1.本发明属于有机化学领域,具体涉及一种苯并咪唑衍生物及其在检测硝基芳香爆炸物中的应用。
背景技术:
2.在现有的技术中,文献和专利已公开的检测芳香爆炸物的方法或材料主要包括:表面增强拉曼光谱法、质谱分析法、电化学法、毛细管电泳法、新型爆炸物检测仪和利用金属配合物、金属有机框架(mof)、有机聚合物、共价有机框架(cof)等荧光传感器检测。值得注意的是,上述检测芳香爆炸物的方法主要分为三种,其一是大型仪器检测法,这类方法存在成本高、耗时长、检测步骤繁琐、无法实现现场实时检测;其二是聚合物和重金属类荧光传感器,这类方法具有难以降解、重金属污染、无法实现多检测等一系列问题;其三是新型检测仪,这类检测仪存在灵敏度低,分析时间长以及无法可视化的问题。
3.近年来,有许多有机小分子发展为爆炸物荧光传感器,它们具有多样的发光特性、高灵敏度和高选择性等优点。例如,具有全色聚集诱导发光和高效固态发射的二苯基富马腈在水溶液中实现苦味酸的传感应用;gunnlaugsson等人设计并成功合成了具有聚集诱导发射活性的4
‑
氨基
‑
1,8
‑
萘二甲酰亚胺衍生物,并进一步用作荧光传感器,用于硝基爆炸物的传感,在水性介质中探针对苦味酸显示出最大的荧光猝灭和高选择性。上述的小分子荧光传感器只能在水溶液中单一检测苦味酸,在实际的检测应用中,被污染的水源往往是同时存在多种硝基芳香爆炸物,它们同样对环境和动植物造成重大威胁,如苦味酸、2,4
‑
二硝基苯酚和对硝基苯酚等。以上这些小分子荧光传感器并不能适应实际污染水样中多种芳香爆炸物的快速简单可视化的检测,因此,有必要开发一种能实现快速简单检测多种芳香爆炸物的小分子荧光传感器。
技术实现要素:
4.为了克服现有技术存在的上述问题,本发明的目的之一在于提供一种苯并咪唑衍生物;本发明的目的之二在于提供这种苯并咪唑衍生物的制备方法;本发明的目的之三在于提供这种苯并咪唑衍生物的检测硝基芳香爆炸物中的应用;本发明的目的之四在于提供一种荧光材料;本发明的目的之五在于提供一种荧光传感器;本发明的目的之六在于提供一种试纸;本发明的目的之七在于提供一种薄膜。
5.为了实现上述目的,本发明所采取的技术方案是:
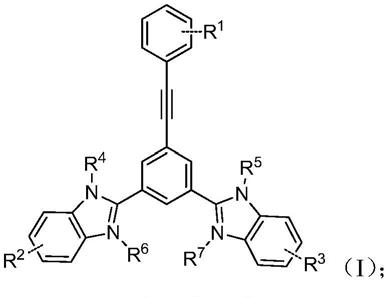
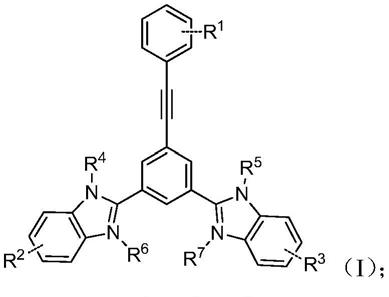
6.本发明的第一方面提供一种苯并咪唑衍生物,所述的苯并咪唑衍生物结构如式(ⅰ)所示:
[0007][0008]
式(ⅰ)中r1、r2、r3分别独立选自卤素、取代或未取代的烷基、取代或未取代的烷氧基、取代或未取代的胺基、取代或未取代的芳基;r4、r5分别独立选自氢、取代或未取代的烷基;r6、r7分别独立选自取代或未取代的烷基,或者没有r6和r7。
[0009]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,没有r6和r7,所述的苯并咪唑衍生物结构如式(ⅱ)所示:
[0010][0011]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,r6选自取代或未取代的烷基,没有r7,所述的苯并咪唑衍生物结构如式(ⅲ)所示:
[0012][0013]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,r7选自取代或未取代的烷基,没有r6,所述的苯并咪唑衍生物结构如式(ⅳ)所示:
[0014][0015]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,r6、r7分别独立选自取代或未取代的烷基,所述的苯并咪唑衍生物结构如式(
ⅴ
)所示:
[0016][0017]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,r1选自取代或未取代的胺基、取代或未取代的烷氧基。
[0018]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,r2选自卤素、取代或未取代的烷基。
[0019]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,r3选自卤素、取代或未取代的烷基。
[0020]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,r4选自氢、甲基、乙基。
[0021]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,r5选自氢、甲基、乙基。
[0022]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,r6选自甲基、乙基,或者没有r6。
[0023]
优选的,所述式(ⅰ)所示的苯并咪唑衍生物中,r7选自甲基、乙基,或者没有r7。
[0024]
优选的,所述的苯并咪唑衍生物结构如式(1)
‑
式(6)所示:
[0025][0026]
本发明的第二方面提供根据本发明第一方面提供的苯并咪唑衍生物的制备方法,包括以下步骤:
[0027]
1)将5
‑
卤代间苯二甲酸与邻苯二胺衍生物混合,反应,得到中间体5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物;
[0028]
2)将中间体5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物与苯乙炔衍生物混合,反应,得到所述的苯并咪唑衍生物。
[0029]
优选的,所述的邻苯二胺衍生物结构式如式(
ⅵ
)所示:
[0030][0031]
式(
ⅵ
)中r8选自卤素、取代或未取代的烷基、取代或未取代的烷氧基、取代或未取代的胺基、取代或未取代的芳基。
[0032]
优选的,所述式(
ⅵ
)所示的邻苯二胺衍生物中,r8选自卤素、取代或未取代的烷基。
[0033]
优选的,所述的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物结构式如式(
ⅶ
)所示:
[0034]
[0035]
式(
ⅶ
)中r9、r
10
分别独立选自卤素、取代或未取代的烷基、取代或未取代的烷氧基、取代或未取代的胺基、取代或未取代的芳基;x选自卤素。
[0036]
优选的,所述式(
ⅶ
)所示的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物中,r9选自卤素、取代或未取代的烷基。
[0037]
优选的,所述式(
ⅶ
)所示的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物中,r
10
选自卤素、取代或未取代的烷基。
[0038]
优选的,所述式(
ⅶ
)所示的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物中,x为溴原子。
[0039]
优选的,所述的苯乙炔衍生物结构式如式(
ⅷ
)所示:
[0040][0041]
式(
ⅷ
)中r
11
选自卤素、取代或未取代的烷基、取代或未取代的烷氧基、取代或未取代的胺基、取代或未取代的芳基。
[0042]
优选的,所述式(
ⅷ
)所示的苯乙炔衍生物生物中,r
11
选自取代或未取代的胺基、取代或未取代的烷氧基。
[0043]
优选的,步骤1)还包括将5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物进行烷基化的步骤。
[0044]
优选的,所述的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物烷基化包括如下步骤:
[0045]
将5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物与卤代烷烃混合,反应,得到烷基化的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物。
[0046]
优选的,所述的卤代烷烃为碘代烷烃或溴代烷烃;进一步优选的,所述的卤代烷烃为碘代c1
‑
c4的烷烃;再进一步优选的,所述的卤代烷烃为碘甲烷。
[0047]
优选的,所述的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物与卤代烷烃的摩尔比为1:(1.5
‑
3);进一步优选的,所述的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物与卤代烷烃的摩尔比为1:(2
‑
2.5)。
[0048]
优选的,步骤2)还包括将苯并咪唑衍生物进行烷基化的步骤。
[0049]
优选的,所述的苯并咪唑衍生物烷基化包括如下步骤:
[0050]
将苯并咪唑衍生物与卤代烷烃混合,反应,得到烷基化的苯并咪唑衍生物。
[0051]
优选的,所述的卤代烷烃为碘代烷烃或溴代烷烃;进一步优选的,所述的卤代烷烃为碘代c1
‑
c4的烷烃;再进一步优选的,所述的卤代烷烃为碘甲烷。
[0052]
优选的,所述的苯并咪唑衍生物与卤代烷烃摩尔比为1:(1.5
‑
4);进一步优选的,所述的苯并咪唑衍生物与卤代烷烃摩尔比为1:(2
‑
3);再进一步优选的,所述的苯并咪唑衍生物与卤代烷烃摩尔比为1:(2.3
‑
2.7)。
[0053]
优选的,所述的5
‑
卤代间苯二甲酸与邻苯二胺衍生物的摩尔比为1:(1.5
‑
4);进一步优选的,所述的5
‑
卤代间苯二甲酸与邻苯二胺衍生物的摩尔比为1:(2
‑
3);再进一步优选的,所述的5
‑
卤代间苯二甲酸与邻苯二胺衍生物的摩尔比为1:(2
‑
2.5)。
[0054]
优选的,步骤1)所述的反应温度为140℃
‑
200℃;进一步优选的,步骤1)所述的反应温度为150℃
‑
190℃;再进一步优选的,步骤1)所述的反应温度为160℃
‑
180℃。
[0055]
优选的,步骤1)所述的反应时间为24h
‑
48h;进一步优选的,步骤1)所述的反应时间为28h
‑
44h;再进一步优选的,步骤1)所述的反应时间为32h
‑
40h。
[0056]
优选的,所述的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物与苯乙炔衍生物的摩尔比为
1:(0.8
‑
1.6);进一步优选的,所述的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物与苯乙炔衍生物的摩尔比为1:(1
‑
1.4);再进一步优选的,所述的5
‑
卤代
‑
1,3
‑
二苯并咪唑基苯衍生物与苯乙炔衍生物的摩尔比为1:(1.1
‑
1.3)。
[0057]
优选的,步骤2)所述的反应温度为90℃
‑
130℃;进一步优选的,步骤2)所述的反应温度为100℃
‑
120℃;再进一步优选的,步骤2)所述的反应温度为105℃
‑
115℃。
[0058]
优选的,步骤2)所述的反应时间为16h
‑
32h;进一步优选的,步骤2)所述的反应时间为20h
‑
28h;再进一步优选的,步骤2)所述的反应时间为22h
‑
26h。
[0059]
优选的,步骤2)所述的反应还包括加入催化剂参与反应;进一步优选的,所述的催化剂包括碘化亚铜和二氯二(三苯基膦)钯(ii)中的至少一种。
[0060]
本发明的第三方面提供根据本发明第一方面提供的苯并咪唑衍生物在检测硝基芳香爆炸物中的应用。
[0061]
优选的,所述的检测硝基芳香爆炸物的方法包括如下步骤:
[0062]
用荧光光谱仪测试苯并咪唑衍生物的荧光发射光谱;
[0063]
将苯并咪唑衍生物与硝基芳香爆炸物混合;
[0064]
用荧光光谱仪测试混合后溶液的荧光发射光谱。
[0065]
优选的,所述的硝基芳香爆炸物包括2,4,6
‑
三硝基苯酚/苦味酸(pa)、2,4二硝基苯酚(dnp)、4
‑
硝基苯酚(np)、2
‑
硝基苯胺(na)、4
‑
硝基苯甲醛(nba)、4
‑
硝基苯甲酸(nbac)中的至少一种。
[0066]
本发明的第四方面提供一种荧光材料,所述的荧光材料包括本发明第一方面提供的苯并咪唑衍生物。
[0067]
本发明的第五方面提供一种荧光传感器,所述的荧光传感器包括本发明第一方面提供的苯并咪唑衍生物。
[0068]
本发明的第六方面提供一种试纸,所述的试纸包括本发明第一方面提供的苯并咪唑衍生物。
[0069]
优选的,所述的试纸为检测硝基芳香爆炸物包的试纸。
[0070]
本发明的第七方面提供一种薄膜,所述的薄膜包括本发明第一方面提供的苯并咪唑衍生物。
[0071]
优选的,所述的薄膜为检测硝基芳香爆炸物包的薄膜。
[0072]
本发明的有益效果是:
[0073]
本发明提供的苯并咪唑衍生物结构新颖且具有优良的荧光发射特性。本发明公开的制备方法具有简单高效,原料便宜易得,条件温和安全的优点。本发明提供的苯并咪唑衍生物可应用于实际水样中多种硝基芳香爆炸物的快速可视化识别,是一种优良的荧光传感器,可进一步将其制备成用于可视化检测的试纸和薄膜。
[0074]
具体来说,本发明具有如下优点:
[0075]
1.本发明提供的苯并咪唑衍生物结构新颖且具有优良的荧光发射特性,具体原因为本发明通过选定1,3
‑
双苯并咪唑基苯为刚性共轭荧光团作为硝基芳香爆炸物的识别单元,然后采用乙炔基作为π桥,延长1,3
‑
双苯并咪唑基苯的共轭长度,调控分子的荧光发射波长;最后通过引入供体单元、转子单元或者大位阻单元,调控分子多样的发光特性。
[0076]
2.本发明公开的制备方法简单高效,原料便宜易得,条件温和安全。本发明通过5
‑
溴间苯二甲酸和邻苯二胺反应,将得到的中间体进行c
‑
c偶联反应,得到的苯并咪唑衍生物在固态和液态都具有高亮、稳定的荧光发射,是优良的荧光传感材料。
[0077]
3.本发明提供的苯并咪唑衍生物可应用于实际水样中多种芳香爆炸物的快速可视化识别,且检测浓度下限低,是一种优良的荧光传感器;该苯并咪唑衍生物具有优良的稳定性和对硝基芳香爆炸物检测的准确性,可进一步将其制备成用于可视化检测的试纸和薄膜。
附图说明
[0078]
图1为实施例1制备苯并咪唑衍生物化合物1的化学反应式图。
[0079]
图2为实施例2制备苯并咪唑衍生物化合物2的化学反应式图。
[0080]
图3为实施例3制备苯并咪唑衍生物化合物3的化学反应式图。
[0081]
图4为实施例4制备苯并咪唑衍生物化合物4的化学反应式图。
[0082]
图5为实施例5制备苯并咪唑衍生物化合物5的化学反应式图。
[0083]
图6为实施例6制备苯并咪唑衍生物化合物6的化学反应式图。
[0084]
图7为化合物1的红外光谱测试图。
[0085]
图8为化合物1的核磁共振氢谱图。
[0086]
图9为化合物1的核磁共振碳谱图。
[0087]
图10为化合物1的高分辨质谱图。
[0088]
图11为化合物1与不同芳香爆炸物作用后的荧光发射强度图。
[0089]
图12为化合物1与芳香爆炸物混合后荧光强度随时间变化图。
[0090]
图13为附着有化合物1的试纸条和薄膜板在滴加芳香爆炸物后的样品图。
具体实施方式
[0091]
以下结合附图和实例对本发明的具体实施作进一步说明,但本发明的实施和保护不限于此。需指出的是,以下若有未特别详细说明之过程,均是本领域技术人员可参照现有技术实现或理解的。所用试剂或仪器末注明生产厂商者,视为可以通过市售购买得到的常规产品。
[0092]
实施例1
[0093]
图1为实施例1制备苯并咪唑衍生物化合物1的化学反应式图,以下根据图1,结合本例具体实验步骤作进一步的说明。
[0094]
称量2mmol的5
‑
溴间苯二甲酸(a),4.2mmol邻苯二胺(b),置于50ml圆底烧瓶中,加入多聚磷酸(ppa)15ml,将温度升至170℃,反应36h,反应结束后,将反应液冷却至室温,然后用naoh溶液调节ph至9
‑
10,抽滤,得到固体粗品,随后选用石油醚:乙酸乙酯=6:1作为洗脱剂,通过柱层析分离法得到纯品中间体5
‑
溴
‑
1,3
‑
二苯并咪唑基苯(c)。
[0095]
称量0.5mmol中间体5
‑
溴
‑
1,3
‑
二苯并咪唑基苯(c),0.6mmol 4
‑
乙炔基
‑
n,n
‑
二苯基苯胺(d),0.05mmol碘化亚铜,0.05mmol二氯二(三苯基膦)钯(ii),3ml三乙胺,3ml四氢呋喃作为溶剂,置于25ml的schlenk反应管中。抽真空,灌氮气,110℃下回流反应24h。反应结束后,将反应液冷却至室温,加入2ml饱和的氯化铵溶液猝灭反应,用二氯甲烷和氯化铵水溶液萃取,收集有机相,用无水硫酸钠干燥,旋干有机溶剂后得到固体粗品,随后通过柱层
析分离法得到纯品化合物,记为化合物1。
[0096]
实施例2
[0097]
图2为实施例2制备苯并咪唑衍生物化合物2的化学反应式图,以下根据图2,结合本例具体实验步骤作进一步的说明。
[0098]
称量2mmol的5
‑
溴间苯二甲酸(a),4.2mmol邻苯二胺(b),置于50ml圆底烧瓶中,加入多聚磷酸(ppa)15ml,将温度升至170℃,反应36h,反应结束后,将反应液冷却至室温,然后用naoh溶液调节ph至9
‑
10,抽滤,得到固体粗品,随后选用石油醚和乙酸乙酯作为洗脱剂,通过柱层析分离法得到纯品中间体5
‑
溴
‑
1,3
‑
二苯并咪唑基苯(c)。
[0099]
称量1mmol的化合物中间体5
‑
溴
‑
1,3
‑
二苯并咪唑基苯(c),4mmol的氢氧化钾,量取10ml的丙酮,置于50ml圆底烧瓶中,室温下搅拌30min,然后加入2.5mmol的碘甲烷,70℃下回流2h。反应结束后,将反应液冷却至室温,用二氯甲烷和氯化铵水溶液萃取,收集有机相,用无水硫酸钠干燥,减压浓缩后得到固体3b粗品,随后选用石油醚:乙酸乙酯=6:1作为洗脱剂,通过柱层析分离法得到纯品中间体e。
[0100]
称量0.5mmol化合物中间体e,0.6mmol 4
‑
乙炔基
‑
n,n
‑
二苯基苯胺(d),0.05mmol碘化亚铜,0.05mmol二氯二(三苯基膦)钯(ii),3ml三乙胺,3ml四氢呋喃作为溶剂,置于25ml的schlenk反应管中。抽真空,灌氮气,110℃下回流反应24h。反应结束后,将反应液冷却至室温,加入2ml饱和的氯化铵溶液猝灭反应,用二氯甲烷和氯化铵水溶液萃取,收集有机相,用无水硫酸钠干燥,旋干有机溶剂后得到固体粗品,随后通过柱层析分离法得到纯品化合物,记为化合物2。
[0101]
实施例3
[0102]
图3为实施例3制备苯并咪唑衍生物化合物3的化学反应式图,以下根据图3,结合本例具体实验步骤作进一步的说明。
[0103]
称量2mmol的5
‑
溴间苯二甲酸(a),4.2mmol 4,5
‑
二氟邻苯二胺(f),置于50ml圆底烧瓶中,加入多聚磷酸(ppa)15ml,将温度升至170℃,反应36h,反应结束后,将反应液冷却至室温,然后用naoh溶液调节ph至9
‑
10,抽滤,得到固体粗品,随后选用石油醚:乙酸乙酯=6:1作为洗脱剂,通过柱层析分离法得到纯品中间体g。
[0104]
称量0.5mmol化合物中间体g,0.6mmol 4
‑
乙炔基
‑
n,n
‑
二苯基苯胺(d),0.05mmol碘化亚铜,0.05mmol二氯二(三苯基膦)钯(ii),3ml三乙胺,3ml四氢呋喃作为溶剂,置于25ml的schlenk反应管中。抽真空,灌氮气,110℃下回流反应24h。反应结束后,将反应液冷却至室温,加入2ml饱和的氯化铵溶液猝灭反应,用二氯甲烷和氯化铵水溶液萃取,收集有机相,用无水硫酸钠干燥,旋干有机溶剂后得到固体粗品,随后通过柱层析分离法得到纯品化合物,记为化合物3。
[0105]
实施例4
[0106]
图4为实施例4制备苯并咪唑衍生物化合物4的化学反应式图,以下根据图4,结合本例具体实验步骤作进一步的说明。
[0107]
称量0.5mmol的化合物1,1.25mmol的碘甲烷,量取3ml干燥的四氢呋喃作为溶剂,置于25ml的schlenk反应管中,80℃下回流2d。反应结束后,将反应液冷却至室温,析出黄色固体,减压抽滤后得到滤饼化合物4粗品,随后选用四氢呋喃清洗滤饼3~4次,得到纯品化合物4。
[0108]
实施例5
[0109]
图5为实施例5制备苯并咪唑衍生物化合物5的化学反应式图,以下根据图5,结合本例具体实验步骤作进一步的说明。
[0110]
称量0.5mmol中间体5
‑
溴
‑
1,3
‑
二苯并咪唑基苯(c),0.6mmol 9
‑
(4
‑
乙炔基苯基)
‑
9h
‑
咔唑(h),0.05mmol碘化亚铜,0.05mmol二氯二(三苯基膦)钯(ii),3ml三乙胺,3ml四氢呋喃作为溶剂,置于25ml的schlenk反应管中。抽真空,灌氮气,110℃下回流反应24h。反应结束后,将反应液冷却至室温,加入2ml饱和的氯化铵溶液猝灭反应,用二氯甲烷和氯化铵水溶液萃取,收集有机相,用无水硫酸钠干燥,旋干有机溶剂后得到固体粗品,随后通过柱层析分离法得到纯品化合物,记为化合物5。
[0111]
实施例6
[0112]
图6为实施例6制备苯并咪唑衍生物化合物6的化学反应式图,以下根据图6作进一步的说明。
[0113]
本例与实施例5的区别是将反应原料9
‑
(4
‑
乙炔基苯基)
‑
9h
‑
咔唑(h)换成同摩尔量的4
‑
甲氧基苯乙炔(i),其余步骤以及原料完全一样。本例所得产物记为化合物6。
[0114]
对实施例1制备得到的化合物1进行分析测试,结果如下:
[0115]
实施例1所制备的化合物1为白色固体,熔点测试结果为m.p.=169.8
‑
171.4℃。
[0116]
将实施例1所制备的化合物1进行红外光谱测试,图7为化合物1的红外光谱测试图。由图7的ft
‑
ir分析(kbr,ν,cm
‑1):3409cm
‑1,杂环上n
‑
h伸缩振动;3057cm
‑1,芳香环上不饱和c
‑
h的伸缩振动吸收峰;2207cm
‑1,不饱和c≡c键伸缩振动吸收峰;1585,1508,1440cm
‑1,芳香环骨架伸缩振动吸收峰;1335,1274cm
‑1,c
‑
n键伸缩振动吸收峰;739cm
‑1,苯环1,2
‑
二取代;694cm
‑1,苯环单取代,1,3,5
‑
三取代。
[0117]
将实施例1所制备的化合物1进行核磁共振氢谱测试,图8为化合物1的核磁共振氢谱图。由图8分析1h nmr(cdcl3,600mhz):δ=6.97(d,j=8.4hz,2h),7.07(t,j=7.2hz,2h),7.12(d,j=7.8hz,4h),7.21
‑
7.23(m,4h),7.27
‑
7.30(m,6h),7.65
‑
7.67(m,4h),8.46(s,2h),9.15(s,1h)。
[0118]
将实施例1所制备的化合物1进行核磁共振碳谱测试,图9为化合物1的核磁共振碳谱图。由图9分析
13
c nmr(cdcl3,150mhz):δ=87.5,91.3,115.5,122.0,122.9,123.7,124.0,124.7,125.1,125.6,129.4,129.9,130.6,130.8,132.7,147.1,148.2,150.7。
[0119]
将实施例1所制备的化合物1进行高分辨质谱测试,图10为化合物1的高分辨质谱图。由图10的hrms分析m/z(%):calcd for c
40
h
28
n
5
([m h]
):578.2339(100),found:578.2335(100)。
[0120]
上述测试数据证明实施例1所制备的产物即为化合物1。
[0121]
应用例1
[0122]
本例研究将化合物1与芳香爆炸物混合后荧光强度的测试,具体实验步骤如下:
[0123]
配置14组相同的2ml浓度为1
×
10
‑5mol/l的化合物1的水溶液;在其中的第2
‑
14组中分别加入20当量不同的芳香爆炸物;选用荧光光谱仪,设置合适的参数,测试该14组溶液的荧光发射光谱。图11为化合物1与不同芳香爆炸物作用后的荧光发射强度图。从图11可以发现,化合物1的水溶液具有较强的荧光发射强度,当加入pa(2,4,6
‑
三硝基苯酚/苦味酸)、dnp(2,4二硝基苯酚)、np(4
‑
硝基苯酚)、na(2
‑
硝基苯胺)、nba(4
‑
硝基苯甲醛)、nbac(4
‑
硝
基苯甲酸)会导致溶液荧光发生强烈的猝灭,而加入tnt(2,4,6
‑
三硝基甲苯)、dnt(2,4
‑
二硝基甲苯)、nt(4
‑
硝基甲苯)、nb(2
‑
硝基苯)、hbac(4
‑
羟基苯甲酸)、phenol(苯酚)、nm(硝基甲烷)时溶液荧光发射强度没有发生明显变化。这说明,荧光材料能够在水溶液中有效识别多种芳香爆炸物。
[0124]
应用例2
[0125]
本例研究将化合物1与芳香爆炸物混合后荧光强度随时间变化的关系,具体测试步骤如下:
[0126]
制备6组2ml浓度为1
×
10
‑5mol/l化合物1的水溶液,将其中一组溶液加入石英比色皿中并置于荧光光谱仪中,快速加入20当量的pa溶液,并记录最大荧光发射强度与时间之间的关系。依次以相同方法测试化合物1与dnp、np、na、nba和nbac混合后的荧光发射强度与时间的关系。测试数据如图12所示,图12为化合物1与芳香爆炸物混合后荧光强度随时间变化图。由图12可知,化合物1与芳香爆炸物混合后溶液的荧光强度随时间快速减小,并在第6分钟后荧光发射强度减弱至最小且不再发生明显变化,这说明化合物1能够对图12中的6种芳香爆炸物进行快速检测。
[0127]
应用例3
[0128]
负载化合物1的便携试纸制备步骤以及可视化识别,具体步骤如下:
[0129]
裁剪7块大小一致空白的滤纸条备用;配制10ml浓度为1
×
10
‑3mol/l的化合物1的thf溶液,将空白的滤纸条浸泡在溶液中1分钟,取出晾干即完成便携试纸条的制备。在6组附着有化合物1的滤纸条上分别滴加3滴不同的芳香爆炸物水溶液(浓度为1
×
10
‑7mol/l),然后在365nm的紫外灯下观察。
[0130]
负载化合物1的便携薄膜制备步骤以及可视化识别,具体步骤如下:
[0131]
配制5ml浓度为1
×
10
‑3mol/l的化合物1的二氯甲烷溶液,称取0.5g的可降解的pbat(聚己二酸/对苯二甲酸丁二酯)加入上述溶液中,常温搅拌放置12h;将配制好的聚合物溶液涂抹在干净的玻璃板上,50℃烘干,待冷却后裁剪7块大小一致的薄膜板备用。在6组附着有化合物1的薄膜板上分别滴加3滴不同的芳香爆炸物(浓度为1
×
10
‑7mol/l),然后在365nm的紫外灯下观察。
[0132]
图13为附着有化合物1的试纸条和薄膜板在滴加芳香爆炸物后的样品图。从图13可以发现,在负载化合物1的试纸条和薄膜都能呈现明亮的蓝绿色荧光,在分别滴加入pa、dnp、np、na、nba和nbac后试纸条和薄膜都能迅速猝灭。这说明,负载化合物1的试纸条和薄膜均可应用于快速高灵敏可视化识别实际污染水样中的多种芳香爆炸物。
[0133]
上述实例为本发明较佳的实施方式,但发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。