低分子量聚(亚芳基醚)及其制备方法
技术领域
1.本发明属于聚(亚芳基醚)树脂技术领域,具体涉及低分子量聚(亚芳基醚)及其制备方法。
背景技术:
2.聚(亚芳基醚)是五大通用工程塑料之一,在电子电器、汽车、家用电器、办公室设备和工业机械等方面有着广泛的应用。近年来,随着通讯技术迅速发展,第5代(5g)通讯技术已经在全球范围内推广使用,5g通讯是高频通讯技术,对材料的电性能特别是介电性能要求高,对通讯设备的基础材料特别是覆铜板的介电损耗因数有很高的要求,在一定范围内介电损耗因数越小越有利于信号的传输。在覆铜板应用领域中,传统的环氧树脂基覆铜板电性能已经满足不了当前通讯技术的要求。
3.聚(亚芳基醚)树脂由于其分子链本身对称性高、极性低,具有优良的电性能、相对低的介电损耗因素和相对小而稳定的介电常数。因此,有利于聚(亚芳基醚)树脂应用在5g通讯中。但是大分子量聚(亚芳基醚)(数均分子量超过10000g/mol)因其具有熔体粘度大、溶液粘度大等缺陷,很难直接应用在覆铜板等领域,这需要降低分子量,得到低分子量聚(亚芳基醚)。低分子量聚(亚芳基醚)的制备技术主要包括再分配方法和单体直接合成法。
4.再分配方法如下:以大分子量聚(亚芳基醚)(数均分子量在10000g/mol以上)为原料,加入双酚类或多酚类再分配单体,溶于聚(亚芳基醚)的良溶剂中形成溶液,加入自由基引发剂,在一定温度下引发反应,形成低分子量聚(亚芳基醚)。例如,中国专利cn101389691a公开了一种低分子量聚(亚芳基醚)的制造方法,该制造方法通过使用数均分子量10000g/mol以上的聚(亚芳基醚)、多酚性化合物和自由基引发剂进行再分配反应,制造出数均分子量在4000g/mol以下的低分子量聚(亚芳基醚)。还有通过单体合成法先合成大分子量聚(亚芳基醚)树脂,然后在合成的溶液中通过加入再分配酚类单体进行再分配反应,形成小分子量聚(亚芳基醚)树脂。又例如,中国专利cn1140565c公开了一种通过再分配生产低分子量聚亚苯醚树脂的方法,首先通过氧化耦合方式合成大分子量聚(亚芳基醚),形成聚(亚芳基醚)溶液,然后将功能化的酚类单体加入该溶液中,不必再添加另外的再分配催化剂,制备出了低分子量聚(亚芳基醚)。另外,中国专利cn1188450c也公开了一种利用再分配方法制备低分子量聚(亚芳基醚)的方法。然而,采用再分配方法制备低分子量聚(亚芳基醚)时,反应过程中难以控制分子链的断链长度,并且引入了油溶性自由基催化剂,在后续的处理过程中难以除去,处理过程复杂。因此,利用再分配方法来制备低分子量聚(亚芳基醚)没有商品化。
5.直接合成方法是一种利用单体酚在铜胺络合催化剂的催化条件下、在聚(亚芳基醚)的溶剂中合成的方法。例如,中国专利cn1142965c公开了制备特性粘度为0.08dl/g~0.16dl/g的低分子量聚(亚芳基醚)树脂的方法,使用含氧气体和配合物金属催化剂在反应溶液中氧化耦合至少一种单价酚,制备低分子量聚(亚芳基醚)树脂溶液,水洗除去催化剂,将反应溶液脱挥除去有机溶剂,得到低分子量聚(亚芳基醚)树脂。例如,中国专利
cn100352848c公开了双官能亚苯基醚低聚物的制造方法,其中加入联苯酚和单酚2,6-二甲基苯酚单体,通过氧化聚合方法制备的低聚物聚(亚芳基醚)。再例如,中国专利cn101305030b公开了制备特性粘度为0.04~0.3dl/g的多官能度聚(亚芳基醚),制备的聚(亚芳基醚)经过精制水洗除去催化剂杂质后,再通过完全分离方法比如脱气挤出、喷雾干燥、转膜蒸发等方法脱去溶剂,进而制备出低分子量聚(亚芳基醚)产品。另外,wo2017105682a1、cn101479319b和cn1125107c也公开类似的低分子量聚(亚芳基醚)的制备方法。但是,对于已经公开的直接合成法制备低分子量聚(亚芳基醚),大都采用完全脱离法得到低分子量聚(亚芳基醚),完全脱离法具有制备工序短、易生产等特点,但是完全脱离法保留了聚合过程中的杂质(例如,红色的醌),同时具有分子量分布宽、耐高温分解性能差等缺陷。
技术实现要素:
6.因此,本发明的目的是针对现有技术所存在的缺陷,提供低分子量聚(亚芳基醚)及其制备方法,本发明的制备方法制备的低分子量聚(亚芳基醚)的醌含量低,分子量分布窄,玻璃化转变温度高。
7.本发明的目的是通过以下技术方案实现的。
8.一方面,本发明提供了低分子量聚(亚芳基醚)的制备方法,其中,所述制备方法包括以下步骤:
9.(1)在氧化剂和催化剂的存在下,酚单体在聚(亚芳基醚)良溶剂中氧化聚合,得到在25℃氯仿溶液中的特性粘度为0.05~0.3dl/g的低分子量聚(亚芳基醚)的混合溶液;
10.(2)对低分子量聚(亚芳基醚)的混合溶液进行水洗,得到水洗后的低分子量聚(亚芳基醚)的混合溶液;
11.(3)搅拌下及在惰性气氛下将水洗后的低分子量聚(亚芳基醚)的混合溶液加入到聚(亚芳基醚)不良溶剂中形成低分子量聚(亚芳基醚)料浆,过滤得到低分子量聚(亚芳基醚)湿物料;其中,水洗后的低分子量聚(亚芳基醚)的混合溶液中低分子量聚(亚芳基醚)的固含量控制为30~80wt%,聚(亚芳基醚)不良溶剂与水洗后的聚(亚芳基醚)混合溶液的重量比为3或以上;
12.(4)干燥低分子量聚(亚芳基醚)湿物料,得到低分子量聚(亚芳基醚)。
13.采用诸如喷雾干燥或脱挥发等完全脱离法处理由氧化耦合法制得的低分子量聚(亚芳基醚)混合液时,得到的低分子量聚(亚芳基醚)产品醌杂质含量高,颜色偏红,并且分子量分布宽,耐高温分解性能差。本技术发明人发现,对于去除残余催化剂的水洗后的低分子量聚(亚芳基醚)的混合溶液,通过控制其浓度、聚(亚芳基醚)不良溶剂的用量以及特定的沉淀条件,能够有效地降低低分子量聚(亚芳基醚)中的醌含量,同时低分子量聚(亚芳基醚)的分子量分布窄,玻璃化转变温度高。不希望受理论限制,认为,在形成料浆过程中,在诸如氮气的惰性气体的保护下,醌至少部分被还原成可溶于聚(亚芳基醚)良溶剂和/或聚(亚芳基醚)不良溶剂中的酚而被去除,由此本发明方法制备得到的低分子量聚(亚芳基醚)具有降低的醌含量。
14.根据本发明提供的制备方法,其中,可以采用本领域中已知的原料和氧化聚合方法(氧化耦合方法)来制备在25℃氯仿溶液中的特性粘度为0.05~0.3dl/g的低分子量聚
(亚芳基醚)的混合溶液。
15.根据本发明提供的制备方法,其中,所述步骤(1)中所述酚单体可以为一元酚、多元酚或其混合物。
16.本发明中,可以采用式(i)所示的一元酚:
[0017][0018]
其中,m1、m2、m3和m4各自独立地为氢原子、烷基(例如,c1-6烷基)、卤素、卤代烷烃或烷氧基。具体地,一元酚的实例包括但不限于:2,6-二甲基苯酚和2,3,6-三甲基苯酚。
[0019]
本发明中,所述多元酚可以为酚羟基数为2~7的多元酚,优选为二元酚。
[0020]
适合用于本发明的二元酚可以是式(ii)所示的二元酚:
[0021][0022]
其中,n1、n2、n3和n4各自独立地为氢原子或诸如甲基、乙基、烯丙基的c原子数为1~8的饱和或不饱和烷基;w为诸如乙基、异丙基或亚甲基的c原子个数1~4的烷基。另外,本发明中也可以采用式(ii)所示的二元酚中缺失w基团的那些二元酚。具体地,所述二元酚的实例包括但不限于:四甲基双酚a、双酚a和四甲基联苯酚。
[0023]
在一些优选实施方案中,所述酚单体为2,6-二甲基苯酚或2,6-二甲基苯酚与2,3,6-三甲基苯酚的混合物。本发明对2,6-二甲基苯酚与2,3,6-三甲基苯酚的混合物中2,6-二甲基苯酚与2,3,6-三甲基苯酚的比例没有特殊要求。在一些实施方案中,2,6-二甲基苯酚与2,3,6-三甲基苯酚的物质的量的比例可以为1:0.00001~0.1;在一些实施方案中为1:0.0001~0.01;以及在一些实施方案中为1:0.0005~0.003。
[0024]
根据本发明提供的制备方法,其中,通过选择不同的酚单体种类和/或它们的比例,可以得到具有不同主链结构的低分子量聚(亚芳基醚)。
[0025]
在一些实施方案中,所述低分子量聚(亚芳基醚)具有如式(iv)所示的结构:
[0026][0027]
其中,k1和k2各自独立地为c1-c8烃基,优选为甲基;n为5~100的整数。
[0028]
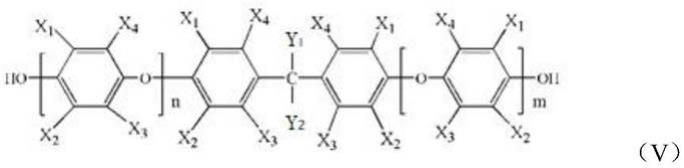
在另一些实施方案中,所述低分子量聚(亚芳基醚)具有如式(v)所示的结构:
[0029][0030]
其中,n、m可以单独为0或大于1的整数,且n m的范围为5~100的整数;x1、x2、x3和x4各自独立地为氢原子、烷基、卤素、卤代烷烃或烷氧基,x1、x2、x3和x4相同或不同;y1和y2各自独立地为氢原子、烷基、卤素、卤代烷烃、酚羟基或烷氧基,y1和y2相同或不同。
[0031]
在一些优选实施方案中,x1和x2各自独立地为氢或甲基,x3和x4各自独立地为甲基,y1和y2为甲基。
[0032]
根据本发明提供的制备方法,其中,所述低分子量聚(亚芳基醚)在25℃氯仿溶液中的特性粘度为0.07~0.15dl/g。特别地,本发明人发现,通过控制固含量和聚(亚芳基醚)不良溶剂比例,可以将特性粘度在0.07~0.15dl/g范围内的低分子量聚(亚芳基醚)在析出过程中的产率提高到90%以上。
[0033]
根据本发明提供的制备方法,其中,所述氧化剂为氧气。通常情况下,氧气可以由空气提纯制备得到,通常包含氮气等空气中所含有的组份。氧气也可以是通过电解水等其他方法制备的氧气。在一些实施方案中,氧气的浓度为5体积%~100体积%;以及在一些实施方案中,为80体积%~100体积%。
[0034]
根据本发明提供的制备方法,其中,所述催化剂为金属胺复合催化剂。所述金属胺复合催化剂含有由金属盐和胺化合物络合形成的络合剂。所述金属盐的金属离子包括铬、锰、钴、或铜离子,优选为铜离子。适合用于本发明的金属盐(铜盐)的实例包括但不限于:氯化亚铜、溴化亚铜、硫酸亚铜、硫酸亚铜四胺、乙酸亚铜、氯化铜、溴化铜、硫酸铜、硫酸铜四胺和乙酸铜。在一些实施方案中,所述金属盐与所述酚单体的物质的量的比例可以为0.005~2:100。
[0035]
通常地,所述胺化合物可以为一胺化合物,例如,伯胺、叔胺、仲胺等;二胺化合物;或者它们的混合物。适合用于本发明的伯胺的实例包括但不限于:正丙胺、异丙胺、正丁胺、仲丁胺、叔丁胺、正戊胺、正己胺和环己胺。适合用于本发明的仲胺的实例包括但不限于:二正丙胺,二正丁胺、二叔丁胺、正丁基正戊胺和二正己基胺。适合用于本发明的叔胺的实例包括但不限于:三乙胺、三正丙基胺、三正丁基胺、二甲基正丁基胺和二甲基正戊基胺。
[0036]
适合用于本发明的二胺化合物的结构如式(iii)所示:
[0037][0038]
其中,r1、r2、r4和r5各自独立地为氢原子、c1-6直链烷基或c3-6支链烷基;r3是碳原子数量为2个以上的饱和烷基。具体地,适合用于本发明的二胺化合物的实例包括但不限于:n,n,n’,n
’-
四甲基-1,3-二氨基丙烷和n,n
’-
二叔丁基乙二胺。
[0039]
在一些实施方案中,所述胺化合物与所述金属盐的物质的量的比例为1~100:1,优选为10~60:1。
[0040]
在一些实施方案中,所述催化剂为铜胺催化剂,所述铜胺催化剂包含由金属铜盐
和胺化合物络合形成的络合剂。
[0041]
在一些具体实施方案中,所述铜胺催化剂包含物质的量的比例为1:20:10:5的溴化亚铜、n,n-二甲基丁胺、二正丁胺和n,n,n’,n
’-
四甲基-1,3-二氨基丙烷。
[0042]
根据本发明提供的制备方法,其中,所述聚(亚芳基醚)良溶剂是指能够溶解聚(亚芳基醚)特别是数均分子量在10000g/mol以下的低分子量聚(亚芳基醚)的有机溶剂。适合用于本发明的聚(亚芳基醚)良溶剂的实例包括但不限于限于:苯、甲苯、二甲苯、氯仿和四氢呋喃。本发明中,聚(亚芳基醚)良溶剂可以是上述溶剂中的一种,也可以是由这些溶剂组成的混合溶剂。在一个优选实施方案中,所述聚(亚芳基醚)良溶剂为甲苯。
[0043]
根据本发明提供的制备方法,其中,所述聚(亚芳基醚)良溶剂与所述酚单体的质量比为1~10:1,优选为2~7:1。
[0044]
根据本发明提供的制备方法,其中,步骤(1)中所述氧化聚合在15~80℃的温度下进行。在一些实施方案中,步骤(1)中所述氧化聚合在25~45℃的温度下进行。
[0045]
根据本发明提供的制备方法,其中,步骤(2)中采用包含铜离子螯合剂的水溶液对低分子量聚(亚芳基醚)的混合溶液进行水洗。
[0046]
适合用于本发明的铜离子螯合剂的实例包括但不限于:edta、edta-2na、edta-3na、edta-4na、柠檬酸钠和氮川三乙酸三钠。
[0047]
本发明中,对包含铜离子螯合剂的水溶液没有特殊要求,只要能够有效螯合催化剂的金属离子即可。在一些实施方案中,所述铜离子螯合剂与所述金属盐中金属离子的物质的量的比例为1.1~3:1。
[0048]
根据本发明提供的制备方法,其中,步骤(2)中水洗的操作如下:向低分子量聚(亚芳基醚)的混合溶液中加入包含铜离子螯合剂的水溶液,分相,液液分离。
[0049]
本发明中,可以通过离心分离的方式进行液液分离。当然,也可以采用本领域中其它任何已知的油水分离方法进行液液分离。
[0050]
根据本发明提供的制备方法,其中,步骤(3)中所述惰性气氛为氮气气氛、氦气气氛或氩气气氛。
[0051]
根据本发明提供的制备方法,其中,步骤(3)中水洗后的低分子量聚(亚芳基醚)的混合溶液中低分子量聚(亚芳基醚)的固含量范围控制在30~80wt%。对于具有不同特性粘度的低分子量聚(亚芳基醚),其混合溶液的固含量范围可以不同。通常情况下,对于特性粘度较低的低低分子量聚(亚芳基醚),混合溶液的固含量可以相对较高。例如,对于在25℃氯仿溶液中的特性粘度为0.07~0.15dl/g的低分子量聚(亚芳基醚),步骤(3)中水洗后的低分子量聚(亚芳基醚)的混合溶液中低分子量聚(亚芳基醚)的固含量可以为60~80wt%。
[0052]
根据本发明提供的制备方法,其中,步骤(3)中可以通过诸如减压蒸馏、常压蒸馏、闪蒸或刮膜蒸发的方法来提高水洗后的低分子量聚(亚芳基醚)的混合溶液中低分子量聚(亚芳基醚)的固含量。当然,也可以向水洗后的低分子量聚(亚芳基醚)的混合溶液中加入聚(亚芳基醚)良溶剂来降低浓度,进而将固含量控制在目标范围内。
[0053]
根据本发明提供的制备方法,其中,所述聚(亚芳基醚)不良溶剂为c1-c5醇或其混合物。适合用于本发明的聚(亚芳基醚)不良溶剂的实例包括但不限于:甲醇、乙醇、正丙醇、正丁醇和正戊醇。在一些优选实施方案中,所述聚(亚芳基醚)不良溶剂为甲醇。
[0054]
根据本发明提供的制备方法,其中,聚(亚芳基醚)不良溶剂比例低于3:1时,产品
析出产后率很低;反之,比例高则消耗大,后处理成本高。在一些实施方案中,所述聚(亚芳基醚)不良溶剂与所述水洗后的低分子量聚(亚芳基醚)的混合溶液的重量比为3~10:1;以及在一些实施方案中为5~8:1。
[0055]
根据本发明提供的制备方法,其中,步骤(3)中在10~30min,优选地在20~30min内将水洗后的低分子量聚(亚芳基醚)的混合溶液匀速加入到聚(亚芳基醚)不良溶剂中。加入速度过快,容易出现难以搅拌开的大块状聚合物沉淀,沉淀中可能包含杂质,进而影响产品的品质;反之,加入速度过慢,生产效率低。
[0056]
根据本发明提供的制备方法,其中,步骤(3)中形成低分子量聚(亚芳基醚)料浆的操作是在0~65℃,优选地30~55℃,更优选地,35~45℃的温度下进行的。
[0057]
在一些实施方案中,所述低分子量聚(亚芳基醚)在25℃氯仿溶液中的特性粘度优选为0.05~0.3dl/g,优选为0.07~0.15dl/g;步骤(3)中水洗后的低分子量聚(亚芳基醚)的混合溶液中低分子量聚(亚芳基醚)的固含量为60~80wt%,步骤(3)中所述聚(亚芳基醚)不良溶剂与所述水洗后的低分子量聚(亚芳基醚)的混合溶液的重量比为3~10:1,优选为5~8:1,步骤(3)形成低分子量聚(亚芳基醚)料浆的操作是在35~45℃的温度下进行的。
[0058]
根据本发明提供的制备方法,其中,步骤(3)中所述搅拌为剪切搅拌,其转速为50~1000rpm,优选为400~800rpm。另外,搅拌的形式可以是螺旋式、叶片式或桨叶式。
[0059]
根据本发明提供的制备方法,其中,步骤(3)中形成低分子量聚(亚芳基醚)料浆的操作如下:
[0060]
(301)向析出釜中加入聚(亚芳基醚)不良溶剂,通入氮气以排出空气;
[0061]
(302)在转速为400~800rpm的螺旋式剪切搅拌下,在20~30min内向装有聚(亚芳基醚)不良溶剂的析出釜中匀速加入水洗后的低分子量聚(亚芳基醚)的混合溶液,继续搅拌5~10min,形成低分子量聚(亚芳基醚)料浆;
[0062]
(303)过滤低分子量聚(亚芳基醚)料浆,得到低分子量聚(亚芳基醚)湿物料。
[0063]
根据本发明提供的制备方法,其中,所述过滤是在固液两相过滤器中进行的。适合用于本发明的固液两相过滤器的实例包括但不限于:抽滤桶、洗涤过滤一体机、离心式过滤机和转鼓式过滤机。
[0064]
根据本发明提供的制备方法,其中,步骤(4)中所述干燥是在负压条件下、在30~120℃的温度范围内进行的。在一些实施方案中,所述干燥可以采用逐步升温的方式进行,例如,第一阶段,升温至40~50℃,保持1~5h;第二阶段,升温至70~80℃,保持1~3h;第三阶段,升温至100~120℃,直至挥发分降低到0.5wt%以下。更具体地,升温速度可以为5~20℃/h。
[0065]
根据本发明提供的制备方法,其中,步骤(4)中所述干燥可以在诸如真空干燥器、耙式干燥机和转鼓干燥机的工业干燥机或干燥器中进行。
[0066]
另一方面,本发明还提供了由上述方法制得的低分子量聚(亚芳基醚)。
[0067]
本发明具有以下优势:
[0068]
(1)本发明的制备方法制备的低分子量聚(亚芳基醚)的醌含量低,分子量分布窄,玻璃化转变温度高;
[0069]
(2)通过控制诸如低分子量聚(亚芳基醚)的特性粘度、固含量、聚(亚芳基醚)不良溶剂比例以及形成低分子量聚(亚芳基醚)料浆的温度等参数,可以提高低分子量聚(亚芳
基醚)收率,同时醌含量低,分子量分布窄,玻璃化转变温度高。
[0070]
(3)本发明的制备方法工序短,易于生产,易于推广应用。
具体实施方式
[0071]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。以下对至少一个示例性实施例的描述实际上仅仅是说明性的,决不作为对本发明及其应用或使用的任何限制。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例中未注明具体技术或者条件,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购买得到的常规产品。
[0072]
特性粘度
[0073]
将聚(亚芳基醚)样品溶解于氯仿中,形成浓度为0.5g/dl的溶液,在25℃下,用乌氏粘度计进行测定,单位为dl/g。
[0074]
分子量分布
[0075]
采用showa denko k.k.公司的商品名为“shodex gpc system 21”凝胶渗透色谱仪分析低分子量聚(亚芳基醚)的分子量分布。其中,淋洗剂为氯仿,流量为1.0ml/min,塔柱温度为40℃。
[0076]
玻璃化转变温度
[0077]
采用pyrisl差式扫描量热计测量玻璃化转变温度。其中,气氛为氮气气氛,升温速度为20℃/min。
[0078]
聚(亚芳基醚)收率
[0079]
以得到的最终产品与所用酚单体的重量百分比表示聚(亚芳基醚)收率。
[0080]
醌含量
[0081]
利用聚(亚芳基醚)与醌的电子吸收光谱的差异,以氯仿为溶剂分别将聚(亚芳基醚)样品和基准样品配制成0.3g/100ml的溶液,在25℃静置2小时,然后以基准样品作为参比,在可见紫外分光光度仪中测定波长为421nm的吸光度,计算含量。
[0082]
以下实施例中所采用的主要原料如下:
[0083]
酚单体:2,6-二甲基苯酚;四甲基双酚a;
[0084]
聚(亚芳基醚)良溶剂:甲苯;
[0085]
氧化剂:99.99%氧气;
[0086]
铜离子螯合剂:edta-2na;
[0087]
铜铵复合催化剂组成:溴化亚铜、n,n-二甲基丁胺、二正丁胺和n,n,n’,n
’-
四甲基-1,3-二氨基丙烷,它们的物质的量的比例为1:20:10:5;
[0088]
聚(亚芳基醚)不良溶剂:甲醇。
[0089]
实施例1
[0090]
(1)取10kg的2,6-二甲基苯酚单体、150kg的甲苯和2kg的铜铵复合催化剂配制成溶液,注入反应釜中,开启搅拌,在20
±
5℃的温度范围内向反应釜中通入99.99%氧气,在40
±
2℃下开始氧化聚合反应,然后在60分钟内向反应釜中匀速加入40kg的2,6二甲基苯
酚,滴加完毕后继续反应,在线取样检测直到聚合产品的特性粘度达到0.12dl/g,停止聚合。
[0091]
(2)向低分子量聚(亚芳基醚)的混合溶液中加入铜离子螯合剂的水溶液,搅拌萃取15分钟,然后静置20~30分钟,分离去掉下方的水相。其中,铜离子螯合剂的水溶液的浓度为0.1mol/l,用量为35l。
[0092]
(3)将水洗后的低分子量聚(亚芳基醚)的混合溶液转移到脱苯釜中,在负压条件下加热脱苯,于80℃下浓缩到固含量65wt%。
[0093]
(4)将450kg甲醇注入到析出釜中,升温至45℃,通入氮气以排出空气,在转速为500rpm的叶片剪切搅拌下通过泵在30分钟内匀速加入浓缩后的低分子量聚(亚芳基醚)的混合溶液,然后继续搅拌10min,形成低分子量聚(亚芳基醚)料浆。
[0094]
(5)用抽滤桶抽滤低分子量聚(亚芳基醚)料浆,得到低分子量聚(亚芳基醚)湿物料。
[0095]
(6)将低分子量聚(亚芳基醚)湿物料转入到转鼓干燥器中在负压条件下,采用逐步升温的方式进行干燥,第一阶段,升温至45℃,保持1h;第二阶段,升温至80℃,保持2h;第三阶段,升温至110℃,直至挥发分降低到0.5wt%以下,其中升温速度为20℃/h。得到类白色低分子量聚(亚芳基醚)产品。
[0096]
对比例1
[0097]
按照实施例1中的步骤(1)~(3)制备得到方法制备得到固含量为65wt%的浓缩的聚(亚芳基醚)溶液,将浓缩的聚(亚芳基醚)溶液置于真空刮膜蒸发器中脱除甲苯,降温后得到红棕色低分子量聚(亚芳基醚)产品。
[0098]
实施例2
[0099]
(1)取10kg的摩尔比为1:0.8的2,6-二甲基苯酚单体和四甲基双酚a的混合单体、150kg的甲苯和2kg的铜铵复合催化剂配制成溶液,注入反应釜中,开启搅拌,在20
±
5℃的温度范围内向反应釜中通入99.99%氧气,在40
±
2℃下开始氧化聚合反应,然后在60分钟内向反应釜中匀速加入40kg的2,6二甲基苯酚,滴加完毕后继续反应,在线取样检测直到聚合产品的特性粘度达到0.09dl/g,停止聚合。
[0100]
(2)向低分子量聚(亚芳基醚)的混合溶液中加入铜离子螯合剂的水溶液,搅拌萃取15分钟,然后静置20~30分钟,分离去掉下方的水相。其中,铜离子螯合剂的水溶液的浓度为0.1mol/l,用量为35l。
[0101]
(3)将水洗后的低分子量聚(亚芳基醚)的混合溶液转移到脱苯釜中,在负压条件下加热脱苯,于80℃下浓缩到固含量70wt%。
[0102]
(4)将450kg甲醇注入到析出釜中,升温至45℃,通入氮气以排出空气,在转速为500rpm的叶片剪切搅拌下通过泵在30分钟内匀速加入浓缩后的低分子量聚(亚芳基醚)的混合溶液,然后继续搅拌5min,形成低分子量聚(亚芳基醚)料浆。
[0103]
(5)用抽滤桶抽滤低分子量聚(亚芳基醚)料浆,得到低分子量聚(亚芳基醚)湿物料。
[0104]
(6)将低分子量聚(亚芳基醚)湿物料转入到转鼓干燥器中在负压条件下,采用逐步升温的方式进行干燥,第一阶段,升温至45℃,保持1h;第二阶段,升温至80℃,保持2h;第三阶段,升温至110℃,直至挥发分降低到0.5wt%以下,其中升温速度为10℃/h。得到类白
色低分子量聚(亚芳基醚)产品。
[0105]
对比例2
[0106]
按照实施例2中的步骤(1)~(3)制备得到方法制备得到固含量为70wt%的浓缩的聚(亚芳基醚)溶液,将浓缩的聚(亚芳基醚)溶液置于真空刮膜蒸发器中脱除甲苯,降温后得到红棕色低分子量聚(亚芳基醚)产品。
[0107]
实施例3
[0108]
(1)取10kg的2,6-二甲基苯酚单体、150kg的甲苯和3kg的铜铵复合催化剂配制成溶液,注入反应釜中,开启搅拌,在20
±
5℃的温度范围内向反应釜中通入99.99%氧气,在40
±
2℃下氧化聚合反应,然后在60分钟内向反应釜中匀速加入40kg的2,6二甲基苯酚,滴加完毕后继续反应,在线取样检测直到聚合产品的特性粘度达到0.28dl/g,停止聚合。
[0109]
(2)向低分子量聚(亚芳基醚)的混合溶液中加入铜离子螯合剂的水溶液,搅拌萃取15分钟,然后静置20~30分钟,分离去掉下方的水相。其中,铜离子螯合剂的水溶液的浓度为0.1mol/l,用量为52.5l。
[0110]
(3)将水洗后的低分子量聚(亚芳基醚)的混合溶液转移到脱苯釜中,在负压条件下加热脱苯,于80℃下浓缩到固含量40wt%。
[0111]
(4)将450kg甲醇注入到析出釜中,升温至45℃,通入氮气以排出空气,在转速为500rpm的叶片剪切搅拌下通过泵在30分钟内匀速加入浓缩后的低分子量聚(亚芳基醚)的混合溶液,然后继续搅拌3min,形成低分子量聚(亚芳基醚)料浆。
[0112]
(5)用抽滤桶抽滤低分子量聚(亚芳基醚)料浆,得到低分子量聚(亚芳基醚)湿物料。
[0113]
(6)将低分子量聚(亚芳基醚)湿物料转入到转鼓干燥器中在负压条件下,采用逐步升温的方式进行干燥,第一阶段,升温至45℃,保持1h;第二阶段,升温至80℃,保持2h;第三阶段,升温至110℃,直至挥发分降低到0.5wt%以下,其中升温速度为10℃/h。得到浅黄色低分子量聚(亚芳基醚)产品。
[0114]
对比例3
[0115]
按照实施例3中的步骤(1)~(3)制备得到方法制备得到固含量为65wt%的浓缩的聚(亚芳基醚)溶液,将浓缩的聚(亚芳基醚)溶液置于真空刮膜蒸发器中脱除甲苯,降温后得到红棕色低分子量聚(亚芳基醚)产品。
[0116]
实施例4
[0117]
基本按照实施例1的方法制备低分子量聚(亚芳基醚),不同之处在于:步骤(3)中将水洗后的低分子量聚(亚芳基醚)的混合溶液浓缩至聚(亚芳基醚)固含量为60wt%。
[0118]
最后得到类白色低分子量聚(亚芳基醚)产品。
[0119]
实施例5
[0120]
基本按照实施例2的方法制备低分子量聚(亚芳基醚),不同之处在于:步骤(3)中将水洗后的低分子量聚(亚芳基醚)的混合溶液浓缩至聚(亚芳基醚)固含量为80wt%;步骤(4)中加入甲醇的用量为400kg。
[0121]
最后得到类白色低分子量聚(亚芳基醚)产品。
[0122]
实施例6
[0123]
基本按照实施例1的方法制备低分子量聚(亚芳基醚),不同之处在于:步骤(4)是
在35℃的温度下进行的。
[0124]
最后得到类白色低分子量聚(亚芳基醚)产品。
[0125]
实施例7
[0126]
基本按照实施例1的方法制备低分子量聚(亚芳基醚),不同之处在于:步骤(4)是在25℃的温度下进行的。
[0127]
最后得到棕黄色低分子量聚(亚芳基醚)产品。
[0128]
实施例8
[0129]
基本按照实施例1的方法制备低分子量聚(亚芳基醚),不同之处在于:步骤(3)中将水洗后的低分子量聚(亚芳基醚)的混合溶液浓缩至聚(亚芳基醚)固含量为35wt%。
[0130]
最后得到类白色低分子量聚(亚芳基醚)产品。
[0131]
实施例9
[0132]
基本按照实施例1的方法制备低分子量聚(亚芳基醚),不同之处在于:步骤(4)采用乙醇作为低分子量聚(亚芳基醚)不良溶剂。
[0133]
最后得到棕黄色低分子量聚(亚芳基醚)产品。
[0134]
对比例4
[0135]
基本按照实施例1的方法制备低分子量聚(亚芳基醚),不同之处在于:步骤(4)中在空气气氛中加入浓缩后的低分子量聚(亚芳基醚)的混合溶液,然后继续搅拌3min。
[0136]
最后得到棕黄色低分子量聚(亚芳基醚)产品。
[0137]
对比例5
[0138]
基本按照实施例2的方法制备低分子量聚(亚芳基醚),不同之处在于:步骤(4)中加入甲醇量为200kg,然后继续搅拌5min。
[0139]
最后得到类白色低分子量聚(亚芳基醚)产品。
[0140]
实施例1~7和对比例1~5制备的低分子量聚(亚芳基醚)的性质如表1所示。
[0141]
表1低分子量聚(亚芳基醚)性质
[0142]
[0143][0144]
从表1可以看出,本发明制备的低分子量聚(亚芳基醚)产品醌含量相对更低,分子量分布相对更窄,玻璃化转变温度相对更高,收率高。特别地,由实施例1与对比例1的比较可知,搅拌下及在惰性气氛下将水洗后的低分子量聚(亚芳基醚)的混合溶液加入到聚(亚芳基醚)不良溶剂中形成低分子量聚(亚芳基醚)料浆再脱除甲苯,相比于置于真空刮膜蒸发器中脱除甲苯,具有降低的醌含量;由实施例1与对比例4可知,在形成料浆过程中,在采用氮气作为惰性气体进行保护下,本发明方法制备的低分子量聚(亚芳基醚)具有降低的醌含量。由实施例1、6和7的比较可知,形成料浆过程中体系的温度对醌含量也具有较大的影响,在35~45℃下,更有利于降低醌含量。
[0145]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。