一种醋酸卡泊芬净杂质g的制备方法
技术领域
1.本发明属于化学领域,具体涉及一种醋酸卡泊芬净杂质g的制备方法。
背景技术:
2.醋酸卡泊芬净是第一个棘白菌素类抗真菌药,首先于2001年1月26日获美国食品药品管理局(fda)批准上市,主要用于治疗侵袭性念珠菌病、对其他治疗无效或不能耐受的侵袭性曲霉菌病以及经验性治疗中性粒细胞减少、伴发热病人的可疑真菌感染,作为新一类棘白菌素类抗菌药物的代表,,醋酸卡泊芬净具有选择性高、抗菌活性好、安全性高、耐药性少等方面的明显优势。
3.醋酸卡泊芬净是先由glarea lozoyensis 进行发酵,得到发酵产物,然后发酵产物经过固液分离、浸提、吸附解析、浓缩干燥等过程得到纽莫康定b0粗品,再以纽莫康定b0作为起始原料,经过一系列合成得到醋酸卡泊芬净粗品,粗品再经过结晶干燥,得到醋酸卡泊芬净原料药成品。
4.在粗品醋酸卡泊芬净的合成过程中,会产生卡泊芬净杂质g的异构体前体,再经下一步反应产生卡泊芬净杂质g,卡泊芬净杂质g的结构如式 (ⅲ)所示:在药品检测领域,需要醋酸卡泊芬净杂质g作为对照品,文献wleonard w r, belyk k m, conlon d a, et al. synthesis of the antifungal β-1, 3-glucan synthase inhibitor cancidas (caspofungin acetate) from pneumocandin b0[j]. the journal of organic chemistry, 2007, 72(7): 2335-2343.中提到羟基异构有助于中间体的稳定,故会产生该构型,并最终生成卡泊芬净杂质g。
[0005]
目前无其他参考文献对卡泊芬净杂质g或醋酸卡泊芬净杂质g有具体的报道,现有技术中也关于没有制备高纯度醋酸卡泊芬净杂质g的相关报道,因此,需要开发出一种制备高纯度的醋酸卡泊芬净杂质g的方法,以满足企业自身和市场上对醋酸卡泊芬净杂质g对照
品的需要。
技术实现要素:
[0006]
为了解决上述技术问题,本发明提供一种制备如式(i)所示的醋酸卡泊芬净杂质g的方法,具体包含以下步骤:1)以纽莫康定b0为原料,通过化学合成得到卡泊芬净杂质g粗品;2)用溶剂将卡泊芬净杂质g粗品溶解,配制成粗品溶液并过滤;3)过滤后的粗品溶液通过制备系统中的色谱柱进行分离纯化,并收集洗脱液;4)将合格的洗脱液进行冷冻干燥,得到纯度达95%以上的醋酸卡泊芬净杂质g固体粉末。
[0007]
进一步地,所述步骤1)中通过还原剂将纽莫康定b0进行还原,得到杂质g的粗品,合成工艺如下列方程式所示:进一步地,所述步骤2)中的溶剂为0.1%醋酸水溶液,过滤采用0.45μm滤膜。
[0008]
进一步地,所述步骤3)中首先将过滤后的卡泊芬净杂质g粗品溶液上色谱柱,所述色谱柱为中低压液相色谱柱,色谱柱直径为30mm或50mm,所述色谱柱的填料为sp-100-8-ods-p;然后用含有10%乙醇的流动相平衡色谱柱,流动相用量为2倍的柱体积;接着用含有10%-45%乙醇的流动相进行梯度洗脱,流动相用量为4倍柱体积;最后用45%-50%乙醇梯度洗脱,流动相用量为8倍柱体积,收集该阶段梯度洗脱液,该阶段梯度洗脱液中的成分为醋酸卡泊芬净杂质g,并进行hplc检测。
[0009]
进一步地,所述步骤3)中洗脱液的流速为3-4倍柱体积/小时。
[0010]
进一步地,所述步骤3)中流动相由流动相a和流动相b组成,二者体积之和为100%,其中流动相a为0.1%醋酸水溶液,流动相b为乙醇。
[0011]
进一步地,所述步骤4)中将合格的洗脱液进行先冷冻,然后在真空状态下使固态洗脱液直接升华,从而得到醋酸卡泊芬净杂质g固体粉末。
[0012]
与现有技术相比,本发明的有益效果如下:1)本发明以醋酸卡泊芬净的中间体纽莫康定b0原料,在还原剂的作用下,对特定位置的基团进行还原直接得到卡泊芬净杂质g粗品,提供了卡泊芬净杂质g粗品的制备方法;2)本发明以杂质g粗品为原料,通过制备色谱的方法,采用0.1%醋酸和乙醇的混合液作为流动相进行等度和梯度洗脱,最后得到纯度大于95%的杂质g固体粉末,提供了高纯度醋酸卡泊芬净杂质g的制备方法,能够满足企业自身和市场的要求;3)本发明的制备方法步骤简单,条件稳定,收率高,适合规模化生产,能够满足市场需求。
附图说明
[0013]
图1 是醋酸卡泊芬净杂质g制备层析示意图;图2是实施例1的醋酸卡泊芬净杂质g成品检测图;图3是实施例2的醋酸卡泊芬净杂质g成品检测图;图4是实施例3的醋酸卡泊芬净杂质g成品检测图;图5是实施例4的醋酸卡泊芬净杂质g成品检测图;图6是实施例5的醋酸卡泊芬净杂质g成品检测图。
具体实施方式
[0014]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明的部分实施例,而不是全部。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。以下实施例所用的试剂均为市售商品。
[0015]
实施例1首先,确定还原剂、粗品溶剂、填料、流动相和洗脱梯度。
[0016]
1.还原剂的确定取8g纽莫康定b0、1.2g苯硼酸(phenylboronic acid )和200ml四氢呋喃(thf)混合溶解后,加热脱水,加入bstfa(n,o-双(三甲基硅烷基)三氟乙酰胺)5ml,再使用还原剂还原,反应结束使用5ml盐酸淬灭,得到卡泊芬净杂质g粗品溶液,反应中最关键的是还原剂的种类,对几种还原剂进行筛选,包括:硼烷四氢呋喃、二甲基硫醚硼烷、硼氢化钠、醋酸硼氢化
钠,以不同还原剂进行还原后底物b0的产物的纯度为考察指标,还原1h后,不同还原剂的情况下杂质g纯度(%)结果如表1所示:表1 不同还原剂得到的卡泊芬净杂质g纯度从以上实验结果可以看到,硼烷四氢呋喃(bh3/thf)作为还原剂时,卡泊芬净杂质g的纯度最高,可达到41%,因此将还原剂确定为硼烷四氢呋喃。
[0017]
2.粗品溶剂的确定。
[0018]
由于抗生素类产品稳定性不好,所以在溶解粗品时中需要选择合适的溶剂,本技术通过筛选了不同酸碱性的溶剂,包括:0.1%醋酸水溶液,0.5%醋酸水溶液,0.1%磷酸二氢钠,0.5%磷酸二氢钠,0.1%磷酸氢二钠,0.5%磷酸氢二钠,将粗品杂质分别溶解在上述溶剂中,室温放置48h后发现,杂质g在不同的酸碱性条件下表现出不同的稳定性,在酸性条件下,表现出较强的稳定性,其中在0.1%醋酸水溶液中,稳定性最强,因此选择0.1%醋酸水溶液作为粗品溶剂。
[0019]
3.填料的确定根据产品特性,采用反相制备色谱进行分离,通过对下面几种c18填料的筛选,如:sp-120-10-ods-bp、sp-100-8-ods-p、sp-120-30/50-ods-bp,发现sp-100-8-ods-p的分离效果最好,可以获得95%以上的馏分,因此确定sp-100-8-ods-p作为制备填料。
[0020]
4.流动相的确定(1)流动相a的确定由于超级抗生素类产品稳定性普遍不佳,需要进一步考察产品的制备稳定性,因此,将含有较高杂质g(41%)的原料置于不同ph的流动相中,室温放置48h后观察结果,结果见表2所示:表2 流动a相稳定性考察
根据表2结果所示:当放置于0.1%醋酸水溶液时,杂质g的归一化面积与原料相差无几,纯度无改变,因此确定使用0.1%醋酸水溶液为流动相a。
[0021]
(2)流动b相的确定结合稳定性的考察结果,采用0.1% 醋酸水溶液作为a相之后,采用同一上柱液,统一收集标准为95%,然后采用不同的反相进行有机溶剂(流动相b)进行考察,包括:乙腈、乙醇、甲醇,以收集馏分的最高纯度及回收率作为考察指标,结果如表3所示:表3 流动b相稳定性考察结果如表3所示:流动相b为乙醇时,馏分中最高纯度达到97%,回收率达到48%,高于乙腈和甲醇,因此确定有机相采用乙醇体系对杂质分离效果最佳。
[0022]
5.洗脱梯度的确定在选定填料和流动相组成后,首先进行梯度粗试,发现杂质g在乙醇浓度46%左右时出峰;将洗脱程序设置为10% ~ 45%乙醇洗脱4倍柱体积后,再采用45% ~ 50%乙醇梯度洗脱8bv,在30mm柱和50mm柱上分别进行实验,发现杂质g在该洗脱梯度下均能得到很好的分离,制备层析过程如图1所示。
[0023]
综上,确定了还原剂、溶剂、填料、流动相和洗脱梯度,实施例一的色谱条件如表4所示。
[0024]
表4:实施例1的色谱条件
卡泊芬净杂质g粗品制备:取40g纽莫康定b0、6g苯硼酸、1l四氢呋喃混合后加入到3l圆底烧瓶中,85℃油浴加热,去除水分;当水分低于0.15%时,向体系中加入25ml bstfa,反应1h;将体系温度降至-15℃以下,缓慢滴加200ml硼烷四氢呋喃,控制温度-10℃以下反应1h;反应结束后,向体系中加入50ml hcl(1mol/l),终止反应,将体系除去溶剂,得到纯度为41%的卡泊芬净杂质g粗品(固体粉末)18g。
[0025]
重复以上反应,粗品纯度均在41%左右,以纯度为41%的卡泊芬净杂质g粗品作为下一步高纯度制备的原料。
[0026]
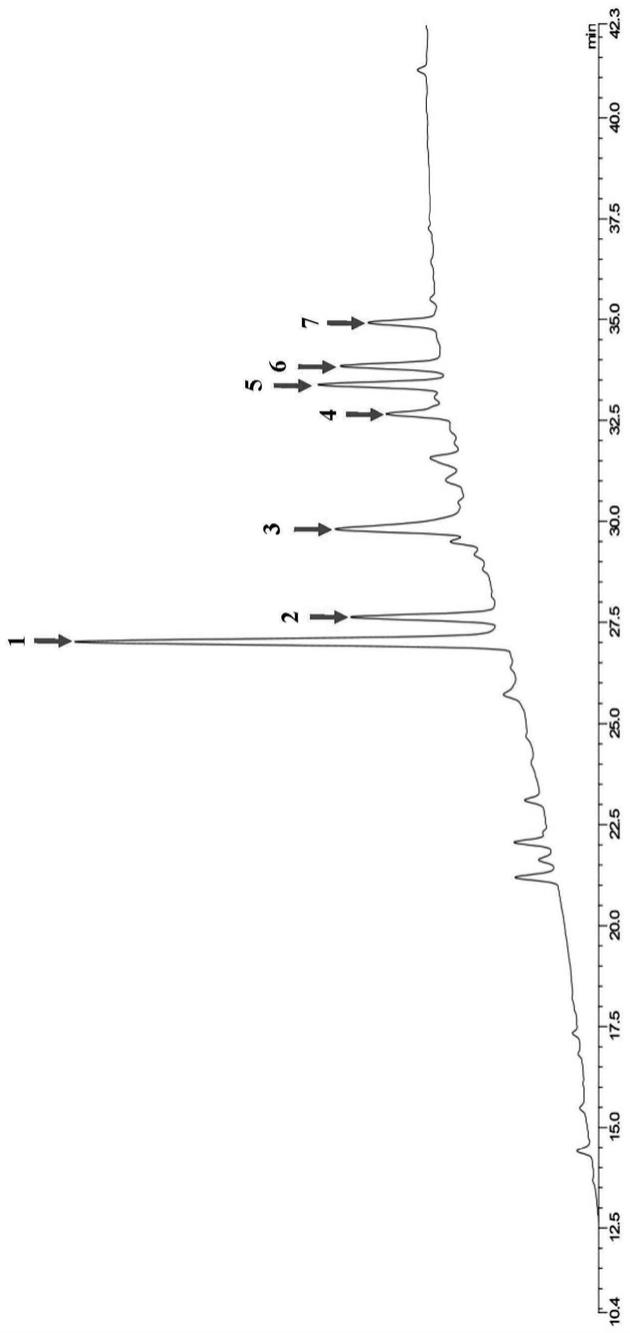
高纯度醋酸卡泊芬净杂质g的制备:取3g纯度为41%的卡泊芬净杂质g粗品,溶解于0.1%醋酸水溶液,用0.45μm滤膜过滤除去不溶物,将过滤后的粗品g溶液泵入制备色谱的色谱柱,色谱柱填料为sp-100-8-ods-p,柱尺寸为50mm*500mm,填料粒径为8μm,上样量为5g/l,流动相a为0.1%醋酸水溶液,流动相b为乙醇,首先采用含有10%乙醇、2倍柱体积的流动相去平衡色谱柱;接着用含有10%-45%乙醇、4倍柱体积的流动相进行梯度洗脱;然后用45%-50%乙醇梯度、8倍柱体积的流动相洗脱,收集该洗脱段46%乙醇以后的组分洗脱液,经hplc检测后发现,收集到的梯度洗脱液中醋酸卡泊芬净杂质g的含量在96%以上,冻干后得到0.65g高纯度醋酸卡泊芬净杂质g的固体粉末, 产品总收率为54%,如图2检测图谱所示,产品纯度为95.09%。
[0027]
实施例2根据实施例1相同的方法和色谱条件,取3.5g、纯度为41%的粗品g溶解于0.1%醋酸水溶液,最终,收集到的梯度洗脱液中醋酸卡泊芬净杂质g的含量在纯度95%以上,冻干后得到0.7g醋酸卡泊芬净杂质g的固体粉末,产品收率为50%,如图3检测图谱所示,产品纯度为97.85%。
[0028]
实施例3
根据实施例1相同的方法和色谱条件,取4g、纯度为41%粗品g溶解于0.1%醋酸水溶液,最终,收集到的梯度洗脱液中醋酸卡泊芬净杂质g的含量在纯度95%以上,冻干后得到0.85g醋酸卡泊芬净杂质g的固体粉末,产品收率为52%,如图4检测图谱所示,产品纯度为98.08%。
[0029]
实施例4根据实施例1相同的方法和色谱条件,取2.5g、纯度为41%粗品g溶解于0.1%醋酸水溶液,最终,收集到的梯度洗脱液中醋酸卡泊芬净杂质g的含量在纯度95%以上,冻干后得到0.48g醋酸卡泊芬净杂质g的固体粉末,产品收率为47%,如图5检测图谱所示,产品纯度为97.70%。
[0030]
实施例5根据实施例1相同的方法和色谱条件,取5g、纯度为41%粗品g溶解于0.1%醋酸水溶液,最终,收集到的梯度洗脱液中醋酸卡泊芬净杂质g的含量在纯度95%以上,冻干后得到1.05g醋酸卡泊芬净杂质g的固体粉末,产品收率为51%,如图6检测图谱所示,产品纯度为97.05%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。