1.本发明属于材料技术领域,尤其涉及一种改性硅胶材料及其制备方法和用途。
背景技术:
2.高效液相色谱(hplc)在分离分析领域一直扮演着重要的作用,由于其具有高分离特性、良好的选择性和优异的重现性等优点,已经成为分析化学中应用最广泛的工具之一。色谱柱作为高校液相色谱的“心脏”,其状态好坏直接影响着结果的准确性和可靠性。目前,反相色谱、亲水作用色谱、离子交换色谱、体积排阻色谱和手性色谱等模式的色谱分离材料已被广泛应用于药物分析、环境监测、食品检测、农产品检测和精细化工分离纯化等诸多领域。
3.其中,反相色谱凭借其柱效高、分离能力强、保留机理清楚,是目前使用最广泛的色谱分离模式,占整个hplc应用的80%左右,现已有几百种商品化的c18色谱柱。尤其在食品、药品检测中,采用c18填料的反相色谱柱可适应大部分目标物的分析要求。但对于一些极性较大的化合物,由于流动相基本上90%以上的水相,如果误使用常规的非耐水性的c18色谱填料进行分析或在实验结束后的冲洗步骤中误采用纯水相流动相进行冲洗,则可能造成“疏水塌陷”现象的发生。此外,目前最为常用的反相色谱分离材料为c18固定相,虽然可分离大多数有机化合物,但其在分离碱性化合物或易解离物质时,也易出现峰拖尾现象。
4.目前,解决该问题的一种方式是通过合成反相亲水作用模式的色谱分离材料来减弱填料“疏水塌陷”问题。反相亲水作用色谱,也被称为富水液相色谱(per aqueous liquid chromatography,palc),1982年bidlingmeyer等完成了这项工作,他们在色谱中使用高比例水作为流动相,采用反相色谱分离模式可以分离极性化合物和可离子化的化合物。该命名工作是由sandra等在2009年完成的,通过考察儿茶酚胺类、碱性、酸性物质和氨基酸等来区别于其他色谱,且基本说明了palc的特性。同年,bidlingmeryer等通过评价富水固定相的色谱行为,研究在高浓度水流动相条件下,反相硅胶键合固定相表现出非极性的色谱行为。目前,主要是采用对硅胶进行c18烷基链不饱和修饰的方式来提高所制备的反相c18填料的耐水性能力,但是,该方式往往会造成低密度键合的c18烷基链容易水解,或者极性基团容易脱落,造成填料的寿命低,从而影响目标物的分离。
5.另一个解决方式是在反相键合相中引入极性基团,目前,在反相固定相中引入极性基团的制备技术主要由两种,分别为极性包埋技术(polar
‑
embeded)和极性封尾技术(polar
‑
endcapped)。通过这两种技术在反相固定相中引入酰胺、氰基、脲基和醇基等极性基团,能有效改善反相固定相的极性选择性。但是,这两种引入极性基团的方式也存在一定的问题。极性包埋技术的弱点在于:(1)极性基团嵌入c18固定相虽解决了碱性物质峰形差的问题,但由于官能团中碳链较短,导致其择形性较差,在分离结构相似的物质时无法达到理想的分离效果;(2)极性包埋的方式是在每非极性基团和硅胶之间插入极性基团,即极性基团和非极性基团之间的配比为1:1,很难调整极性基团和非极性基团的配比,另外,极性基团和非极性基团的空间位置相对固定,很难进行调控,因而造成分离选择性。极性封尾的
方法是在硅胶表面键合极性小分子基团,该方法虽然能够极大程度地增大填料的亲水性,但是同样存在极性基团的位置和数量不固定的问题,从而使得填料的重现性难以得到保证。
6.cn111001188a公开了一种反相分离介质及其制备方法,包括在固定相以及在固定相表面包覆中性的亲水层,提供的反相分离介质具有耐水性好、离子选择性高、与离子交换作用干扰低等特性,但耐用性不够。cn101987293b公开了一种硅胶表面共聚反应的色谱分离材料的制备工艺,该分离材料键合相结构新颖,同时具有非极性基团和极性基团,能同时提供非极性作用力和多种形式的极性作用力,可以提高反相色谱选择性,但是耐水性不够。
7.因此,提供一种具有良好的耐水性、分离选择性好、且寿命长的反相色谱分离材料非常有必要。
技术实现要素:
8.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种改性硅胶材料及其制备方法和用途,本技术的材料具有耐水性好、选择性高和使用寿命长的特点,用于解决现有技术中的问题。
9.为实现上述目的及其他相关目的,本发明是通过以下技术方案获得的。
10.本发明的目的之一在于提供一种改性硅胶材料的制备方法,包括如下步骤:
11.1)硅烷偶联剂改性硅胶与亲水性聚合物进行交联反应,得到亲水改性硅胶;
12.2)所述亲水改性硅胶与非极性硅烷试剂进行取代反应,得到疏水改性硅胶;
13.3)所述疏水改性硅胶与极性硅烷试剂进行取代反应,得到所述的改性硅胶材料。
14.本技术的合成路线如下所示:
[0015][0016]
根据本技术的制备方法,步骤1)中,所述亲水聚合物选自琼脂糖、糊精、可溶性淀粉、纤维素、茶多糖、香菇多糖、灵芝多糖、灰树花多糖、海藻多糖或葡聚糖中的一种或多种。具体地,所述亲水性聚合物选自琼脂糖、糊精、可溶性粉、纤维素或葡聚糖中的一种或多种。
[0017]
本技术步骤1)中,通过引入亲水聚合物与硅烷偶联剂改性硅胶表面的环氧基发生交联反应,在硅胶表面形成致密的亲水性聚合物层,避免硅羟基的裸露,从而降低硅羟基对碱性待分离物不利的电荷相互作用的影响,同时对改性硅胶材料的亲水性至关重要。
[0018]
根据本技术的制备方法,步骤1)中,所述硅烷偶联剂改性硅胶与亲水聚合物的质量比为1:(1~10)。具体地,所述硅烷偶联剂改性硅胶与亲水聚合物的质量比1:3、1:4、1:5、1:6、1:7、1:8、1:10。
[0019]
根据本技术的制备方法,步骤1)中,所述交联反应的温度为70~110℃。
[0020]
根据本技术的制备方法,步骤1)中,所述交联反应的时间为6~24h。具体地,所述交联反应的时间为6h、7h、8h、10h、12h、15h、18h、20h。较佳的,所述交联反应的时间为20h。
[0021]
根据本技术的制备方法,步骤1)中,所述交联反应在介质中进行。具体地,所述介质为甲苯或四氢呋喃中的一种或两种。
[0022]
根据本技术的制备方法,步骤1)中,所述交联反应后还包括后处理。具体地,所述后处理包括洗涤和固液分离。更具体地,所述固液分离为抽滤;所述洗涤依次采用水、四氢呋喃或乙腈进行。
[0023]
根据本技术的制备方法,步骤1)中,所述硅烷偶联剂改性硅胶的制备方法为:硅胶与硅烷偶联剂进行硅烷化反应,得到所述的硅烷偶联剂改性硅胶。
[0024]
优选地,所述硅胶与硅烷偶联剂的质量比为1:(1~20)。
[0025]
优选地,所述硅烷偶联剂选自选自γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷、γ
‑
(2,3
‑
环氧丙氧)丙基甲基二甲氧基硅烷、γ
‑
(2,3
‑
环氧丙氧)丙基二甲基甲氧基硅烷、γ
‑
(2,3
‑
环氧丙氧)丙基三乙氧基硅烷、γ
‑
(2,3
‑
环氧丙氧)丙基甲基二乙氧基硅烷和γ
‑
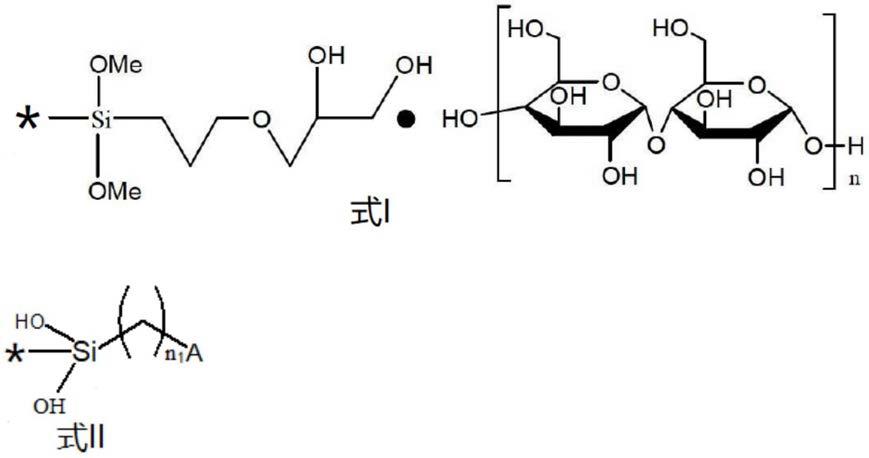
(2,3
‑
环氧丙氧)丙基二甲基乙氧基硅烷中的一种或多种。具体地,所述硅烷偶联剂为γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷。
[0026]
优选地,所述硅烷化反应的温度为70~110℃。
[0027]
优选地,所述硅烷化反应的时间为0.5~6h。
[0028]
优选地,所述反应在介质中进行。具体地,所述介质选自甲苯或四氢呋喃。
[0029]
优选地,所述硅胶为含有硅羟基基团的硅胶。
[0030]
优选地,所述硅胶可以为颗粒物、块状、片层状和球状等目前现有技术中常用的各种形状。
[0031]
优选地,所述硅胶的粒径为1.2~100μm,例如1.8μm、3.0μm、3.5μm、5.0μm、10μm、15μm、20μm、40μm、50μm、80μm、100μm。
[0032]
优选地,所述硅胶的比表面积为50~1000m2/g,例如:50m2/g、100m2/g、200m2/g、300m2/g、500m2/g、1000m2/g。
[0033]
优选地,所述硅胶为全多孔硅胶,平均孔径为例如例如和
[0034]
优选地,所述硅胶为球状和无定型状。更具体地,所述硅胶为硅胶微球。硅胶微球具有优越的机械强度。
[0035]
根据本技术的制备方法,步骤2)中,所述非极性硅烷试剂的结构式为:
[0036][0037]
其中,x选自氯、甲氧基或乙氧基;
[0038]
n1为5~29;
[0039]
a为甲基或苯基。
[0040]
本技术步骤2)中,通过非极性硅烷试剂与含醇羟基或硅羟基的亲水改性硅胶之间的取代反应,引入疏水性基团,此反应条件温和,简便,且无副产物。
[0041]
本技术步骤中,非极性硅烷试剂应为过量,用量少会导致材料疏水基团密度低,从而导致的保留能力不够。
[0042]
根据本技术的制备方法,所述非极性硅烷试剂选自十八烷基三氯硅烷、辛烷基三氯硅烷、苯基三氯硅烷、丁基三氯硅烷中的一种或多种。
[0043]
根据本技术的制备方法,步骤2)中,所述取代反应的温度为70~110℃。
[0044]
根据本技术的制备方法,步骤2)中,所述取代反应的时间为6~24h。
[0045]
根据本技术的制备方法,步骤2)中,所述取代反应在介质中进行。具体地,所述介质选自甲苯、二甲苯和四氢呋喃中的一种或多种。
[0046]
根据本技术的制备方法,步骤2)中,所述取代反应后还包括后处理。具体地,所述后处理包括固液分离、洗涤和干燥。更具体地,所述固液分离为抽滤;所述洗涤采用水或甲醇进行。
[0047]
根据本技术的制备方法,步骤3)中,所述极性硅烷试剂的结构式为:
[0048][0049]
其中,x为氯、二甲胺基、甲氧基或乙氧基;
[0050]
n2为1~10;
[0051]
b选自氯、溴、碘、氰基、氨基、苯磺酸基、磺酸基、酚羟基、羧基、季铵基和醇羟基。
[0052]
本技术步骤3)中,通过极性硅烷试剂对疏水改性硅胶进行封端处理,屏蔽了硅胶表面残余硅羟基,并且极性基团的引入进一步增大了材料的亲水性,从而提高了材料的耐水能力;此外,通过进一步的极性封端,有效地保持了硅胶基质的稳定性。
[0053]
本技术步骤中,极性硅烷试剂应为过量,用量少会导致硅胶表面的硅羟基的封尾掩蔽效果不完全,从而导致某些碱性目标物的峰型拖尾,不够理想。
[0054]
根据本技术的制备方法,所述极性硅烷试剂选自氨丙基三甲氧基硅烷、氰丙基三甲氧基硅烷、氯丙基三氯硅烷和溴丙基三氯硅烷中的一种或多种。
[0055]
根据本技术的制备方法,步骤3)中,所述取代反应的温度为70~110℃。
[0056]
根据本技术的制备方法,步骤3)中,所述取代反应的时间为6~24h。
[0057]
根据本技术的制备方法,步骤3)中,所述取代反应在介质中进行。具体地,所述介质选自甲苯、二甲苯和四氢呋喃中的一种或多种。
[0058]
根据本技术的制备方法,步骤3)中,所述取代反应后还包括后处理。具体地,所述后处理包括固液分离、洗涤和干燥。更具体地,所述固液分离为抽滤;所述洗涤采用水或甲醇进行;所述干燥为真空干燥。
[0059]
根据本技术的制备方法,所述硅烷偶联剂改性硅胶、非极性硅烷试剂和极性硅烷试剂的质量之比为1:(1~10):(1~10)。
[0060]
根据本技术的制备方法,步骤2)和步骤3)中,所述取代反应在催化剂催化下进行反应,所述催化剂选自二乙胺、三乙胺、吡啶、2,6
‑
二甲基吡啶、2,4
‑
二甲基吡啶或氨水中的一种或多种。
[0061]
本发明目的之二在于提供一种改性硅胶材料,由上述所述的制备方法制备得到。
[0062]
本发明目的之三在于提供一种改性硅胶材料,包括硅胶,所述硅胶的表面修饰有如式(i)所示基团、如式(ii)所示基团和如式(iii)所示基团;
[0063][0064][0065]
其中,在式(i)、式(ii)和(式ii)中,*表示修饰位点,
·
表示氢键或偶极作用;
[0066]
在式(i)中,me表示甲烷基,n为10~1000;
[0067]
在式(ii)中,a选自甲基或苯基,n1为5~29;
[0068]
在式(iii)中,b选自氟、氯、溴、碘、氰基、氨基、苯磺酸基、磺酸基、酚羟基、羧基、季铵基和醇羟基,n2为1~10。
[0069]
根据本技术的改性硅胶材料,所述硅胶的部分硅羟基中的h被如式(i)所示的基团所取代,所述硅胶的部分硅羟基中的h被如式(ii)所示的基团所取代,所述硅胶的部分硅羟基中的h被如式(iii)所示的基团所取代。
[0070]
根据本技术的改性硅胶材料,所述硅胶的部分硅羟基中的h被如式(i)所示的基团所取代,所述硅胶的部分硅羟基中的h被如式(ii)所示的基团所取代,所述如式(i)所示的基团的羟基中的h被如式(iii)所示的基团所取代。
[0071]
根据本技术的改性硅胶材料,所述硅胶的部分硅羟基中的h被如式(i)所示的基团所取代,所述如式(i)所示的基团的羟基中的h被如式(ii)所示的基团所取代,所述硅胶的部分硅羟基中的h被如式(iii)所示的基团所取代。
[0072]
根据本技术的改性硅胶材料,所述硅胶的部分硅羟基中的h被如式(i)所示的基团所取代,所述如式(i)所示的基团的羟基中的h被如式(ii)所示的基团所取代,所述如式(i)所示的基团的羟基中的h被如式(iii)所示的基团所取代。
[0073]
根据本技术的改性硅胶材料,以硅胶的总质量为基准计,所述如式(i)所示基团的取代率为40~60%。
[0074]
根据本技术的改性硅胶材料,以硅胶的总质量为基准计,所述如式(ii)所示基团的取代率为20~40%。
[0075]
根据本技术的改性硅胶材料,以硅胶的总质量为基准计,所述如式(iii)所示基团的取代率为5~25%。
[0076]
本技术通过在硅胶表面引入致密的亲水性聚合物包覆层形成亲水改性硅胶,增大了硅胶的亲水性,并且采用非极性基团对亲水改性硅胶进行改性,进一步用极性基团进行改性,极大程度地屏蔽了硅胶表面残余的硅羟基,从而拓宽了材料耐受高比例水相、增大了材料的水解稳定性、改善碱性化合物的峰型、增强了材料的使用寿命。
[0077]
本发明的目的之三在于提供上述所述的改性硅胶材料作为反相色谱材料的用途。
[0078]
本发明改性硅胶材料作为色谱材料时,对中性、酸性和碱性化合物具有良好的峰形;与纯水流动相具有良好的兼容性;与传统的c18相比,具有互补的选择性。
[0079]
本技术通过硅胶表面的活性硅羟基与含有环氧基的硅烷偶联剂进行反应,将环氧基团键合到粒径均一的多孔硅胶的表面,形成硅烷偶联剂改性硅胶,然后通过硅胶表面的环氧基与亲水性聚合物交联从而在硅胶表面包覆一层亲水性聚合物,最后将亲水改性硅胶与非极性硅烷试剂和极性硅烷试剂进行反应,制得高耐水性材料。本技术的材料具有较大的孔体积,较低的疏水性,较高的理论塔板数和较大的峰容量。
[0080]
与现有技术相比,本发明具有以下有益效果:
[0081]
1)本发明提供一种耐水性好、立体选择性高使用寿命长的材料,本技术的材料用作色谱柱可适用于多种混合作用分离保留模式,可作为反相或亲水作用色谱模式用于液相色谱的分离分析,特别适用于食品中防腐剂和甜味剂的等食品添加剂的分离。
[0082]
2本发明在硅胶表面引入致密的中性亲水包覆层,制备的材料具有优越的耐水性,用作色谱柱时,不会出现“疏水塌陷”等问题。
[0083]
3)本发明的硅胶表面包覆的亲水性聚合物克服了色谱柱存在亲水性不理想的问题,并且增强了其稳定性。
[0084]
4)本发明的改性硅胶材料通过极性基团的进一步引入,确保了材料的封尾完全,防止碱性化合物拖尾,同时又进一步增加了其亲水性,提升了材料的耐水性能力。
[0085]
5)本发明通过在硅胶的表面引入亲水性高分子聚合物包覆层,使硅胶表面的羟基被极大程度地掩蔽,因此,本发明提供的材料作为色谱材料能够避免常规填料产生的不利的离子交换现象,从而改善酸性化合物和碱性化合物的峰形。
附图说明
[0086]
图1为本技术中采用实施例1的改性硅胶材料作为反相色谱柱进行分离咖啡因的色谱图。
[0087]
图2为本技术中采用对比例1的硅胶材料作为反相色谱柱进行分离咖啡因的色谱图。
[0088]
图3为本技术中采用对比例2的硅胶材料作为反相色谱柱进行分离咖啡因的色谱图。
[0089]
图4为本技术中采用对比例3的硅胶材料作为反相色谱柱进行分离咖啡因的色谱图。
[0090]
图5为本技术中采用实施例1的改性硅胶材料以及对比例1、对比例2和对比例3的硅胶材料分别作为反相色谱柱进行分离防腐剂和甜味剂的色谱图。
[0091]
图6为本技术中采用实施例1的改性硅胶材料作为反相色谱柱进行加标回收率试验的色谱图。
[0092]
图7为本技术中连续合成三个批次的实施例1的改性硅胶材料作为反相色谱柱对饼干基质样品进行加标寿命测试效果色谱图。
[0093]
图5、6和7中附图标记如下:
[0094]
1、安赛蜜,2、苯甲酸,3、山梨酸,4、糖精钠,5、脱氢乙酸。
具体实施方式
[0095]
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。
[0096]
在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。下列实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。
[0097]
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
[0098]
本技术提供一种改性硅胶材料的制备方法,以解决现在有技术中的硅胶材料作为色谱柱时,耐水性差、选择性差和寿命短的问题。改性硅胶材料的制备方法,包括如下步骤:
[0099]
1)硅烷偶联剂改性硅胶与亲水性聚合物进行交联反应,得到亲水改性硅胶;
[0100]
2)所述亲水改性硅胶与非极性硅烷试剂进行取代反应,得到疏水改性硅胶;
[0101]
3)所述疏水改性硅胶与极性硅烷试剂进行取代反应,得到所述的改性硅胶材料。
[0102]
本技术中改性硅材料的制备方法的具体路线如下所示:
[0103][0104]
本技术通过硅胶表面的活性硅羟基与含有环氧基的硅烷偶联剂进行反应,将环氧基团键合到粒径均一的多孔硅胶的表面,形成硅烷偶联剂改性硅胶,然后通过硅胶表面的环氧基与亲水性聚合物交联从而在硅胶表面包覆一层亲水性聚合物,最后将亲水改性硅胶与非极性硅烷试剂和极性硅烷试剂进行反应,制得高耐水性材料。本技术的材料具有较大的孔体积,较低的疏水性,较高的理论塔板数和较大的峰容量。
[0105]
本技术的下述实施例中,硅胶采用球形硅胶为原料制备改性硅胶材料。球形硅胶的粒径为5μm,孔径为比表面为300m2/g。
[0106]
实施例1
[0107]
本实施例中,改性硅胶材料及其制备方法,包括如下:
[0108]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g的硅烷偶联剂改性硅胶。
[0109]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g可溶性淀粉,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0110]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷、2g
的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0111]
4)在上述的疏水改性硅胶,加入250ml的甲苯、10g的氯丙基三氯硅烷、2g的三乙胺于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料1。
[0112]
实施例2
[0113]
本实施例中,改性硅胶材料及其制备方法,包括如下:
[0114]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g的硅烷偶联剂改性硅胶。
[0115]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g可溶性淀粉,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0116]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的辛烷基三氯硅烷、2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0117]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氯丙基三氯硅烷、2g的三乙胺于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料2。
[0118]
实施例3
[0119]
本实施例中,硅胶材料及其制备方法,包括如下:
[0120]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0121]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g可溶性淀粉,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0122]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷、2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0123]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氨丙基三氯硅烷、2g的三乙胺于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料3。
[0124]
实施例4
[0125]
本实施例中,硅胶材料及其制备方法,包括如下:
[0126]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0127]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g可溶性淀粉,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0128]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷、2g
的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0129]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氰丙基三氯硅烷、2g的三乙胺于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料4。
[0130]
实施例5
[0131]
本实施例中,硅胶材料及其制备方法,包括如下:
[0132]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0133]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g可溶性淀粉,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0134]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷、2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0135]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的溴丙基三氯硅烷、2g的三乙胺于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料5。
[0136]
实施例6
[0137]
本实施例中,硅胶材料及其制备方法,包括如下:
[0138]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应30min,得到53g硅烷偶联剂改性硅胶。
[0139]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g可溶性淀粉,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0140]
3)在上述的亲水改性硅胶中依次加入500ml的甲苯和50g的丁基三氯硅烷,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0141]
4)在上述的疏水改性硅胶中,加入500ml的甲苯和50g的氯丙基三氯硅烷,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料6。
[0142]
实施例7
[0143]
本实施例中,硅胶材料及其制备方法,包括如下:
[0144]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0145]
2)在上述53g硅烷偶联剂改性硅胶中加入100g可溶性淀粉,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0146]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的丁基三氯硅烷和2g的三
乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0147]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氯丙基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料7。
[0148]
实施例8
[0149]
本实施例中,硅胶材料及其制备方法,包括如下:
[0150]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0151]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g微晶纤维素,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0152]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0153]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氯丙基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料8。
[0154]
实施例9
[0155]
本实施例中,硅胶材料及其制备方法,包括如下:
[0156]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0157]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g琼脂糖,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0158]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0159]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氯丙基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料9。
[0160]
实施例10
[0161]
本实施例中,硅胶材料及其制备方法,包括如下:
[0162]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0163]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g海藻多糖,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0164]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷和2g
的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0165]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氯丙基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料10。
[0166]
实施例11
[0167]
本实施例中,硅胶材料及其制备方法,包括如下:
[0168]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0169]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g葡聚糖,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0170]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0171]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氯丙基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料11。
[0172]
实施例12
[0173]
本实施例中,硅胶材料及其制备方法,包括如下:
[0174]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0175]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g环糊精,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0176]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0177]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氯丙基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料12。
[0178]
实施例13
[0179]
本实施例中,硅胶材料及其制备方法,包括如下:
[0180]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0181]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g香菇多糖,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0182]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷和2g
的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0183]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氯丙基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料13。
[0184]
实施例14
[0185]
本实施例中,硅胶材料及其制备方法,包括如下:
[0186]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0187]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g麦芽糊精,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0188]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0189]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氯丙基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料14。
[0190]
实施例15
[0191]
本实施例中,硅胶材料及其制备方法,包括如下:
[0192]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g硅烷偶联剂改性硅胶。
[0193]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g麦芽糊精,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0194]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的苯基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到疏水改性硅胶。
[0195]
4)在上述的疏水改性硅胶中,加入250ml的甲苯、10g的氯丙基三氯硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到改性硅胶材料,标记为材料15。
[0196]
对比例1
[0197]
本实施例中,硅胶材料及其制备方法,包括如下:
[0198]
1)50g的球形硅胶和100g的γ
‑
(2,3
‑
环氧丙氧)丙基三甲氧基硅烷分散于250ml甲苯溶液,于80℃反应2h,得到53g的硅烷偶联剂改性硅胶。
[0199]
2)在上述53g的硅烷偶联剂改性硅胶中加入100g可溶性淀粉,于97℃下回流进行交联反应20h;反应结束后冷却回流12h;依次用200ml蒸馏水、200ml四氢呋喃和200ml乙腈清洗、过滤,得到亲水改性硅胶。
[0200]
3)在上述的亲水改性硅胶中依次加入250ml的甲苯、50g的十八烷基三氯硅烷、2g
的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到硅胶材料,标记为材料16。
[0201]
对比例2
[0202]
本实施例中,硅胶材料及其制备方法,包括如下:
[0203]
1)50g的球形硅胶、2g的三乙胺和50g的十八烷基三氯硅烷,分散于400ml甲苯溶液,于105℃下回流进行反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到十八烷基基团改性硅胶。
[0204]
2)在上述的十八烷基基团改性硅胶中,加入500ml的甲苯、50g的3
‑
氨丙基三甲氧基硅烷和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到硅胶材料,标记为材料17。
[0205]
对比例3
[0206]
本实施例中,硅胶材料及其制备方法,包括如下:
[0207]
1)50g的球形硅胶和和50g的3
‑
氨丙基三甲氧基硅烷,分散于400ml甲苯溶液,于105℃下回流进行交联反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于105℃真空干燥2h,得到十八烷基基团改性硅胶。
[0208]
2)在上述十八烷基基团改性硅胶中,加入500ml的甲苯、50g的硬脂酰氯和2g的三乙胺,于105℃下回流搅拌反应1h;反应结束后用400ml蒸馏水、400ml甲醇清洗,过滤、并重复该步骤一次;晾干,于110℃真空干燥2h,得到硅胶材料,标记为材料18。
[0209]
性能测试
[0210]
对实施例1和对比例1
‑
3提供的材料进行性能测试,方法如下:将材料进行装柱,柱长250mm,柱内径4.6mm。然后进行如下测试:
[0211]
(1)亲水性测试
[0212]
将实施例1中制备的改性硅胶材料进行装柱,柱长250mm,柱内径4.6mm。
[0213]
将色谱柱用ph=7.0的20mmol的磷酸二氢钠缓冲盐为洗脱液,在1ml/min流速下冲洗2h后,以咖啡因为测试对象进行测定。
[0214]
测试条件如下:以甲醇
‑
水=75:25为流动相,流速为1ml/min,检测波长为254nm,进样量20μl,温度30℃。
[0215]
图1为采用实施例1的改性硅胶材料作为反相色谱柱分离咖啡因的色谱图。从图1可知,使用本技术的改性硅胶材料作为色谱柱,能够得到较好的咖啡因峰型和较高的柱效,理论塔板数为18058,拖尾因子为1.06。而且经过反复冲洗300次,均呈现优越的保留时间重现性,表明本发明提供的改性硅胶材料具有优异的耐水性。
[0216]
图2为采用对比例1的硅胶材料作为反相色谱柱分离咖啡因的色谱图。从图2可知,使用对比例1的硅胶材料作为色谱柱,峰型拖尾且柱效低,拖尾因子为2.59,其理论塔板数仅为5823,硅胶材料经过纯水相条件冲洗后,对咖啡因显示了极其不理想的峰型,原因大概是由于c18烷基链的收缩,导致咖啡因目标物不能很好的在色谱柱上被有效保留,因而峰型不够理想。
[0217]
图3为采用对比例2的硅胶材料作为反相色谱柱分离咖啡因的色谱图。从图3可知,开始时具有良好的峰型和分离效果,但随时间的推移保留时间和分离效果持续降低,300次冲洗后保留时间降低至初始时的55%,出现典型的“疏水塌陷”现象。
[0218]
图4为采用对比例3的硅胶材料作为反相色谱柱分离咖啡因的色谱图。从图4可知,分离一开始具有良好的峰型,但是冲洗100次进样后,咖啡因峰型出现了开裂,表明了合成的材料的寿命不够理想。
[0219]
(2)对五种防腐剂和甜味剂的分离效果测试
[0220]
将实施例1中的改性硅胶材料进行装柱,柱长250mm,柱内径4.6mm。应用于食品中安赛蜜、苯甲酸、山梨酸、糖精钠和脱氢乙酸等五种防腐剂和甜味剂的测定。
[0221]
测试条件为:流动相:20mm乙酸铵(ph=6.7):甲醇=93:7;流速:1.0ml/min;柱温:30℃;检测波长:230nm。
[0222]
图5中a为采用实施例1的改性硅胶材料作为反相色谱柱分离五种防腐剂和甜味剂的色谱图。从图5可知,五种防腐剂和甜味剂在93%的高比例水相流动相条件下,对五种目标物能够实现较好的分离效果,五种目标物的分离度均在1.5以上,具体地,安赛蜜、苯甲酸、山梨酸、糖精钠和脱氢乙酸的分离度分别为9.09、11.92、7.07和4.30。五种目标物均具有理想的峰型,其拖尾因子分别为:1.06、1.07、1.01、1.02和1.25。
[0223]
图5中b为采用对比例1的硅胶材料作为反相色谱柱分离五种防腐剂和甜味剂的色谱图。
[0224]
图5中c为采用对比例2的硅胶材料作为反相色谱柱分离五种防腐剂和甜味剂的色谱图。
[0225]
图5中d为采用对比例3的硅胶材料作为反相色谱柱分离五种防腐剂和甜味剂的色谱图。
[0226]
从图5b,图5c和图5d可知,采用对比例1、对比例2和对比例3制备的硅胶材料作为色谱柱均无法将糖精钠和脱氢乙酸进行有效的分离。原因可能是,这三种类型的材料的亲水性不够,导致在高比例水相的流动相的条件下有轻微“相塌陷”的情况发生,从而使得c18烷基链无法伸展开来,从而造成这三种填料类型色谱柱的分离效果不够理想,无法对五种目标物进行有效的基线分离。其中,采用对比例1制备的材料作为色谱柱对糖精钠和脱氢乙酸的分离度为1.27,采用对比例2制备的材料作为色谱柱分离糖精钠和脱氢乙酸时两个峰完全重叠,无法分离;采用对比例3制备的材料作为色谱柱对糖精钠和脱氢乙酸的分离度1.43。
[0227]
(3)寿命测试
[0228]
以饼干为基质进行加标回收率实验。
[0229]
图6为采用实施例1的改性硅胶材料作为反相色谱柱进行加标回收率试验的色谱图。从图6可知,混合溶液的浓度为200μg/ml时,五种防腐剂和甜味剂连续运行1000针以上仍旧拥有理想的峰形和优异的分离效果,其分离度仍然没有明显的变化、且峰型良好。其中,脱氢乙酸拖尾因子从一开始第1针0.682变为1000针后的1.499,脱氢乙酸的理论塔板数从一开始第1针11295变为1000针后的16380;糖精钠和脱氢乙酸分离度从一开始第1针3.848变为1000针后的5.784,充分说明了经过亲水性高分子聚合物包覆的材料应用于五种组分的分离具有优异的耐用性。
[0230]
(4)分离材料的批次重现性测试
[0231]
图7是连续合成三个批次的实施例1的改性硅胶材料作为色谱柱对饼干基质样品进行加标寿命测试效果色谱图。以脱氢乙酸为例,三批次材料作为色谱柱的保留时间分别
为20.826min、20.927min、20.987min;拖尾因子分别为0.689、0.692和0.695;脱氢乙酸和糖精钠之之间的分离度度为3.396、3.416和3.433。
[0232]
上述结果充分表明了本技术制备的三批色谱柱具有良好的批次稳定性,进而说明本研究所采用的方法具有优异的稳定性和重现性。
[0233]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。