反应性亲电基团,更优选每个单体含有0.04至0.35个侧基反应性亲电基团,甚至更优选每个单体含有0.05至0.25个侧基反应性亲电基团。
[0085]
根据优选实施方案,亲电聚噁唑啉的反应性亲电基团选自羧酸酯、磺酸酯、膦酸酯、五氟苯基酯、对硝基苯基酯、对硝基噻吩基酯、酰卤基团、酸酐、酮、醛、异氰酸根合、硫代异氰酸根合、异氰基、环氧化物、活化羟基基团、烯烃、缩水甘油醚、羧基、琥珀酰亚胺基酯、磺基琥珀酰亚胺基酯、马来酰亚氨基(马来酰亚胺基)、乙烯磺酰基、酰亚氨基酯、乙酰乙酸酯、卤代缩醛、邻二硫吡啶、二羟基-苯基衍生物、乙烯基、丙烯酸酯、丙烯酰胺、碘乙酰胺及其组合。更优选地,反应性亲电基团选自羧酸酯、磺酸酯、膦酸酯、五氟苯基酯、对硝基苯基酯、对硝基噻吩基酯、酰卤基团、酸酐、酮、醛、异氰酸根合、硫代异氰酸根合、异氰基、环氧化物、活化羟基基团、缩水甘油醚、羧基、琥珀酰亚胺基酯、磺基琥珀酰亚胺基酯、酰亚氨基酯、二羟基-苯基衍生物及其组合。甚至更优选地,反应性亲电基团选自卤代缩醛、邻二硫吡啶、马来酰亚胺、乙烯砜、二羟基苯基衍生物、乙烯基、丙烯酸酯、丙烯酰胺、碘乙酰胺、琥珀酰亚胺基酯及其组合。最优选地,反应性亲电基团选自马来酰亚胺、乙烯基、丙烯酸酯、丙烯酰胺、琥珀酰亚胺基酯、磺基琥珀酰亚胺基酯及其组合。
[0086]
可使用的琥珀酰亚胺基酯的实例包括琥珀酰亚胺基戊二酸酯、琥珀酰亚胺基丙酸酯、琥珀酰亚胺基琥珀酰胺、琥珀酰亚胺基碳酸酯、二琥珀酰亚胺基辛二酸酯、双(磺基琥珀酰亚胺基)辛二酸酯、二硫代双(琥珀酰亚胺基丙酸酯)、双(2-琥珀酰亚氨基氧基羰氧基)乙基砜、3,3'-二硫代双(磺基琥珀酰亚胺基丙酸酯)、琥珀酰亚胺基氨基甲酸酯、磺基琥珀酰亚胺基(4-碘乙酰基)氨基苯甲酸酯、双(磺基琥珀酰亚胺基)辛二酸酯、磺基琥珀酰亚胺基-4-(n-马来酰亚氨基甲基)-环己烷-1-甲酸酯、二硫代双-磺基琥珀酰亚胺基丙酸酯、二磺基琥珀酰亚胺基酒石酸酯;双[2-(磺基-琥珀酰亚胺基氧羰基氧乙基砜)]、乙二醇双(磺基琥珀酰亚胺基琥珀酸酯)、二硫代双(琥珀酰亚胺基丙酸酯)。
[0087]
可以使用的二羟基苯基衍生物的实例包括二羟基苯基丙氨酸、3,4-二羟基苯基丙氨酸(dopa)、多巴胺、3,4-二羟基氢化肉桂酸(doha)、去甲肾上腺素、肾上腺素和儿茶酚。
[0088]
包含在亲核聚合物颗粒中的水溶性亲核聚合物优选在3至7的ph下在20℃的脱盐水中的溶解度为至少100g/l,更优选至少150g/l,最优选至少200g/l。如果亲电聚噁唑啉与血液组分、组织和/或亲核聚合物反应时释放酸性物质,则本发明可以适当地使用在酸性ph下可溶的亲核聚合物。例如,当亲电聚噁唑啉含有n-羟基琥珀酰亚胺基团时就是这种情况。
[0089]
亲核聚合物颗粒优选含有至少30wt.%,更优选至少50wt.%和最优选至少80wt.%的亲核聚合物。
[0090]
根据特别优选的实施方案,在附聚物颗粒中使用的水溶性亲核聚合物在水中相对缓慢地溶解。如本文前面所解释的,据信当亲电聚噁唑啉与水溶性亲核聚合物以相对慢的速率反应时,亲电聚噁唑啉也可以与出血部位的血液和组织中的蛋白质反应。通过使用相对缓慢溶解的水溶性亲核聚合物,溶解的亲电聚噁唑啉具有与血液和组织中的蛋白质以及与(缓慢)水溶性亲核聚合物反应的机会,从而产生强的均匀的密封凝胶。
[0091]
具有高分子量的水溶性亲核聚合物倾向于在水中相对缓慢地溶解。因此,在非常优选的实施方案中,包含在亲核聚合物颗粒中的水溶性亲核聚合物具有至少3kda,更优选至少10kda,最优选20kda至3,000kda的分子量。
[0092]
亲核聚合物优选含有至少4个反应性亲核基团,更优选至少8个反应性亲核基团,最优选至少10个反应性亲核基团。最优选地,这些反应性亲核基团是胺基团,最优选伯胺基团。
[0093]
可适用于亲核聚合物颗粒的水溶性亲核聚合物的实例包括蛋白质、壳聚糖、亲核聚噁唑啉、亲核聚乙二醇、聚乙烯亚胺及其组合。更优选地,亲核聚合物选自明胶、胶原、壳聚糖、亲核聚噁唑啉及其组合。
[0094]
根据特别有利的实施方案,根据本发明使用的亲核聚合物颗粒通过加速凝血过程提供止血。胶原能够活化凝血级联的固有途径。胶原具有大的表面积,其充当血小板活化、附聚和血栓形成的基质。明胶颗粒具有限制血流和提供用于凝块形成的基质的能力。因此,在非常优选的实施方案中,亲核聚合物选自明胶、胶原及其组合。最优选地,亲核聚合物是明胶,甚至更优选交联明胶。
[0095]
交联明胶的分子量优选为30kda至3,000kda,更优选400kda至2,000kda,最优选500kda至1,500kda。
[0096]
交联明胶的平均伯胺含量为5
×
10-4
μmol至2
×
10-2
μmol,更优选1.0
×
10-3
μmol至1.0
×
10-2
μmol的伯胺/μg的减少交联的明胶。
[0097]
根据另一个有利的实施方案,亲核聚合物是亲核聚乙二醇(peg)。优选地,亲核peg含有至少3个,更优选至少5个,最优选8个反应性亲核基团。
[0098]
根据另一个优选实施方案,亲核聚合物是壳聚糖。壳聚糖是甲壳素(聚-n-乙酰基-d-葡糖胺)的可生物降解的、无毒的、复杂的碳水化合物衍生物,甲壳素是天然存在的物质。壳聚糖是脱乙酰化形式的甲壳素。根据本发明应用的壳聚糖优选具有大于70%的脱乙酰度。根据本发明使用的壳聚糖优选具有至少5kda,更优选10kda至10,000kda的分子量。
[0099]
根据另一个非常有利的实施方案,亲核聚合物是亲核聚噁唑啉。优选地,亲核聚噁唑啉含有至少3个,更优选至少5个,最优选8至20个反应性亲核基团。
[0100]
与天然存在的亲核生物聚合物如明胶、胶原和壳聚糖相比,使用合成的亲核聚合物如亲核聚噁唑啉和亲核peg提供了以下优点:含有这些合成的亲核聚合物的颗粒附聚物表现出更可预测的(可再现的)止血性质。
[0101]
水溶性亲核聚合物的反应性亲核基团优选选自胺基团、硫醇基团及其组合。
[0102]
根据优选实施方案,水溶性亲核聚合物含有两个或更多个胺基团,并且亲电聚噁唑啉中的反应性亲电基团选自羧酸酯、磺酸酯、膦酸酯、五氟苯基酯、对硝基苯基酯、对硝基噻吩基酯、酰卤基团、酸酐、酮、醛、异氰酸根合、硫代异氰酸根合、异氰基、环氧化物、活性羟基基团、缩水甘油醚、羧基、琥珀酰亚胺基酯、磺基琥珀酰亚胺基酯、酰亚氨基酯、二羟基-苯基衍生物及其组合。
[0103]
根据另一个优选实施方案,水溶性亲核聚合物含有两个或更多个硫醇基团,并且亲电聚噁唑啉的反应性亲电基团选自卤代缩醛、邻二硫吡啶、马来酰亚胺、乙烯砜、二羟基苯基衍生物、乙烯基、丙烯酸酯、丙烯酰胺、碘乙酰胺、琥珀酰亚胺基酯、磺基琥珀酰亚胺基酯及其组合。更优选地,反应性亲电基团选自琥珀酰亚胺基酯、磺基琥珀酰亚胺基酯、卤代缩醛、马来酰亚胺或二羟基苯基衍生物及其组合。最优选地,反应性亲电基团选自马来酰亚胺或二羟基苯基衍生物及其组合。
[0104]
根据特别优选的实施方案,由亲电聚噁唑啉提供的反应性亲电基团的总数与由亲核聚合物提供的反应性亲核基团的总数之间的比率为1:1.5至1.5:1,更优选1:1.2至1.2:1,最优选1:1.1至1.1:1。
[0105]
本发明人出乎意料地发现,如果颗粒附聚物含有1wt.%至20wt.%,更优选2.5wt.%至15wt.%,最优选5wt.%至10wt.%的非反应性非离子聚合物,则可获得对器官如脾脏和肾脏显示出优异的粘附的颗粒附聚物。该非反应性非离子聚合物不包含反应性亲电基团或反应性亲核基团。
[0106]
在非常优选的实施方案中,用非反应性非离子聚合物涂覆附聚的颗粒。
[0107]
非反应性非离子聚合物的熔点优选为40℃至70℃,更优选45℃至65℃,最优选50℃至60℃。在此,熔点是指聚合物完全熔融时的温度。
[0108]
可适用于本发明附聚物颗粒的非反应性非离子聚合物的实例包括泊洛沙姆、聚乙二醇及其组合。泊洛沙姆是非离子三嵌段共聚物,其由聚氧丙烯(聚(环氧丙烷))的中心疏水链与聚氧乙烯(聚(环氧乙烷))的两个亲水链侧接组成,并且由式(i)表示:
[0109][0110]
其中a是10至110的整数并且b是20至60的整数。当a是80且b是27时,该聚合物被称为泊洛沙姆188。可用于本发明的其它已知的泊洛沙姆是泊洛沙姆237(a=64;并且b=37)、泊洛沙姆338(a=141;并且b=44)和泊洛沙姆407(a=101;并且b=56)。其它已知的且可用于本发明的泊洛沙姆包括泊洛沙姆108、泊洛沙姆182、泊洛沙姆183、泊洛沙姆212、泊洛沙姆217、泊洛沙姆238、泊洛沙姆288、泊洛沙姆331、泊洛沙姆338和泊洛沙姆335。
[0111]
根据特别优选的实施方案,非反应性非离子聚合物是泊洛沙姆,甚至更优选平均
分子量为2,000至18,000的泊洛沙姆、最优选平均分子量为7,000至10,000的泊洛沙姆。
[0112]
应用于颗粒附聚物中的泊洛沙姆优选在室温下是固体。
[0113]
在另一个有利的实施方案中,本发明的止血粉末是生物可吸收的,允许该粉末用于腹部手术。
[0114]
本发明的另一方面涉及制备止血颗粒附聚物的方法,所述止血颗粒具有:
[0115]
(a)亲电聚噁唑啉颗粒,其含有携带至少3个反应性亲电基团的亲电聚噁唑啉,所述反应性亲电基团能够在形成共价键的情况下与血液中的胺基团反应;以及
[0116]
(b)亲核聚合物颗粒,其含有携带至少3个反应性亲核基团的水溶性亲核聚合物,所述反应性亲核基团能够在亲电聚噁唑啉与亲核聚合物之间形成共价键的情况下与亲电聚噁唑啉的反应性亲电基团反应;
[0117]
所述方法包括在非水性制粒液的存在下将100重量份的亲电聚噁唑啉颗粒与10重量份至1000重量份的亲核聚合物颗粒组合。
[0118]
在本方法中使用的亲电聚噁唑啉颗粒优选与本文以上所述的亲电聚合物颗粒相同。同样,所使用的亲核聚合物颗粒优选与本文以上所述的亲核聚合物颗粒相同。
[0119]
根据优选实施方案,在本方法中使用的亲电聚噁唑啉颗粒和亲核聚合物颗粒两者具有小于2wt.%,更优选小于1wt.%的水含量。为了获得这样低的水含量,可能需要在制粒之前干燥聚合物颗粒。
[0120]
亲电聚噁唑啉在20℃下在非水性制粒液中的溶解度优选小于1mg/l,更优选小于0.5mg/l,甚至更优选小于0.2mg/l,最优选小于0.1mg/l。
[0121]
亲核聚合物在20℃下在非水性制粒液中的溶解度优选为至少5mg/l,更优选至少10mg/l,甚至更优选为20mg/l至200mg/l,最优选为40mg/l至150mg/l。
[0122]
在优选实施方案中,该方法包括在非水性制粒液的存在下将100重量份的亲电聚噁唑啉颗粒与20重量份至400重量份,更优选50重量份至200重量份的亲核聚合物颗粒组合。
[0123]
在本方法中使用的非水性制粒液的量优选为在该方法中使用的亲电聚噁唑啉颗粒和亲核聚合物颗粒的组合量的0.5重量%至5重量%。更优选地,所使用的非水性制粒液的量为亲电聚噁唑啉颗粒和亲核聚合物颗粒的组合量的1重量%至4重量%,最优选1.5重量%至3重量%。
[0124]
根据特别优选的实施方案,该方法包括用非水性制粒液润湿亲电聚噁唑啉颗粒的步骤,随后是将润湿的聚噁唑啉颗粒与亲核聚合物颗粒组合的步骤。该具体实施方案提供的优点在于其产生在粒度和组成方面非常均匀的颗粒。
[0125]
用于制备粉末共混物的亲电聚噁唑啉颗粒的体积加权平均直径(d[4,3])优选为1μm至100μm,更优选50μm至50μm,最优选10μm至40μm。
[0126]
用于制备粉末共混物的亲核聚合物颗粒的体积加权平均直径(d[4,3])优选为10μm至300μm,更优选15μm至200μm,最优选20μm至100μm。
[0127]
通过本发明方法获得的附聚粉末的体积加权平均直径(d[4,3])优选为12μm至1000μm,更优选20μm至500μm,最优选30μm至300μm。
[0128]
用于湿法制粒的非水性制粒液优选含有至少60wt.%的有机溶剂,所述有机溶剂选自异丙醇、乙醇、甲醇、乙醚、庚烷、己烷、戊烷、环己烷、二氯甲烷、丙酮及其混合物。更优
选地,非水性制粒液含有至少60wt.%,最优选至少85wt.%的有机溶剂,所述有机溶剂选自异丙醇、乙醇及其混合物。甚至更优选地,非水性制粒液含有至少60wt.%,最优选至少85wt.%的异丙醇。
[0129]
非水性制粒液优选含有不超过1wt.%的水,更优选不超过0.1wt.%的水。
[0130]
本方法提供的优点在于不需要使用制粒粘合剂。因此,在该方法的优选实施方案中,不使用制粒粘合剂。根据特别优选的实施方案,在制备方法中使用的仅有材料是亲电聚噁唑啉颗粒、亲核聚合物颗粒和非水性制粒液。
[0131]
本发明的颗粒附聚物可以有利地应用于止血片以改善其止血性能。因此,本发明的又一方面涉及生物相容性柔性止血片,其包括:
[0132]
·
包含三维互连的间隙空间的粘性纤维载体结构;以及
[0133]
·
分布在间隙空间内的如本文以上所述的止血粉末。
[0134]
根据特别优选的实施方案,粘性纤维载体结构是耐水性的。
[0135]
当片与可以渗透间隙空间的血液或其它水性体液接触时,分布在整个纤维载体结构中的止血粉末中的亲电聚噁唑啉迅速溶解。因此,一旦将片应用于伤口部位上,在一方面亲电聚噁唑啉与另一方面亲核聚合物颗粒中的亲核聚合物、血液蛋白和组织之间发生快速共价交联,导致凝胶的形成,该凝胶密封伤口表面并阻止出血,并进一步导致止血片与组织的强粘附。根据本发明应用的壳聚糖优选具有大于70%的脱乙酰度。耐水性纤维载体结构在应用期间和应用之后提供机械强度并且防止过度溶胀。
[0136]
由于亲电聚噁唑啉和亲核聚合物包含在单个颗粒中,因此确保了这两种反应性组分可以均匀地分布在整个止血片中,在运输和处理期间不发生分离,并且当颗粒附聚物与血液接触时这些组分可以立即相互反应。
[0137]
根据特别优选的实施方案,本发明的止血片是生物可吸收的,这意味着载体结构、颗粒附聚物和止血片的任何其它组分最终被吸收在体内。载体结构和颗粒附聚物的吸收通常需要其中包含的聚合物的化学分解(例如水解)。人体对止血片的完全生物吸收通常在1至10周内,优选在2至8周内实现。
[0138]
本发明的止血片通常具有0.5mm至25mm的非压缩平均厚度。更优选地,非压缩平均厚度为1mm至10mm,最优选1.5mm至5mm。
[0139]
止血片的尺寸优选使得片的顶部和底部的表面积各自为至少2cm2,更优选至少10cm2,最优选25cm2至50cm2。通常,片的形状是矩形的,并且具有25mm至200mm的长度,25mm至200mm的宽度。
[0140]
止血片优选具有小于200mg/cm3,更优选小于150mg/cm3和最优选10mg/cm3至100mg/cm3的非压缩密度。
[0141]
本发明的止血片优选基本上是无水的。通常,止血片的水含量不超过5wt.%,更优选不超过2wt.%,最优选不超过1wt.%。
[0142]
止血片的吸水能力优选为至少50%,更优选100%至800%,最优选200%至500%。
[0143]
本发明的止血片优选是无菌的。
[0144]
在本发明的止血片中使用纤维载体结构提供的优点在于止血粉末可以毫无困难地均匀地分布在整个载体结构中。这种均匀分布在例如发泡载体结构中更难实现。
[0145]
纤维载体结构中的纤维优选具有1μm至500μm,更优选2μm至300μm,最优选5μm至
200μm的平均直径。纤维的平均直径可以适当地使用显微镜来测定。
[0146]
典型地,纤维载体结构中的至少50wt.%,更优选至少80wt.%的纤维具有1μm至300μm的直径和至少1mm的长度。
[0147]
优选地,纤维载体结构中的至少50wt.%,更优选至少80wt.%的纤维具有至少1000的纵横比(长度与直径的比率)。
[0148]
根据本发明使用的纤维载体结构优选为毡结构、织造结构或针织结构。最优选地,纤维载体结构是毡结构。在此,术语“毡结构”是指通过将纤维编织和压制在一起以形成粘性材料而产生的结构。
[0149]
纤维载体结构优选包含至少50wt.%,更优选至少80wt.%,最优选至少90wt.%的纤维,所述纤维含有明胶、胶原、纤维素、改性纤维素、羧甲基葡聚糖、plga、透明质酸钠/羧甲基纤维素、聚乙烯醇、壳聚糖或其组合。
[0150]
在本发明的实施方案中,纤维载体结构不包含氧化的再生纤维素。
[0151]
优选的纤维载体结构具有开孔结构,其对空气的渗透率为至少0.1l/min
×
cm2,更优选至少0.5l/min
×
cm2。根据en iso 9237:1995(纺织品-织物对空气的渗透率的测定)测定空气渗透率。
[0152]
纤维载体结构中的纤维可以通过本领域已知的方法生产,例如电纺、电吹纺和高速旋转喷雾器纺丝。us 2015/0010612中描述了通过高速旋转喷雾器纺丝生产纤维载体结构。也可以使用市售的止血纤维片作为纤维载体结构。
[0153]
止血粉末优选以纤维载体结构的5重量%至90重量%,更优选10重量%至80重量%,甚至更优选20重量%至75重量%,最优选50重量%至70重量%的量存在于本发明的止血片中。
[0154]
通过以下非限制性实施例进一步说明本发明。
[0155]
实施例
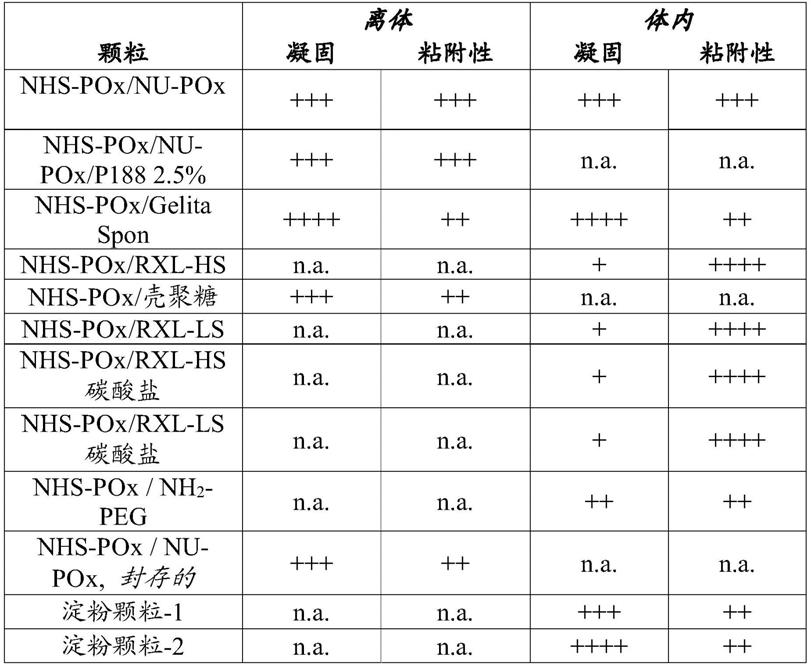
[0156]
通常:无论何处未明确提及干燥后的残余水分(即,干燥粉末、颗粒和/或粘性纤维载体结构中的残余水),水平低于2.0%w/w。
[0157]
nhs-pox的制备
[0158]
含有20%nhs-酯基团的nhs侧链活化的聚[2-(乙基/羟基-乙基-酰胺-乙基/nhs-酯-乙基-酯-乙基-酰胺-乙基)-2-噁唑啉]三元共聚物(=el-pox,20%nhs)如下合成:
[0159]
使用60%2-乙基-2-噁唑啉(etox)和40%2-甲氧基羰基-乙基-2-噁唑啉(mestox)通过crop合成聚[2-(乙基/甲氧基-羰基-乙基)-2-噁唑啉]共聚物(dp= /-100)。获得含有40%2-甲氧基羰基-乙基的统计共聚物(1h-nmr)。其次,含有40%2-甲氧基羰基-乙基的聚合物与乙醇胺反应,得到具有40%2-羟基-乙基-酰胺-乙基的共聚物(1h-nmr)。此后,根据1h-nmr,使一半的2-羟基-乙基-酰胺-乙基与琥珀酸酐反应,得到具有60%2-乙基、20%2-羟基-乙基-酰胺-乙基和20%2-羧基-乙基-酯-乙基-酰胺-乙基的三元共聚物。最后,用n-羟基琥珀酰亚胺(nhs)和二异丙基碳二亚胺(dic)活化2-羧基-乙基-酯-乙基-酰胺-乙基,得到el-pox,20%nhs。根据1h-nmr,nhs-pox含有20%nhs-酯基团。将nhs-pox在2℃至8℃下溶解在水中(60g在300ml中),在-80℃冷却半小时并冷冻干燥。将如此获得的冷冻干燥的粉末在旋转蒸发器中在40℃下干燥直至通过karl fischer滴定测定的水含量低于0.8%w/w。使用球磨机(retch mm400)研磨该干燥(白色)粉末,直至平均粒度不超过40μm(d[4,3]),并
且真空密封在alu-alu袋中。
[0160]
nhs-pox粉末的染色
[0161]
将20g的nhs-pox粉末溶解在水中,并且使用高性能分散仪器(ultra-turrax,ika)与50mg brilliant blue fcf(sigma aldrich)混合。在混合(2分钟)后立即将溶液在-78℃下冷冻,随后冷冻干燥过夜。将如此获得的冷冻干燥的粉末在旋转蒸发器中在40℃下干燥直至通过karl fischer滴定测定的残余水含量低于0.8%w/w。然后,使用球磨机(retch mm400)研磨干燥的(蓝色)粉末,直至蓝色染色的nhs-pox粉末具有不超过40μm(d[4,3])的平均粒度,并且真空密封在alu-alu袋中。
[0162]
nhs-pox的交联对pdi(多分散性指数)的影响
[0163]
通过将80mg的edea溶解在10ml的dcm/ipa 95:5(v/v)中,在二氯甲烷(dcm)和异丙醇(ipa)的混合物中制备2,2'-(亚乙基二氧基)-双-(乙胺)(edea,aldrich,mw 148.2)的80μg/ml溶液。将该溶液用dcm/ipa 95:5(v/v)稀释10倍,并且将26.8μl的edea溶液添加到100μl的在dcm/ipa 95:5(v/v)中的nhs-pox的0.5g/ml溶液中,对应于相对于nhs基团有0.05摩尔%胺基团。在添加edea溶液后立即使用涡旋混合器彻底混合反应混合物。将反应混合物在40℃下搅拌3小时,并通过在减压下旋转蒸发除去所有挥发物。
[0164]
将干燥的样品在含有50mm氯化锂的尺寸排阻色谱(sec)流动相n,n-二甲基乙酰胺中重构。相对于聚(甲基丙烯酸甲酯)标准物测量sec。根据获得的尺寸排阻色谱图,测定mn、mw和pdi。pdi大于2.5。
[0165]
在没有添加edea的情况下,在dcm/ipa 95:5(v/v)中的nhs-pox的0.5g/ml溶液遵循相同的sec程序得到1.5的pdi,这表明nhs基团的仅0.05摩尔%交联已经导致聚合物的pdi的显著增加。
[0166]
nu-pox的制备
[0167]
通过etox和mestox的crop合成在烷基侧链中含有乙基和胺基团的聚噁唑啉,随后用乙二胺对甲基酯侧链进行酰胺化以得到聚(2-乙基/氨基乙基酰氨基乙基-2-噁唑啉)共聚物(nu-pox)。根据1h-nmr,nu-pox含有10%nh2。将nu-pox在2℃至8℃下溶解在水中(60g在300ml中),在-80℃冷却半小时并冷冻干燥。将如此获得的冷冻干燥的粉末在旋转蒸发器中在40℃下干燥直至通过karl fischer滴定测定的水含量低于0.8%w/w。将该干燥粉末在台式研磨机中研磨直至平均粒度不超过100μm(d[4,3]),并且真空密封在alu-alu袋中。
[0168]
反应性nhs-pox/nu-pox颗粒的制备
[0169]
在高剪切混合器中用异丙醇(ipa)润湿蓝色或白色(未染色的)nhs-pox粉末,直至获得含有约1至2%w/w ipa的均匀雪花状粉末。此后,添加nu-pox粉末并且混合。将润湿的蓝色nhs-pox粉末与nu-pox粉末以1:0.6的摩尔比混合,所述摩尔比是指由nhs-pox提供的nhs基团的数量与由nu-pox提供的胺基团的数量的比率。还将润湿的未染色nhs-pox粉末与nu-pox粉末以其它摩尔比(1:0.8;1:1和1:1.2)混合。
[0170]
混合后,将湿颗粒在减压下干燥直至ipa含量小于0.1%w/w,如经由1h-nmr测定的。使用球磨机(retch mm400)研磨干燥的颗粒,直至平均粒度不超过50μm(d[4,3]),并且真空密封在alu-alu袋中。
[0171]
如此获得的颗粒的粒度分布为约:90vol.%《90μm、50vol.%《45μm并且10vol.%《15μm。
[0172]
使用1h-nmr光谱分析nhs-pox/nu-pox颗粒(1:1)。将25mgd的颗粒通过超声20分钟溶解在三氟乙酸(0.20ml)中。在颗粒完全溶解后,用含有马来酸(2.5mg/ml)作为内标的氘化二甲亚砜(dmso-d6)(0.80ml)稀释样品,转移至nmr管并且记录1h-nmr光谱。根据获得的光谱,可以计算与nhs-pox结合的nhs的量,以及颗粒中存在的nhs和胺基团的摩尔比。颗粒中与nhs-pox结合的nhs的量等于与nhs-pox起始材料结合的nhs的量,表明在制粒期间没有衰变或交联。
[0173]
使用已知量的内标(马来酸)和由不同浓度的nhs-pox和nu-pox记录的1h-nmr光谱构建的校准曲线测定nmr样品中的总聚合物回收率,即nhs-pox和nu-pox的组合。测得总聚合物回收率为99%,表明没有形成不溶性交联材料。
[0174]
通过尺寸排阻色谱法进一步分析nhs-pox/nu-pox颗粒(1:1)。将20mg的颗粒用乙酸酐(1.00ml)在50℃下处理1小时。随后,添加甲醇(2.00ml),并且将混合物在50℃下再搅拌1小时。取等分试样(0.75ml),并且在减压下去除所有挥发物。将样品吸收在含有50mm氯化锂的n,n-二甲基乙酰胺(2.50ml)中,其为用于sec分析的洗脱液。相对于聚(甲基丙烯酸甲酯)标准物测量sec,并且根据获得的尺寸排阻色谱图确定mn、mw和pdi。pdi不超过1.5,表明在制粒期间没有发生交联。该尺寸排阻色谱方法的分析验证表明0.05mol%水平的nhs-pox与nu-pox的有意交联将pdi增加至超过2.5。
[0175]
通过共冷冻干燥制备nhs-pox/nu-pox混合物
[0176]
如下制备共冷冻干燥的nhs-pox/nu-pox粉末:将2.4g的nu-pox溶解在40ml的冰醋酸中。在聚合物完全溶解后,添加2.0g的nhs-pox,并将样品超声30分钟。将溶液在液氮中快速冷冻并冷冻干燥。将所得粘性固体在减压(《1kpa)下进一步干燥并真空密封在alu-alu袋中。
[0177]
共冷冻干燥的nhs-pox/nu-pox样品不能通过1h-nmr光谱和尺寸排阻色谱的手段进行分析,因为由于nhs-pox与nu-pox之间的高度交联,样品既不溶于三氟乙酸也不溶于乙酸酐。
[0178]
通过干混制备nhs-pox/nu-pox混合物
[0179]
如下制备nhs-pox/nu-pox混合物:将2g的nhs-pox和2g的nu-pox通过翻滚混合的手段干混30分钟。将所得粉末在减压(《1kpa)下干燥并真空密封在alu-alu袋中。
[0180]
使用1h-nmr光谱分析粉末。将25mg的粉末通过超声20分钟溶解在三氟乙酸(0.20ml)中。在粉末完全溶解后,用含有马来酸(2.5mg/ml)作为内标物的氘化二甲亚砜(dmso-d6)(0.80ml)稀释样品,转移至nmr管并记录1h-nmr光谱。根据获得的光谱,与nhs-pox相比计算未反应的nhs的量为99%。
[0181]
使用已知量的内标物(马来酸)和由不同浓度的nhs-pox和nu-pox记录的1h-nmr光谱构建的校准曲线测定nmr样品中的总聚合物回收率,即nhs-pox和nu-pox的组合。测得总聚合物回收率为101%,表明没有形成不溶性交联材料。
[0182]
通过尺寸排阻色谱法的手段进一步分析粉末。将20mg的粉末用乙酸酐(1.00ml)在50℃下处理1小时。随后,添加甲醇(2.00ml),并将混合物在50℃下再搅拌1小时。取等分试样(0.75ml),并在减压下除去所有挥发物。将样品放入含有50mm氯化锂的n,n-二甲基乙酰胺(其为用于sec分析的洗脱液)(2.50ml)中。相对于聚(甲基丙烯酸甲酯)标准物测量sec,并且根据获得的尺寸排阻色谱图确定mn、mw和pdi。pdi不超过1.5,表明在干混期间没有发生
交联。
[0183]
nhs-pox/nu-pox(封存的)颗粒的制备
[0184]
通过冷冻干燥生产以下粉末:
[0185]
磷酸盐粉末
[0186]
将42.66g的磷酸氢二钠二水合物和26.56g的磷酸一氢钠一水合物溶解在170ml超纯水中。在完全溶解后,通过添加62ml的0.1摩尔氢氧化钠水溶液将ph调节至7。将溶液在液氮中冷冻并冷冻干燥。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0187]
碳酸盐粉末
[0188]
通过将25.31g的碳酸钠和20.06g的碳酸氢钠溶解在350ml超纯水中来制备碳酸钠和碳酸氢钠的1:1摩尔/摩尔混合物。将溶液在液氮中快速冷冻并冷冻干燥。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0189]
nhs-pox/柠檬酸粉末
[0190]
将3.08g的柠檬酸溶解在10ml超纯水中。将溶液在2至8℃冷却。随后,添加7.00g的nhs-pox并借助于高性能分散仪器(ultra-turrax,ika)将其溶解。在混合后(2分钟)立即使用液氮将溶液快速冷冻并冷冻干燥过夜。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0191]
nu-pox/碳酸盐粉末
[0192]
接着,如下制备含碳酸盐的nu-pox粉末:将9.60g的nu-pox和0.80g的碳酸盐粉末溶解在40ml超纯水中。使用液氮快速冷冻溶液并冷冻干燥。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0193]
如下制备nhs-pox/nu-pox(封存的)颗粒:在高剪切混合器中混合6.50g的nhs-pox/柠檬酸粉末和5.50g的磷酸盐粉末。在获得均匀混合物后,缓慢添加0.40ml ipa,同时继续混合,直到形成均匀的雪花状粉末。接着,添加5.41g的nu-pox/碳酸盐粉末,一旦形成均匀的颗粒就停止混合。将颗粒在减压下干燥直至ipa含量小于0.1%w/w。将干燥的颗粒在咖啡研磨机中研磨直至平均粒度不超过100μm(d[4,3])并真空密封在alu-alu袋中。
[0194]
反应性nhs-pox/nu-pox/p188颗粒的制备
[0195]
如前所述用pluronic p188涂覆反应性nhs-pox/nu-pox颗粒。通过在高剪切混合器中在65℃下将nhs-pox/nu-pox颗粒与p188粉末一起加热10分钟,然后冷却至环境条件来制备2.5%w/w p188涂覆的反应性nhs-pox/nu-pox颗粒。使用球磨机(retch mm400)研磨涂覆的颗粒,直至平均粒度不超过40μm(d[4,3]),并且真空密封在alu-alu袋中。
[0196]
如此获得的颗粒的粒度分布为约:90vol.%《80μm,50vol.%《40μm并且10vol.%《10μm。
[0197]
使用1h-nmr光谱分析nhs-pox/nu-pox/p188颗粒。将25mg的粉末通过超声20分钟溶解在三氟乙酸(0.20ml)中。在颗粒完全溶解后,用氘化二甲亚砜(dmso-d6)(0.80ml)稀释样品,转移至nmr管并且记录1h-nmr光谱。根据获得的光谱,与nhs-pox相比计算未反应的nhs的量为98%。
[0198]
通过尺寸排阻色谱法进一步分析nhs-pox/nu-pox/p188颗粒。将20mg的颗粒用乙酸酐(1.00ml)在50℃下处理1小时。随后,添加甲醇(2.00ml),并且将混合物在50℃下再搅拌1小时。取等分试样(0.75ml),并且在减压下去除所有挥发物。将样品吸收在含有50mm氯
化锂的n,n-二甲基乙酰胺(2.50ml)中,其为用于sec分析的洗脱液。相对于聚(甲基丙烯酸甲酯)标准物测量sec,并且从获得的尺寸排阻色谱图确定mn、mw和pdi。pdi不超过1.5,表明在制粒期间没有发生交联。
[0199]
减少交联的明胶(rxl)的制备
[0200]
根据三个程序制备减少交联的明胶(rxl):
[0201]
·
通过在40℃下搅拌2小时,将12g的明胶粉末(gelita-gelita medical gmbh)溶解在350ml的0.1摩尔氢氧化钠水溶液中。获得澄清溶液后,使混合物冷却至环境温度,并且通过添加32.5ml的1.0摩尔盐酸水溶液将ph调节至7。使用液氮快速冷冻溶液并且冷冻干燥。随后,将粉末在咖啡研磨机中研磨,在减压下干燥并且真空密封在alu-alu袋中。此后,这种减少交联的明胶将被称为rxl-ls(低盐)。
[0202]
·
通过在40℃下搅拌10分钟,将12g的明胶粉末(gelita-gelita medical gmbh)溶解在360ml的1.0摩尔氢氧化钠水溶液中。将获得的澄清溶液冷却至环境温度,并且通过添加30ml的浓盐酸溶液(37%w/w)将ph降低至7。使用液氮快速冷冻溶液并且冷冻干燥过夜。随后,将粉末在咖啡研磨机中研磨,在减压下干燥并且真空密封在alu-alu袋中。此后,这种减少交联的明胶将被称为rxl-hs(高盐)。
[0203]
·
通过在40℃下搅拌2小时,将12g的明胶粉末(gelita-gelita medical gmbh)溶解在350ml的0.1摩尔氢氧化钠水溶液中。获得澄清溶液后,使混合物冷却至环境温度,并通过添加32.5ml的1.0摩尔盐酸水溶液将ph调节至7。随后,将400ml的甲醇添加到溶液中,将其置于-20℃的冷冻器中16小时,产生rxl沉淀,通过倾析液相分离rxl。将rxl用100ml部分的甲醇洗涤三次,然后在真空下干燥。将粗产物溶解在200ml超纯水中,在液氮中快速冷冻并冷冻干燥。将粉末在咖啡研磨机中研磨,在减压下干燥并真空密封在alu-alu袋中。此后,这种减少交联的明胶将被称为rxl(无盐)。
[0204]
反应性nhs-pox/rxl颗粒(无盐、低盐和高盐)的制备
[0205]
如下制备nhs-pox/rxl反应性颗粒:在高剪切混合器中用ipa润湿5g的蓝色nhs-pox粉末,直至获得含有约1至2%w/w ipa的均匀雪花状粉末。此后,添加5g的rxl、rxl-ls或rxl-hs粉末并且混合。混合后,将湿颗粒在减压下干燥直至ipa含量小于0.1%w/w,如经由1h-nmr测定的。将干燥的颗粒在咖啡研磨机中研磨,直至平均粒度不超过90μm(d[4,3]),并且真空密封在alu-alu袋中。
[0206]
如此获得的颗粒的粒度分布为约:90vol.%《190μm,50vol.%《60μm并且10vol.%《15μm。
[0207]
通过1h-nmr光谱分析来分析含有rxl的颗粒。为此,将含有5%(v/v)乙酸的氘化氯仿(cdcl3)(1.0ml)添加至25mg的颗粒中。通过超声处理样品20分钟选择性地提取nhs-pox。使分散体通过0.22μm过滤器,转移至nmr管并且记录1h-nmr光谱。根据获得的光谱,与nhs-pox相比计算未反应的nhs的量为98%。
[0208]
使用三甲基硅烷作为内标和由不同浓度的nhs-pox的1h-nmr光谱构建的校准曲线测定nmr样品中nhs-pox的回收率。测得总nhs-pox回收率为100%,表明没有形成不溶性交联材料。
[0209]
通过尺寸排阻色谱法进一步分析nhs-pox/rxl颗粒。因此,从用于1h-nmr光谱分析
的溶液中取得等分试样(0.15ml)。将该溶液用含有50mm氯化锂(1.00ml)的n,n-二甲基乙酰胺稀释,其为用于sec分析的洗脱液。相对于聚(甲基丙烯酸甲酯)标准物测量sec,并且根据获得的尺寸排阻色谱图确定mn、mw和pdi。pdi不超过1.5,再次表明在制粒期间没有发生交联。
[0210]
通过共冷冻干燥制备nhs-pox/rxl混合物
[0211]
如下制备共冷冻干燥的nhs-pox/rxl粉末:将2.5g的rxl粉末溶解在200ml的超纯水中。通过添加乙酸将ph调节至4.5,并将溶液冷却至4℃。随后,添加2.5g的nhs-pox,并借助于高剪切搅拌将其溶解。在nhs-pox完全溶解后立即将溶液在液氮中快速冷冻并冷冻干燥。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0212]
通过1h-nmr光谱分析共冷冻干燥的nhs-pox/rxl粉末。为此,将含有5%(v/v)乙酸的氘化氯仿(cdcl3)(1.0ml)添加到25mg的颗粒中。通过超声处理样品20分钟选择性地提取nhs-pox。使分散体通过0.22μm过滤器,转移至nmr管并记录1h-nmr光谱。根据获得的光谱,与nhs-pox相比计算未反应的nhs的量为93%。
[0213]
使用三甲基硅烷作为内标和由不同浓度的nhs-pox的1h-nmr光谱构建的校准曲线测定nmr样品中nhs-pox的回收率。测得总nhs-pox回收率为81%,表明在某种程度上形成了不溶性交联材料。
[0214]
通过尺寸排阻色谱法进一步分析nhs-pox/rxl粉末。从用于1h-nmr光谱分析的溶液中取等分试样(0.15ml)。将该溶液用含有50mm氯化锂的n,n-二甲基乙酰胺(1.00ml)(其为用于sec分析的洗脱液)稀释。相对于聚(甲基丙烯酸甲酯)标准物测量sec,并且根据获得的尺寸排阻色谱图确定mn、mw和pdi。pdi为3.3,表明在两种组分的共冷冻干燥期间发生了交联。
[0215]
通过干混制备nhs-pox/rxl混合物
[0216]
如下制备nhs-pox/rxl混合物:通过翻滚混合将2g的rxl粉末和2g的nhs-pox干混合30分钟。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0217]
通过1h-nmr光谱分析分析粉末。为此,将含有5%(v/v)乙酸(1.0ml)的氘化氯仿(cdcl3)添加到25mg的颗粒中。通过超声处理样品20分钟选择性地提取nhs-pox。使分散体通过0.22μm过滤器,转移至nmr管并记录1h-nmr光谱。根据获得的光谱,与nhs-pox相比计算未反应的nhs的量为100%。
[0218]
使用三甲基硅烷作为内标物和由不同浓度的nhs-pox的1h-nmr光谱构建的校准曲线测定nmr样品中nhs-pox的回收率。测得总nhs-pox回收率为104%,表明没有形成不溶性交联材料。
[0219]
通过尺寸排阻色谱法进一步分析nhs-pox/rxl粉末。因此,从用于1h-nmr光谱分析的溶液中取等分试样(0.15ml)。将该溶液用含有50mm氯化锂的n,n-二甲基乙酰胺(1.00ml)(其为用于sec分析的洗脱液)稀释。相对于聚(甲基丙烯酸甲酯)标准物测量sec,并且根据获得的尺寸排阻色谱图确定mn、mw和pdi。pdi不超过1.5,表明在干混期间没有发生交联。
[0220]
含碳酸盐的反应性nhs-pox/rxl颗粒的制备
[0221]
首先,通过将25.31g的碳酸钠和20.06g的碳酸氢钠溶解在350ml超纯水中,来制备碳酸钠和碳酸氢钠的1:1摩尔/摩尔混合物。将溶液在液氮中快速冷冻并且冷冻干燥。将所得粉末在减压下干燥并且真空密封在alu-alu袋中。
[0222]
如下制备nhs-pox/rxl/碳酸盐颗粒:使用高剪切混合器混合5g的rxl-ls或rxl-hs和0.178g的碳酸钠/碳酸氢钠。然后,添加含有约1至2%w/w ipa的5g的蓝色nhs-pox并且混合直至获得均匀粉末。混合后,将湿颗粒在减压下干燥直至ipa含量小于0.1%w/w,如经由1h-nmr测定的。将干燥的颗粒在咖啡研磨机中研磨,直至平均粒度不超过100μm(d[4,3]),并且真空密封在alu-alu袋中。
[0223]
反应性nhs-pox/nh
2-peg颗粒的制备
[0224]
在高剪切混合器中用ipa润湿nhs-pox(6,9g),直至获得含有约1至2%w/w ipa的均匀雪花状粉末。随后,添加8.1g的胺-peg-胺(2-臂,mw 2k)(购自creative pegworks)(摩尔比为1:1.16,所述摩尔比是指nhs-pox提供的nhs基团的数量与peg-胺提供的胺基团的数量的比率)。将形成的颗粒在减压下干燥直至ipa含量小于0.1%w/w,如经由1h-nmr测定的。将干燥的颗粒在咖啡研磨机中研磨,直至平均粒度不超过100μm(d[4,3]),并且真空密封在alu-alu袋中。
[0225]
使用1h-nmr光谱分析nhs-pox/nh
2-peg颗粒。将25mg的颗粒通过超声20分钟溶解在三氟乙酸(0.20ml)中。在颗粒完全溶解后,用氘化二甲亚砜(dmso-d6)(0.80ml)稀释样品,转移至nmr管并且记录1h-nmr光谱。根据获得的光谱,与nhs-pox相比计算未反应的nhs的量为97%。
[0226]
通过尺寸排阻色谱法进一步分析nhs-pox/nh
2-peg颗粒。将20mg的颗粒用乙酸酐(1.00ml)在50℃下处理1小时。随后,添加甲醇(2.00ml),并且将混合物在50℃下再搅拌1小时。取等分试样(0.75ml),并且在减压下去除所有挥发物。
[0227]
将样品吸收在含有50mm氯化锂的n,n-二甲基乙酰胺(2.50ml)中,其为用于sec分析的洗脱液。相对于聚(甲基丙烯酸甲酯)标准物测量sec,并且根据获得的尺寸排阻色谱图确定mn、mw和pdi。pdi不超过1.5,表明在制粒期间没有发生交联。
[0228]
淀粉/nhs-pox/nu-pox颗粒的制备
[0229]
通过冷冻干燥生产以下淀粉粉末:
[0230]
nhs-pox/淀粉粉末
[0231]
使用高剪切混合器将2.79g的淀粉(arista
tm
ah,bard)分散在50ml的超纯水中。将分散体在2至8℃冷却,添加0.71g的nhs-pox,并借助于高剪切混合将其溶解。
[0232]
在nhs-pox溶解后立即使用液氮快速冷冻溶液并冷冻干燥。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0233]
nu-pox/淀粉粉末
[0234]
使用高剪切混合器将1.45g的淀粉(arista
tm
ah,bard)分散在30ml的超纯水中。随后,添加0.36g的nu-pox并使其溶解。在nu-pox完全溶解后,使用液氮快速冷冻分散体并冷冻干燥。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0235]
nu-pox/淀粉/碳酸钠粉末
[0236]
使用高剪切混合器将2.25g的淀粉(arista
tm
ah,bard)分散在50ml的超纯水中。随后,添加0.58g的nu-pox和0.25g的碳酸钠并使其溶解。在nu-pox完全溶解后,使用液氮快速冷冻分散体并冷冻干燥。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0237]
由这三种粉末制备两种颗粒。如下制备淀粉/nhs-pox/nu-pox颗粒:
[0238]
淀粉颗粒1:
[0239]
使用研杵和研钵混合1.41g的nhs-pox/淀粉粉末、1.68g的nu-pox/淀粉粉末和0.20ml的ipa。在获得均匀的颗粒后,在减压下除去残留的ipa,直到ipa含量小于0.1%w/w。将干燥的颗粒在咖啡研磨机中研磨直至平均粒度不超过100μm(d[4,3])并真空密封在alu-alu袋中。
[0240]
淀粉颗粒2:
[0241]
使用研杵和研钵混合1.20g的nhs-pox/淀粉粉末、1.45g的nu-pox/淀粉/碳酸钠粉末3和0.15ml的ipa。在形成均匀的颗粒后,在减压下除去残留的ipa,直到ipa含量小于0.1%w/w。将干燥的颗粒在咖啡研磨机中研磨直至平均粒度不超过100μm(d[4,3])并真空密封在alu-alu袋中
[0242]
反应性nhs-pox/gelita spon颗粒的制备
[0243]
使用高剪切混合器在20,000rpm下操作20分钟,将水含量小于0.2%w/w的7.01g的预干燥明胶粉末(gelita-购自gelita medical gmbh)分散在二氯甲烷(200ml)中。随后,添加nhs-pox(7.02g)并且继续搅拌5分钟。nhs-pox不溶解。在减压下从悬浮液中去除所有挥发物。使用咖啡研磨机研磨获得的粉末直至平均粒度不超过95μm(d[4,3])并且真空密封在alu-alu袋中,进一步在减压下干燥并且真空密封在alu-alu袋中。
[0244]
如此获得的颗粒的粒度分布为约:90vol.%《190μm,50vol.%《80μm并且10vol.%《15μm。
[0245]
通过1h-nmr光谱分析来分析颗粒。为此,将含有5%(v/v)乙酸的氘化氯仿(cdcl3)(1.0ml)添加至25mg的颗粒中。通过超声处理样品20分钟选择性地提取nhs-pox。使分散体通过0.22μm过滤器,转移至nmr管并且记录1h-nmr光谱。根据获得的光谱,与nhs-pox相比计算未反应的nhs的量为97%。
[0246]
通过尺寸排阻色谱(sec)分析进一步分析颗粒。将上述过滤的nhs-pox提取物的等分试样(0.15ml)用含有50mm氯化锂的n,n-二甲基乙酰胺(1.00ml)稀释,其为用于sec分析的洗脱液。通过相对于聚(甲基丙烯酸甲酯)标准物的sec分析样品,并且pdi为1.45,表明没有发生交联。
[0247]
反应性nhs-pox/壳聚糖颗粒的制备
[0248]
如下制备nhs-pox/壳聚糖反应性颗粒:在高剪切混合器中用ipa润湿5g的nhs-pox粉末,直到获得含有约1至2%w/w ipa的均匀雪花状粉末。此后,添加5g的壳聚糖粉末(shanghai waseta international,85%dac度)并混合。混合后,在减压下干燥湿颗粒,直到通过1h-nmr测定ipa含量小于0.1%w/w。将干燥的颗粒在咖啡研磨机中研磨直至平均粒度不超过200μm(d[4,3])并真空密封在alu-alu袋中。
[0249]
如此获得的颗粒的粒度分布为约:90vol.%《350μm,50vol.%《180μm和10vol.%《60μm。
[0250]
通过1h-nmr光谱分析分析颗粒。为此,将含有5%(v/v)乙酸的氘化氯仿(cdcl3)(1.0ml)添加到25mg的颗粒中。通过超声处理样品20分钟选择性地提取nhs-pox。使分散体通过0.22μm过滤器,转移至nmr管并记录1h-nmr光谱。根据获得的光谱,与nhs-pox相比计算未反应的nhs的量为95%。
[0251]
使用三甲基硅烷作为内标和由不同浓度的nhs-pox的1h-nmr光谱构建的校准曲线测定nmr样品中nhs-pox的回收率。测得总nhs-pox回收率为103%,表明没有形成不溶性交
联材料。
[0252]
通过尺寸排阻色谱法进一步分析nhs-pox/壳聚糖颗粒。因此,从用于1h-nmr光谱分析的溶液中取等分试样(0.15ml)。将该溶液用含有50mm氯化锂的n,n-二甲基乙酰胺(1.00ml)(其为用于sec分析的洗脱液)稀释。相对于聚(甲基丙烯酸甲酯)标准物测量sec,并且根据获得的尺寸排阻色谱图确定mn、mw和pdi。pdi不超过1.5,表明在制粒期间没有发生交联。
[0253]
通过共冷冻干燥制备nhs-pox/壳聚糖混合物
[0254]
如下制备共冷冻干燥的nhs-pox/壳聚糖粉末:将2.5g的壳聚糖粉末(shanghai waseta international,85%dac度)溶解在200ml的0.2%v/v乙酸超纯水溶液中。通过添加乙酸将ph调节至4.5,并将溶液冷却至4℃。随后,添加2.5g的nhs-pox,并借助于高剪切搅拌将其溶解。在nhs-pox完全溶解后立即将溶液在液氮中快速冷冻并冷冻干燥。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0255]
通过1h-nmr光谱分析分析颗粒。为此,将含有5%(v/v)乙酸的氘化氯仿(cdcl3)(1.0ml)添加到25mg的颗粒中。通过超声处理样品20分钟选择性地提取nhs-pox。使分散体通过0.22μm过滤器,转移至nmr管并记录1h-nmr光谱。根据获得的光谱,与nhs-pox相比计算未反应的nhs的量为12%,这意味着发生交联和/或水解。
[0256]
使用三甲基硅烷作为内标物和由不同浓度的nhs-pox的1h-nmr光谱构建的校准曲线测定nmr样品中nhs-pox的回收率。测得总nhs-pox回收率为11%(与nmr结果一致),表明形成了不溶性交联材料。
[0257]
通过尺寸排阻色谱法进一步分析nhs-pox/壳聚糖颗粒。因此,从用于1h-nmr光谱分析的溶液中取等分试样(0.15ml)。将该溶液用含有50mm氯化锂的n,n-二甲基乙酰胺(1.00ml)(其为用于sec分析的洗脱液)稀释。相对于聚(甲基丙烯酸甲酯)标准物测量sec,并且根据获得的尺寸排阻色谱图确定mn、mw和pdi。pdi测定为5.4,表明发生交联。
[0258]
干混法制备nhs-pox/壳聚糖混合物
[0259]
如下制备nhs-pox/壳聚糖混合物:通过翻滚混合将2g的壳聚糖粉末(shanghai waseta international,85%dac度)和2g的nhs-pox干混合30分钟。将所得粉末在减压下干燥并真空密封在alu-alu袋中。
[0260]
通过1h-nmr光谱分析分析粉末。为此,将含有5%(v/v)乙酸的氘化氯仿(cdcl3)(1.0ml)添加到25mg的颗粒中。通过超声处理样品20分钟选择性地提取nhs-pox。使分散体通过0.22μm过滤器,转移至nmr管并记录1h-nmr光谱。根据获得的光谱,与nhs-pox相比计算未反应的nhs的量为99%。
[0261]
使用三甲基硅烷作为内标物和由不同浓度的nhs-pox的1h-nmr光谱构建的校准曲线测定nmr样品中nhs-pox的回收率。测得总nhs-pox回收率为96%,表明没有形成不溶性交联材料。
[0262]
通过尺寸排阻色谱法进一步分析nhs-pox/壳聚糖粉末。因此,从用于1h-nmr光谱分析的溶液中取等分试样(0.15ml)。将该溶液用含有50mm氯化锂的n,n-二甲基乙酰胺(1.00ml)(其为用于sec分析的洗脱液)稀释。相对于聚(甲基丙烯酸甲酯)标准物测量sec,并且根据获得的尺寸排阻色谱图确定mn、mw和pdi。pdi不超过1.5,表明在干混期间没有发生交联。
[0263]
粘性纤维载体结构
[0264]
选择以下市售止血产品用作用于制备根据本发明的组织粘附性片的纤维载体结构:
[0265]
gelita tuft-由八层减少交联的凝胶泡沫纤维组成的粘性纤维载体结构。每层约2mm厚度的八个层具有50mm
×
75mm的尺寸。gelita tuft-的水含量不超过15%。将产物在真空烘箱中在40℃下干燥数小时以将水含量降低至不超过2.0%w/w(通过重量分析法测定),然后将其用附聚物颗粒进行浸渍。
[0266]
出血实验
[0267]
使用标准化的离体和体内猪出血模型来评估止血功效。所有模型使用肝素将血液的凝固时间增加至活化凝固时间(act)的约2至3倍。
[0268]
离体模型:将来自屠宰场的肝素化新鲜血液灌注具有新鲜肝脏的活的离体猪模型,以尽可能接近地模拟真实的体内情况。将肝脏安装在灌注机上,通过灌注机进行氧合作用,将血液的ph、温度和血压保持在体内边界内。在屠宰场收集两个肝脏和10升肝素化血液(5000单位/l)。肝脏用冰运输;血液在环境温度下运输。在收集后2小时内,针对用手套和氰基丙烯酸酯胶封闭的肝脏检查损伤。
[0269]
·
灌注参数:流速600ml/min;压力10至12mmhg;温度37℃( /-1℃);卡波原0.25升/分钟
[0270]
·
使用平的圆形旋转磨具,在肝脏表面上形成圆形出血伤口(8mm直径),使用橡胶外置物,使得穿孔出血的深度总是3mm
[0271]
·
在肝脏适当灌注(颜色和温度检查)后,根据以下程序测试样品:将样品切割成正确的尺寸(2.7
×
2.7cm);照相机打开;照相机上的位点数量;活检穿孔8mm;切下活组织;用纱布(2x)从出血中去除血液;在预称重的纱布中收集血液30秒;对出血进行评分(由2位研究人员评分);将止血粉末倾倒在出血部位上并且使用不锈钢刮刀均匀地分布粉末;观察5分钟(检查并评分粘附性和凝固)并且在30分钟后重复。
[0272]
体内模型:在经麻醉的猪(家猪,雄性,体重范围:40kg,成年)中造成标准化组合的穿透性脾脏破裂。进行中线剖腹术以通达脾脏和其它器官。使用手术刀,进行n=3(s1
…
s3)囊下标准化损伤(10mm
×
10mm)。通过预润湿的纱布(盐水)在温和的压力下应用止血产品并保持1分钟。在应用产品后,评估止血时间(tth)。如果tth等于零,这意味着在1分钟压力后已经实现了止血。
[0273]
用于颗粒的评分体系:凝固
[0274]
在应用后立即实现
[0275]
在应用后《10秒实现
[0276]
在应用后《30秒实现
[0277]
在应用后3分钟内实现
[0278]-未实现
[0279]
用于颗粒的评分体系:应用后10分钟的粘附性
[0280]
非常强的粘附性(几乎不能移除凝固的颗粒)
[0281]
强粘附性(难以移除凝固的颗粒)
[0282]
强粘附性(可以移除凝固的颗粒)
[0283]
中等粘附性(容易移除凝固的颗粒)
[0284]
/-轻微粘附性(凝固的颗粒被血流移除)
[0285]-未实现
[0286]
用于贴片的评分体系:凝固
[0287]
在压塞后立即实现
[0288]
在压塞后《10秒实现
[0289]
在压塞后《30秒实现
[0290]
在压塞后3分钟内实现
[0291]
/-在3分钟后,施加第二次压塞实现
[0292]-未实现
[0293]
用于贴片的评分体系:应用后10分钟的粘附性
[0294]
非常强的粘附性(在移除时贴片断裂)
[0295]
强粘附性(在移除时贴片断裂)
[0296]
强粘附性(贴片可以在不断裂的情况下移除)
[0297]
中等粘附性(贴片可以在不断裂的情况下移除)
[0298]
/-轻微粘附性(贴片可以在不断裂的情况下移除)
[0299]-未实现
[0300]
实施例1
[0301]
在上述离体和体内出血试验中评价不同反应性颗粒的止血性质。结果总结在表1和表2中。
[0302]
表1
[0303]
[0304]
表2
[0305][0306][0307]
注意:
[1]
未测定(粉末由于高交联度而不可溶)
[0308]
实施例2
[0309]
用颗粒附聚物浸渍载体结构
[0310]
使用机械摇动,用蓝色染色的nhs-pox/nu-pox(1:0.6)颗粒浸渍gelita tuft-贴片(50
×
75mm,约0.71g)。使用涂料摇动机(collomix gmbh的viba pro v)将粉末(约0.75g)引入到贴片中。将具有载体结构保持器的阵列夹在机器中。将阵列垂直振动。
[0311]
将浸渍的样品置于pmma板上并置于烘箱中,在烘箱中对样品进行不同的热处理。为了评价粉末固定,在白色pmma板上轻击样品两次。如果没有释放蓝色粉末,则认为结果是固定的。结果显示在表3中。
[0312]
表3
[0313]
[0314][0315]
nhs-pox/nu-pox颗粒是吸湿的。在环境温度和低于40%的相对湿度(rh)下,纤维载体结构可以可再现地在暴露的半小时内被浸渍。然而,如果在例如rh75%和25℃下进行浸渍,则颗粒在数分钟内变粘,产生不可再现的不均匀浸渍特性。
[0316]
实施例3
[0317]
将止血贴片(gelita tuft-50x75mm,约0.7g)用先前所述的反应性nhs-pox/nu-pox(1:0.6)颗粒浸渍。如实施例2中所述,将1克的颗粒分布在整个贴片中。然后,将止血贴片包装在含有1g的二氧化硅的alu-alu袋中并且真空密封。
[0318]
将贴片切成2cm
×
2cm的片并且在离体肝脏灌注模型中一式三份地测试。止血时间(tth)为0分钟(在1分钟压力后),并且在30分钟观察时间期间未观察到再出血。发现贴片具有很大的柔性和弯曲性质。
[0319]
还在体内猪肝素化模型中评估贴片。发现它们具有非常好的凝固和粘附性质。在以下各种器官的切除中有效地停止活动性出血:脾脏、肝脏和肾脏。表3中示出了结果的概述。
[0320]
表3
[0321][0322]
实施例4
[0323]
将止血贴片(gelita tuft-50x75mm,约0.7g)用nhs-pox/nu-pox/p188 2.5%浸渍。如实施例2中所述,将1克的颗粒分布在整个贴片中。然后,将止血贴片包装在含有1g的二氧化硅的alu-alu袋中并且真空密封。
[0324]
在体内猪肝素化模型中评估贴片。发现它们具有非常好的凝固和足够的粘附性质—与不包括p188的相同贴片相反,降低的粘附性质使得贴片能够整体移除。在以下各种器官的切除中有效地停止活动性出血:脾脏、肝脏和肾脏。表4中示出了结果的概述。
[0325]
表4
[0326][0327]
实施例5
[0328]
将gelita tuft-(50
×
75mm,约0.7g)用不同的反应性聚合物粉末浸渍。如此获得的贴片的止血性质在本文上述离体和体内猪出血模型中测试。
[0329]
使用气动摇动设备用1.4g的粉末浸渍纤维状载体结构。将纤维状载体的片垂直振动。长冲程类型的发动机(ntk 25al l,购自netter vibration gmbh)在6巴、11hz和30mm的振幅下操作。使用4个15秒的循环将粉末分散到片中。颗粒被分布在片的整个厚度上。片的表面上的分布也是均匀的。
[0330]
测试了7种不同的反应性颗粒。这些颗粒含有与水溶性亲核聚合物组合的nhs-pox。本文以上已经描述了这些颗粒的制备。
[0331]
测试的颗粒列举如下:
[0332]
·
nhs-pox/gelita spon
[0333]
·
nhs-pox/rxl(高盐)
[0334]
·
含碳酸盐的nhs-pox/rxl(高盐)
[0335]
·
nhs-pox/rxl(低盐)
[0336]
·
nhs-pox/nh
2-peg
[0337]
测试的纤维载体结构和反应性聚合物粉末的不同组合显示在表5中。
[0338]
表5
[0339][0340]
用这些贴片在离体和体内猪出血模型中获得的结果总结在表6中。
[0341]
表6
[0342]
[0343]
比较例a
[0344]
将7.03g的nhs-pox分散在50ml二氯甲烷(dcm)中。形成混浊的混合物并缓慢添加ipa(4ml)以获得澄清溶液;使用在室温下以20,000rpm操作20分钟的高剪切混合器,将7.00g的预干燥的明胶(gelita-gelita medical gmbh)分散在dcm/ipa(150ml/12ml)中。添加制备的nhs-pox溶液,并将分散体在20,000rpm下搅拌5分钟。随后,在减压下除去所有挥发物。使用咖啡研磨机研磨所形成的颗粒,在减压下干燥并真空密封在alu-alu袋中。
[0345]
使用1h-nmr光谱和尺寸排阻色谱法分析颗粒。如此获得的结果表明,明胶/nhs-pox颗粒中不存在nhs-pox,表明nhs-pox与明胶之间发生了完全交联。
[0346]
实施例6
[0347]
进行实验以确定反应性nhs-pox/nu-pox颗粒的nu-pox含量对止血贴片的体内性能的影响。
[0348]
浸渍方法
[0349]
将止血贴片(gelita tuft-50x75mm,约0.7g)用通过丙酮制粒制备的反应性nhs-pox/nu-pox颗粒以1:0.10、1:0.20和1:0.40的摩尔比浸渍,所述摩尔比是指由nhs-pox提供的nhs基团的数量与由nu-pox提供的胺基团的数量的比率。同样的止血贴片也用反应性nhs-pox粉末浸渍。
[0350]
使用fibroline sl-preg实验室机器将1克颗粒/粉末分布在整个贴片中。然后,将止血贴片固定、干燥和包装在含有1g的二氧化硅的alu-alu袋中并且真空密封。
[0351]
机器
[0352]
fibroline sl-preg实验室机器通过以高达200hz的频率施加高达40kv的电压持续高达60秒的时间在电极之间移动颗粒。两个电极板具有约50
×
40cm的尺寸。顶板接地。
[0353]
使用以下标准设置:40kv,100hz,20秒。
[0354]
阵列
[0355]
在阵列已经安装到底部电极板上之后,将粉末通过重量计量配量到3d印刷的pmma阵列中。使用刮纸盒或金属刮刀用反应性聚合物粉末填充阵列。阵列测量为50
×
75
×
4mm并且含有22
×
33=726个正方形孔(每个孔的内部尺寸:2
×2×
2mm)。726个孔的合并体积为约5.8ml。
[0356]
间隔物
[0357]
将间隔物遮盖物置于阵列的顶部。当经受交变电场时,间隔物用于允许颗粒上下移动。如果不使用间隔物,则通过载体的渗透和分布受到限制。对于tuft-it,这是3mm的遮盖物。这导致电极的3 4mm=7mm的距离。
[0358]
在非肝素化的体内猪模型中评估含有nhs-pox:nu-pox颗粒(来自nu-pox的0%、10%、20%和40%的胺基团,基于由nhs-pox提供的nhs基团的数量计算百分比)或nhs-pox粉末的止血贴片的体内性能。测试的贴片的细节显示在表7中。
[0359]
表7
[0360][0361]
体内测试
[0362]
对成年雌猪(40至50kg)进行测试,没有应用抗凝剂。在脾脏和肝脏上测试贴片性能。随着测试阶段的进行,脾脏或肝脏根据需要被定位和外化,并且通过用盐水浸泡的海绵覆盖它们来保持它们的自然湿度。
[0363]
产生不同类型的损伤:
[0364]
·
肝脏:擦伤、活检穿孔和切除
[0365]
·
脾脏:切除
[0366]
将适当大小的肝脏薄壁组织刮擦/打孔以引起中度至重度出血。通过手术刀和1
×
1cm2的模板以及使用8mm圆形活检穿孔的圆形穿孔产生肝脏擦伤。使用手术刀进行肝脏和脾脏切除。
[0367]
在组织切除或划痕之后立即应用贴片:
[0368]-用于活检穿孔和擦伤的2
×
2cm的片
[0369]-用于切除的完整7.5
×
5cm贴片
[0370]
将测试的贴片应用于出血组织上并使用用盐水溶液预润湿的纱布通过压缩而轻轻地向下压。将施加压塞10秒的初始时间,随后是30秒的间隔,直至总共5分钟。
[0371]
未浸渍的tuft-it贴片用作参考物(称为tuft-it)。
[0372]
体内测试的结果概述在表8中。
[0373]
表8
[0374][0375]
贴片1至4显示出非常强的组织粘附,而对于tuft-it贴片仅观察到轻微的粘附。
[0376]
贴片1和2在施用后显示出不超过非常有限的溶胀。贴片3至4显示出更多但仍可接受的溶胀。
[0377]
实施例7
[0378]
将止血贴片(gelita tuft-50x75mm,约0.7g)用nhs-pox的溶液、nhs-pox粉末或nhs-pox/nu-pox颗粒浸渍。使用的nhs-pox/nu-pox颗粒通过丙酮制粒以1:0.20的摩尔比制备(参见实施例8)。
[0379]
通过将nhs-pox溶解在异丙醇和二氯甲烷(200g/l)的1:1混合物中来制备含有nhs-pox的喷雾溶液。使用玻璃实验室喷雾器和加压空气在单个喷雾循环中用5ml的这种喷雾溶液浸渍贴片。以这种方式递送的nhs-pox的总量为1克/每贴片。在浸渍之后,使贴片在40℃的烘箱内干燥2小时,然后将它们在干燥器中储存2天,然后包装在含有1g的二氧化硅的alu-alu袋中并且真空密封。
[0380]
此外,使用实施例8中所述的程序,用1克的nhs-pox粉末或1克的nhs-pox/nu-pox颗粒浸渍贴片。
[0381]
如此制备的贴片的性能在离体肝脏灌注模型中在轻微出血(《20ml/min)和严重出血(》50ml/min)条件下一式三份地测试。使用平的圆形旋转磨具,在肝脏表面上产生圆形出血伤口(8mm直径),使用橡胶外置物,使得穿孔出血的深度总是3mm。结果显示在表9中。
[0382]
表9
[0383][0384]
实施例8
[0385]
通过将nhs-pox/nu-pox颗粒与止血淀粉粉末(arista
tm
ah,购自bard)混合来制备止血粉末。所用的nhs-pox/nu-pox颗粒通过丙酮制粒以1:0.20的摩尔比制备(参见实施例8)。
[0386]
将nhs-pox/nu-pox颗粒在研杵和研钵中与淀粉粉末以80/20和90/10w/w的比例混合。
[0387]
如下评价这些粉末共混物、纯淀粉粉末和纯nhs-pox/nu-pox颗粒的凝胶形成能力:
[0388]
·
向试管中装入20mg的粉末样品
[0389]
·
添加250μl的肝素化绵羊血液,并立即使用涡旋混合器将混合物搅拌10秒
[0390]
·
2分钟后,将试管倒置以确定是否形成凝胶
[0391]
·
在形成凝胶的情况下,向凝胶中添加1ml的水,并且在30分钟后确定凝胶是否仍
然完整。
[0392]
结果显示在表10中。
[0393]
表10
[0394]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
