jak抑制剂化合物及其用途
技术领域
1.本发明提供了一种具有药学活性的新颖化合物,所述化合物可用于抑制janus激酶(jak)。本发明还涉及包含所述化合物的组合物,以及所述化合物和所述组合物在医药领域的用途。
背景技术:
2.蛋白质激酶是催化蛋白质中特定残基磷酸化的酶家族,并广义地分类为酪氨酸和丝氨酸/苏氨酸激酶。由于突变、过度表达或不适当调节、调节异常或失调,以及生长因子或细胞因子的过度产生或产生不足所导致的不适当的激酶活性涉及许多疾病,其包括但不限于癌症、心血管疾病、变态反应、哮喘和其它呼吸疾病、自身免疫病、炎症疾病、骨病、代谢紊乱及神经病症和神经变性病症(例如阿尔茨海默病)。不适当的激酶活性触发多种生物细胞反应,所述生物细胞反应与涉及上述和相关疾病的细胞生长、细胞分化、细胞功能、存活、凋亡和细胞运动性有关。因此,蛋白质激酶已成为一类重要的作为治疗性介入的靶点的酶。特别地,细胞蛋白质酪氨酸激酶的jak家族在细胞因子信号转导中扮演重要的角色(kisseleva等人,gene,2002,285,1;yamaoka等人,genome biology 2004,5,253)。
3.从20世纪90年代初首个jak被发现以来,jak抑制剂的开发走过了近30年的历程。jak是一类胞内非受体酪氨酸激酶家族,其通过与信号转导子和转录激活子(signal transducer and activator of transcription;stat)之间的相互作用在细胞因子受体信号通路中发挥着重要作用。jak/stat信号通路参与细胞的增殖、分化、凋亡以及免疫调节等许多重要的生物学过程。与其它信号通路相比,这条信号通路的传递过程相对简单,它主要由三个成分组成,即酪氨酸激酶相关受体(tyrosine kinase associated receptor)、酪氨酸激酶jak和信号转导子和转录激活子stat。
4.许多细胞因子和生长因子通过jak-stat信号通路来传导信号,这包括白介素类(如il-2~7,il-9,il-10,il-15,il-21等)、gm-csf(粒细胞/巨噬细胞集落刺激因子)、gh(生长激素)、egf(表皮生长因子)、prl(催乳素)、epo(促红细胞生成素)、tpo(促血小板生成素)、pdgf(血小板衍生因子)以及干扰素类(包括ifn-α,ifn-β,ifn-γ等)等等。这些细胞因子和生长因子在细胞膜上有相应的受体。这些受体的共同特点是受体本身不具有激酶活性,但胞内段具有酪氨酸激酶jak的结合位点。受体与配体结合后,通过与之相结合的jak的活化,来磷酸化各种靶蛋白的酪氨酸残基以实现信号从胞外到胞内的转递。
5.jak是转导细胞因子信号从膜受体到stat转录因子的细胞质酪氨酸激酶。如上所述,jak是英文janus kinase的缩写,janus在罗马神话中是掌管开始和终结的两面神。之所以称为两面神激酶,是因为jak既能磷酸化与其相结合的细胞因子受体,又能磷酸化多个含特定sh2结构域的信号分子。jak蛋白家族共包括4个成员:jak1、jak2、jak3以及tyk2,它们在结构上有7个jak同源结构域(jak homology domain,jh),其中jh1结构域为激酶区,功能是编码激酶蛋白;jh2结构域是“假”激酶区,对jh1的活性起调节作用;jh3-jh7组成一个四合一结构域,调节jak与受体的结合。
6.stat是一类能与靶基因调控区dna结合的胞质蛋白,是jak的下游底物。目前已发现stat家族的7个成员,即stat1、stat2、stat3、stat4、stat5a、stat5b及stat6。stat蛋白在结构上可分为以下几个功能区段:n-端保守序列、dna结合区、sh3结构域、sh2结构域及c-端的转录激活区。其中,序列上最保守和功能上最重要的区段是sh2结构域,它具有与酪氨酸激酶src的sh2结构域完全相同的核心序列“gtfllrfss”。
7.jak-stat信号通路功能广泛,参与细胞的增殖、分化、凋亡以及免疫调节等许多重要的生物学过程,目前与疾病及药物创新相关的研究大都集中于炎症性疾病及肿瘤性疾病。其中,炎症性疾病主要包括类风湿性关节炎、犬皮炎、银屑病、溃疡性结肠炎及克罗恩病;而肿瘤性疾病则主要涉及骨髓纤维化、真性红细胞增多症及原发性血小板增多症。另外,jak分子自身的突变也会导致急性骨髓细胞性白血病(aml)、急性淋巴细胞性白血病(all)、乳腺导管癌及非小细胞肺癌(nsclc)、真性红细胞增多症(pv)、特发性血小板增多症(et)、特发性骨髓纤维化(imf)、慢性粒细胞白血病(cml)等。此外,据报道jak-stat信号通路与新冠肺炎(covid-19)中的细胞因子风暴密切相关。
8.jak是一类非常重要的药物靶点,针对这一靶点而研发的jak抑制剂主要用于筛选血液系统疾病、肿瘤、类风湿性关节炎及银屑病等治疗药物。jak-1、jak-2和tyk-2在人体各组织细胞中均有表达,jak-3主要表达于各造血组织细胞中,主要存在于骨髓细胞、胸腺细胞、nk细胞及活化的b淋巴细胞、t淋巴细胞中。研究表明:jak2抑制剂适用于骨髓增殖性疾病(santos等人,blood,2010,115:1131;barosi g.和rosti v.,curr.opin.hematol.,2009,16:129;atallah e.和versotvsek s.,2009 exp.rev.anticancer ther.9:663),jak3的抑制剂适用作免疫抑制剂(例如美国专利6,313,129;borie等人,curr.opin.investigational drugs,2003,4:1297)。另外,jak抑制剂被认为有望在新冠肺炎等重症肺炎的治疗中用于抑制炎症反应、降低细胞因子风暴风险从而降低死亡率(wei luo等人,trends in pharmacological science,41(8)
·
june 2020,“targeting jak-stat signaling to control cytokine release syndrome in covid-19”)。还有文献报道了jak抑制剂对于瘙痒症的治疗有一定疗效(landon k.oetjen等人,cell,171,p217
–
228,september 21,2017,“sensory neurons co-opt classical immune signaling pathways to mediate chronic itch”)。
9.目前,获得fda、ema批准的jak抑制剂有tofacitinib(托法替尼)、ruxolitinib(鲁索利替尼)、oclacitinib(奥拉替尼)等。处于临床中后期的jak抑制剂有例如filgotinib、peficitinib等。
10.托法替尼,jak3抑制剂,由辉瑞公司研发,于2012年11月获fda批准上市,用于治疗成人患者的对甲氨喋呤应答不充分或不耐受的中度至重度类风湿性关节炎。系首个获批用于ra治疗的口服jak抑制剂。之后于2013年3月获得日本pmda批准上市,商品名为xeljanz。2017年3月16日,辉瑞中国宣布,cfda已正式批准辉瑞公司的口服jak抑制剂的上市申请。据悉,该药物被批准用于对甲氨蝶呤疗效不足或对其无法耐受的中度至重度类风湿关节炎成年患者的治疗。目前,托法替尼用于银屑病、溃疡性结肠炎、青少年特发性关节炎等适应症已接近获批;治疗克罗恩病、斑秃等适应症的临床试验也已经进入临床中后期。托法替尼的主要副作用有严重感染率和低密度脂蛋白水平提高,最常见的不良反应为上呼吸道感染、头痛、腹泻、鼻充血、咽喉痛和鼻咽炎。此外,有临床研究报道,托法替尼可引起贫血和中性
粒细胞减少症等副作用。
[0011][0012]
filgotinib,jak1抑制剂,已于2018年9月完成三期临床试验,用于治疗风湿性关节炎。同时,filgotinib用于治疗溃疡性结肠炎和克罗恩病的研究目前正处于临床二/三期试验中。filgotinib是一种选择性jak1抑制剂,其对jak1、jak2、jak3和tyk2的ic50据报道分别约为10nm、28nm、810nm和116nm。
[0013][0014]
虽然目前已有一些jak抑制剂获批准上市,还有一些jak抑制剂处于临床研究阶段,但这些jak抑制剂在疗效或安全性方面并不尽如人意。因此,始终存在对疗效更好和/或副作用更少的jak抑制剂的需求。
技术实现要素:
[0015]
本发明的一个目的是提供一类替代现有jak抑制剂的新型jak抑制剂,从而为jak相关疾病的治疗提供更多的选择。
[0016]
本发明的进一步目的是提供一类比现有jak抑制剂功效更好和/或安全性更佳的新型jak抑制剂。
[0017]
本发明的更进一步目的是提供更适宜药剂配制的新型jak抑制剂的不同固体形式。
[0018]
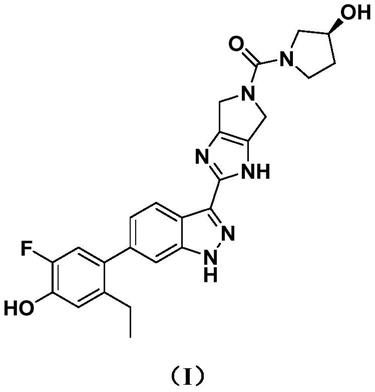
在第一个方面,本发明提供了一种固体形式的化合物,所述化合物是式(i)化合物
[0019][0020]
、式(i)化合物的同位素标记化合物、式(i)化合物的光学异构体、式(i)化合物的几何异构体、式(i)化合物的互变异构体、式(i)化合物的异构体混合物、式(i)化合物的药学上可接受的盐、或这些化合物中任意一种的溶剂化物。优选地,所述化合物的固体形式是晶体。
[0021]
在第二个方面,本发明提供了一种药物组合物,其含有本发明第一方面所述的固体形式的化合物和一种或多种药学上可接受的载体、佐剂或赋形剂。例如,所述药物组合物可以是分散液形式或溶液形式。
[0022]
在第三个方面,本发明提供了本发明第一方面所述的固体形式的化合物或本发明第二方面所述的药物组合物在制备用于治疗和/或预防与jak相关的疾病或病症的药物中的用途。
[0023]
在第四个方面,本发明提供了一种治疗与jak相关的疾病或病症的方法,所述方法包括将治疗有效量的本发明第一方面所述的固体形式的化合物或本发明第二方面所述的药物组合物给予有需要的患者。其中,所述患者优选是哺乳动物,更优选是人类患者。其中给药途径可以是口服、外用、胃肠外施予、支气管施予或鼻施予等。其中,优选地通过口服施予。
[0024]
本发明提供了一种新型抑制jak的化合物,从而为jak相关疾病的预防或治疗提供了更多的选择。另外,本发明提供了该化合物的多种固体形式,包括多种晶型和无定形物,这些不同的固体形式具有不同的化学性质和物理性质、在体内的代谢性能也不相同,因此为药物的生产、存贮和不同药物剂型的选择提供了更多的自由度,从而可以提供生物利用率更高和/或药效更好的新型药物剂型。
具体实施方式
[0025]
下面具体描述本发明技术方案的实施方式。在这些具体描述之中,使用了一些技术术语。在本技术中,除非特别指明,所有的术语具有本领域技术人员所通常理解的含义。
[0026]
本发明的第一方面涉及化合物的固体形式,所述化合物是式(i)化合物
[0027][0028]
、式(i)化合物的同位素标记化合物、式(i)化合物的光学异构体、式(i)化合物的几何异构体、式(i)化合物的互变异构体、式(i)化合物的异构体混合物、式(i)化合物的药学上可接受的盐、或这些化合物中任意一种的溶剂化物。
[0029]
式(i)化合物可以命名为(s)-(2-(6-(2-乙基-5-氟-4-羟基苯基)-1h-吲唑-3-基)-4,6-二氢吡咯并[3,4-d]咪唑-5-(1h)-基)(3-羟基吡咯烷-1-基)甲酮。
[0030]
为了简明起见,本文所使用的术语“本发明的化合物”或“本技术的化合物”涵盖了式(i)化合物、式(i)化合物的同位素标记化合物、式(i)化合物的光学异构体、式(i)化合物的几何异构体、式(i)化合物的互变异构体、式(i)化合物的异构体混合物、或这些化合物中任意一种的溶剂化物。
[0031]
术语“光学异构体”意指,当化合物具有一个或更多个手性中心时,每个手性中心可以存在r构型或s构型,由此构成的各种异构体为光学异构体。光学异构体包括所有的非对映异构体、对映异构体、内消旋体、外消旋体或其混合物形式。例如,通过手性色谱柱或通过手性合成可以分离光学异构体。
[0032]
术语“几何异构体”意指,当化合物中存在双键时,该化合物可以存在顺式异构体、反式异构体、e型异构体和z型异构体。几何异构体包括顺式异构体、反式异构体、e型异构体、z型异构体或其混合物形式。
[0033]
术语“互变异构体”指因分子中某一原子在两个位置迅速移动而产生的异构体。本领域技术人员可以理解:互变异构体之间可以互相转变,在在某一状态下可能会达到一种平衡状态而共存。本文所述“式(i)所示的化合物”也涵盖式(i)化合物的任意互变异构体。
[0034]
除非另有指明,本文提到“式(i)所示的化合物”或“式(i)化合物”或“本发明的化合物”或“本技术的化合物”时也涵盖该化合物中任一个原子被其同位素原子代替而得到的同位素标记化合物。
[0035]
术语“同位素标记化合物”是指化合物中一个或者多个原子被具有与通常在自然界中所发现的原子相同原子序数但是不同原子质量或者质量数的原子所替换。
[0036]
适用于包含在本发明的化合物中的同位素的实例包括氢的同位素,诸如2h(d)和3h(t),碳的同位素,诸如
11
c、
13
c和
14
c,氯的同位素,诸如
36
cl,氟的同位素,诸如
18
f,碘的同位素,诸如
123
i和
125
i,氮的同位素,诸如
13
n和
15
n,氧的同位素,诸如
15
o、
17
o和
18
o,以及硫的同位素,诸如
35
s。
[0037]
式(i)化合物的某些同位素标记化合物(例如包含放射性同位素的那些)可用于药物和/或底物组织分布研究。考虑到引入的容易性和检测手段的方便性,放射性同位素氘(即2h)和碳-14(即
14
c)对于该目的是特别有用的。
[0038]
利用诸如氘(即2h)的较重同位素进行取代可以提供某些治疗方面的好处并且因此在某些情况下可能是优选的,所述治疗方面的好处是由更大的代谢作用稳定性(例如,增长的体内半衰期或者减小的剂量要求)带来的。
[0039]
利用正电子放射同位素(诸如
11
c、
18
f、
15
o和
13
n)进行取代可以用于正电子放射受体图像(positron emission topography(pet))研究,用于检测底物受体占用状态。
[0040]
式(i)化合物的同位素标记化合物一般可以通过本领域技术人员已知的常规技术或者通过使用合适的同位素标记试剂代替先前使用的非标记试剂以类似于在本文所附的实例和制备中所描述的方法,来进行制备。
[0041]
本发明的化合物可以是式(i)化合物的药学上可接受的盐,具体是式(i)化合物的酸加成盐。适当的酸加成盐是由形成无毒性盐的酸所形成的。其实例包括但不限于:粘酸盐、马尿酸盐、抗坏血酸盐、琥珀酸盐、萘-2-磺酸盐、乙酸盐、己二酸盐、天冬氨酸盐、苯甲酸盐、苯磺酸盐、碳酸氢盐/碳酸盐、硫酸氢盐/硫酸盐、硼酸盐、樟脑磺酸盐、柠檬酸盐、环己胺磺酸盐、乙二磺酸盐、甲酸盐、反丁烯二酸盐、葡萄庚糖酸盐、葡萄糖酸盐、葡萄糖醛酸盐、六氟磷酸盐、2-(4-羟苄基)苯甲酸盐、氢氯化物/氯化物、氢溴化物/溴化物、氢碘化物/碘化物、2-羟乙磺酸盐、乳酸盐、苹果酸盐、顺丁烯二酸盐、丙二酸盐、甲磺酸盐、甲基硫酸盐、萘酸盐、2-萘磺酸盐、烟碱酸盐、硝酸盐、乳清酸盐、草酸盐、十六酸盐、磷酸盐/磷酸氢盐/磷酸二氢盐、焦谷氨酸盐、葡萄糖二酸盐、硬脂酸盐、水杨酸盐、单宁酸盐、酒石酸盐、甲苯磺酸盐和三氟乙酸盐。关于合适的盐的综述,可以参见handbook of pharmaceutical salts:properties,selection and use by stahl and wermuth(wiley-vch,2002)。用于制备本文中所述的化合物的药学上可接受的盐的方法是本领域技术人员已知的。
[0042]
特别优选的式(i)化合物的药学上可接受的盐是其盐酸盐、磷酸盐、马来酸盐、l-酒石酸盐、富马酸盐、粘酸盐、柠檬酸盐、对甲苯磺酸盐、甲磺酸盐、苯磺酸盐。最优选的式(i)化合物的药学上可接受的盐是其磷酸盐、马来酸盐、甲磺酸盐。
[0043]
术语“药学上可接受的”是指相应的化合物、载体或分子适于给予哺乳动物(优选人)。优选地,该术语是指由管理机构例如cfda(中国)、emea(欧洲)、fda(美国)等任意国家管理机构认证的用于哺乳动物(优选人)。
[0044]
本文所使用的术语“式(i)化合物的药学上可接受的盐”用于表示任意形式的式(i)化合物的药学上可接受的盐,即涵盖了式(i)化合物、式(i)化合物的同位素标记化合物、式(i)化合物的光学异构体、式(i)化合物的几何异构体、式(i)化合物的互变异构体、式(i)化合物的异构体混合物中任意一种的药学上可接受的盐。
[0045]
本发明的某些化合物可以以非溶剂化形式以及溶剂化形式(包括水合形式)存在。在本技术中,“式(i)化合物”、“式(i)化合物的同位素标记化合物”、“式(i)化合物的光学异构体”、“式(i)化合物的几何异构体”、“式(i)化合物的互变异构体”、“式(i)化合物的异构体混合物”、“式(i)化合物的药学上可接受的盐”等术语均涵盖其溶剂化物。
[0046]
在本技术中,“化合物的固体形式”与“固体形式的化合物”被理解为具有相同的含义并因此可以互换使用,表示化合物主要以固体形式存在。本领域技术人员可以理解,固体
形式的化合物的存在形式可以为晶体(晶型)、无定形物(非晶体)、或者两者的混合物。
[0047]
术语“晶型”或“晶体形式”是指作为晶体的固体形式,例如可以通过x射线衍射测定。在某些实施方案中,物质的晶型可基本上不含非晶形和/或其它晶型。在某些实施方案中,物质的晶型可含有小于约1%、小于约2%、小于约3%、小于约4%、小于约5%、小于约6%、小于约7%、小于约8%、小于约9%、小于约10%重量的一种或多种非晶形和/或其它晶型。在某些实施方案中,在某些实施方案中,物质的晶型可为约99%、约98%、约97%、约96%、约95%、约94%、约93%、约92%、约91%或约90%纯的。
[0048]
术语“非晶形体”或“非晶形式”或“无定形物”(有些现有技术文献称之为“无定型物”)意指基本上不是晶体的固体形式,例如可以通过x射线衍射测定。具体地说,术语“非晶形体”或“非晶形式”或“无定形物”描述无序的固体形式,即固体形式缺乏长程有序性。在某些实施方案中,物质的非晶形式可基本上不含其它非晶形和/或晶型。在某些实施方案中,物质的非晶形式可含有以重量计小于约1%、小于约2%、小于约3%、小于约4%、小于约5%、小于约6%、小于约7%、小于约8%、小于约9%、小于约10%重量的一种或多种其它的非晶形和/或晶型。在某些实施方案中,物质的非晶形式可为约99%、约98%、约97%、约96%、约95%、约94%、约93%、约92%、约91%或约90%纯的。
[0049]
本技术的化合物存在多晶型现象。术语“多晶型”是指具有相同分子结构、但由于分子晶格中的排列或构象不同而形成的不同晶体。本领域公知,分子结构相同但晶形不同的化合物晶体,有可能具有不同的生物利用度、溶解度、溶解速率、化学物理稳定性、熔点、颜色、可滤性、吸湿性、密度等化学物理性质。
[0050]
在本技术的优选实施方案中,所述固体形式是本技术实施例部分所制备的式(i)化合物的晶体或式(i)化合物的盐的晶体。例如,在本技术的一些优选实施方案中,所述固体形式式(i)化合物的磷酸盐晶型a、式(i)化合物的马来酸盐晶型a、式(i)化合物的甲磺酸盐晶型b、式(i)化合物水合物的晶型a、式(i)化合物的晶型b等。
[0051]
在本技术的优选实施方案中,所述固体形式是本技术实施例部分所制备的式(i)化合物的晶体或式(i)化合物的盐的晶体,所述晶体(或晶型)的粉末x-射线衍射图谱优选地具有与本技术实施例部分所测定的对应xrpd谱中衍射角(2θ)位置相同的一个或多个特征峰。所述“一个或多个特征峰”是指至少1个、至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少11个、至少12个、至少13个、至少14个、至少15个、至少16个、至少17个、至少18个、至少19个或至少20个特征峰。这里“特征峰”被理解为xrpd谱中相对强度最大的1个或多个峰。本领域技术人员理解,由于样品纯度和测试条件的差异,不同条件下测得的xrpd峰位置可能有一定的偏离,因此这里“位置相同”被理解为相对于本技术实施例中给出的对应的衍射角(2θ)位置可以有
±
0.50
°
、
±
0.40
°
、
±
0.30
°
、
±
0.20
°
、优选
±
0.10
°
的差异。最优选地,所述晶体(或晶型)的粉末x-射线衍射图谱与本技术实施例部分所测定的对应xrpd图谱基本相同。所述“xrpd图谱基本相同”是指:两个xrpd图谱完全相同,或者虽然有所差异,但是本领域技术人员可以确认所述差异是非实质性的、是由不同的样品纯度、测定条件、仪器误差或操作习惯导致的,并由此可以确认两个xrpd图谱是由同样的晶体获得的。
[0052]
在本技术的优选实施方案中,所述固体形式是本技术实施例部分所制备的式(i)化合物的晶体或式(i)化合物的盐的晶体,所述晶体(或晶型)的tga或dsc曲线优选地具有
与本技术实施例部分所测定的对应tga或dsc曲线位置相同的至少1个、至少2个、至少3个、或至少4个特征峰。这里“特征峰”被理解为tga/dsc曲线中的吸热峰或放热峰。本领域技术人员理解,由于样品纯度和测试条件的差异,不同条件下测得的tga/dsc曲线中表示峰位置的温度读数可能有一定的偏离,因此这里“位置相同”被理解为相对于本技术实施例中给出的对应的tga和/或dsc曲线,峰位置的温度可以有
±
5℃、
±
4℃、
±
3℃、
±
2℃、
±
1℃的差异。最优选地,所述晶体(或晶型)的tga/dsc曲线与本技术实施例部分所测定的对应tga/dsc曲线基本相同。所述“tga/dsc曲线基本相同”是指:两个tga曲线和/或dsc曲线完全相同,或者虽然有所差异,但是本领域技术人员可以确认所述差异是非实质性的、是由不同的样品纯度、测定条件、仪器误差或操作习惯导致的,并由此可以确认两个tga曲线和/或dsc曲线是由同样的晶体获得的。
[0053]
本发明的一个优选实施方案涉及式(i)化合物水合物的晶型a,其粉末x-射线衍射图谱(xrpd)具有选自8.67
±
0.10
°
、13.39
±
0.10
°
、15.05
±
0.10
°
、17.38
±
0.10
°
、21.24
±
0.10
°
、22.86
±
0.10
°
、24.89
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自8.67
±
0.10
°
、11.53
±
0.10
°
、13.39
±
0.10
°
、15.05
±
0.10
°
、17.38
±
0.10
°
、21.24
±
0.10
°
、21.63
±
0.10
°
、22.19
±
0.10
°
、22.86
±
0.10
°
、23.43
±
0.10
°
、24.89
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0054]
进一步优选地,式(i)化合物水合物的晶型a的tga/dsc曲线在约50-290℃范围内存在多个热信号。
[0055]
本发明的一个优选实施方案涉及式(i)化合物的晶型b,其粉末x-射线衍射图谱(xrpd)具有选自7.70
±
0.10
°
、11.20
±
0.10
°
、12.46
±
0.10
°
、15.44
±
0.10
°
、18.17
±
0.10
°
、18.48
±
0.10
°
、19.27
±
0.10
°
、21.84
±
0.10
°
、22.94
±
0.10
°
、24.29
±
0.10
°
、25.40
±
0.10
°
、27.92
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自7.70
±
0.10
°
、11.20
±
0.10
°
、12.46
±
0.10
°
、15.44
±
0.10
°
、18.17
±
0.10
°
、18.48
±
0.10
°
、18.93
±
0.10
°
、19.27
±
0.10
°
、19.50
±
0.10
°
、20.83
±
0.10
°
、21.84
±
0.10
°
、22.94
±
0.10
°
、23.22
±
0.10
°
、24.29
±
0.10
°
、25.40
±
0.10
°
、26.74
±
0.10
°
、27.92
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0056]
进一步优选地,式(i)化合物的晶型b的tga/dsc曲线在约319.1℃(起始温度)处存在吸热峰。
[0057]
本发明的一个优选实施方案涉及式(i)化合物水合物的晶型c,其粉末x-射线衍射图谱(xrpd)具有选自4.96
±
0.10
°
、7.66
±
0.10
°
、10.18
±
0.10
°
、11.07
±
0.10
°
、14.73
±
0.10
°
、22.87
±
0.10
°
衍衍射角(2θ)处的一个或多个特征峰,优选地具有选自4.96
±
0.10
°
、7.66
±
0.10
°
、10.18
±
0.10
°
、11.07
±
0.10
°
、12.43
±
0.10
°
、14.73
±
0.10
°
、16.10
±
0.10
°
、19.24
±
0.10
°
、22.87
±
0.10
°
、23.52
±
0.10
°
、24.27
±
0.10
°
、25.09
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0058]
进一步优选地,式(i)化合物水合物的晶型c的tga/dsc曲线在约78.1℃(起始温度)处存在一个热信号,在约279.4(起始温度)处和303.8℃(起始温度)处存在热信号。
[0059]
本发明的一个优选实施方案涉及式(i)化合物水合物的晶型g,其粉末x-射线衍射图谱(xrpd)具有选自6.42
±
0.10
°
、7.04
±
0.10
°
、7.69
±
0.10
°
、12.52
±
0.10
°
、15.81
±
0.10
°
、18.83
±
0.10
°
、22.85
±
0.10
°
、23.40
±
0.10
°
、衍射角(2θ)处的一个或多个特征峰,
优选地具有选自6.42
±
0.10
°
、7.04
±
0.10
°
、7.69
±
0.10
°
、11.48
±
0.10
°
、12.52
±
0.10
°
、15.81
±
0.10
°
、18.83
±
0.10
°
、19.58
±
0.10
°
、22.85
±
0.10
°
、23.40
±
0.10
°
、25.31
±
0.10
°
、27.85
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0060]
进一步优选地,式(i)化合物水合物的晶型g的tga/dsc曲线在约81.2℃(起始温度)处存在一个热信号,在约209.6℃(起始温度)处、约314.0℃(起始温度)处存在两个热信号。
[0061]
本发明的一个优选实施方案涉及式(i)化合物的磷酸盐晶型a,其粉末x-射线衍射图谱(xrpd)具有选自5.29
±
0.10
°
、7.47
±
0.10
°
、10.61
±
0.10
°
、19.16
±
0.10
°
和21.32
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自5.29
±
0.10
°
、7.47
±
0.10
°
、10.61
±
0.10
°
、15.94
±
0.10
°
、16.77
±
0.10
°
、18.68
±
0.10
°
、19.16
±
0.10
°
、21.32
±
0.10
°
及25.36
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0062]
进一步优选地,式(i)化合物的磷酸盐晶型a的tga/dsc曲线在约279.3℃(起始温度)处有尖锐吸热峰数。
[0063]
本发明的一个优选实施方案涉及式(i)化合物的马来酸盐晶型a,其粉末x-射线衍射图谱(xrpd)具有选自4.55
±
0.10
°
、8.78
±
0.10
°
、12.69
±
0.10
°
、13.96
±
0.10
°
、16.62
±
0.10
°
、17.61
±
0.10
°
、18.32
±
0.10
°
、25.39
±
0.10
°
、26.53
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自4.55
±
0.10
°
、8.78
±
0.10
°
、12.69
±
0.10
°
、13.74
±
0.10
°
、13.96
±
0.10
°
、16.62
±
0.10
°
、17.61
±
0.10
°
、18.32
±
0.10
°
、21.72
±
0.10
°
、25.39
±
0.10
°
、26.53
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0064]
进一步优选地,式(i)化合物的马来酸盐晶型a的tga/dsc曲线在约211.2℃和约278.1℃(起始温度)处有两个热信号。
[0065]
本发明的一个优选实施方案涉及式(i)化合物的甲磺酸盐晶型b,其粉末x-射线衍射图谱(xrpd)具有选自5.91
±
0.10
°
、9.22
±
0.10
°
、15.83
±
0.10
°
、17.73
±
0.10
°
、19.02
±
0.10
°
、25.01
±
0.10
°
、衍射角(2θ)处的一个或多个特征峰,优选地具有选自5.91
±
0.10
°
、9.22
±
0.10
°
、15.83
±
0.10
°
、17.73
±
0.10
°
、19.02
±
0.10
°
、20.61
±
0.10
°
、21.36
±
0.10
°
、23.18
±
0.10
°
、25.01
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0066]
进一步优选地,式(i)化合物的甲磺酸盐晶型b的tga/dsc曲线在约234.4℃(峰值温度)和约264.1℃(起始温度)有两个吸热信号。
[0067]
本发明的一个优选实施方案涉及式(i)化合物的盐酸盐晶型a,其粉末x-射线衍射图谱(xrpd)具有选自8.05
±
0.10
°
、17.11
±
0.10
°
、18.02
±
0.10
°
、20.84
±
0.10
°
、21.09
±
0.10
°
、22.77
±
0.10
°
、23.14
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自8.05
±
0.10
°
、15.04
±
0.10
°
、17.11
±
0.10
°
、18.02
±
0.10
°
、20.44
±
0.10
°
、20.84
±
0.10
°
、21.09
±
0.10
°
、22.07
±
0.10
°
、22.77
±
0.10
°
、23.14
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0068]
进一步优选地,式(i)化合物的盐酸盐晶型a的tga/dsc曲线在约80.1℃、约118.0℃(峰值温度)和约224.5℃(起始温度)处有三个吸热峰,在约231.5℃(起始温度)处有放热信号。
[0069]
本发明的一个优选实施方案涉及式(i)化合物的酒石酸盐晶型a,其粉末x-射线衍射图谱(xrpd)具有选自7.90
±
0.10
°
、8.69
±
0.10
°
、13.12
±
0.10
°
、13.43
±
0.10
°
、18.11
±
0.10
°
、21.28
±
0.10
°
、22.90
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自7.90
±
0.10
°
、8.69
±
0.10
°
、13.12
±
0.10
°
、13.43
±
0.10
°
、15.08
±
0.10
°
、17.17
±
0.10
°
、17.44
±
0.10
°
、18.11
±
0.10
°
、19.40
±
0.10
°
、20.74
±
0.10
°
、21.28
±
0.10
°
、22.90
±
0.10
°
、24.15
±
0.10
°
、24.95
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0070]
进一步优选地,式(i)化合物的酒石酸晶型a的tga/dsc曲线在约125.2℃和约168.8℃(起始温度)处有两个热信号。
[0071]
本发明的一个优选实施方案涉及式(i)化合物的富马酸盐晶型a,其粉末x-射线衍射图谱(xrpd)具有选自4.96
±
0.10
°
、7.66
±
0.10
°
、10.21
±
0.10
°
、11.06
±
0.10
°
、14.74
±
0.10
°
、16.11
±
0.10
°
、22.87
±
0.10
°
、25.10
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自4.96
±
0.10
°
、7.66
±
0.10
°
、10.21
±
0.10
°
、11.06
±
0.10
°
、12.44
±
0.10
°
、14.74
±
0.10
°
、16.11
±
0.10
°
、19.25
±
0.10
°
、22.87
±
0.10
°
、23.54
±
0.10
°
、24.27
±
0.10
°
、25.10
±
0.10
°
、25.37
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0072]
进一步优选地,式(i)化合物的富马酸晶型a的tga/dsc曲线在约71.1℃、约205.3℃、约273.7℃和约299.4℃(起始温度)处有多个热信号。
[0073]
本发明的一个优选实施方案涉及式(i)化合物的粘酸盐晶型a,其粉末x-射线衍射图谱(xrpd)具有选自4.98
±
0.10
°
、10.22
±
0.10
°
、11.07
±
0.10
°
、14.75
±
0.10
°
、14.94
±
0.10
°
、16.13
±
0.10
°
、19.65
±
0.10
°
、30.79
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自4.98
±
0.10
°
、7.69
±
0.10
°
、10.22
±
0.10
°
、11.07
±
0.10
°
、14.75
±
0.10
°
、14.94
±
0.10
°
、16.13
±
0.10
°
、19.65
±
0.10
°
、21.51
±
0.10
°
、22.92
±
0.10
°
、30.79
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0074]
进一步优选地,式(i)化合物的粘酸盐晶型a的tga/dsc曲线在约69.8℃和约210.4℃(起始温度)处可观察到两个热信号。
[0075]
本发明的一个优选实施方案涉及式(i)化合物的柠檬酸盐晶型a,其粉末x-射线衍射图谱(xrpd)具有选自4.85
±
0.10
°
、6.42
±
0.10
°
、14.63
±
0.10
°
、17.12
±
0.10
°
、20.75
±
0.10
°
、25.34
±
0.10
°
、衍射角(2θ)处的一个或多个特征峰,优选地具有选自4.85
±
0.10
°
、6.42
±
0.10
°
、7.64
±
0.10
°
、14.63
±
0.10
°
、15.40
±
0.10
°
、17.12
±
0.10
°
、18.12
±
0.10
°
、18.84
±
0.10
°
、19.37
±
0.10
°
、20.75
±
0.10
°
、25.34
±
0.10
°
、衍射角(2θ)处的一个或多个特征峰。
[0076]
进一步优选地,式(i)化合物的柠檬酸盐晶型a的tga/dsc曲线在约68.2℃(峰值温度)、约154.4℃和164.0℃(起始温度)处有三个吸热峰。
[0077]
本发明的一个优选实施方案涉及式(i)化合物的柠檬酸盐晶型b,其粉末x-射线衍射图谱(xrpd)具有选自4.96
±
0.10
°
、7.67
±
0.10
°
、10.20
±
0.10
°
、11.04
±
0.10
°
、14.73
±
0.10
°
、19.23
±
0.10
°
、22.86
±
0.10
°
、23.54
±
0.10
°
、24.28
±
0.10
°
、25.08
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自4.96
±
0.10
°
、7.67
±
0.10
°
、10.20
±
0.10
°
、11.04
±
0.10
°
、12.42
±
0.10
°
、14.73
±
0.10
°
、16.09
±
0.10
°
、17.55
±
0.10
°
、19.23
±
0.10
°
、22.86
±
0.10
°
、23.54
±
0.10
°
、24.28
±
0.10
°
、25.08
±
0.10
°
、27.91
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0078]
进一步优选地,式(i)化合物的柠檬酸盐晶型b的tga/dsc曲线在约73.1℃、约280.2℃、约301.6℃(起始温度)和约179.7℃(峰值温度)处有四个吸热峰。
[0079]
本发明的一个优选实施方案涉及式(i)化合物的对甲苯磺酸盐晶型a,其粉末x-射线衍射图谱(xrpd)具有选自5.90
±
0.10
°
、9.34
±
0.10
°
、14.87
±
0.10
°
、15.33
±
0.10
°
、17.88
±
0.10
°
、18.76
±
0.10
°
、19.71
±
0.10
°
、24.26
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自5.90
±
0.10
°
、9.34
±
0.10
°
、12.99
±
0.10
°
、14.87
±
0.10
°
、15.33
±
0.10
°
、16.10
±
0.10
°
、17.88
±
0.10
°
、18.76
±
0.10
°
、19.34
±
0.10
°
、19.71
±
0.10
°
、20.39
±
0.10
°
、24.26
±
0.10
°
、24.99
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0080]
进一步优选地,式(i)化合物的对甲苯磺酸晶型a的tga/dsc曲线在约121.2℃和约222.3℃(起始温度)处由两个热信号。
[0081]
本发明的一个优选实施方案涉及式(i)化合物的苯磺酸盐晶型a,其粉末x-射线衍射图谱(xrpd)具有选自5.82
±
0.10
°
、6.74
±
0.10
°
、11.85
±
0.10
°
、16.38
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自5.82
±
0.10
°
、6.74
±
0.10
°
、11.85
±
0.10
°
、16.38
±
0.10
°
、19.46
±
0.10
°
、20.20
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0082]
进一步优选地,式(i)化合物的苯磺酸晶型a的tga/dsc曲线在约60-200℃处有多个热信号。
[0083]
本发明的一个优选实施方案涉及式(i)化合物的苯磺酸盐晶型b,其粉末x-射线衍射图谱(xrpd)具有选自4.93
±
0.10
°
、5.63
±
0.10
°
、9.02
±
0.10
°
、10.89
±
0.10
°
、14.84
±
0.10
°
、17.55
±
0.10
°
、18.84
±
0.10
°
、23.12
±
0.10
°
、25.55
±
0.10
°
、26.14
±
0.10
°
衍射角(2θ)处的一个或多个特征峰,优选地具有选自4.93
±
0.10
°
、5.63
±
0.10
°
、9.02
±
0.10
°
、10.30
±
0.10
°
、10.89
±
0.10
°
、11.43
±
0.10
°
、14.23
±
0.10
°
、14.84
±
0.10
°
、17.04
±
0.10
°
、17.55
±
0.10
°
、18.84
±
0.10
°
、19.66
±
0.10
°
、20.24
±
0.10
°
、22.71
±
0.10
°
、23.12
±
0.10
°
、24.91
±
0.10
°
、25.55
±
0.10
°
、26.14
±
0.10
°
衍射角(2θ)处的一个或多个特征峰。
[0084]
进一步优选地,式(i)化合物的苯磺酸晶型b的tga/dsc曲线在约90.1℃(峰值温度)和约236.7℃(起始温度)处有两个热信号。
[0085]
在本技术的最优选实施方案中,所述固体形式是式(i)化合物的磷酸盐晶型a、式(i)化合物的马来酸盐晶型a、式(i)化合物的甲磺酸盐晶型b、式(i)化合物水合物的晶型a、式(i)化合物的晶型b,它们各自具有与本技术实施例部分所述对应晶型基本上相同的xprd谱。
[0086]
在本技术的一些实施方案中,所述固体形式是式(i)化合物的磷酸盐晶型a、式(i)化合物的马来酸盐晶型a、式(i)化合物的甲磺酸盐晶型b、式(i)化合物水合物的晶型a、式(i)化合物的晶型b,它们各自具有与本技术实施例部分所述对应晶型基本上相同的tga和/或dsc曲线。
[0087]
在第二个方面,本发明提供了一种药物组合物,其含有本发明第一方面所述固体形式的化合物和一种或多种药学上可接受的载体、佐剂或赋形剂。
[0088]
本发明的药物组合物可以是固体组合物或液体组合物(例如分散液或溶液)。
[0089]
本发明的药物组合物可根据需要配制成适用于口服、外用(包括但不限于外敷、喷涂等)、胃肠外(包括皮下、肌肉、皮层和静脉)施予、支气管施予或鼻施予的剂型。其中,优选地,本发明的药物组合物被配制成适用于外用或支气管施予或鼻施予的剂型(制剂)。更优选地,本发明的药物组合物被配制成适用于外用的剂型(制剂)。
[0090]
如果使用固体载剂,则该制剂可以成片,以粉末或颗粒形式置于硬质凝胶胶囊中,
或以糖锭或锭剂形式。固体载剂可以包括常规的赋形剂,诸如粘合剂、填料、成片润滑剂、崩解剂、润湿剂等等。如果需要可以通过常规技术膜包衣该片剂。如果使用液体载剂,则该制剂可以是糖浆、乳液、膏剂、软凝胶胶囊、用于注射的无菌载体、水性或非水性液体悬浮液形式的,或者可以是在使用前用水或其他适当载体复原的干品。液体制剂可以包含常规添加剂,诸如悬浮剂、乳化剂、润湿剂、非水性载体(包括可食用油)、防腐剂以及香味剂和/或着色剂。为了胃肠外施予,通常载体至少大部分包括无菌水,但也可以使用盐水溶液、葡萄糖溶液等。也可以使用可注射悬浮液,在这种情况下,可以使用常规悬浮剂。常规防腐剂、缓冲试剂等也可以添加到胃肠外剂型中。药物组合物通过对包含适量的活性成分(即本发明的式(i)化合物)的所需制剂合适的常规技术制备。
[0091]
适于非肠道注射的组合物可以包括生理学上可接受无菌水性或非水性溶液、分散液(例如悬浮液或乳液)和用于无菌可注射溶液或分散液的无菌粉末。合适的水性和非水性载体、稀释剂、溶剂、分散剂的实例包括水、乙醇、多元醇(丙二醇、聚乙二醇、丙三醇等)、其合适的混合物、植物油(例如,橄榄油)和可注射有机酯(例如,油酸乙酯)。
[0092]
这些组合物还可以包含各种赋形剂,例如,防腐剂、润湿剂、乳化剂和分散剂。可以通过各种抗菌剂和抗真菌剂(例如,对羟基苯甲酸酯、氯丁醇、苯酚、山梨酸等)来确保对微生物的作用的抑制。还可以包括等渗剂,例如,糖、氯化钠等。可以通过使用延迟吸收试剂(例如,单硬脂酸铝和凝胶)来延长可注射药学剂型的吸收。
[0093]
用于口服的固体剂型包括胶囊、药片、药丸、粉末和颗粒。在这种固体剂型中,将活性化合物与至少一种惰性赋型剂(或载体)(例如,柠檬酸钠或磷酸二钙)混合,其中还可以包括:(a)填料或混合剂(例如,淀粉、乳糖、蔗糖、葡萄糖、甘露醇和硅酸);(b)粘结剂(例如,羧基甲基纤维素、褐藻酸酯、凝胶、聚乙烯基吡咯烷酮、蔗糖和阿拉伯树胶);(c)保湿剂(例如,丙三醇);(d)崩解剂(例如,琼脂-琼脂、碳酸钙、马铃薯或木薯淀粉、褐藻酸、某些合成的硅酸酯、碳酸钠);(e)溶液阻滞剂(例如,石蜡);(f)吸收促进剂(例如,季铵化合物);(g)润湿剂(例如,十六烷醇和单硬脂酸丙三醇酯);(h)吸附剂(例如,高岭土和斑脱土)和(i)润滑剂(例如,滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、月桂基硫酸钠)或其混合物混合。
[0094]
类似类型的固体组合物还可以在使用例如乳糖以及高分子量聚乙二醇等作为赋型剂的软填充和硬填充凝胶胶囊中作为填料。
[0095]
固体剂型(例如,药片、糖衣丸、胶囊、药丸和颗粒)可以采用涂层和外壳(例如,肠道涂层和本领域已知的其它)来制备。它们可以包含遮光剂,它们还可以是以延迟方式在肠道的某一部分中释放活性化合物或各种活性化合物的组合物。可用的包埋组合物的实例是聚合物质和蜡。活性组分还可以以微胶囊化形式,如果适当的话,可以具有一种或更多种上述赋型剂。
[0096]
用于口服的液体剂型包括药学上可接受乳液、溶液、分散液、糖浆和酏剂。除了活性化合物以外,液体剂型可以包含本领域中通常所用的惰性稀释剂(例如,水或其它溶剂)、增溶剂和乳化剂(例如,乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苯甲醇、苯甲酸苯甲酯、丙二醇、1,3丁二醇、二甲基甲酰铵)、油(具体为,棉花子油、落花生油、玉米油、橄榄油、蓖麻油、芝麻油)、丙三醇、四氢呋喃醇、聚乙二醇和山梨聚糖的脂肪酸酯或这些物质的混合物等。
[0097]
除了这些惰性稀释剂,组合物还可以包括,例如,润湿剂、乳化和悬浮剂、香化剂、调味剂和加香剂。
[0098]
除了活性化合物,悬浮液可以包含悬浮剂,例如乙氧基化异十八烷醇、聚氧化乙烯山梨醇、山梨聚糖酯、微晶纤维、偏氢氧化铝、斑脱土、琼脂-琼脂和黄芪胶或这些物质的混合物等。
[0099]
本发明的化合物的局部给药用剂型包括膏剂、粉末、喷雾和吸入剂。该活性组分在无菌条件下与生理学上可接受载体和任何所需要的防腐剂、缓冲剂或推进剂混合。眼用配方、眼药膏、粉末和溶液也包括在本发明的范围内。
[0100]
本发明的化合物的外用剂型可以呈油包水(w/o)或水包油(o/w)乳液的形式,多乳液形式,如水包油包水(w/o/w)形式或油包水包油(o/w/o)乳液形式,或者以水分散体或脂分散体、凝胶或气溶胶形式制得。
[0101]
本发明的化合物的外用剂型可以包含添加剂和制剂助剂,如乳化剂、增稠剂、胶凝剂、水固定剂、铺展剂、稳定剂、染料、香料和防腐剂。合适的乳化剂包括硬脂酸、三乙醇胺和peg-40-硬脂酸酯。合适的增稠剂包括单硬脂酸甘油酯,卡波姆和peg600。合适的防腐剂包括对羟苯甲酸丙酯和氯甲酚。适的铺展剂包括二甲聚硅氧烷和聚二甲基环硅氧烷。合适的水固定剂包括聚乙二醇,优选聚乙二醇600。
[0102]
本发明的化合物的外用剂型可以包括膏剂、洗剂、凝胶、乳液、微乳剂、喷雾剂、皮肤贴剂等,其可局部施用,以治疗特应性皮炎、湿疹、银屑病、硬皮病、瘙痒、白癜风、脱发等皮肤疾病。特别地,本发明的化合物的外用剂型是膏剂,其可局部外敷施用,以治疗特应性皮炎、湿疹、银屑病、硬皮病、瘙痒、白癜风、脱发等皮肤疾病。
[0103]
在药物组合物和剂型中式(i)化合物的量可以由本领域技术人员根据需要适当地确定,例如式(i)化合物可以治疗有效量存在于药物组合物或剂型中。
[0104]
在第三个方面,本发明提供了本发明第一方面所述固体形式的化合物、或本发明第二方面所述药物组合物在制备用于治疗和/或预防与jak相关的疾病或病症的药物中的用途。
[0105]“与jak相关的疾病或病症”包括但不限于:
[0106]
关节炎,包括类风湿性关节炎、幼年型关节炎和银屑病关节炎;
[0107]
自身免疫疾病或病症,包括单一器官或单一细胞类型自身免疫病症,例如桥本甲状腺炎、自身性免疫溶血性贫血、恶性贫血的自身免疫性萎缩性胃炎、自身免疫性脑脊髓炎、自身免疫性睾丸炎、古德帕斯彻病、自身免疫性血小板减少症、交感性眼炎、重症肌无力、格雷夫斯病、原发性胆汁性肝硬变、慢性侵袭性肝炎、溃疡性结肠炎和膜性肾小球病、涉及全身性自身免疫病症的那些(例如全身性红斑狼疮、类风湿性关节炎、干燥综合征、莱特尔综合征、多肌炎-皮肌炎、系统性硬化病、结节性多动脉炎、多发性硬化和大疱性类天疱疮)以及其它o-细胞(体液)型或t细胞型自身免疫病(包括寇甘综合征)、强直性脊椎炎、wegener’s氏肉芽肿、自身免疫性脱发、i型糖尿病或幼年发病型糖尿病或甲状腺炎;
[0108]
癌症或肿瘤,包括消化道/胃肠道癌、结直肠癌、肝癌、皮肤癌(包括肥大细胞瘤和鳞状细胞癌)、乳房和乳腺癌、卵巢癌、前列腺癌、淋巴瘤、白血病(包括急性髓性白血病和慢性髓性白血病)、肾癌、肺癌、肌癌、骨癌、膀胱癌、脑癌、黑色素瘤(包含口腔和转移性黑色素瘤)、卡波西肉瘤、骨髓瘤(包括多发性骨髓瘤)、骨髓增殖性病症、增生型糖尿病视网膜病变或与血管生成相关的病症(包括实体瘤);
[0109]
糖尿病,包括i型糖尿病或糖尿病并发症;
[0110]
眼疾病、病症或病况,包括眼的自身免疫病、角膜结膜炎、春季结膜炎、葡萄膜炎(包括与贝切特病相关的葡萄膜炎和晶状体性葡萄膜炎)、角膜炎、疱疹性角膜炎、圆锥形角膜炎、角膜上皮营养障碍、角膜白斑、眼天疱疮、莫伦溃疡、巩膜炎、格雷夫眼病、vogt-koyanagi-harada综合征、干燥性角结膜炎(干眼症)、水疱、虹膜睫状体炎、结节病、内分泌性眼病、交感性眼炎、变应性结膜炎或眼部新生血管形成;
[0111]
肠炎症、变态反应或病况,包括克罗恩氏病和/或溃疡性结肠炎、炎性肠病、乳糜泻、直肠炎、嗜酸细胞性胃肠炎或肥大细胞增多症;
[0112]
神经变性疾病,包括运动神经元病、阿尔茨海默病、帕金森病、肌萎缩侧索硬化、亨廷顿病、脑缺血或创伤性损伤引发的神经变性疾病、卒中、谷氨酸神经毒性或缺氧;卒中的缺血/再灌注损伤、心肌缺血、肾缺血、心脏病发作、心脏肥大、动脉粥样硬化和动脉硬化、器官缺氧或血小板聚集;
[0113]
皮肤疾病、病况或病症,包括特应性皮炎、湿疹、银屑病、硬皮病、瘙痒症或其它瘙痒病况、白癜风、脱发;
[0114]
变态反应,包括哺乳动物的变应性皮炎(包括变应性疾病,例如叮咬过敏)、夏季湿疹、库蚊叮痒综合症(sweet itch)、肺气肿、炎症性气道疾病、复发性气道阻塞、气道反应过度或慢性阻塞性肺疾病;
[0115]
哮喘和其它阻塞性气道疾病,包括慢性或顽固型哮喘、晚期哮喘、支气管炎、支气管性哮喘、变应性哮喘、内源性哮喘、外源性哮喘或粉尘性哮喘;
[0116]
移植排斥,包括胰岛移植排斥、骨髓移植排斥、移植物抗宿主病、器官和细胞移植排斥(例如骨髓、软骨、角膜、心脏、椎间盘、胰岛、肾、四肢、肝、肺、肌肉、成肌细胞、神经、胰脏、皮肤、小肠或气管)或者异种移植;
[0117]
流感病毒或者冠状病毒感染导致的重症肺炎,包括sars、mers和covid-19(新冠肺炎),尤其是在新冠肺炎中抑制炎症反应、抑制或防止细胞因子风暴。
[0118]
在第四个方面,本发明提供了一种治疗与jak相关的疾病或病症的方法,所述方法包括将治疗有效量的本发明第一方面所述固体形式的化合物、或本发明第二方面所述药物组合物给予有需要的患者。其中,所述患者优选是哺乳动物,更优选是人类患者。其中给药途径可以是口服、外用(包括但不限于外敷、喷涂等)、胃肠外(包括皮下、肌肉、皮层和静脉)施予、支气管施予或鼻施予等。其中,优选地通过鼻施予或外用施予。更优选地通过外用施予。
[0119]
为了起到预期效果,通常需要将治疗有效量的本发明的固体化合物或药物组合物或剂型给予患者。短语“治疗有效量”是本领域公认的术语。在某些实施方案中,该术语是指消除、减少或维持特定治疗方案的目标所必需或足够的量。有效量可以根据诸如所治疗的疾病或病症、所施用的特定靶向构建体、受试者的大小或疾病或病症的严重性等因素而变化。本领域普通技术人员或医生可凭经验确定特定化合物的有效量,而无需过多的实验。在某些实施方案中,体内使用的治疗剂的治疗有效量可能取决于许多因素,包括:施用的方式和方法;除了药剂之外还包含在药物中的任何其他材料。可选使用体外或体内试验来帮助确定最佳剂量范围。
[0120]
出人意料地,本发明的化合物在实验中展示出作为jak激酶抑制剂的优异功效(优于现有的jak激酶抑制剂,例如filgotinib或tofacitinib),而且潜在具有良好的安全性。
[0121]
下面结合附图和具体实施例对本发明做进一步的说明和描述。
附图说明
[0122]
图1示出了式(i)化合物的晶型a的xrpd图;
[0123]
图2示出了式(i)化合物的晶型a的tga/dsc曲线;
[0124]
图3示出了式(i)化合物的晶型a的加热前、后的xrpd对比图;
[0125]
图4示出了式(i)化合物的晶型a的变温xrpd图;
[0126]
图5示出了式(i)化合物的晶型b的xrpd图;
[0127]
图6示出了式(i)化合物的晶型b的tga/dsc曲线;
[0128]
图7示出了式(i)化合物的晶型b的变温xrpd图;
[0129]
图8示出了式(i)化合物的晶型c的xrpd图;
[0130]
图9示出了式(i)化合物的晶型c的tga/dsc曲线;
[0131]
图10示出了式(i)化合物的晶型c的加热前、后的xrpd对比图;
[0132]
图11示出了式(i)化合物的晶型d的xrpd图;
[0133]
图12示出了式(i)化合物的晶型d的xrpd对比图;
[0134]
图13示出了式(i)化合物的晶型e的xrpd图;
[0135]
图14示出了式(i)化合物的晶型e的tga/dsc曲线;
[0136]
图15示出了式(i)化合物的晶型e的加热前、后的xrpd对比图;
[0137]
图16示出了式(i)化合物的晶型e的变温xrpd图;
[0138]
图17示出了式(i)化合物的晶型f的xrpd图;
[0139]
图18示出了式(i)化合物的晶型f的tga/dsc曲线;
[0140]
图19示出了式(i)化合物的晶型f的加热前、后的xrpd对比图;
[0141]
图20示出了式(i)化合物的晶型g的xrpd图;
[0142]
图21示出了式(i)化合物的晶型g的tga/dsc曲线;
[0143]
图22示出了式(i)化合物的晶型g的变温xrpd图;
[0144]
图23示出了式(i)化合物的晶型j的xrpd图;
[0145]
图24示出了式(i)化合物的晶型j的xrpd对比图;
[0146]
图25示出了式(i)化合物的晶型h的xrpd图;
[0147]
图26示出了式(i)化合物的晶型i的xrpd图;
[0148]
图27示出了式(i)化合物的磷酸盐晶型a的xrpd图;
[0149]
图28示出了式(i)化合物的磷酸盐晶型a的tga/dsc曲线;
[0150]
图29示出了式(i)化合物的马来酸盐晶型a的xrpd图;
[0151]
图30示出了式(i)化合物的马来酸盐晶型a的tga/dsc曲线;
[0152]
图31示出了式(i)化合物的甲磺酸盐晶型b的xrpd图;
[0153]
图32示出了式(i)化合物的甲磺酸盐晶型b的tga/dsc曲线;
[0154]
图33示出了式(i)化合物的盐酸盐晶型a的xrpd图;
[0155]
图34示出了式(i)化合物的盐酸盐晶型a的tga/dsc曲线;
[0156]
图35示出了式(i)化合物的酒石酸盐晶型a的xrpd图;
[0157]
图36示出了式(i)化合物的酒石酸盐晶型a的tga/dsc曲线;
[0158]
图37示出了式(i)化合物的富马酸盐晶型a的xrpd图;
[0159]
图38示出了式(i)化合物的富马酸盐晶型a的tga/dsc曲线;
[0160]
图39示出了式(i)化合物的粘酸盐晶型a的xrpd图;
[0161]
图40示出了式(i)化合物的粘酸盐晶型a的tga/dsc曲线;
[0162]
图41示出了式(i)化合物的柠檬酸盐晶型a的xrpd图;
[0163]
图42示出了式(i)化合物的柠檬酸盐晶型a的tga/dsc曲线;
[0164]
图43示出了式(i)化合物的柠檬酸盐晶型b的xrpd图;
[0165]
图44示出了式(i)化合物的柠檬酸盐晶型b的tga/dsc曲线;
[0166]
图45示出了式(i)化合物的对甲苯磺酸盐晶型a的xrpd图;
[0167]
图46示出了式(i)化合物的对甲苯磺酸盐晶型a的tga/dsc曲线;
[0168]
图47示出了式(i)化合物的苯磺酸盐晶型a的xrpd图;
[0169]
图48示出了式(i)化合物的苯磺酸盐晶型a的tga/dsc曲线;
[0170]
图49示出了式(i)化合物的苯磺酸盐晶型b的xrpd图;
[0171]
图50示出了式(i)化合物的苯磺酸盐晶型b的tga/dsc曲线;
[0172]
图51示出了式(i)化合物的磷酸盐晶型a的dvs图;
[0173]
图52示出了式(i)化合物的马来酸盐晶型a的dvs图;
[0174]
图53示出了式(i)化合物的甲磺酸盐晶型b的dvs图。
[0175]
实施例
[0176]
本发明的化合物可以用有机合成领域的技术人员所熟悉的多种方法合成。以下具体实施例中给出了示例性的式(i)化合物的合成方法。显然,参照本技术中的示例性方案,本领域技术人员可以适当调整反应物、反应条件和保护基团而容易地设计式(i)化合物的其他合成路线或其它化合物的合成路线。
[0177]
本发明的化合物的适当的晶型或无定形物可以采用本领域技术人员所熟悉的纯化、结晶和/或干燥方法得到。以下具体实施例中给出了示例性的某些晶型和无定形物的制备方法。显然,参照本专利中的示例性方案,本领域技术人员可以适当调整溶剂、设备和工艺条件而容易地设计其它晶型或无定形物的制备方法。
[0178]
下面进一步结合实施例来阐述本发明;但这些实施例并不限制本发明的范围。除非另有声明,各实施例中所用的所有反应物均从商业途径获得;合成实验和产物分析检测中所用仪器设备等均为有机合成中通常使用的常规仪器和设备。
[0179]
实施例1:(s)-(2-(6-(2-乙基-5-氟-4-羟基苯基)-1h-吲唑-3-基)-4,6-二氢吡咯并[3,4-d]咪唑-5-(1h)-基)(3-羟基吡咯烷-1-基)甲酮(i)的合成
[0180]
[0181]
为了合成式(i)化合物,首先合成了化合物中间体1-1至中间体1-15,然后以中间体1-15为原料来合成式(i)化合物。
[0182]
除特别指明外,实施例1的各化学反应中所使用的化学试剂、溶剂和反应设备等均为化学合成中的常规原料和设备,可通过商业途径方便地获得。
[0183]
一、合成中间体1-6:3,4-二氨基吡咯烷基-1-甲酸叔丁酯
[0184]
通过以下合成路线来合成中间体1-6:
[0185][0186]
1.合成中间体1-1:2,5-二氢-1h-吡咯-1-甲酸叔丁酯
[0187]
将3-吡咯啉(10.0g,0.15mol)溶于400ml二氯甲烷和三乙胺(40.6ml,0.29mol)中,冷却到0度,缓慢加入(boc)2o(37.9g,0.17mol),室温反应过夜,加入水,用二氯甲烷萃取2次,合并有机相,用水洗3次,饱和食盐水洗,无水硫酸钠干燥,浓缩,硅胶柱纯化,得中间体1-1,收率91.0%。
[0188]
2.合成中间体1-2:6-氧杂-3-氮杂双环并[3.1.0]己烷-3-甲酸叔丁酯
[0189]
将中间体1-1(24.5g,0.15mol)溶于450ml二氯甲烷中,冷却到0度,分批缓慢加入间氯过氧苯甲酸(37.5g,0.22mol),室温反应过夜,加入饱和硫代硫酸钠(40ml),搅拌30分钟,水相用二氯甲烷萃取2次,用饱和碳酸钾溶液,水和饱和食盐水洗,无水硫酸钠干燥,浓缩,硅胶柱纯化,得中间体1-2,收率84.9%。
[0190]1h nmr(400mhz,cdcl3)δ3.85(d,j=12.0hz,1h),3.77(d,j=12.0hz,1h),3.69-3.67(m,2h),3.36-3.30(m,2h),1.45(s,9h).
[0191]
3.合成中间体1-3:3-叠氮基-4-羟基吡咯烷基-1-甲酸叔丁酯
[0192]
将中间体1-2(20.8g,0.12mol)溶于150ml 1,4-二氧六环和50ml水中,加入叠氮化钠(24.0g,0.37mol),加热到106度反应18小时,冷却到室温,加入饱和食盐水100ml,用二氯甲烷萃取(250ml x 4),合并有机相,用饱和食盐水洗,无水硫酸钠干燥,浓缩,得中间体1-3,收率100%。
[0193]1h nmr(400mhz,cdcl3)δ4.27-4.24(m,1h),3.94(s,1h),3.73-3.59(m,2h),3.41-3.36(m,2h),1.47(s,9h).
[0194]
4.合成中间体1-4:3-叠氮基-4-((甲磺酰基)氧基)吡咯烷基-1-甲酸叔丁酯
[0195]
将中间体1-3(28.0g,0.12mol)溶于350ml二氯甲烷和三乙胺(37.3g,0.37mol)中,冷却到0度,缓慢滴加甲磺酰氯(16.9g,0.15mol),滴加完后,室温反应2小时,加入水淬灭反应,用二氯甲烷萃取2次,合并有机相,用饱和碳酸氢钠溶液,水和饱和食盐水洗,无水硫酸钠干燥,浓缩,得中间体1-4,收率98.0%。
[0196]
5.合成中间体1-5:3,4-二叠氮基吡咯烷基-1-甲酸叔丁酯
[0197]
将中间体1-4(36.9g,0.12mol)溶于250ml dmf中,加入叠氮化钠(23.5g,0.36mol),加热到90度,反应2天,冷却到室温,加入750ml水,用甲基叔丁基醚萃取(400ml*4),合并有机相,用饱和食盐水洗,无水硫酸钠干燥,硅胶柱纯化,得中间体1-5,收率62.2%。
[0198]
6.合成中间体1-6:3,4-二氨基吡咯烷基-1-甲酸叔丁酯
[0199]
将中间体1-5(18.9g,0.08mol)溶于200ml甲醇中,加入10%pd/c,氢气置换3次,加热到40度,反应2天,过滤,浓缩,得中间体1-6,收率78%。
[0200]1h nmr(400mhz,cdcl3)δ3.51-3.49(m,2h),3.40-3.36(m,2h),3.21-3.11(m,2h),1.47(s,9h).
[0201]
二、合成中间体1-10:2-(4-(苄氧基)-2-乙基-5-氟苯基)-4,4,5,5-四甲基-1,3,2-二氧杂硼烷
[0202]
通过以下合成路线来合成中间体1-10:
[0203][0204]
1.合成中间体1-7:5-乙基-2-氟苯酚
[0205]
将5-溴-2-氟苯酚(200.0mg,1.05mmol)和二(三叔丁基磷)钯(10.7mg,0.02mmol)溶于10ml thf。氮气置换3次,降温至10~20℃,缓慢滴加1mol/l的二乙基锌溶液(2.3ml,2.30mmol),滴加完毕,升温至50℃。反应过夜,降温至0℃,加水淬灭,用硅藻土过滤,硅藻土垫用乙酸乙酯洗涤,用乙酸乙酯萃取,合并有机相,用饱和氯化钠溶液洗涤,无水硫酸钠干燥。干燥后浓缩柱层析分离得到油状液体中间体1-7,收率65.1%。1h nmr(400mhz,cdcl3)δ6.97(d,j=8.0hz,1h),6.85(d,j=12.0hz,1h),6.69
–
6.65(m,1h),2.61
–
2.55(m,2h),1.21(t,j=8.0hz,3h).
[0206]
2.合成中间体1-8:4-溴-5-乙基-2-氟苯酚
[0207]
将中间体1-7(200.1mg,1.43mmol)溶于6ml乙腈中,加入cubr2(957.5mg,4.29mmol),室温搅拌3小时,加水淬灭,用乙酸乙酯萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥。浓缩柱层析得到无色油状物中间体1-8,收率:78.1%。1h nmr(400mhz,cdcl3)δ7.25(d,j=12.0hz,1h),6.89(d,j=12.0hz,1h),2.69
–
2.63(m,2h),1.19(t,j=12.0hz,3h).
[0208]
3.合成中间体1-9:1-(苄氧基)-4-溴-5-乙基-2-氟苯
[0209]
将中间体1-8(15.5g,70.8mmol)溶于200ml dmf中,加入碳酸钾(19.5g,141.5mmol),溴化苄(14.5g,84.9mmol),升温到60℃反应。反应完成后,加水淬灭,用ea萃
取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥。浓缩后柱层析得到19.0g中间体1-9,收率:86.8%。1h nmr(400mhz,cdcl3)δ7.44
–
7.31(m,5h),7.27(d,j=8.0hz,1h),6.87(d,j=8.0hz,1h),5.12(s,2h),2.68
–
2.63(m,2h),1.16(t,j=8.0hz,3h).
[0210]
4.合成中间体1-10:2-(4-(苄氧基)-2-乙基-5-氟苯基)-4,4,5,5-四甲基-1,3,2-二氧杂硼烷
[0211]
将中间体1-9(1.0g,3.23mmol),频哪醇硼酸酯(0.82g,3.23mmol),pd(dppf)cl2(0.24g,0.32mmol)和koac(0.95g,9.70mmol)溶于15ml 1,4-二氧六环中,氮气置换3次。加热到100℃反应。反应完成后加水淬灭,用乙酸乙酯萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥。浓缩柱层析得到0.98g中间体1-10,收率:85.1%。1h nmr(400mhz,cdcl3)δ7.50
–
7.30(m,6h),7.82(d,j=8.0hz,1h),5.16(s,2h),2.87
–
2.81(m,2h),1.32(s,12h),1.14(t,j=8.0hz,3h).
[0212]
三、合成中间体1-15:2-(6-溴-1-((2-(三甲基硅基)乙氧基)甲基)-1h-吲唑-3-基)-1-((2-(三甲基硅基)乙氧基)甲基)-4,6-二氢吡咯并[3,4-d]咪唑-5(1h)-甲酸叔丁酯
[0213]
通过以下合成路线来合成中间体1-15:
[0214][0215]
1.合成中间体1-11:6-溴-1h-吲唑-3-甲醛
[0216]
将亚硝酸钠(14.00g,200mmol)溶于75ml dmf和100ml水中,冷却到0度,氮气保护下,缓慢滴加3n hcl(23ml,68.9mmol),滴加完反应10分钟。在0度下,向反应液中缓慢滴加6-溴吲哚(5.00g,25.5mmol)的dmf(35ml)溶液,滴加完,室温反应过夜。用乙酸乙酯萃取3次,合并有机相,用水洗3次,饱和食盐水洗,无水硫酸钠干燥,浓缩,硅胶柱纯化,得中间体1-11,收率83.6%。
[0217]1h nmr(400mhz,cdcl3)δ10.29(s,1h),8.24(d,j=8.0hz,1h),7.80(d,j=4.0hz,1h),7.52(dd,j=8.0hz,j=4.0hz,1h).
[0218]
2.合成中间体1-12:6-溴-1-((2-(三甲基硅基)乙氧基)甲基)-1h-吲唑-3-甲醛
[0219]
将中间体1-11(1.56g,6.93mmol)溶于干燥的四氢呋喃中,冷却到0度,缓慢加入氢化钠(0.33g,8.32mmol),室温反应1小时,冷却到0度,缓慢滴加2-(三甲基硅烷基)乙氧甲基
氯(1.73g,10.40mmol),滴加完室温反应过夜,加入水淬灭反应,用乙酸乙酯萃取2次,合并有机相,用水和饱和食盐水洗,无水硫酸钠干燥,浓缩,硅胶柱纯化,得中间体1-12,收率49.2%。
[0220]1h nmr(400mhz,cdcl3)δ10.25(s,1h),8.22(dd,j=8.0hz,j=4.0hz 1h),7.88(dd,j=4.0hz,j=4.0hz,1h),7.52(dd,j=4.0hz,j=4.0hz,1h),5.81(s,2h),3.63-3.58(m,2h),0.97-0.93(m,2h),0.04(s,9h).
[0221]
3.合成中间体1-13:2-(6-溴-1-((2-(三甲基硅基)乙氧基)甲基)-1h-吲唑-3-基)-3,4,6,6a-四氢吡咯并[3,4-d]咪唑-5(1h)-甲酸叔丁酯
[0222]
将中间体1-12(1.56g,6.93mmol)和3,4-二氨基吡咯啉-1-甲酸叔丁酯(1.56g,6.93mmol)溶于5ml六氟异丙醇中,加热到40度反应2天,浓缩,硅胶柱纯化,得中间体1-13,收率54.7%。
[0223]
4.合成中间体1-14:2-(6-溴-1-((2-(三甲基硅基)乙氧基)甲基)-1h-吲唑-3-基)-4,6-二氢吡咯并[3,4-d]咪唑-5(1h)-甲酸叔丁酯
[0224]
将草酰氯(0.53g,4.20mmol)溶于干燥的15ml二氯甲烷中,氮气保护下,冷却到-78度,缓慢滴加dmso(0.61g,7.84mmol),滴加完,反应30分钟,缓慢滴加中间体1-13(1.00g,1.87mmol)的二氯甲烷溶液,滴加完后,反应30分钟,缓慢滴加干燥的三乙胺(1.89g,18.66mmol),反应10分钟,缓慢升温室温反应2小时,加入饱和氯化铵溶液淬灭反应,用二氯甲烷萃取2次,合并有机层,用水和饱和食盐水洗,无水硫酸钠干燥,浓缩,硅胶柱纯化,得中间体1-14,收率36.3%。
[0225]1h nmr(400mhz,cdcl3)δ8.36(d,j=4.0hz,1h),7.78(d,j=4.0hz,1h),7.44(dd,j=8.0hz,j=4.0hz,1h),5.69(s,2h),4.64-4.52(m,4h),3.67-3.56(m,2h),1.56(s,9h),0.95-0.89(m,2h),0.03(s,9h).
[0226]
5.合成中间体1-15:2-(6-溴-1-((2-(三甲基硅基)乙氧基)甲基)-1h-吲唑-3-基)-1-((2-(三甲基硅基)乙氧基)甲基)-4,6-二氢吡咯并[3,4-d]咪唑-5(1h)-甲酸叔丁酯
[0227]
将中间体1-14(110mg,0.21mmol)溶于干燥的四氢呋喃中,冷却到0,加入氢化钠(12.3mg,0.31mmol),室温反应30分钟,冷却到0度,缓慢滴加2-(三甲基硅烷基)乙氧甲基氯(41.2mg,0.25mmol),室温反应4小时,加入水淬灭反应,用乙酸乙酯萃取2次,合并有机相,用水和饱和食盐水洗,无水硫酸钠干燥,浓缩,硅胶柱纯化,得中间体1-15,收率73.1%。
[0228]1h nmr(400mhz,cdcl3)δ8.41-8.36(m,1h),7.79(s,1h),7.44(dd,j=8.0hz,j=4.0hz,1h),5.94(d,j=12.0hz,2h),5.73(s,2h),4.65-4.52(m,4h),3.63-3.57(m,4h),1.56(s,9h),0.96-0.91(m,4h),0.03(s,18h).
[0229]
四、合成式(i)化合物
[0230]
通过以下合成路线,从中间体1-15出发来合成式(i)化合物:
[0231][0232]
1.合成中间体1-16:6-(4-(苄氧基)-2-乙基-5-氟苯基)-1-((2-(三甲基硅基)乙氧基)甲基)-3-(1-((2-(三甲基硅基)乙氧基)甲基)
-ꢀ
1,4,5,6-四氢吡咯并[3,4-d]咪唑-2-基)-1h-吲唑
[0233]
将2-(6-溴1-((2-(三甲基硅基)乙氧基)甲基)-1h-吲哚-3-基)-1-((2-(三甲基硅基)乙氧基)甲基)-4,6-二氢吡咯并[3,4-d]咪唑-5(1h)-甲酸叔丁酯(500mg,0.75mmol),2-(4-(苄氧基)-2-乙基-5-氟苯基)-4,4,5,5-四甲基-1,3,2-二氧杂硼烷(401mg,1.13mmol),pd(dppf)cl2(75mg,0.075mmol)和磷酸钾(495mg,2.25mmol)溶于1,4-二氧六环(30ml)和水(6ml)中,氮气置换3次,加热到100度,反应16h,冷却到室温,加入水,用乙酸乙酯萃取2次,合并有机相,用水和饱和食盐水洗,无水硫酸钠干燥,浓缩,硅胶柱纯化,纯化所得产品溶于25ml二氯甲烷,滴加5ml三氟乙酸,室温搅拌30分钟,浓缩,用二氯甲烷带3次三氟乙酸,浓缩,硅胶柱纯化,得中间体1-16共210mg,收率39.2%。
[0234]1h nmr(400mhz,cdcl3)δ8.48(d,j=8.3hz,1h),7.52(d,j=7.4hz,1h),7.49
–
7.37(m,5h),7.25(d,j=8.4hz,1h),7.23-6.96(m,2h),5.93(s,2h),5.77(s,2h),5.23(s,2h),4.21(d,j=35.1hz,4h),3.66
–
3.52(m,4h),2.54(q,j=7.6hz,2h),1.05(t,j=7.5hz,3h),0.95
–
0.89(m,4h),0.02(s,9h),-0.05(d,j=3.4hz,9h).
[0235]
2.合成中间体1-17:(s)-(2-(6-(4-(苄氧基)-2-乙基-5-氟苯基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1h-吲唑-3-基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-4,6-二氢吡咯并[3,4-d]咪唑-5-(1h)-基)(3-羟基吡咯烷-1-基)甲酮
[0236]
将三光气(91.5mg,0.31mmol)溶于10ml干燥二氯甲烷中,0度滴加中间体6-(4-(苄氧基)-2-乙基-5-氟苯基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-3-(1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1-1,4,5,6-四氢吡咯并[3,4-d]咪唑-2-基)-1h-吲唑(220.0mg,0.31mmol)的二氯甲烷(5ml)溶液,加完后缓慢滴加干燥三乙胺(312.2mg,3.09mmol),室温
搅拌10分钟,tlc监测原料消失,室温加入(s)-吡咯烷-3-醇(40.3mg,0.46mmol)的二氯甲烷(2ml)溶液,室温搅拌20分钟反应完毕,加水淬灭,用二氯甲烷萃取2次,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,浓缩,硅胶柱纯化,得192mg中间体1-17,收率75.3%。
[0237]1h nmr(400mhz,cdcl3)δ8.47(d,j=8.3hz,1h),7.53
–
7.51(m,2h),7.47
–
7.35(m,4h),7.25(d,j=8.4hz,1h),7.06
–
6.96(m,2h),5.96(s,2h),5.78(s,2h),5.23(s,2h),4.79
–
4.56(m,4h),4.49
–
4.45(m,1h),3.81
–
3.72(m,2h),3.68
–
3.58(m,5h),3.46
–
3.43(m,1h),2.56(q,j=7.6hz,2h),2.09
–
1.96(m,2h),1.16(t,j=7.5hz,3h),0.99
–
0.89(m,4h),0.02(s,9h),-0.05(s,9h).
[0238]
3.合成式(i)化合物:(s)-(2-(6-(2-乙基-5-氟-4-羟基苯基)-1h-吲唑-3-基)-4,6-二氢吡咯并[3,4-d]咪唑-5-(1h)-基)(3-羟基吡咯烷-1-基)甲酮
[0239]
将中间体1-17(22.00g,26.60mmol,1.00eq)加入到1l的三口瓶中,然后再加入dcm(330ml,15v)溶解后降温到-70℃至-60℃,磁力搅拌,用n2置换三次,缓慢滴加bcl3(133ml,132.99mmol,5.00eq,1n in dcm),约15min滴加完毕,滴加过程保持内温不超过-55℃,滴加完之后继续在-70℃至-60℃搅拌反应1h。向反应体系缓慢滴加50ml甲醇淬灭反应,淬灭温度不超过-50℃,约15min滴加完毕,体系自然回温至15-20℃,反应液在38℃真空下浓缩,残渣加入200ml甲醇和氨水,在40℃搅拌反应30min后,旋蒸除去甲醇,体系(水相)中有大量浅黄色固体析出,将体系过滤得到固体,固体烘干得到粗品(15.00g)。粗品送prep-hplc纯化(正相,0.1%氨水碱性体系,乙醇体系),所得馏分在40℃浓缩至200ml时有大量固体析出,过滤,得到浅黄色固体粉末5.90g,即式(i)化合物。
[0240]1h nmr(400mhz,dmso-d6)δ13.25(s,1h),12.69(s,1h),9.84(s,1h),8.32(d,j=8.4hz,1h),7.39(s,1h),7.11(d,j=9.2hz,1h),7.03(d,j=12hz,1h),6.92(d,j=9.2hz,1h),4.92(s,1h),4.74-4.53(m,2h),4.51-4.35(m,2h),4.27(s,1h),3.58-3.49(m,2h),3.45-3.37(m,1h),3.23(m,1h),2.49-2.45(m,2h,被dmso-d6的溶剂峰部分遮挡),1.92-1.71(m,2h),1.02(t,j=7.6hz,3h)
[0241]
lc-ms:c
25h26
fn6o3[m h]
m/z计算值为477.2,检测值为477.1.
[0242]
实施例2:式(i)化合物的药理学活性评价i
[0243]
1.实验原理
[0244]
通过基于jak1、jak2、jak3、tyk2激酶的药物筛选体系来检测化合物分别对于激酶活性的抑制能力。激酶与其底物irs1,igf1rtide,poly(4:1glu,tyr)进行酶学反应,消耗atp产生adp,利用adp-glo试剂和发光的方法检测产物的量用以反映激酶的活性。
[0245]
2.实验方案
[0246]
2.1实验材料和仪器
[0247]
序号名称来源货号1hepeslife technologies15630-0802brij 35detergent(10%)sigma10189401003mgcl2sigmam10284egtasigmae38895adp-glo kinase assaypromegav91016jak1carna08-144
7jak2carna08-0458jak3carna08-0469tyk2carna08-14710atppromegav915b11irs1signalchemi40-58-100012igf1rtidesignalchemi15-5813poly(4:1glu,tyr)sigmap027515384polystyrene shallow flat whitegreiner78407516384-well polypropylene microplatelabcytepp-020017biotek酶标仪bioteksynergy 418微孔板低速离心机湘智td5b
[0248]
2.2实验方法
[0249]
2.2.1激酶反应试剂配方
[0250]
2.2.1.1 1x激酶反应缓冲液(400ml)
[0251]
名称储液浓度体积终浓度hepes1m(20x)20ml50mmmgcl21m(100x)4ml10mmbrij-3510%(1000x)400μl0.01%egta粉末152mg1mmddh2o 375.6ml [0252]
2mm dtt,现用现配
[0253]
2.2.1.2 2x激酶配方
[0254][0255][0256]
[0257][0258]
2.2.1.3 4x底物混合物配方
[0259][0260][0261][0262][0263]
2.2.1.4待测化合物
[0264]
化合物名称质量/mg分子量浓度/mmfilgotinib5.0420.510式(i)化合物1.5476.5110
[0265]
2.2.2激酶反应实验步骤
[0266]
2.2.2.1 jak1&jak2激酶反应实验步骤
[0267]
a)用100%dmso将filgotinib(10mm储液)原倍,待测化合物稀释5倍,在96孔稀释板中进行4倍等比稀释,取1μl的化合物加入49μl的激酶反应缓冲液中,在微孔板振荡器上震荡20min。
[0268]
b)转移2μl的激酶(2.2.1.2步骤中制备)到384反应板中,加入1μl的待测化合物(a步骤中准备)到384反应板中(greiner,784075),1000rpm/min,离心1min,25℃孵育10min。
[0269]
c)转移1μl底物混合物(2.2.1.3步骤中制备)到384反应板中,1000rpm/min,离心1min,25℃孵育60min。在反应体系中,filgotinib终浓度50,12.5,3.125,0.7812,0.1953,0.0488,0.0122,0.003,0.00076,0.00019,0.000047μm。待测化合物终浓度:10,2.5,0.625,0.15625,0.039,0.0097,0.0024,0.0006,0.0015,0.000038,0.0000095μm。dmso终浓度均为0.5%。
[0270]
d)转移4μl adp-glo到384反应板中1000rpm/min,离心1min,25℃孵育40min。
[0271]
e)转移8μl detection溶液到384反应板中1000rpm/min,离心1min,25℃孵育40min。
[0272]
f)使用biotek多功能读板机读取rlu(relative luminescence unit)信号。信号强度用于表征激酶的活性程度。
[0273]
2.2.2.2 jak3&tyk2激酶反应实验步骤
[0274]
a)用100%dmso将filgotinib(10mm储液)原倍,待测化合物稀释5倍,在96孔稀释板中进行3倍等比稀释,取1μl的化合物加入49μl的激酶反应缓冲液中,在微孔板振荡器上震荡20min。
[0275]
b)转移2μl的激酶(2.2.1.2步骤中制备)到384反应板中,加入1μl的待测化合物(a步骤中准备)到384反应板中(greiner,784075),1000rpm/min,离心1min,25℃孵育10min。
[0276]
c)转移1μl底物混合物(2.2.1.3步骤中制备)到384反应板中,1000rpm/min,离心1min,25℃孵育60min。在反应体系中,filgotinib终浓度50,16.67,5.555,1.851,0.617,0.205,0.0686,0.0228,0.00762,0.0025μm。待测化合物终浓度:10,3.33,1.11,0.37,0.12,0.04,0.014,0.0046,0.0015,0.0005μm。dmso终浓度均为0.5%。
[0277]
d)转移4μl adp-glo到384反应板中1000rpm/min,离心1min,25℃孵育40min。
[0278]
e)转移8μl detection溶液到384反应板中1000rpm/min,离心1min,25℃孵育40min。
[0279]
f)使用biotek多功能读板机读取rlu(relative luminescence unit)信号。信号强度用于表征激酶的活性程度。
[0280]
2.2.3实验数据处理方法
[0281]
化合物抑制率(%inh)=(阴性对照-化合物)/(阴性对照-阳性对照)
×
100%
[0282]
阴性对照:dmso
[0283]
阳性对照:10um/100um/30um filgotinib
[0284]
利用以下非线性拟合公式来得到化合物的ic50(半数抑制浓度):
[0285]
y=bottom (top-bottom)/(1 10^((logic50-x)*hillslope))
[0286]
x:化合物浓度log值
[0287]
y:化合物抑制率(%inh)
[0288]
z’因子计算方程:
[0289]
z’=1-3(sdmin sdmax)/(avemax-avemin)
[0290]
其中:
[0291]
min为阳性对照10um/100um/30umfilgotinib rlu值,max为阴性对照dmso rlu值。
[0292]
sd为标准误差,ave为rlu平均值。
[0293]
3.结果
[0294]
化合物检测结果见下表:
[0295][0296]
测试结果表明:实施例1得到的式(i)化合物的抑制活性远远高于filgotinib(高两个数量级以上),能够在极低的浓度下有效抑制jak1、jak2、jak3、tyk2。
[0297]
实施例3:式(i)化合物的药理学活性评价ii
[0298]
1.实验目的
[0299]
本实施例旨在检测化合物在jak细胞活性实验-人t细胞增殖实验中的活性。
[0300]
2.实验方案
[0301]
2.1实验材料和仪器
[0302][0303]
2.2实验方法
[0304]
2.2.1待测化合物储液的配制
[0305]
化合物储液的配制
[0306][0307]
2.2.2激酶反应实验步骤
[0308]
a)根据人总t细胞分选试剂盒从人pbmc中分选出t细胞。
[0309]
b)用anti-cd3抗体和anti-cd28抗体刺激t细胞,在37℃、5%co2培养箱中孵育72h。
[0310]
c)收集t细胞,并用pbs洗涤细胞。
[0311]
d)用echo 550转移40nl稀释好的待测化合物至384孔板。
[0312]
e)将t细胞(c步骤中准备)种植到384孔反应板(d步骤中准备),35μl/孔,1000rpm离心1min,37℃、5%co2孵育。
[0313]
f)加入5μl/孔人重组il-2蛋白,终浓度为10ng/ml,1000rpm离心1min,37℃、5%co2孵育72h。
[0314]
g)加入20μl/孔celltiter glo缓冲液,1000rpm离心1min,350g混匀2min,在室温孵育30min。
[0315]
h)在envision多功能读板机上读取lumilencence信号值。
[0316]
2.2.3实验数据处理方法
[0317]
化合物抑制率(%inh)=(阴性对照-化合物)/(阴性对照-阳性对照)
×
100%
[0318]
阴性对照:dmso的读值
[0319]
阳性对照:10um tofacitinib的读值
[0320]
利用以下非线性拟合公式来得到化合物的ic50(半数抑制浓度):
[0321]
y=bottom (top-bottom)/(1 10^((logic50-x)*hillslope))
[0322]
x:化合物浓度log值
[0323]
y:化合物抑制率(%inh)
[0324]
3.结果
[0325]
化合物检测结果见下表:
[0326]
化合物ic50(nm)tofacitinib21.74式(i)化合物13.01
[0327]
以上结果表明:式(i)化合物在活性实验-il-2诱导的人t细胞增殖实验中显示出优于tofacitinib的活性。
[0328]
实施例4:式(i)化合物的晶型研究
[0329]
通过不同的结晶方法来研究实施例1中的式(i)化合物(因为其为碱性化合物,在后文有时也称为“游离碱”)的多晶型现象,通过x射线粉末衍射(xrpd)、热重分析(tga)、差示扫描量热(dsc)、核磁共振氢谱(1h nmr)等方法对得到各种晶型进行表征和鉴定,以便为药物制剂的生产和配制提供基础和依据。
[0330]
一、仪器和测定方法
[0331]
1)x射线粉末衍射(xrpd)
[0332]
xrpd图在panalytical x射线粉末衍射分析仪上采集,扫描参数如下表所示。
[0333]
表1:xrpd测试参数
[0334][0335][0336]
2)热重分析(tga)和差示扫描量热(dsc)
[0337]
tga和dsc图分别在ta discovery tga 5500热重分析仪和ta q2000/discovery dsc 2500差示扫描量热仪上采集,下表列出了测试参数。
[0338]
表2:tga和dsc测试参数
[0339]
参数tgadsc方法线性升温线性升温样品盘铝盘,敞开铝盘,压盖/不压盖温度范围室温-设置终点温度室温/25℃-设置终点温度扫描速率(℃/分钟)1010保护气体氮气氮气
[0340]
3)液态核磁(1h nmr)
[0341]
液态核磁谱图在bruker 400m核磁共振仪上采集,dmso-d6作为溶剂。
[0342]
二、具体实验过程和结果
[0343]
1.晶型a的制备和表征
[0344]
将实施例1获得的式(i)化合物5.00g加入到三口瓶中,加入100ml甲醇升温至50~55℃搅拌,体系变得粘稠,又加入50ml甲醇,继续搅拌5h。过滤,滤饼用20ml甲醇淋洗,45℃下真空干燥8h,最终得到3.7g式(i)化合物的一种晶体,命名为晶型a。
[0345]
晶型a的xrpd测试结果见图1,具体数据如下:
[0346]
表3:游离碱晶型a的xrpd数据
[0347][0348]
晶型a的tga/dsc测试结果见图2,从图中可知,将晶型a加热至200℃失重4.4%,推测来自样品中溶剂或水的脱除,在50-290℃范围内可观察到多个热信号。
[0349]
晶型a的1h nmr中未观察到相应溶剂meoh残留,说明tga中4.4%的失重来自水的脱除,提示晶型a可能为水合物。
[0350]
为了研究晶型a的属性,对晶型a开展了加热实验。氮气保护下将晶型a加热至190℃并降至室温后,取出样品暴露在空气中用于xrpd测试,测试结果见图3,显示样品晶型未变,同时可观察到部分衍射峰相对强度降低。结合tga/nmr结果,加热后结晶度的变化推测由水的脱除引起,晶型a可能为水合物。
[0351]
为进一步确定晶型a的属性,对晶型a开展了变温xrpd测试,结果见图4。显示在30℃条件下,样品经过氮气吹扫20分钟后晶型未变;在氮气吹扫下加热至190℃并降至30℃后,均得到仅在氮气保护下存在的无水晶型d(不同温度下衍射峰位置的偏移可能与高温引起的晶格膨胀相关)。综合以上晶型a的tga/dsc/nmr结果,可以认定晶型a加热至190℃后的晶型变化由样品中水的脱除引起,晶型a为水合物。
[0352]
2.晶型b的制备和表征
[0353]
将晶型a在etoh中室温悬浮搅拌三天后,离心分离并将固体置于室温下真空干燥约两小时后得到晶体,命名为晶型b。
[0354]
晶型b的xrpd结果如图5所示,具体数据如下:
[0355]
表4:游离碱晶型b的xrpd数据
[0356][0357]
晶型b的tga/dsc结果如图6所示。晶型b加热至150℃失重6.0%,推测来自样品中溶剂或水的脱除;在319.1℃(起始温度)处可观察到一个吸热信号,推测来自样品熔融。
[0358]
晶型b的1h nmr显示晶型b中etoh与化合物(i)的摩尔比为0.12:1.00(1.1wt%)。
[0359]
为研究晶型b的属性,对晶型b开展了变温xrpd测试,结果如图7所示。晶型b样品在氮气下吹扫20分钟后晶型未变;在氮气吹扫下加热至150℃,与晶型b相比,样品衍射峰位置略有偏移,推测与高温下晶格膨胀相关;降温至30℃后,样品的衍射峰位置与晶型b一致,且衍射峰的偏移消失,结合tga/dsc结果,推测tga失重来自样品表面的水分或溶剂残留,判断晶型b为无水晶型。
[0360]
3.晶型c的制备和表征
[0361]
将水合物晶型a在含有l-抗坏血酸的丙酮/h2o(v/v,19:1)体系中室温悬浮搅拌三天,离心分离并将固体置于室温下真空干燥约两小时后得到晶体,命名为晶型c。
[0362]
晶型c的xrpd结果如图8所示,具体数据如下:
[0363]
表5:游离碱晶型c的xrpd数据
[0364][0365]
晶型c的tga/dsc结果如图9所示:样品加热至100℃失重6.4%,相应地在78.1℃(起始温度)处可观察到一个热信号,推测来自样品中溶剂或水的脱除;在279.4和303.8℃(起始温度)处可观察到重叠的热信号。
[0366]
晶型c的1h nmr显示晶型c中丙酮与化合物(i)的摩尔比为0.06:1.00(0.7wt%)。结合tga结果,推测tga中6.4%的失重基本来自水的脱除,晶型c可能为水合物。
[0367]
为了研究晶型c的属性,对晶型c开展了加热实验。氮气保护下将晶型c加热至150℃并降至室温后,取出样品暴露在空气中用于xrpd测试,结果(图10),显示加热后样品结晶度明显降低。结合核磁及tga结果,推测加热后结晶度的降低由水的脱除引起,判断晶型c为水合物。
[0368]
4.晶型d的制备和表征
[0369]
将晶型a在氮气保护下加热至190℃并降至30℃后得到,xrpd结果如图11所示。
[0370]
将晶型d暴露在空气中约2小时后测试xrpd,结果(图12)显示已转变为晶型a。推测晶型d仅能在氮气保护下存在,暴露在空气中将快速吸收环境中的水份转为晶型a。由于晶型d不稳定,未开展进一步研究。
[0371]
5.晶型e的制备和表征
[0372]
将水合物晶型a在etoh中50℃悬浮搅拌一天后,离心分离并将固体置于室温,敞口干燥过夜后得到晶体,命名为晶型e。
[0373]
晶型e的xrpd结果如图13所示,具体数据如下:
[0374]
表6:游离碱晶型e的xrpd数据
[0375][0376]
晶型e的tga/dsc结果如图14所示:样品加热至150℃失重7.9%,相应地在43.5℃(起始温度)处可观察到一个热信号,推测来自样品中溶剂或水的脱除;在187.1℃(起始温度)处可观察到一个吸热信号。
[0377]
晶型e的1h nmr显示etoh与化合物(i)的摩尔比为0.62:1.00(5.7wt%),结合tga结果,推测tga中失重来自etoh和水的脱除。
[0378]
为了研究晶型e的属性,对晶型e开展了加热实验。氮气保护下将晶型e加热至120℃并降至室温后,取出样品暴露在空气中用于xrpd测试,结果如图15所示,结果显示样品晶型未变。核磁结果显示:加热后样品中etoh与化合物(i)的摩尔比为0.65:1.00(5.7wt%)。
[0379]
为进一步研究晶型e的属性,对晶型e开展了变温xrpd测试。结果如图16所示:样品在氮气下吹扫20分钟后,晶型未变;在氮气吹扫下加热至120℃,样品已转变为仅在氮气保护下存在的无水晶型d(少数衍射峰位置与晶型d相比有偏移,可能与不同温度下晶格膨胀程度不同相关,参比晶型d的xrpd图谱为30℃测得);继续加热至210℃,样品已转为无定形;降温至30℃后,样品仍为无定形,且观察到样品成胶,推测样品已熔融。与将晶型e加热至120℃后暴露在空气中的实验结果(晶型未变)相比,推测两次加热结果的差异与样品加热后是否暴露在空气中相关,即无水晶型d暴露在空气中加热后能快速吸附环境中的水份转为晶型e,推测水分子参与晶型e的晶格组成。
[0380]
6.晶型f的制备和表征
[0381]
将水合物晶型a及无水晶型b的晶种在丙酮中50℃悬浮搅拌一天后,离心分离并将固体置于室温敞口干燥过夜后,得到晶体,命名为晶型f。
[0382]
晶型f的xrpd结果见图17,具体数据如下:
[0383]
表7:游离碱晶型f的xrpd数据
[0384][0385]
晶型f的tga/dsc结果(图18)显示,样品加热至170℃失重10.5%,相应地在119.7℃(起始温度)处可观察到一个热信号,推测来自样品中溶剂或水的脱除;在209.3、316.3℃(起始温度)处可观察两个热信号。晶型f的1h nmr显示,晶型f中丙酮与化合物(i)的摩尔比为0.68:1.00(7.7wt%),结合tga结果,推测tga中失重大部分来自丙酮的脱除。
[0386]
为了研究晶型f的属性并研究dsc中热信号可能的来源,对晶型f开展了加热实验。氮气保护下将晶型f加热至170℃并降至室温后,取出样品暴露在空气中用于xrpd测试,结果(图19)显示样品已转为水合物晶型g,核磁结果显示加热后的样品中未观察到明显丙酮残留。综合加热前、后的数据,xrpd中可观察到晶型的变化,tga/nmr显示加热后脱除的组分主要为丙酮,加热后晶型f转为晶型g可能由丙酮的脱除引起,判断晶型f可能为丙酮溶剂合物。
[0387]
7.晶型g的制备和表征
[0388]
将丙酮溶剂合物晶型f在氮气保护下加热至170℃并降至室温后,取出样品暴露在空气中,得到晶体,命名为晶型g。
[0389]
晶型g的xrpd结果如图20所示,具体数据如下:
[0390]
表8:游离碱晶型g的xrpd数据
[0391][0392]
晶型g的tga/dsc结果(图21)显示:晶型g加热至100℃失重4.7%,相应地在81.2℃(起始温度)处可观察到一个热信号,推测来自样品中溶剂或水的脱除;在209.6、314.0℃(起始温度)处可观察到两个热信号。晶型g的核磁结果显示,样品中未观察到明显丙酮残留,晶型g可能为水合物或无水晶型。
[0393]
通过变温xrpd对晶型g开展了进一步研究(以晶型f为起始原料进行研究)。结果(图22)显示:将晶型f在氮气下吹扫20分钟后,晶型未变;在氮气吹扫下加热至170℃,已转变为仅在氮气保护下存在的无水晶型j;降温至30℃后将样品开盖暴露在空气中测试xrpd,结果显示已转为晶型g,推测晶型j暴露在空气中后吸收环境中的水份转为晶型g,晶型g为水合物。
[0394]
8.晶型j的制备和表征
[0395]
通过将丙酮溶剂合物晶型f在氮气保护下加热至170℃并降至30℃,得到晶体,命名为晶型j。晶型j的xrpd结果如图23所示。
[0396]
将晶型j暴露在空气中,再次测试xrpd,结果(图24)显示已转变为水合物晶型g,推测晶型j仅能在氮气保护下存在,暴露在空气中将快速吸收环境中的水份转为晶型g。由于晶型j不稳定,未开展进一步研究。
[0397]
9.晶型h的制备和表征
[0398]
将晶型e在氮气保护下加热至150℃并降至室温后,取出样品暴露在空气中,得到晶体,命名为晶型h,其xrpd结果如图25所示。
[0399]
10.晶型i的制备和表征
[0400]
通过变温xrpd将水合物晶型c在氮气保护下加热至150℃并降至室温后得到晶体,命名为晶型i,其xrpd结果如图26所示。
[0401]
实施例5:式(i)化合物的盐型研究
[0402]
以实施例1得到的式(i)化合物为起始物料,选用不同的酸对其进行成盐研究,并在不同条件下进行结晶,研究各盐的多晶型现象,通过x射线粉末衍射(xrpd)、热重分析(tga)、差示扫描量热(dsc)、核磁共振氢谱(1h nmr)、高效液相色谱/离子色谱(hplc/ic)等方法对得到各种晶型进行表征和鉴定,以便为药物制剂的生产和配制提供基础和依据。
[0403]
一、研究中所用的主要试剂、仪器和测定方法
[0404]
1.实验中所用主要溶剂的缩写与相应中文名称对照如下表所示。
[0405]
表9:试验中选用溶剂的中英文名称对照表
[0406]
简写中文简写中文meoh甲醇mtbe甲基叔丁基醚etoh乙醇anisole苯甲醚ipa异丙醇n-heptane正庚烷acetone丙酮toluene甲苯mibk甲基异丁基酮dcm二氯甲烷etoac乙酸乙酯acn乙腈ipac乙酸异丙酯dmso二甲基亚砜thf四氢呋喃chcl3氯仿2-methf2-甲基四氢呋喃dmfn,n-二甲基甲酰胺1,4-dioxane1,4-二氧六环h2o水
[0407]
2.x射线粉末衍射(xrpd)
[0408]
xrpd图在panalytical x射线粉末衍射分析仪上采集,扫描参数如表10所示。
[0409]
表10:xrpd测试参数
[0410][0411]
3.热重分析(tga)和差示扫描量热(dsc)
[0412]
tga和dsc图分别在ta q5000/discovery tga 5500热重分析仪和ta discovery dsc 2500差示扫描量热仪上采集,表11列出了测试参数。
[0413]
表11:tga和dsc测试参数
[0414]
参数tgadsc方法线性升温线性升温样品盘铝盘,敞开铝盘,压盖温度范围室温-设置终点温度25℃-设置终点温度
扫描速率(℃/分钟)1010保护气体氮气氮气
[0415]
4.动态水分吸附(dvs)
[0416]
动态水分吸附(dvs)曲线在sms(surface measurement systems)的dvs intrinsic上采集。在25℃时的相对湿度用licl,mg(no3)2和kcl的潮解点校正。dvs测试参数列于表12。
[0417]
表12:dvs测试参数
[0418][0419]
5.液态核磁(1h nmr)
[0420]
液态核磁谱图在bruker 400m核磁共振仪上采集,dmso-d6作为核磁溶剂。
[0421]
6.ph计(ph)
[0422]
ph通过sartorius pb-10ph计采集。
[0423]
7.高效液相色谱(hplc)及离子色谱(ic)
[0424]
样品纯度及溶解度通过高效液相色谱采集,无机酸体系的盐型样品摩尔比通过高效液相色谱及离子色谱采集,高效液相色谱在agilent 1290 hpl上测试,离子色谱在thermo ics1100上采集。具体仪器和测试参数见表13及表14。
[0425]
表13:溶解度,摩尔比及纯度hplc测试参数
[0426][0427]
*:配置时加硫酸助溶。
[0428]
表14:ic测试参数
[0429]
参数设定值色谱柱ionpac as18 analytical column(4
×
250mm)流动相20mm氢氧化钠进样量25微升流速1.0毫升/分钟样品室温度35℃柱温35℃电流80ma运行时间40分钟
[0430]
二、盐的制备和表征
[0431]
1.磷酸盐晶型a的制备和表征
[0432]
以式(i)化合物水合物晶型a为原料制备磷酸盐,制备具体步骤为:
[0433]
1)量取24μl浓磷酸(约85%)至20ml玻璃小瓶中;
[0434]
2)称取约200mg的式(i)化合物水合物晶型a至20ml玻璃小瓶中,并加入10ml acetone/h2o(v/v,19:1);
[0435]
3)25℃悬浮搅拌约3天后得到乳白色悬浊液,室温减压抽滤得到固体,将固体转移至室温真空干燥3小时得到白色粉末状样品,xrpd结果显示样品为晶体,同时hplc/ic结果显示式(i)化合物与磷酸根的摩尔比为1.00:1.25(推测磷酸根含量偏高可能由未反应的磷酸残留导致);
[0436]
4)为除去可能残留的磷酸,室温下向第3步得到的样品中加入总计2ml etoac进行悬浮搅拌,约5小时后离心分离,固体转至室温真空干燥约13小时,得到磷酸盐晶型a。
[0437]
磷酸盐晶型a的xrpd图如图27所示,具体数据见下表:
[0438]
表15:磷酸盐晶型a的xrpd数据
[0439][0440]
磷酸盐晶型a的tga/dsc数据如图28所示:样品加热至190℃时失重为1.8%,推测来自样品中水或溶剂的脱除,在279.3℃(起始温度)处可观察到一个尖锐吸热峰,推测来自样品熔融。基于tga中样品熔融前少量的失重以及dsc中仅有单一熔融吸热峰,判断磷酸盐晶型a可能为无水晶型。hplc/ic结果显示,样品中式(i)化合物与磷酸根的摩尔比为1:1。
[0441]
2.马来酸盐晶型a的制备和表征
[0442]
以式(i)化合物水合物晶型a为原料制备马来酸盐,制备具体步骤为:
[0443]
1)称取约200mg式(i)化合物水合物晶型a至20ml玻璃小瓶中;
[0444]
2)加入53mg马来酸至20ml玻璃小瓶中,并加入10ml acetone/h2o(v/v,19:1);
[0445]
3)25℃悬浮搅拌约3天后得到乳白色悬浊液,室温减压抽滤得到固体,将固体转移至室温真空干燥3小时得到白色粉末状样品,xrpd结果显示样品为晶体,同时核磁结果显示样品中式(i)化合物与马来酸的摩尔比为1.00:0.86,推测样品中存在少量未反应的式(i)化合物,样品成盐可能不充分;
[0446]
4)为促进样品充分成盐,向第3步得到的163mg样品中加入10mg马来酸,在2ml etoac中50℃搅拌约14小时后,固体转至室温真空干燥约2小时,干燥后得到晶体,命名为马来酸盐晶型a,进行xrpd及核磁测试。
[0447]
马来酸盐晶型a的xrpd结果见图29,具体数据如下:
[0448]
表16:马来酸盐晶型a的xrpd数据
[0449][0450]
核磁结果显示式(i)化合物与马来酸的摩尔比为1:1。
[0451]
马来酸盐晶型a的tga/dsc见图30,结果显示样品加热至150℃失重3.3%,在211.2和278.1℃(起始温度)处可观察到两个热信号。
[0452]
3.甲磺酸盐晶型b的制备和表征
[0453]
以式(i)化合物水合物晶型a为原料制备甲磺酸盐,制备具体步骤为:
[0454]
1)称取约200mg式(i)化合物水合物晶型a至20ml玻璃小瓶中;
[0455]
2)加入54mg甲磺酸至20ml玻璃小瓶中,并加入10ml etoac;
[0456]
3)25℃悬浮搅拌约3天后得到乳白色悬浊液,室温减压抽滤得到固体,将固体转移至室温真空干燥3小时得到白色粉末状样品,即甲磺酸盐晶型b。
[0457]
由此获得的甲磺酸盐晶型b的xrpd结果如图31所示,具体数据如下:
[0458]
表17:甲磺酸盐晶型b的xrpd数据
[0459][0460]
甲磺酸盐晶型b的核磁结果显示样品中式(i)化合物与甲磺酸的摩尔比为1:1。
[0461]
甲磺酸盐晶型b的tga/dsc数据如图32所示,样品加热至190℃时失重为2.2%,推测来自样品中水或溶剂的脱除,在234.4℃(峰值温度)和264.1℃(起始温度)处可观察到两个吸热信号。基于tga中样品分解前少量缓慢的失重以及dsc中190℃之前较平滑的曲线,推测甲磺酸盐晶型b为无水晶型。
[0462]
4.盐酸盐晶型a的制备和表征
[0463]
以式(i)化合物水合物晶型a为原料制备盐酸盐,制备具体步骤为:将式(i)化合物水合物晶型a与盐酸按照摩尔比1:1在etoac中室温悬浮搅拌约3天后,离心分离,将固体转移至室温真空干燥2小时后获得盐酸盐晶型a。
[0464]
盐酸盐晶型a的xrpd结果如图33所示,具体数据如下:
[0465]
表18:盐酸盐晶型a的xrpd数据
[0466][0467]
盐酸盐晶型a的tga/dsc见图34,结果显示样品加热至240℃失重13.7%,在80.1、118.0℃(峰值温度)和224.5℃(起始温度)处可观察到三个吸热峰,在231.5℃(起始温度)处可观察到一个放热信号。hplc/ic结果显示,盐酸盐晶型a中式(i)化合物与氯离子的摩尔比为1:0.7(推测样品成盐可能不充分)。
[0468]
5.酒石酸盐晶型a的制备和表征
[0469]
以式(i)化合物水合物晶型a为原料制备酒石酸盐,制备具体步骤为:将式(i)化合物水合物晶型a与酒石酸按照摩尔比1:1在etoac中室温悬浮搅拌约3天后,离心分离,将固体转移至室温真空干燥2小时后获得酒石酸盐晶型a。
[0470]
酒石酸盐晶型a的xrpd结果如图35所示,具体数据如下:
[0471]
表19:酒石酸盐晶型a的xrpd数据
[0472][0473]
酒石酸晶型a的tga/dsc见图36,结果显示样品加热至200℃失重9.1%,在125.2和168.8℃(起始温度)处可观察到两个热信号。1h nmr结果显示,酒石酸盐晶型a中式(i)化合物与酒石酸的摩尔比为1:1(已扣除与式(i)化合物重叠的5个h)。
[0474]
6.富马酸盐晶型a的制备和表征
[0475]
以式(i)化合物水合物晶型a为原料制备富马酸盐,制备具体步骤为:将式(i)化合物水合物晶型a与富马酸按照摩尔比1:1在acetone/h2o(19:1,v/v)中室温悬浮搅拌约3天后,离心分离,将固体转移至室温真空干燥2小时后获得富马酸盐晶型a。
[0476]
富马酸盐晶型a的xrpd结果如图37所示,具体数据如下:
[0477]
表20:富马酸盐晶型a的xrpd数据
[0478][0479]
富马酸晶型a的tga/dsc见图38,结果显示样品加热至110℃失重7.5%,继续加热至250℃失重8.0%;样品在71.1、205.3、273.7和299.4℃(起始温度)处可观察到多个热信号。1h nmr结果显示,富马酸盐晶型a中式(i)化合物与富马酸的摩尔比为1:0.6。
[0480]
7.粘酸盐晶型a的制备和表征
[0481]
以式(i)化合物水合物晶型a为原料制备粘酸盐,制备具体步骤为:将式(i)化合物水合物晶型a与粘酸按照摩尔比1:1在acetone/h2o(19:1,v/v)中室温悬浮搅拌约3天后,离心分离,将固体转移至室温真空干燥2小时后获得粘酸盐晶型a。
[0482]
粘酸盐晶型a的xrpd结果如图39所示,具体数据如下:
[0483]
表21:粘酸盐晶型a的xrpd数据
[0484][0485][0486]
粘酸盐晶型a的tga/dsc见图40,结果显示样品加热至100℃失重4.4%,继续加热至260℃失重26.4%,在69.8和210.4℃(起始温度)处可观察到两个热信号。1h nmr结果显示,粘酸盐晶型a中式(i)化合物与粘酸的摩尔比为1:1.4。
[0487]
8.柠檬酸盐晶型a/b的制备和表征
[0488]
以式(i)化合物水合物晶型a为原料制备柠檬酸盐,制备具体步骤为:将式(i)化合物水合物晶型a与柠檬酸按照摩尔比1:1分别在etoac、acetone/h2o(19:1,v/v)中室温悬浮搅拌约3天后,离心分离,将固体转移至室温真空干燥2小时后获得柠檬酸盐晶型a/b。
[0489]
柠檬酸盐晶型a的xrpd结果如图41所示,具体数据如下:
[0490]
表22:柠檬酸盐晶型a的xrpd数据
[0491][0492]
柠檬酸盐晶型a的tga/dsc见图42,结果显示样品加热至150℃失重9.1%,在68.2℃(峰值温度)、154.4和164.0℃(起始温度)处可观察到三个吸热峰。1h nmr结果显示,柠檬酸盐晶型a中式(i)化合物与柠檬酸的摩尔比为1:0.5。
[0493]
柠檬酸盐晶型b的xrpd结果如图43所示,具体数据如下:
[0494]
表23:柠檬酸盐晶型b的xrpd数据
[0495][0496]
柠檬酸盐晶型b的tga/dsc见图44,结果显示样品加热至100℃失重7.1%,继续加热至250℃失重6.8%;在73.1、280.2、301.6℃(起始温度)和179.7℃(峰值温度)处可观察到四个吸热峰。1h nmr结果显示,柠檬酸盐晶型b中式(i)化合物与柠檬酸的摩尔比为1:0.5。
[0497]
9.对甲苯磺酸盐晶型a的制备和表征
[0498]
以式(i)化合物水合物晶型a为原料制备对甲苯磺酸盐,制备具体步骤为:将式(i)化合物水合物晶型a与对甲苯磺酸按照摩尔比1:1在acetone/h2o(19:1,v/v)中室温悬浮搅拌约3天后,离心分离,将固体转移至室温真空干燥2小时后获得对甲苯磺酸盐晶型a。
[0499]
对甲苯磺酸盐晶型a的xrpd结果如图45所示,具体数据如下:
[0500]
表24:对甲苯磺酸盐晶型a的xrpd数据
[0501][0502]
对甲苯磺酸晶型a的tga/dsc见图46,结果显示样品加热至150℃失重5.1%,在121.2和222.3℃(起始温度)处可观察到两个热信号。1h nmr结果显示,对甲苯磺酸盐晶型a中式(i)化合物与对甲苯磺酸的摩尔比为1:1。
[0503]
10.苯磺酸盐晶型a/b的制备和表征
[0504]
以式(i)化合物水合物晶型a为原料制备苯磺酸盐,制备具体步骤为:将式(i)化合物水合物晶型a与苯磺酸按照摩尔比1:1分别在etoh、acetone/h2o(v/v,19:1)中室温悬浮搅拌约3天后,离心分离,将固体转移至室温真空干燥2小时后获得苯磺酸盐晶型a/b。
[0505]
苯磺酸晶型a的xrpd结果如图47所示,具体数据如下:
[0506]
表25:苯磺酸盐晶型a的xrpd数据
[0507][0508]
苯磺酸晶型a的tga/dsc见图48,结果显示样品加热至150℃失重9.0%,在60-200℃处可观察到多个热信号。1h nmr的结果显示,苯磺酸晶型a中式(i)化合物与苯磺酸的摩尔比为1:1。
[0509]
苯磺酸晶型b的xrpd结果如图49所示,具体数据如下:
[0510]
表26:苯磺酸盐晶型b的xrpd数据
[0511][0512]
苯磺酸晶型b的tga/dsc见图50,结果显示样品加热至100℃失重4.6%,在90.1℃(峰值温度)和236.7℃(起始温度)处可观察到两个热信号。1h nmr结果显示,苯磺酸晶型b中式(i)化合物与苯磺酸的摩尔比为1:0.5。
[0513]
三、盐型的筛选
[0514]
根据以上制备所得盐型的物理表征数据,选择xrpd衍射峰强度较高(峰形尖锐)、tga失重较小、dsc熔融温度较高、配体酸安全等级较高的磷酸盐晶型a、马来酸盐晶型a和甲磺酸盐晶型b进行下一步研究和评估。
[0515]
表27:盐型样品表征数据汇总
[0516][0517]
*:峰值温度;
[0518]
#
:通过ic及hplc测定。
[0519]
四、盐型的评估
[0520]
对三种盐型样品从溶解度、固体稳定性、引湿性三个方面开展了体外评估。
[0521]
1.试剂与耗材
[0522]
表28:溶解度实验所用试剂
[0523][0524]
2.溶解度
[0525]
为评估三种盐型在不同介质中的溶解度,考察了室温条件下三批样品在生物溶媒(模拟胃液sgf和模拟肠液fessif)中的动态溶解度(1、2、4、24小时),以及它们在乙醇、水、ph=7.4磷酸盐缓冲液中的24小时的溶解度。。具体步骤为:
[0526]
1)称取约10mg盐型样品至hplc小瓶中,分别加入1ml乙醇、水、ph=7.4磷酸盐缓冲液。
[0527]
2)另外称取约40mg盐型样品至5ml离心管中,分别加入4mlsgf、fessif;
[0528]
3)以~750rpm转速在25℃条件下磁力悬浮搅拌;
[0529]
4)平衡相应时间后,室温离心分离固体及上清液,上清液用于ph及溶解度测试。
[0530]
实验结果如表29所示,按照10mg/ml的盐型投料浓度,在sgf及fessif中,三种盐型在sgf中的溶解度在0.05-0.30mg/ml之间,其中甲磺酸盐晶型b的溶解度相对较高;在fessif中的溶解度在0.08-6.21mg/ml之间,其中甲磺酸盐晶型b、马来酸盐晶型a的溶解度相对较高。在水、乙醇及ph=7.4磷酸盐缓冲液中,三种盐型在水中的溶解度在0.04-1.30mg/ml之间,其中磷酸盐晶型a溶解度相对较高;在乙醇中的溶解度在1.14-2.14mg/ml之间,其中马来酸盐晶型a溶解度相对较高;在ph=7.4介质中的溶解度在0.02-0.15mg/ml之间,其中甲磺酸盐晶型b溶解度相对较高。
[0531]
表29:三种盐型在不同介质中25℃溶解度汇总1
[0532][0533]
s:api溶解度(mg/ml);
[0534]
na:未测试ph。
[0535]
表30:三种盐型在不同介质中25℃溶解度汇总2
[0536][0537]
s:api溶解度(mg/ml);
[0538]
na:未测试ph。
[0539]
3.固体稳定性
[0540]
为评估盐型的固体稳定性,将三批样品分别在25℃/60%rh和40℃/75%rh(封口膜包裹并扎6个小孔)条件下放置1周以评估其物理化学稳定性。对放置后的样品进行hplc测试和xrpd表征,以检测样品纯度及晶型的变化,xrpd结果及hplc结果显示,样品晶型及纯度均无明显变化,测试结果汇总在表31。
[0541]
表31:三种盐型的稳定性测试结果小结
[0542][0543]
#
:相对纯度为稳定性样品纯度与起始样品纯度的比值。
[0544]
4.引湿性
[0545]
为评估盐型的引湿性,对三批样品开展dvs测试。dvs测试结果如图51、图52和图53所示,磷酸盐晶型a在80%rh/25℃吸湿增重0.6%,马来酸盐晶型a在80%rh/25℃下吸湿增重0.8%,甲磺酸盐晶型b在80%rh/25℃下吸湿增重1.3%,三批样品均略有引湿性。随着湿度进一步提高,可观察到三批样品吸湿增重相对更明显。另外,xrpd结果显示,三批样品测试dvs前、后晶型均无变化。
[0546]
五、结论
[0547]
1)对(s)-(2-(6-(2-乙基-5-氟-4-羟基苯基)-1h-吲唑-3-基)-4,6-二氢吡咯并[3,4-d]咪唑-5-(1h)-基)(3-羟基吡咯烷-1-基)甲酮的式(i)化合物进行了成盐和盐型筛选试验,基于xrpd、tga、dsc、nmr或hplc/ic表征结果,共发现10种不同的盐型(涉及12种晶型)。
[0548]
2)根据xrpd衍射峰强度较高、tga失重较小、dsc熔融温度较高、配体酸安全等级较高等标准,选择了式(i)化合物的磷酸盐晶型a、马来酸盐晶型a和甲磺酸盐晶型b进行了引湿性、溶解度、固体稳定性等方面的体外评估。
[0549]
3)磷酸盐晶型a、马来酸盐晶型a和甲磺酸盐晶型b在sgf中溶解度为0.05-0.30mg/ml,在fessif中溶解度为0.08-6.21mg/ml;在水中溶解度为0.04-1.30mg/ml,在乙醇中溶解度为1.14-2.14mg/ml,在ph=7.4磷酸盐缓冲液中溶解度为0.02-0.15mg/ml。三种样品在40℃/75%rh、25℃/60%rh条件下放置1周后,纯度及晶型均未发生明显变化。三种样品dvs结果显示吸湿增重0.6-1.3%,样品均略有引湿性,完成dvs测试后晶型均未变。综上所述,三批盐型在引湿性及固体稳定性方面均具有较好的性质。
[0550]
实施例6:式(i)化合物的盐的药理学活性评价
[0551]
1.实验原理
[0552]
通过基于jak1、jak2、jak3、tyk2激酶的药物筛选体系来检测小分子化合物分别对于激酶活性的抑制能力。激酶与其底物irs1,igf1rtide,poly(4:1glu,tyr)进行酶学反应,消耗atp产生adp,利用adp-glo试剂和发光的方法检测产物的量用以反映激酶的活性。
[0553]
2.实验方案
[0554]
2.1实验材料和仪器
[0555]
序号名称来源货号1tris国药301883362dttsigma438163mgcl2sigmam10284bsapecr84-1005adp-glo kinase assaypromegav91016jak1thermofisherpv47747jak2carna08-0458jak3carna08-0469tyk2carna08-14710atppromegav915b11irs1signalchemi40-58-100012igf1rtidesignalchemi15-5813poly(4:1glu,tyr)sigmap027515384polystyrene shallow flat whitegreiner78407516384-well polypropylene microplatelabcytepp-020017biotek酶标仪bioteksynergy 418微孔板低速离心机湘智td5b
[0556]
2.2实验方法
[0557]
2.2.1激酶反应试剂配方
[0558]
2.1.1.1 1x激酶反应缓冲液(6ml)
[0559]
名称储液浓度体积终浓度tris1m(25x)240μl40mmmgcl21m(50x)120μl20mmbsa7.5%(75x)80μl0.1%dtt1m(500x)3μl0.5mmddh2o 5557μl [0560]
2.2.1.2 2x激酶配方
[0561][0562]
[0563][0564][0565]
2.2.1.3 4x底物混合物配方
[0566][0567][0568][0569]
2.2.1.4待测化合物
[0570]
[0571]
2.2.2激酶反应实验步骤
[0572]
2.2.2.1激酶反应实验步骤
[0573]
a)用100%dmso将tofacitinib及待测化合物(10mm储液)50倍稀释,在96孔稀释板中进行4倍等比稀释,共10个浓度,取1μl的化合物加入49μl的激酶反应缓冲液中,在微孔板振荡器上震荡20min。
[0574]
b)转移2μl的激酶(2.2.1.2步骤中制备)到384反应板中,加入1μl的待测化合物(a步骤中准备)到384反应板中(greiner,784075),1000rpm/min,离心1min,25℃孵育10min。
[0575]
c)转移1μl底物混合物(2.2.1.3步骤中制备)到384反应板中,1000rpm/min,离心1min,25℃孵育60min。在反应体系中,tofacitinib及待测化合物的终浓度1000,250,62.5,15.625,3.906,0.977,0.244,0.061,0.015,0.0038,nm。dmso终浓度均为0.5%。
[0576]
d)转移4μl adp-glo到384反应板中1000rpm/min,离心1min,25℃孵育40min。
[0577]
e)转移8μl detection溶液到384反应板中1000rpm/min,离心1min,25℃孵育40min。
[0578]
f)使用biotek多功能读板机读取luminescence信号。信号强度用于表征激酶的活性程度。
[0579]
2.2.3实验数据处理方法
[0580]
化合物抑制率(%inh)=(阴性对照-化合物)/(阴性对照-阳性对照)
×
100%
[0581]
阴性对照:dmso
[0582]
阳性对照:1000nm tofacitinib
[0583]
利用以下非线性拟合公式来得到化合物的ic50(半数抑制浓度):
[0584]
y=bottom (top-bottom)/(1 10^((logic50-x)*hillslope))
[0585]
x:化合物浓度log值
[0586]
y:化合物抑制率(%inh)
[0587]
z’因子计算方程:
[0588]
z’=1-3(sdmin sdmax)/(avemax-avemin)
[0589]
其中:
[0590]
min为阳性对照10um/100um/30umfilgotinib rlu值,max为阴性对照dmso rlu值。
[0591]
sd为标准误差,ave为rlu平均值。
[0592]
3.结果
[0593]
化合物检测结果见下表:
[0594][0595]
测试结果表明:与式(i)化合物类似,与式(i)化合物的甲磺酸盐也具有jak抑制活性,且其抑制活性远高于tofacitinib(对jak1和tyk2的抑制高一到个数量级以上),能够在极低的浓度下有效抑制jak1、jak2、jak3、tyk2。
[0596]
虽然已经阐明并描述了本发明的特定实施方式,但并不意味着这些实施方式阐明了并描述了本发明的所有可能形式。更确切地,用在本说明书中的文字仅仅是描述性的文字并非限制性的。对于本领域技术人员明显的是,在不脱离本公开的一般范围的情况下,可以进行各种其他改变和修改。因此,在所附权利要求中,旨在包括在本发明范围内的所有这些改变和修改。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。