用于序列判定的方法和系统
交叉引用
1.本技术要求于2019年3月10日提交的第62/816,145号美国专利申请的权益,其全部内容以引用方式并入本文。
背景技术:
2.阐明整个人类基因组这一目标引起了对用于小规模和大规模应用的快速核酸(例如,dna)测序技术的兴趣。随着对人类疾病遗传基础知识的增加,高通量dna测序已被用于各种各样的临床应用。尽管核酸测序方法和系统在广泛的分子生物学和诊断应用中普遍存在,但此类方法和系统在准确的碱基判定(base calling)方面可能会遇到挑战,诸如当测序信号包含被称为同聚物的重复核苷酸碱基区域时。特别地,基于指示核苷酸掺入的量化特征性信号执行碱基判定的测序方法可能具有测序错误(例如,在量化同聚物长度时),其源于信号水平的随机和不可预测的系统性变化,以及对于每个序列都可能不同的邻近序列(context)依赖性信号。此类信号变化和邻近序列依赖性信号可能导致序列(例如,同聚物)判定方面的问题。
技术实现要素:
3.本文认识到需要改进的对序列(诸如包含同聚物的序列)的碱基判定。本文提供的方法和系统可显著减少或去除量化同聚物长度中的误差以及与邻近序列依赖性相关的误差。这样的方法和系统可实现对序列(诸如包含同聚物的序列)的准确和有效的碱基判定、同聚物长度的量化以及序列信号中邻近序列依赖性的量化。
4.在一个方面,本公开提供了一种用于生成训练集的方法,该方法包括:获得第一经训练的算法,该算法包括在实际参考测序信号与可信参考测序信号之间的第一映射,其中实际参考测序信号和可信参考测序信号代表与第二属的第二基因组不同的第一属的参考基因组的部分,其中参考基因组小于第二基因组;获得对应于第二基因组的实际测序信号;以及生成用于训练第二经训练的算法的训练集,该第二经训练的算法包括在对应于第二基因组的实际测序信号与对应于第二基因组的可信测序信号之间的第二映射,其中训练集是基于第一映射采用对应于第二基因组的实际测序信号生成的。
5.在一些实施方案中,第一经训练的算法不同于第二经训练的算法。在一些实施方案中,第一经训练的算法与第二经训练的算法相同。在一些实施方案中,生成第一映射包括训练第一神经网络。在一些实施方案中,第二基因组是人类基因组。
6.在另一方面,本公开提供了一种用于生成训练集的方法,该方法包括:训练第一神经网络以生成实际参考测序信号到可信参考测序信号之间的第一映射,其中实际参考测序信号和可信参考测序信号代表不同于人类基因组且小于人类基因组的参考基因组的部分;接收或生成实际人类测序信号;以及生成用于训练第二神经网络以提供实际人类测序信号到可信人类测序信号之间的第二映射的人类训练集,其中生成人类训练集是基于第一映射,并且包括向第二神经网络馈送实际人类测序信号。
7.在一些实施方案中,第一神经网络不同于第二神经网络。在一些实施方案中,第一神经网络与第二神经网络相同。在一些实施方案中,该方法还包括使用人类训练集训练第二神经网络以将实际人类测序信号映射到可信人类测序信号。在一些实施方案中,生成人类训练集包括将实际人类测序信号与代表整个参考基因组的可信参考测序信号进行比对(align)。在一些实施方案中,训练第一神经网络包括将实际参考测序信号与代表整个参考基因组的可信参考测序信号进行比对。在一些实施方案中,第一神经网络的训练包括使用第一比对过程将实际参考测序信号与代表整个参考基因组的可信参考测序信号进行比对;并且其中生成人类训练集包括使用第二比对过程将实际人类测序信号与代表整个参考基因组的可信参考测序信号进行比对;其中第一比对过程比第二比对过程消耗更少的资源。在一些实施方案中,第一比对过程包括计算实际参考测序信号与代表整个参考基因组的可信参考测序信号的不同部分之间的相关性。在一些实施方案中,第二比对过程包括使用基于散列的搜索来执行比对。在一些实施方案中,训练第一神经网络包括执行以下各项的一次或多次迭代:选择实际参考测序信号的部分和与参考测序信号的选定部分相关的可信参考测序信号的部分;使用第一神经网络处理实际参考测序信号的选定部分以产生第一神经网络输出信号;计算代表第一神经网络输出信号与可信参考测序信号的选定部分之间的差异的误差;以及通过反向传播误差来调整第一神经网络。在一些实施方案中,第一神经网络是回归网络。在一些实施方案中,回归网络是完全连接的回归网络。在一些实施方案中,回归网络包括输入层,该输入层包括实际参考信号的每个值一个神经元。在一些实施方案中,回归网络包括多个大于输入层的中间层。在一些实施方案中,回归网络包括包含约一百个神经元的输入层、包含约一百个神经元的输出层以及多个中间层,每个中间层包含约八百个神经元。在一些实施方案中,生成人类训练集包括将截短的实际人类测序信号与代表整个参考基因组的截短的可信参考测序信号进行比对。在一些实施方案中,该方法还包括使用第二神经网络处理实际人类测序信号和不同于实际人类测序信号的类型的附加信息。在一些实施方案中,附加信息包括关于测光背景噪声的信息。在一些实施方案中,附加信息包括从前导码获得的测序信号。在一些实施方案中,附加信息包括对应于读数附近的本地信息。在一些实施方案中,附加信息包括指示流基准和流位置中的至少一个的流信息。
8.在另一方面,本公开提供了一种用于基于第一属评估第二属的基因组的方法(a method for first genus
‑
based estimation of a genome of a second genus),该方法包括:对于第二属的基因组的多个部分中的每一个:接收或生成代表第二属的基因组的部分的实际测序信号;以及基于实际测序信号评估第二属的基因组的部分;其中评估包括将第二机器学习过程应用于实际测序信号;其中训练第二机器学习过程以提供对应于第二基因组的实际测序信号到对应于第二基因组的可信测序信号之间的第二映射;其中第二映射是基于实际参考测序信号到可信参考测序信号之间的第一映射生成的;并且其中实际参考测序信号和可信参考测序信号代表第一属的参考基因组的不同于包括第二基因组的第二属的部分,其中参考基因组小于第二基因组。
9.在一些实施方案中,该方法还包括通过训练第一神经网络生成第一映射。在一些实施方案中,第二基因组是人类基因组。在一些实施方案中,评估人类基因组的部分包括计算人类基因组的部分的至少一个评估的核苷酸的置信水平。在一些实施方案中,该方法还包括基于与至少一个评估的核苷酸相关的置信水平确定实际人类测序信号的有效性。
10.在另一方面,本公开提供了一种用于评估属的基因组的方法,该方法包括:(a)接收或生成代表该属的基因组的第一部分的实际测序信号;(b)对至少部分实际测序信号应用当前模型以提供部分当前结果;其中当前模型是由经训练的算法生成的;(c)评价部分当前结果的准确度;(d)基于部分当前结果的准确度,确定是否继续使用当前模型完成基因组评估;(e)在确定继续使用当前模型后,使用当前模型完成基因组的评估;以及(f)在确定不继续使用当前模型后,获得具有足够准确度的第二模型,并使用第二模型评估基因组。
11.在一些实施方案中,该模型是基于对应于参考基因组的信息生成的,该参考基因组小于该属的基因组。在一些实施方案中,该评估由计算机系统执行,并且其中在使用当前模型之前由计算机系统使用的至少一个模型是基于对应于参考基因组的信息生成的,该参考基因组小于该属的基因组。在一些实施方案中,该方法还包括执行(a)
‑
(f)的多次迭代。
12.在另一方面,本公开提供了一种用于评估属的多个生物体的基因组的计算机实现的方法,该方法包括:执行用于评估多个生物体的基因组的多个不同的评估过程,其中执行多个不同的评估过程包括使用多个不同的评估模型。
13.在一些实施方案中,多个不同模型中的至少一个是通过再训练经训练的算法生成的。在一些实施方案中,再训练是至少部分地基于对应于参考基因组的信息执行的,该参考基因组小于该属的基因组。在一些实施方案中,多个不同模型中的至少一个是基于对应于参考基因组的信息生成的,该参考基因组小于该属的基因组。在一些实施方案中,该方法还包括在多个预定持续时间的每一个期间用第二模型替换多个不同模型中的模型。在一些实施方案中,该方法还包括在多个预定数目的评估过程的每一个期间用第二模型替换多个不同模型中的模型。在一些实施方案中,该方法还包括基于模型的准确度的评价,用第二模型替换多个不同模型中的模型。
14.在另一方面,本公开提供了一种用于评估属的基因组的方法,该方法包括:评估该属的基因组,其中该评估包括从多个不同模型中选择模型,并使用选定的模型评估该属的基因组。
15.在一些实施方案中,该选择是基于与对应于多个模型的评估的准确度相关的评估。在一些实施方案中,评估是基于对基因组的部分进行的测试。在一些实施方案中,评估由计算机系统执行。
16.在另一方面,本公开提供了一种用于评估属的基因组的计算机实现的方法,该方法包括:接收或生成代表该属的基因组的至少部分的实际测序信号;其中实际测序信号是通过对包括多个基底区段的基底进行成像而生成的;以及通过将第一模块应用于与多个基底区段中的第一基底区段相关的实际测序信号中的信号,并将不同于第一模块的第二模块应用于与多个基底区段中的第二基底区段相关的实际测序信号中的信号来评估该属的基因组。
17.在一些实施方案中,基于多个基底区段的照度之间的预期或实际的差异确定多个基底区段。在一些实施方案中,基于来自多个基底区段的辐射的收集或测量之间的预期或实际的差异确定多个基底区段。在一些实施方案中,基于多个基底区段上的化学材料的预期或实际的分布确定多个基底区段。在一些实施方案中,多个基底区段包括相同的形状和/或大小。在一些实施方案中,多个基底区段中的至少两个在至少一种形状和大小上不同。
18.在另一方面,本公开提供了一种用于评估属的基因组的计算机实现的方法,该方
法包括:接收或生成代表该属的基因组的至少部分的实际测序信号,其中实际测序信号属于与多个dna珠相连的基底的至少一部分的至少一幅图像;以及通过将至少一个模型应用于实际测序信号来评估该属的基因组。
19.本公开的另一方面提供了包含机器可执行代码的非暂时性计算机可读介质,所述机器可执行代码在由一个或多个计算机处理器执行时实现上述或本文其他地方的任何方法。
20.本公开的另一方面提供了包含一个或多个计算机处理器和与之耦合的计算机存储器的系统。所述计算机存储器包含机器可执行代码,所述机器可执行代码在由一个或多个计算机处理器执行时实现上述或本文其他地方的任何方法。
21.通过以下在其中仅示出和描述了本公开的说明性实施方案的详细描述,本公开的其他方面和优点对于本领域技术人员将变得显而易见。将会认识到,本公开能够具有其他和不同的实施方案,并且其若干细节能够在各个明显的方面进行修改,所有这些都不偏离本公开。因此,附图和说明书在本质上将被认为是说明性而非限制性的。援引并入
22.本说明书中提及的所有出版物、专利和专利申请均通过引用并入本文,其程度如同特别地且单独地指出每个单独的出版物、专利或专利申请通过引用而并入。在通过引用并入的出版物和专利或专利申请与本说明书中包含的公开内容相抵触的程度下,本说明书旨在取代和/或优先于任何此类矛盾的材料。
附图说明
23.本发明的新颖特征在所附权利要求中具体阐述。通过参考以下对其中利用到本发明原理的说明性实施方案加以阐述的详细描述以及附图(本文也称为“图”),将会获得对本发明特征和优点的更好理解,在这些附图中:
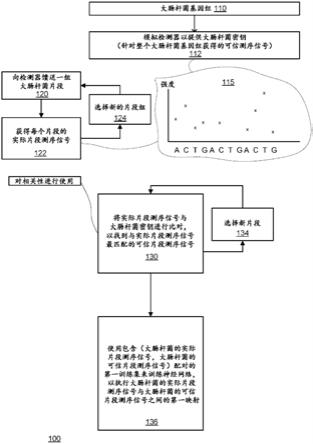
24.图1示出了用于训练神经网络以执行大肠杆菌的实际片段测序信号与大肠杆菌的可信片段测序信号之间的第一映射的方法100的示例;
25.图2示出了使用神经网络(经训练以应用第一映射)生成第二训练集的方法200的示例,该第二训练集可用于将某个人的实际片段测序信号映射到参考人类基因组的可信片段测序信号;
26.图3示出了用于评估某个人的基因组的方法300的示例;
27.图4示出了用于基于散列的比对的方法的示例(例如,根据操作322);
28.图5示出了可以在方法100和/或方法200期间训练并且可以在方法300期间使用的神经网络500的示例;
29.图6示出了用于生成训练集的方法600的示例;
30.图7示出了用于评估第二属的某个实体的基因组的方法700的示例。该评估基于第一属并且方法700可以被称为用于基于第一属评估第二属的基因组的方法;
31.图8示出了经训练以评估第二属的某个实体的基因组的u
‑
net型神经网络的示例;
32.图9示出了被编程或以其他方式配置用于实现本文提供的方法的计算机系统;
33.图10示出了图1000的示例,其图示了经训练以评估第二属的某个实体的基因组的神经网络的输入信号1001和输出信号1002;
34.图11示出了经训练以评估第二属的某个实体的基因组的神经网络的输入信号直方图1010和输出信号直方图1020的示例;
35.图12示出了用于评估属的基因组的方法的示例;
36.图13示出了用于评估属的多个生物体的基因组的方法的示例;
37.图14示出了用于评估属的基因组的方法的示例;
38.图15示出了用于评估属的基因组的方法的示例;
39.图16示出了基底(例如,晶片)及其区段的两个示例—具有其区段的晶片1610(例如,以网格状图案排列)和具有其区段的晶片1620(例如,以同心圆图案排列);和
40.图17示出了具有给定幅度的每一个原始测序信号的碱基数目的绘制直方图(左)和经处理信号的直方图(右)的示例,该经处理信号的直方图示出了具有约0、1、2和3的幅度的经处理序列的碱基数目的窄分布。
具体实施方式
41.虽然本文已经示出和描述了本公开的各种实施方案,但对于本领域技术人员显而易见的是,这样的实施方案仅以示例的方式提供。本领域技术人员可在不偏离本发明的情况下想到许多变化、改变和替代。应当理解,可以使用本文中所述的本发明的实施方案的各种替代方案。
42.在值被描述为范围的情况下,应当理解,此公开包括此范围内所有可能的子范围以及落入这些范围内的特定数值的公开,无论特定数值或特定子范围是否被明确陈述。
43.如在说明书和权利要求中使用的,单数形式“一个”、“一种”和“该”包括复数形式,除非上下文另有明确规定。
44.如本文所用,术语“至少部分”通常是指总量的任何分数。例如,“至少部分”可以指总量的至少约1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、99.9%或更多。
45.如本文所用,术语“测序”通常是指用于生成或鉴别生物分子(诸如核酸分子或多肽)的序列的过程。这样的序列可以是核酸序列,其可包括核酸碱基序列(例如,核碱基)。测序方法可以是大规模平行阵列测序(例如,illumina测序),其可使用固定在支持物如流动细胞或珠上的模板核酸分子来执行。测序方法可包括但不限于:高通量测序、下一代测序、合成测序、流式测序、大规模平行测序、鸟枪法测序、单分子测序、纳米孔测序、焦磷酸测序、半导体测序、连接测序、杂交测序、rna
‑
seq(illumina)、数字基因表达(helicos)、单分子合成测序(smss)(helicos)、克隆单分子阵列(solexa)和maxim
‑
gilbert测序。
46.如本文所用,术语“流式测序”通常是指合成测序(sbs)过程,在该过程中,循环或非循环地引入的单核苷酸溶液产生被感测到(例如,通过检测来自dna延伸的荧光信号的检测器)的分离的dna延伸。
47.如本文所用,术语“读数”通常是指核酸序列,例如测序读数。测序读数可以是通过核酸测序法获得的核酸碱基(例如,核苷酸)或碱基对的推断序列。测序读数可以由核酸测序仪生成,例如大规模平行阵列测序仪(例如,illumina或pacific biosciences of california)。测序读数可对应于受试者基因组的一部分,或在某些情况下对应于全部。测
序读数可以是测序读数集合的部分,其可以通过例如比对(例如,与参考基因组)被组合以产生受试者的基因组序列。
48.如本文所用,术语“受试者”通常是指可以从中获得生物样品(例如,正在经受或将经受加工或分析的生物样品)的个体或实体。受试者可以是动物(例如哺乳动物或非哺乳动物)或植物。受试者可以是人、狗、猫、马、猪、鸟、非人灵长类动物、猿猴、农场动物、宠物、运动动物或啮齿动物。受试者可以是患者。受试者可以患有或意思患有疾病或病症,例如癌症(例如,乳腺癌、结肠直肠癌、脑癌、白血病、肺癌、皮肤癌、肝癌、胰腺癌、淋巴瘤、食道癌或宫颈癌)或传染病。可选地或另外,受试者可以已知先前患有疾病或病症。受试者可患有或疑似患有遗传病症,诸如软骨发育不全、α
‑
1抗胰蛋白酶缺乏症、抗磷脂综合征、孤独症、常染色体显性多囊肾病、进行性神经性腓骨肌萎缩征(charcot
‑
marie
‑
tooth)、猫叫综合征、克罗恩病、囊性纤维化、痛性脂肪病(dercum disease)、唐氏综合征、杜安综合征(duane syndrome)、杜氏肌营养不良、莱顿第五因子血栓形成倾向、家族性高胆固醇血症、家族性地中海热、脆性x综合征、戈谢病、血色素沉着症、血友病、前脑无裂畸形、亨廷顿病、克林费尔特综合征、马方综合征、强直性肌营养不良、神经纤维瘤病、努南综合征、成骨不全、帕金森病、苯丙酮尿症、波伦异常、卟啉症、早衰、色素性视网膜炎、重度联合免疫缺陷、镰状细胞病、脊髓性肌萎缩、泰
‑
萨克斯病(tay
‑
sachs)、地中海贫血、三甲基胺尿症、特纳综合征、腭帆心脏面部综合征、wagr综合征或威尔逊病。受试者可以正在经受疾病或病症的治疗。受试者可以对给定的疾病或病症有症状或无症状。受试者可以是健康的(例如,未意思患有疾病或病症)。受试者可以具有一种或多种针对给定疾病的风险因素。受试者可以具有给定的体重、身高、体重指数或其他身体特征。受试者可以具有给定的民族或种族、出生地或居住地、国籍、疾病或缓解状态、家族病史或其他特征。
49.如本文所用,术语“样品”通常是指生物样品。如本文所用,术语“生物样品”通常是指从受试者获得的样品。生物样品可以直接或间接地从受试者获得。可以通过任何合适的方法,包括但不限于啐唾沫、擦拭、抽血、活检、获得排泄物(例如,尿液、粪便、痰、呕吐物或唾液)、切除、刮擦和穿刺从受试者获得样品。样品可以通过例如静脉内或动脉内进入循环系统、收集分泌的生物样品(例如粪便、尿液、唾液、痰等)、呼吸或手术提取组织(例如,活检)获得。样品可以通过非侵入性方法(包括但不限于:刮皮肤或子宫颈、擦拭脸颊或收集唾液、尿液、粪便、月经、眼泪或精液)获得。可选地,样品可以通过侵入性程序(例如活检、针吸或放血)获得。样品可以包括体液,例如但不限于血液(例如全血、红细胞、白细胞或白血球、血小板)、血浆、血清、汗液、眼泪、唾液、痰、尿液、精液、黏液、滑液、母乳、初乳、羊水、胆汁、骨髓、间质或细胞外液或脑脊液。例如,可以通过穿刺方法获得样品,以获得包含血液和/或血浆的体液。这样的样品可以包含细胞和无细胞核酸材料。可选地,样品可以从任何其他来源(包括但不限于血液、汗液、毛囊、口腔组织、眼泪、月经、粪便或唾液)获得。生物样品可以是组织样品,例如肿瘤活检物。样品可以从本文提供的任何组织获得,包括但不限于皮肤、心脏、肺、肾、乳腺、胰腺、肝脏、肠、脑、前列腺、食道、肌肉、平滑肌、膀胱、胆囊、结肠或甲状腺。本文提供的获得方法包括活检法,包括细针抽吸、空芯针活检、真空辅助活检、粗芯针活检、切口活检、切除活检、穿孔活检、刮取活检或皮肤活检。生物样品可包含一种或多种细胞。生物样品可包含一种或多种核酸分子,例如一种或多种脱氧核糖核酸(dna)和/或核糖核酸(rna)分子(例如,包含在细胞内或未包含在细胞内)。核酸分子可被包含在细胞内。可
选地或另外,核酸分子可不被包含在细胞内(例如,无细胞核酸分子)。生物样品可以是无细胞样品。
50.如本文所用,术语“无细胞样品”通常是指基本上不含细胞的样品(例如,以体积计小于10%的细胞)。无细胞样品可以来自任何来源(例如,如本文所述)。例如,无细胞样品可以来自血液、汗液、尿液或唾液。例如,无细胞样品可以来自组织或体液。无细胞样品可以来自多种组织或体液。例如,来自第一组织或流体的样品可以与来自第二组织或流体的样品组合(例如,在获得样品时或获得样品之后)。在一个示例中,可以从受试者收集第一流体和第二流体(例如,在相同或不同时间)并且可以将第一流体和第二流体组合以提供样品。无细胞样品可包含一种或多种核酸分子,例如一种或多种dna或rna分子。
51.不是无细胞样品的样品(例如,包含一个或多个细胞的样品)可以经处理以提供无细胞样品。例如,可以从受试者获得包括一种或多种细胞以及未包含在细胞内的一种或多种核酸分子(例如,dna和/或rna分子)(例如,无细胞核酸分子)的样品。样品可以经处理(例如,如本文所述)以将细胞和其他材料与未包含在细胞内的核酸分子分离,从而提供无细胞样品(例如,包括未包含在细胞内的核酸分子)。然后可以对无细胞样品进行进一步分析和处理(例如,如本文提供的)。未包含在细胞内的核酸分子(例如,无细胞核酸分子)可以源自细胞和组织。例如,无细胞核酸分子可源自肿瘤组织或降解细胞(例如身体组织的)。无细胞核酸分子可包含任何类型的核酸分子(例如,如本文所述)。无细胞核酸分子可以是双链、单链或其组合。无细胞核酸分子可以通过分泌或细胞死亡过程,例如细胞坏死、凋亡等释放到体液中。无细胞核酸分子可以从癌细胞(例如,循环肿瘤dna(ctdna))释放到体液中。无细胞核酸分子也可以是在母体血流中自由循环的胎儿dna(例如,无细胞胎儿核酸分子,例如cffdna)。可选地或另外,无细胞核酸分子可以从健康细胞释放到体液中。
52.生物样品可以直接从受试者获得并被分析而无需任何干预处理,例如样品纯化或提取。例如,可以通过进入受试者的循环系统,从受试者取出血液(例如,通过针)并将取出的血液转移到容器中而直接从受试者获得血液样品。容器可包含试剂(例如抗凝剂),使得血液样品可用于进一步分析。此类试剂可用于在分析之前处理在该容器或另一容器中的样品或源自样品的分析物。在另一个示例中,可以使用拭子取得受试者口咽表面上的上皮细胞。在从受试者获得生物样品之后,包含生物样品的拭子可以与流体(例如缓冲液)接触以从拭子收集生物流体。
53.可以从受试者获得包含一种或多种核酸分子的任何合适的生物样品。适合根据本文提供的方法使用的样品(例如,生物样品或无细胞生物样品)可以是待测个体的任何材料,包括组织、细胞、降解的细胞、核酸、基因、基因片段、表达产物、基因表达产物和/或基因表达产物片段。生物样品可以是固体物质(例如,生物组织)或可以是流体(例如,生物流体)。一般而言,生物流体可包括与活生物体相关的任何流体。生物样品的非限制性示例包括从受试者的任何解剖位置(例如,组织、循环系统、骨髓)获得的血液(或血液成分—例如,白细胞、红细胞、血小板)、从受试者的任何解剖位置获得的细胞、皮肤、心脏、肺、肾、呼吸气体、骨髓、粪便、精液、阴道液、源自肿瘤组织的间质液、乳房、胰腺、脑脊髓液、组织、咽拭子、活检物、胎盘液、羊水、肝脏、肌肉、平滑肌、膀胱、胆囊、结肠、肠、脑、腔液、痰、脓、微生物群、胎粪、母乳、前列腺、食道、甲状腺、血清、唾液、尿液、胃液和消化液、眼泪、眼液、汗液、黏液、耳垢、油、腺体分泌物、脊髓液、毛发、指甲、皮肤细胞、血浆、鼻拭子或鼻咽冲洗液、脊髓液、
脐带血、重点液(emphatic fluid)和/或其他排泄物或身体组织。提供了用于确定样品适用性和/或充分性的方法。样品可包括但不限于血液、血浆、组织、细胞、降解的细胞、无细胞核酸分子和/或来自细胞或源自个体的细胞的生物材料,例如无细胞核酸分子。样品可以是细胞、组织或无细胞生物材料的异质或同质群体。可以使用能够提供适用于本文所述的分析方法的样品的任何方法获得生物样品。
54.样品(例如,生物样品或无细胞生物样品)可以经受一个或多个为分析做制备的过程,包括但不限于过滤、离心、选择性沉淀、透化、分离、搅拌、加热、纯化和/或其他过程。例如,可以过滤样品以去除污染物或其他材料。在一个示例中,可以处理包含细胞的样品以将细胞与样品中的其他材料分离。这种过程可用于制备包含仅无细胞核酸分子的样品。这种过程可以由多步离心过程组成。可以获得多个样品,例如来自同一受试者的多个样品(例如,以相同或不同方式从相同或不同身体位置获得,和/或在相同或不同时间(例如,相距数秒、分钟、小时、天、周、月或年)获得)或来自不同受试者的多个样品,以用于如本文所述的分析。在一个示例中,第一样品在受试者经受治疗方案或程序之前从受试者获得,而第二样品在受试者经受治疗方案或程序之后从受试者获得。可选地或另外,可同时或大约同时从同一受试者获得多个样品。从同一受试者获得的不同样品可以以相同或不同的方式获得。例如,第一样品可以通过活检获得,而第二样品可以通过抽血获得。以不同方式获得的样品可以由不同的医疗专业人员、使用不同的技术、在不同的时间和/或在不同的位置获得。从同一受试者获得的不同样品可以从身体的不同区域获得。例如,第一样品可以从身体的第一区域(例如,第一组织)获得并且第二样品可以从身体的第二区域(例如,第二组织)获得。
55.如本文所用,生物样品(例如,包含一种或多种核酸分子的生物样品)当在反应容器中被提供时,可以不被纯化。此外,对于包含一种或多种核酸分子的生物样品,当将生物样品提供给反应容器时,可以不提取一种或多种核酸分子。例如,当将生物样品提供给反应容器时,可以不从生物样品中提取生物样品的核糖核酸(rna)和/或脱氧核糖核酸(dna)分子。此外,当将生物样品提供给反应容器时,生物样品中存在的靶核酸(例如,靶rna或靶dna分子)可以不被浓缩。可选地,可以纯化生物样品,并且/或者可以将核酸分子与生物样品中的其他材料分离。
56.如本文所用,术语“核酸”或“多核苷酸”通常是指包含一个或多个核酸亚单位或核苷酸的分子。核酸可包含选自腺苷(a)、胞嘧啶(c)、鸟嘌呤(g)、胸腺嘧啶(t)和尿嘧啶(u)或其变体的一种或多种核苷酸。核苷酸通常包含核苷和至少1、2、3、4、5、6、7、8、9、10个或更多个磷酸(po3)基团。核苷酸可包含核碱基、五碳糖(核糖或脱氧核糖)以及一个或多个磷酸基团。
57.核糖核苷酸是其中的糖为核糖的核苷酸。脱氧核糖核酸是其中的糖为脱氧核糖的核苷酸。核苷酸可以是核苷单磷酸或核苷多磷酸。核苷酸可以是脱氧核糖核苷多磷酸,例如,脱氧核糖核苷三磷酸(dntp),其可选自脱氧腺苷三磷酸(datp)、脱氧胞苷三磷酸(dctp)、脱氧鸟苷三磷酸(dgtp)、尿苷三磷酸盐(dutp)和脱氧胸苷三磷酸(dttp)dntp,包含可检测的标签,诸如发光标签或标记(例如,荧光团)。核苷酸可包括任何可掺入生长的核酸链的亚单位。这样的亚单位可以是a、c、g、t或u,或者特定于一个或多个互补a、c、g、t或u,或者互补于嘌呤(即a或g,或其变体)或嘧啶(即c、t或u,或其变体)的任何其他亚单位。在一些示例中,核酸是脱氧核糖核酸(dna)、核糖核酸(rna)或其衍生物或变体。核酸可以是单链的
或双链的。在一些情况下,核酸分子是环形的。
58.如本文所用,术语“核酸分子”、“核酸序列”、“核酸片段”、“寡核苷酸”和“多核苷酸”通常是指具有各种长度的多核苷酸,诸如脱氧核糖核酸或核糖核苷酸(rna)或其类似物。核酸可以具有任何三维结构,并且可以执行任何已知或未知的功能。核酸分子可具有至少约10个碱基、20个碱基、30个碱基、40个碱基、50个碱基、100个碱基、200个碱基、300个碱基、400个碱基、500个碱基、1千碱基(kb)、2kb、3kb、4kb、5kb、10kb、50kb或更大的长度。寡核苷酸通常由四种核苷酸碱基:腺嘌呤(a);胞嘧啶(c);鸟嘌呤(g);和胸腺嘧啶(t)(当多核苷酸为rna时,用尿嘧啶(u)替代胸腺嘧啶(t))的特测序列组成。因此,术语“寡核苷酸序列”是多核苷酸分子的字母表示;或者,该术语可适用于多核苷酸分子本身。这种字母表示可输入到具有中央处理单元的计算机中的数据库中,并用于生物信息学应用,如功能基因组学和同源性搜索。寡核苷酸可包含一个或多个非标准核苷酸、核苷酸类似物和/或修饰核苷酸。核酸的非限制性示例包括dna、rna、基因组dna(例如,gdna,例如剪切的gdna)、无细胞dna(例如,cfdna)、合成的dna/rna、基因或基因片段的编码或非编码区、从连锁分析定义的位点(多个位点)、外显子、内含子、信使rna(mrna)、转移rna、核糖体rna、短干扰rna(sirna)、短发夹rna(shrna)、微rna(mirna)、核糖酶、互补dna(cdna)、重组核酸、分支核酸、质粒、载体、任何序列的分离dna、任何序列的分离rna、核酸探针和引物。核酸可以包含一种或多种修饰的核苷酸,例如甲基化的核苷酸和核苷酸类似物。对核苷酸结构的修饰如果存在,则可以在核酸组装之前或之后进行。核酸的核苷酸序列可以被非核苷酸成分中断。核酸可以在聚合后进一步被修饰,例如通过与报道剂缀合或结合。
59.如本文所述的靶核酸或样品核酸可以被扩增以生成扩增产物。靶核酸可以是靶rna或靶dna。当靶核酸是靶rna时,靶rna可以是任何类型的rna,包括本文别处描述的rna类型。靶rna可以是病毒rna和/或肿瘤rna。病毒rna可以对受试者具有致病性。致病病毒rna的非限制性示例包括人类免疫缺陷病毒i(hiv i)、人类免疫缺陷病毒n(hiv 11)、正黏病毒、埃博拉病毒、登革病毒、流感病毒(例如h1n1、h3n2、h7n9或h5n1)、疱疹病毒、甲型肝炎病毒、乙型肝炎病毒、丙型肝炎(例如,装甲rna
‑
hcv病毒)病毒、丁型肝炎病毒、戊型肝炎病毒、庚型肝炎病毒、爱泼斯坦
‑
巴尔病毒、单核细胞增多症病毒、巨细胞病毒、sars病毒、西尼罗热病毒、脊髓灰质炎病毒和麻疹病毒。
60.生物样品可包含多个靶核酸分子。例如,生物样品可以包含来自单个受试者的多个靶核酸分子。在另一个示例中,生物样品可以包含来自第一受试者的第一靶核酸分子和来自第二受试者的第二靶核酸分子。
61.如本文所用,“双链”分子是包含双链核酸分子区域的分子。在一些实施方案中,双链是100%双链。在一些实施方案中,双链是至少50%、55%、60%、65%、70%、75%、80%、85%、90%、92%、95%、97%、99%或100%双链。每种可能性代表本发明的单独实施方案。在一些实施方案中,双链分子包含长度为至少1、2、3、4、5、6、7、8、9、10、12、14、15、16、18、20、25、30、35、40、45或50个碱基的一段双链核苷酸。每种可能性代表本发明的单独实施方案。在一些实施方案中,双链分子包含单链突出端。在一些实施方案中,突出端的长度不超过1、2、3、4、5、6、7、8、9或10个碱基。每种可能性代表本发明的单独实施方案。
62.如本文所用,术语“核苷酸”通常是指包括碱基(例如,核碱基)、糖部分和磷酸部分的物质。核苷酸可包含具有连接的磷酸基团的游离碱。包括具有三个连接的磷酸基团的碱
基的物质可被称为核苷三磷酸。当核苷酸被添加到正在生长的核酸分子链中时,核苷酸的近端磷酸酯与生长链之间的磷酸二酯键的形成可以伴随着高能磷酸键的水解和作为焦磷酸酯释放的两个远端磷酸酯。核苷酸可以是天然存在的或非天然存在的(例如,修饰的或工程化的核苷酸)。
63.如本文所用,术语“核苷酸类似物”可包括但不限于二氨基嘌呤、5
‑
氟尿嘧啶、5
‑
溴尿嘧啶、5
‑
氯尿嘧啶、5
‑
碘尿嘧啶、次黄嘌呤、黄嘌呤(xantine)、4
‑
乙酰胞嘧啶、5
‑
(羧基羟甲基)尿嘧啶、5
‑
羧甲基氨基甲基
‑2‑
硫尿苷、5
‑
羧甲基氨基甲基尿嘧啶、二氢尿嘧啶、β甲基氨半乳糖基辫苷、肌苷、n6
‑
异戊烯基腺嘌呤、1
‑
甲基鸟嘌呤、1
‑
甲基肌苷、2,2
‑
二甲基鸟嘌呤、2
‑
甲基腺嘌呤、2
‑
甲基鸟嘌呤、3
‑
甲基胞嘧啶、5
‑
甲基胞嘧啶、n6
‑
腺嘌呤、7
‑
甲基鸟嘌呤、5
‑
甲基氨基甲基尿嘧啶、5
‑
甲氧基氨基甲基
‑2‑
硫尿嘧啶、β嘧啶氨甘露糖基辫苷、5'
‑
甲氧基羧甲基尿嘧啶、5
‑
甲氧基尿嘧啶、2
‑
甲基硫
‑
d46
‑
异戊烯基腺嘌呤、尿嘧啶
‑5‑
氧乙酸(v)、怀丁氧苷(wybutoxosine)、假尿嘧啶、辫苷(queuosine)、2
‑
硫胞嘧啶、5
‑
甲基
‑2‑
硫尿嘧啶、2
‑
硫尿嘧啶、4
‑
硫尿嘧啶、5
‑
甲基尿嘧啶、尿嘧啶
‑5‑
氧乙酸甲酯、尿嘧啶
‑5‑
氧乙酸(v)、5
‑
甲基
‑2‑
硫尿嘧啶、3
‑
(3
‑
氨基
‑3‑
n
‑2‑
羧基丙基)尿嘧啶、(acp3)w、2,6
‑
二氨基嘌呤、硒代磷酸(phosphoroselenoate)核酸等。在一些情况下,核苷酸可包括其磷酸部分的修饰,包括对三磷酸部分的修饰。另外,修饰的非限制性示例包括更大长度的磷酸链(例如,具有4、5、6、7、8、9、10或多于10个磷酸部分的磷酸链)、具有巯基部分的修饰(例如,α硫代三磷酸和β硫代三磷酸)或具有硒部分的修饰(例如,硒代磷酸核酸)。核酸分子还可在碱基部分(例如,在通常可用于与互补核苷酸形成氢键的一个或多个原子处和/或在通常不能与互补核苷酸形成氢键的一个或多个原子处)、糖部分或磷酸骨架处进行修饰。核酸分子还可含有胺修饰基团,诸如氨基烯丙基dutp(aa
‑
dutp)和氨基己基丙烯酰胺dctp(aha
‑
dctp),以允许胺反应性部分(诸如n
‑
羟基琥珀酰亚胺酯(nhs))的共价连接。本公开的寡核苷酸中的标准dna碱基对或rna碱基对的替代物可提供更高的密度(单位为每立方毫米(mm)的比特数)、更高的安全性(例如,对天然毒素的意外或有意合成的抗性)、更容易的光程序化聚合酶辨别或更低的二级结构。核苷酸类似物可能够与用于核苷酸检测的可检测部分反应或结合。可切割碱基的类似物可以是碱基的不可切割替代物。例如,胸腺嘧啶是尿嘧啶的不可切割类似物,腺嘌呤是肌苷的不可切割类似物。
64.如本文所用,术语“游离核苷酸类似物”通常是指未与另外的核苷酸或核苷酸类似物偶联的核苷酸类似物。游离核苷酸类似物可通过引物延伸反应掺入生长的核酸链中。
65.如本文所用,术语“引物”或“引物分子”通常是指与模板核酸分子的部分互补的多核苷酸。例如,引物可以与模板核酸分子的链的部分互补。引物可以是用作核酸合成(例如可以是核酸反应(例如,核酸扩增反应,例如pcr)的组成部分的引物延伸反应)的起点的核酸链。引物可以与模板链杂交,然后有时在聚合化酶例如聚合酶的帮助下可以将核苷酸(例如,规范核苷酸或核苷酸类似物)添加到引物的末端。因此,在dna样品的复制过程中,催化复制的酶可以在连接到dna样品的引物的3'端开始复制并复制相反的链。引物(例如,寡核苷酸)可以具有可用于将引物偶联至支持物或运载体例如珠或颗粒的一个或多个官能团。引物的长度可以是8个核苷酸碱基至50个核苷酸碱基。引物的长度可大于或等于6个核苷酸碱基、7个核苷酸碱基、8个核苷酸碱基、9个核苷酸碱基、10个核苷酸碱基、11个核苷酸碱基、12个核苷酸碱基、13个核苷酸碱基、14个核苷酸碱基、15个核苷酸碱基、16个核苷酸碱基、17
个核苷酸碱基、18个核苷酸碱基、19个核苷酸碱基、20个核苷酸碱基、21个核苷酸碱基、22个核苷酸碱基、23个核苷酸碱基、24个核苷酸碱基、25个核苷酸碱基、26个核苷酸碱基、27个核苷酸碱基、28个核苷酸碱基、29个核苷酸碱基、30个核苷酸碱基、31个核苷酸碱基、32个核苷酸碱基、33个核苷酸碱基、34个核苷酸碱基、35个核苷酸碱基、37个核苷酸碱基、40个核苷酸碱基、42个核苷酸碱基、45个核苷酸碱基、47个核苷酸碱基或50个核苷酸碱基。
66.引物可以与模板核酸完全或部分互补。引物可表现出与模板核酸的序列同一性或同源性或互补性。引物与模板核酸之间的同源性或序列同一性或互补性可基于引物的长度。例如,若引物长度为约20个核酸,则其可包含10个或更多个与模板核酸互补的连续核酸碱基。
67.在本文中术语“%序列同一性”可以与术语“%同一性”互换使用,并且可以指当使用序列比对程序比对时两个或多个核苷酸序列之间的核苷酸序列同一性水平。如本文所用,80%同一性可以与通过定义的算法确定的80%序列同一性相同,并且意味着给定序列与另一长度的另一序列具有至少80%同一性。%同一性可以选自与给定序列具有例如至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、或至少99%或更高的序列同一性。%同一性可以在例如约60%至约70%、约70%至约80%、约80%至约85%、约85%至约90%、约90%至约95%、或约95%至约99%的范围内。
68.在本文中术语“%序列同源性”或“百分比序列同源性”或“百分比序列同一性”可以与术语“%同源性”、“%序列同一性”或“%同一性”互换使用,并且可以指当使用序列比对程序比对时,两个或多个核苷酸序列之间的核苷酸序列同源性水平。例如,如本文所用,80%同源性可以与通过定义的算法确定的80%序列同源性相同,因此给定序列的同源物在给定序列的长度上具有大于80%的序列同源性。%同源性可以选自例如与给定序列具有至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、或至少99%或更高的序列同源性。%同源性可以在例如约60%至约70%、约70%至约80%、约80%至约85%、约85%至约90%、约90%至约95%、或约95%至约99%的范围内。
69.如本文所用,术语“引物延伸”通常是指引物与模板核酸链的结合,然后是引物的延伸。其还可包括双链核酸的变性以及引物链与变性的模板核酸链中的一个或两个的结合,然后是引物的延伸。引物延伸反应可用于通过使用酶(聚合酶)以模板指导的方式将核苷酸或核苷酸类似物掺入引物。
70.如本文所用,术语“聚合化酶”或“聚合酶”通常是指能够催化聚合反应的任何酶。聚合化酶可用于通过掺入核苷酸或核苷酸类似物来延伸与模板链配对的核酸引物。聚合化酶可以通过延伸现有核苷酸链的3'端,经由创建磷酸二酯键而一次一个地添加与模板链匹配的新核苷酸来添加新的dna链。本文使用的聚合酶可具有链置换活性或非链置换活性。聚合酶的示例包括但不限于核酸聚合酶。聚合酶可以是天然存在的或是合成的。在一些情况下,聚合酶具有相对较高的持续合成能力,即聚合酶将核苷酸连续掺入核酸模板而不释放核酸模板的能力。示例性的聚合酶是φ29聚合酶或其衍生物。聚合酶可以是聚合作用的酶。在一些情况下,使用转录酶或连接酶(即催化键形成的酶)。聚合酶的示例包括但不限于,dna聚合酶、rna聚合酶、热稳定聚合酶、野生型聚合酶、修饰聚合酶、大肠杆菌(e.coli)dna聚合酶i、t7 dna聚合酶、噬菌体t4 dna聚合酶φ29(phi29)dna聚合酶、taq聚合酶、tth聚合酶、tli聚合酶、pfu聚合酶、pwo聚合酶、vent聚合酶、deepvent聚合酶、ex
‑
taq聚合酶、la
‑
taq聚合酶、sso聚合酶、poc聚合酶、pab聚合酶、mth聚合酶、es4聚合酶、tru聚合酶、tac聚合酶、tne聚合酶、tma聚合酶、tea聚合酶、tih聚合酶、tfi聚合酶、platinum taq聚合酶、tbr聚合酶、tfl聚合酶、pfutubo聚合酶、pyrobest聚合酶、pwo聚合酶、kod聚合酶、bst聚合酶、sac聚合酶、klenow片段、具有3'至5'外切核酸酶活性的聚合酶及其变体、修饰产物和衍生物。在一些情况下,聚合酶是单亚单位聚合酶。聚合酶可具有高持续合成能力,即聚合酶在不释放核酸模板的情况下连续地将核苷酸掺入核酸模板的能力。在一些情况下,聚合酶是经修饰以接受双脱氧核苷酸三磷酸的聚合酶,例如具有667y突变的taq聚合酶(参见例如,tabor等人,pnas,1995,92,6339
‑
6343,其为了所有目的通过引用整体并入本文)。在一些情况下,聚合酶是具有修饰的核苷酸结合的聚合酶,其可能对核酸测序有用,非限制性示例包括thermosequenas聚合酶(ge life sciences)、amplitaq fs(thermofisher)聚合酶和sequencing pol聚合酶(jena bioscience)。在一些情况下,聚合酶被基因工程化为对双脱氧核苷酸具有辨别性,例如测序酶dna聚合酶(thermofisher)。
71.聚合酶可以是家族a聚合酶或家族b dna聚合酶。家族a聚合酶包括例如taq、klenow和bst聚合酶。家族b聚合酶包括,例如,vent(exo
‑
)和therminator聚合酶。已知家族b聚合酶比家族a聚合酶接受更多不同的核苷酸底物。可能由于它们的高持续合成能力和保真度,家族a聚合酶广泛用于合成测序。
72.如本文所用,术语“互补序列”通常是指与另一序列杂交的序列。两条单链核酸分子之间的杂交可以涉及形成在某些条件下稳定的双链结构。如果两条单链多核苷酸通过两个或更多个顺序相邻的碱基配对彼此结合,则可以认为它们是杂交的。双链结构的一条链中的大部分核苷酸可以与另一条链上的核苷经历沃森
‑
克里克碱基配对。杂交还可包括核苷类似物例如脱氧肌苷、具有2
‑
氨基嘌呤碱基的核苷等的配对,其可用于降低探针的简并性,无论这种配对是否涉及氢键的形成。
73.如本文所用,术语“支持物或基底”通常是指其上可固定诸如核酸分子等试剂的固体或半固体支持物,例如载玻片、珠、树脂、芯片、阵列、基质、膜、纳米孔或凝胶。核酸分子可以被合成、附接、连接或以其他方式固定。核酸分子可以通过任何方法固定在基底上,包括但不限于物理吸附、通过离子键或共价键形成或其组合。基底可以是二维的(例如,平面2d基底)或三维的。在一些情况下,基底可以是流动池的组件并且/或者可以被包含在测序仪器内或适于被测序仪器接收。基底可包括聚合物、玻璃或金属材料。基底的示例包括膜、平面基底、微量滴定板、珠(例如磁珠)、过滤器、测试条、载玻片、盖玻片和试管。基底可包括有机聚合物,例如聚苯乙烯、聚乙烯、聚丙烯、聚氟乙烯、聚氧乙烯和聚丙烯酰胺(例如聚丙烯酰胺凝胶)以及其共聚物和接枝物。基底可包括胶乳或葡聚糖。基底也可以是无机物,例如玻璃、二氧化硅、金、可控孔度玻璃(cpg)或反相二氧化硅。支持物的构造可以是例如珠、球、微粒、颗粒、凝胶、多孔基质或基底的形式。在一些情况下,基底可以是单个固体或半固体制品(例如,单个颗粒),而在其他情况下,基底可以包括多个固体或半固体制品(例如,颗粒的集合)。基底可以是平面的、基本平面的或非平面的。基底可以是多孔的或无孔的,并且可以具有溶胀或非溶胀特性。基底可以成形为包括一个或多个孔、凹陷或其他容器、器皿、特征或位置。多个基底可以在不同位置配置在阵列中。基底可以是可寻址的(例如,用于试剂的机器人递送),或通过检测方法,例如通过激光照射和共焦或偏转光收集进行扫描。例如,基底可以与检测器光学和/或物理连通。或者,基底可以与检测器物理分离一定距离。可将扩
增基底(例如珠)放置在另一基底内或另一基底上(例如,第二支持物的孔内)。基底可具有表面特性,例如纹理、图案、微结构涂层、表面活性剂或其任何组合,以将扩增基底(例如,珠)保留在所需位置(例如在与检测器可操作连通的位置)。基于珠的支持物的检测器可以被配置为保持基本相同的读取速率,而与珠的大小无关。支持物可与检测器进行光通信、可与检测器物理接触、可与检测器相隔一定距离或其任何组合。支持物可具有多个可独立寻址的位置。核酸分子可在多个可独立寻址位置的给定可独立寻址位置处固定至支持物。多个核酸分子中的每一个与支持物的固定可借助于衔接物的使用。支持物可与检测器光学耦合。在支持物上的固定可借助于衔接物。
74.术语“固体支持物”是指任何人造固体结构,包括任何固体支持物或基底。固体支持物的示例包括但不限于珠、树脂、凝胶、水凝胶、胶体、颗粒或纳米颗粒。例如,固体支持物可以是珠。可选地,固体支持物可以是表面。例如,固体支持物可以包括与表面偶联的珠。可选地,固体支持物可以是树脂。固体支持物可以是可分离的。固体支持物可以被标记。固体支持物可以是磁性的并且可以用磁体隔离。可选地或另外,固体支持物可通过离心或一些其他力按重量、大小或一些其他可测量的量进行分离。
75.支持物(例如,固体支持物)可以是颗粒或包含颗粒。颗粒可以是珠。珠可以包括任何合适的材料,例如玻璃或陶瓷、一种或多种聚合物和/或金属。合适的聚合物的示例包括但不限于尼龙、聚四氟乙烯、聚苯乙烯、聚丙烯酰胺、琼脂糖、纤维素、纤维素衍生物或葡聚糖。合适的金属的示例包括顺磁性金属,例如铁。珠可以是磁性的或非磁性的。例如,珠可以包括带有一种或多种磁性标记的一种或多种聚合物。可以使用电磁力操纵磁珠(例如,在位置之间移动或物理地限制到例如反应容器诸如流动池室的给定位置)。珠可以具有任何有用的形状,包括例如近似立方体、球形、椭圆体、哑铃形或任何其他形状的形状。例如,珠的形状可以近似为球形。珠可以具有一个或多个不同的尺寸,包括直径。珠的尺寸(例如,珠的直径)可以小于约1mm、小于约0.1mm、小于约0.01mm、小于约0.005mm、小于约1nm、小于约1μm或更小。珠的尺寸(例如,珠的直径)可以为约1nm至约100nm、约1μm至约100μm、约1mm至约100mm。珠的集合可以包括一个或多个具有相同或不同特征的珠。例如,珠的集合的第一珠可以具有第一直径,珠的集合的第二珠可以具有第二直径。第一直径可以与第二直径相同或近似相同或不同。类似地,第一珠可具有与第二珠相同或不同的形状和组成。
76.如本文所用,术语“标记”通常是指能够与诸如核苷酸类似物等物种偶联的部分。在一些情况下,标记可以是发射可被检测的信号(或减少已发射的信号)的可检测标记。在一些情况下,这样的信号可指示一个或多个核苷酸或核苷酸类似物的掺入。在一些情况下,标记可与核苷酸或核苷酸类似物偶联,其中核苷酸或核苷酸类似物可用于引物延伸反应。在一些情况下,标记可在引物延伸反应后与核苷酸类似物偶联。在一些情况下,标记可与核苷酸或核苷酸类似物特异性反应。偶联可以是共价的或非共价的(例如,通过离子相互作用、范德华力等)。在一些情况下,偶联可经由可切割的接头,该接头可以是可切割的,诸如可光切割(例如,在紫外光下可切割)、可化学切割(例如,经由还原剂,诸如二硫苏糖醇(dtt)、tris(2
‑
羧基乙基)膦(tcep))或可酶切割(例如,经由酯酶、脂肪酶、肽酶或蛋白酶)。
77.在一些情况下,标记可以是光学活性的。(例如,发冷光的,例如荧光的或磷光的)。在一些实施方案中,光学活性标记是光学活性染料(例如,荧光染料)。染料和标记可以掺入核酸序列中。染料和标记也可掺入接头,例如用于将一个或多个珠彼此连接的接头中。例
如,标记如荧光部分可以通过接头连接到核苷酸或核苷酸类似物。染料的非限制性示例包括sybr绿、sybr蓝、dapi、碘化丙锭、hoechst、sybr金、溴化乙锭、吖啶、原黄素、吖啶橙、吖啶黄素、荧光香豆素(fluorcoumanin)、椭圆玫瑰树碱、道诺霉素、氯喹、偏端霉素d、色霉素、乙菲啶(homidium)、光神霉素、多吡啶钌、氨茴霉素、菲啶和吖啶、碘化丙锭、碘化己锭、二氢乙锭、乙锭同型二聚体
‑
1和乙锭同型二聚体
‑
2、单叠氮化乙锭、acma、hoechst 33258、hoechst 33342、hoechst 34580、dapi、吖啶橙、7
‑
aad、放线菌素d、lds751、羟脒(hydroxystilbamidine)、sytox blue、sytox green、sytox orange、popo
‑
1、popo
‑
3、yoyo
‑
1、yoyo
‑
3、toto
‑
1、toto
‑
3、jojo
‑
1、lolo
‑
1、bobo
‑
1、bobo
‑
3、po
‑
pro
‑
1、po
‑
pro
‑
3、bo
‑
pro
‑
1、bo
‑
pro
‑
3、to
‑
pro
‑
1、to
‑
pro
‑
3、to
‑
pro
‑
5、jo
‑
pro
‑
1、lo
‑
pro
‑
1、yo
‑
pro
‑
1、yo
‑
pro
‑
3、picogreen、oligreen、ribogreen、sybr gold、sybr green i、sybr green ii、sybr dx、syto标记(例如syto
‑
40、syto
‑
41、syto
‑
42、syto
‑
43、syto
‑
44和syto
‑
45(蓝色);syto
‑
13、syto
‑
16、syto
‑
24、syto
‑
21、syto
‑
23、syto
‑
12、syto
‑
11、syto
‑
20、syto
‑
22、syto
‑
15、syto
‑
14和syto
‑
25(绿色);syto
‑
81、syto
‑
80、syto
‑
82、syto
‑
83、syto
‑
84和syto
‑
85(橙色);以及syto
‑
64、syto
‑
17、syto
‑
59、syto
‑
61、syto
‑
62、syto
‑
60和syto
‑
63(红色))、荧光素、异硫氰酸荧光素(fitc)、四甲基异硫氰酸罗丹明(tritc)、罗丹明、四甲基罗丹明、r
‑
藻红蛋白、cy
‑
2、cy
‑
3、cy
‑
3.5、cy
‑
5、cy5.5、cy
‑
7、德克萨斯红(texas red)、phar
‑
red、别藻蓝蛋白(apc)、sybr green i、sybr green ii、sybr gold、celltracker green、7
‑
aad、乙锭同型二聚体i、乙锭同型二聚体ii、乙锭同型二聚体iii、溴化乙锭、伞形酮、曙红、绿色荧光蛋白、赤藓红、香豆素、甲基香豆素、芘、孔雀绿、茋、萤光黄、级联蓝(cascade blue)、二氯三嗪胺荧光素、丹磺酰氯、荧光镧系络合物(如包含铕和铽的那些络合物)、羧基四氯荧光素、5
‑
羧基荧光素和/或6
‑
羧基荧光素(fam)、vic、5
‑
碘乙酰胺基荧光素或6
‑
碘乙酰胺基荧光素、5
‑
{[2
‑5‑
(乙酰基巯基)
‑
琥珀酰基]氨基}荧光素和5
‑
{[3
‑5‑
(乙酰基巯基)
‑
琥珀酰基]氨基}荧光素(samsa
‑
荧光素)、丽丝胺罗丹明b磺酰氯、5
‑
羧基罗丹明和/或6
‑
羧基罗丹明(rox)、7
‑
氨基
‑
甲基
‑
香豆素、7
‑
氨基
‑4‑
甲基香豆素
‑3‑
乙酸(amca)、bodipy荧光团、8
‑
甲氧基芘
‑
1,3,6
‑
三磺酸三钠盐、3,6
‑
二磺酸
‑4‑
氨基
‑
萘二甲酰亚胺、藻胆蛋白、alexafluor标记(例如alexafluor 350、alexafluor405、alexafluor 430、alexafluor 488、alexafluor 532、alexafluor 546、alexafluor 555、alexafluor 568、alexafluor 594、alexafluor 610、alexafluor 633、alexafluor 635、alexafluor 647、alexafluor 660、alexafluor 680、alexafluor 700、alexafluor 750和alexafluor 790染料)、dylight染料(例如dylight 350、dylight 405、dylight 488、dylight 550、dylight 594、dylight 633、dylight 650、dylight 680、dylight 755和dylight 800染料)、黑洞淬灭染料(biosearch technologies)(例如bh1
‑
0、bhq
‑
1、bhq
‑
3和bhq
‑
10)、qsy染料荧光猝灭剂(molecular probes/invitrogen)(例如qsy7、qsy9、qsy21和qsy35)、dabcyl、dabsyl、cy5q、cy7q、深花青染料(ge healthcare)、dy
‑
猝灭剂(dyomics)(例如,dyq
‑
660和dyq
‑
661)、atto荧光猝灭剂(atto
‑
tec gmbh)(例如,atto 540q、atto580q、atto 612q、atto532[例如,atto532琥珀酰亚胺酯]和atto633)和其他荧光团和/或猝灭剂。荧光染料可以通过施加对应于电磁光谱的可见区域(例如,约430
‑
770纳米(nm)之间)的能量来激发。可以使用任何有用的设备例如激光器和/或发光二极管进行激发。包括但不限于反射镜、波片、滤光器、单色仪、光栅、分束器和透镜的光学元件可用于将光引导至荧光染料或从荧光染料引导光。荧光染料可以在电磁
光谱的可见区域(例如,约430
‑
770nm之间)发射光(例如,荧光)。荧光染料可以在单个波长或波长范围内被激发。荧光染料可以被电磁光谱的可见部分的红色区域(约625
‑
740nm)中的光激发(例如,在电磁光谱的可见部分的红色区域中具有激发最大值)。可选地或另外,荧光染料可以被电磁光谱的可见部分的绿色区域(约500
‑
565nm)中的光激发(例如,在电磁光谱的可见部分的绿色区域中具有激发最大值)。荧光染料可在电磁光谱的可见部分的红色区域(约625
‑
740nm)中发射信号(例如,在电磁光谱的可见部分的红色区域中具有发射最大值)。可选地或另外,荧光染料可在电磁光谱的可见部分的绿色区域(约500
‑
565nm)中发射信号(例如,在电磁光谱的可见部分的绿色区域中具有发射最大值)。
[0078]
在一些示例中,标记可以是核酸嵌入剂染料。示例包括但不限于溴化乙锭、yoyo
‑
1、sybr绿和evagreen。能量供体与能量受体之间、嵌入剂与能量供体之间或者嵌入剂与能量受体之间的近场相互作用可导致独特信号的生成或信号幅度的改变。例如,这样的相互作用可导致猝灭(即导致非辐射能量衰减的从供体到受体的能量转移)或福斯特共振能量转移(即导致辐射能量衰减的从供体到受体的能量转移)。标记的其他示例包括电化学标记、静电标记、比色标记和质量标签。
[0079]
标记可以是猝灭剂分子。如本文所用,术语“猝灭剂”是指可以是能量受体的分子。猝灭剂可以是可以减少发射信号的分子。例如,模板核酸分子可被设计成发射可检测信号。包含猝灭剂的核苷酸或核苷酸类似物的掺入可减少或消除信号,然后检测到该减少或消除。来自标记(例如,荧光部分,例如与核苷酸或核苷酸类似物连接的荧光部分)的发光也可以被猝灭(例如,通过掺入可以包含或不包含标记的其他核苷酸)。在一些情况下,如本文其他部分所述,在核苷酸或核苷酸类似物掺入后,可发生使用猝灭剂的标记。在一些情况下,标记可以是不自猝灭或表现出邻近猝灭的类型。不自猝灭或表现出邻近猝灭的标记类型的非限制性示例包括(bimane)衍生物,例如溴代双满(monobromobimane)。如本文所用,术语“邻近猝灭”通常是指一种或多种彼此靠近的染料可以表现出与它们单独表现出的荧光相比较低的荧光的现象。在一些情况下,染料可经受邻近猝灭,其中供体染料和受体染料彼此相距1nm至50nm。猝灭剂的示例包括但不限于黑洞猝灭剂染料(biosearch technologies)(例如bh1
‑
0、bhq
‑
1、bhq
‑
3和bhq
‑
10)、qsy染料荧光猝灭剂(molecular probes/invitrogen)(例如,qsy7、qsy9、qsy21和qsy35)、dabcyl、dabsyl、cy5q、cy7q、深花青染料(ge healthcare)、dy
‑
猝灭剂(dyomics)(例如,dyq
‑
660和dyq
‑
661)和atto荧光猝灭剂(atto
‑
tec gmbh)(例如atto 540q、atto 580q和atto 612q)。荧光团供体分子可以与猝灭剂结合使用。可与猝灭剂结合使用的荧光团供体分子的示例包括但不限于荧光团诸如cy3b、cy3或cy5;dy
‑
猝灭剂(dyomics)(例如,dyq
‑
660和dyq
‑
661);和atto荧光猝灭剂(atto
‑
tec gmbh)(例如atto 540q、580q和612q)。
[0080]
如本文所用,术语“检测器”通常是指能够检测信号的装置,该信号包括指示存在或不存在掺入的核苷酸或核苷酸类似物的信号。在一些情况下,检测器可包括可检测信号的光学和/或电子组件。术语“检测器”可用于检测方法中。检测方法的非限制性示例包括光学检测、光谱检测、静电检测、电化学检测等。光学检测方法包括但不限于荧光测定法和紫外
‑
可见光吸收。光谱检测方法包括但不限于质谱、核磁共振(nmr)波谱和红外光谱。静电检测方法包括但不限于基于凝胶的技术,例如凝胶电泳。电化学检测方法包括但不限于在对扩增产物进行高效液相色谱分离后对扩增产物的电化学检测。
[0081]
如本文所用,术语“衔接子(adapter)”或“衔接物(adaptor)”通常是指适于允许测序仪器对靶多核苷酸进行测序的分子(例如,多核苷酸),例如通过与靶核酸分子相互作用以便于测序(例如,下一代测序(ngs))。测序衔接子可以允许靶核酸分子被测序仪器测序。例如,测序衔接子可包含与附着于测序系统的固体支持物(例如珠或流动池)的捕获多核苷酸杂交或结合的核苷酸序列。测序衔接子可包含与多核苷酸杂交或结合以产生发夹环的核苷酸序列,这允许通过测序系统对靶多核苷酸进行测序。测序衔接子可以包括测序仪基序,该基序可以是与另一分子(例如,多核苷酸)的流动池序列互补并且可由测序系统用于对靶多核苷酸进行测序的核苷酸序列。测序仪基序还可包括用于测序例如合成测序的引物序列。测序仪基序可包括用于将文库衔接子偶联到测序系统和对靶多核苷酸(例如样品核酸)进行测序的序列。衔接子可以包括条形码。
[0082]
如本文所用,术语“条形码”或“条形码序列”通常指可用于鉴别一种或多种特定核酸的一种或多种核苷酸序列(例如,基于它们与特定样品关联、源自特定来源例如特定细胞、包含在特定分区或其他隔室中等)。条形码可包含至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或更多个核苷酸(例如,连续的核苷酸)。条形码可包含至少约10、约20、约30、约40、约50、约60、约70、约80、约90、约100或更多个连续核苷酸。用于扩增和/或测序过程(例如,ngs)的所有条形码可以不同。包含条形码的核酸群体中不同条形码的多样性可以随机生成或非随机生成。例如,可以根据分离
‑
合并(split
‑
pool)方案以组合方式组装包含多个区段的条形码序列,其中多个不同的第一区段被分布在多个第一分区中,然后内容物被合并并分布在多个第二分区中。
[0083]
如本文所述,条形码的使用可允许使用下一代测序技术对多个样品进行高通量分析。包含多个核酸分子的样品可以分布遍及多个分区(例如,乳液中的液滴),其中每个分区包含含有独特条形码序列的核酸条形码分子。样品可以被分区,使得多个分区中的所有或大部分分区包含多个核酸分子中的至少一个核酸分子。然后给定分区的核酸分子和核酸条形码分子可用于生成至少核酸分子序列的一个或多个拷贝和/或互补序列(例如,通过核酸扩增反应),其拷贝和/或互补序列包括核酸条形码分子的条形码序列或其互补序列。然后各种分区的内容物(例如,扩增产物或其衍生物)可以被合并并经历测序。在一些情况下,核酸条形码分子可以与珠偶联。在这种情况下,拷贝和/或互补序列也可以与珠偶联。核酸条形码分子和拷贝和/或互补序列可以从分区内的珠释放或在汇集之后释放,以便于使用测序仪器进行核酸测序。因为多个核酸分子中的核酸分子的拷贝和/或互补序列各自包括独特的条形码序列或其互补序列,所以使用核酸测序法获得的测序读数可以与它们对应的多个核酸分子中的核酸分子相关。该方法可以应用于包含在划分于多个分区之中的细胞内的核酸分子,和/或源自多个不同样品的核酸分子。
[0084]
如本文所用,术语“信号”、“信号序列”和“序列信号”通常是指与dna分子或dna的克隆群体相关的一系列信号(例如,荧光测量值),包括原始数据。可使用高通量测序技术(例如,流式sbs)获得此类信号。可对此类信号进行处理以获得估算序列(例如,在初步分析期间)。
[0085]
如本文所用,术语“序列”或“序列读取”通常是指在测序过程中进行的一系列核苷酸定位(assignment)(例如,通过碱基判定)。这样的序列可衍生自信号序列(例如,在初步分析期间)。
[0086]
如本文所用,术语“同聚物”通常是指包含相同单体单元的聚合物或聚合物部分,例如0,1,2,
…
,n个顺序的核苷酸的序列。例如,含有顺序的a核苷酸的同聚物可表示为a、aa、aaa,
…
,最多n个顺序的a核苷酸。同聚物可以具有同聚物序列。核酸同聚物可以指包含相同核苷酸或其任何核苷酸变体的连续重复的多核苷酸或寡核苷酸。例如,同聚物可以是聚(da)、聚(dt)、聚(dg)、聚(dc)、聚(ra)、聚(u)、聚(rg)或聚(rc)。同聚物可以是任何长度。例如,同聚物可具有至少2、3、4、5、10、20、30、40、50、100、200、300、400、500或更多个核酸碱基的长度。同聚物可具有10至500、或15至200、或20至150个核酸碱基。同聚物可具有至多500、400、300、200、100、50、40、30、20、10、5、4、3或2个核酸碱基的长度。分子,例如核酸分子,可以包括一个或多个同聚物部分和一个或多个非同聚物部分。分子可以完全由同聚物、多种同聚物或同聚物与非同聚物的组合形成。在核酸测序中,可以将多个核苷酸并入核酸链的同聚化区域中。此类核苷酸可以是非终止的,以允许掺入连续的核苷酸(例如,在单核苷酸流期间)。
[0087]
如本文所用,术语“hpn截短”通常是指处理一个或多个序列的集合的方法,使得具有大于或等于整数n的长度的一个或多个序列的集合中的每个同聚物被截短为长度n的同聚物。例如,序列“agggggt”到3个碱基的hpn截短可导致“agggt”的截短序列。
[0088]
如本文所用,术语“类似物比对”通常是指将信号序列与参考信号序列进行比对。
[0089]
术语“扩增”和“核酸扩增”可互换使用,并且如本文所用一般指核酸分子拷贝的产生。例如,dna的“扩增”通常是指生成一个或多个dna分子拷贝。扩增子可以是通过扩增程序从起始模板核酸分子产生的单链或双链核酸分子。这种扩增程序可以包括延伸或连接程序的一个或多个循环。扩增子可包含核酸链,其至少部分可与起始模板的至少部分基本相同或基本互补。当起始模板是双链核酸分子时,扩增子可以包含与一条链的至少部分基本相同并且与任一链的至少部分基本互补的核酸链。无论初始模板是单链还是双链,扩增子都可以是单链或双链的。核酸的扩增可以是线性的、指数的或其组合。扩增可以是基于乳液的或可以是基于非乳液的。核酸扩增方法的非限制性示例包括逆转录、引物延伸、聚合酶链反应(pcr)、连接酶链反应(lcr)、解旋酶依赖性扩增、不对称扩增、滚环扩增和多重置换扩增(mda)。扩增反应可以是例如聚合酶链反应(pcr),例如乳液聚合酶链反应(empcr;例如,在微反应器例如孔或液滴内进行的pcr)。在使用pcr的情况下,可以使用任何形式的pcr,非限制性示例包括实时pcr、等位基因特异性pcr、组装pcr、不对称pcr、数字pcr、乳液pcr、拨出(dial
‑
out)pcr、解旋酶依赖性pcr、巢式pcr、热启动pcr、反向pcr、甲基化特异性pcr、微型引物pcr、多重pcr、巢式pcr、重叠延伸pcr、热不对称交错pcr和降落式pcr。此外,扩增可以在包含参与或促进扩增的各种组分(例如,引物、模板、核苷酸、聚合酶、缓冲液组分、辅因子等)的反应混合物中进行。在一些情况下,反应混合物包含允许核苷酸的独立于背景的掺入的缓冲液。非限制性示例包括镁离子、锰离子和异柠檬酸缓冲液。tabor,s.等人,c.c.pnas,1989,86,4076
‑
4080和美国专利号5,409,811和5,674,716描述了此类缓冲液的其他示例,它们中的每一个通过引用整体并入本文。
[0090]
扩增可以是克隆扩增。如本文所用,术语“克隆”通常是指其成员的相当部分(例如,大于50%、60%、70%、80%、90%、95%或99%)具有基本相同的序列(例如,彼此具有至少约50%、60%、70%、80%、90%、95%或99%同一性的序列)的核酸群体。核酸分子克隆群体的成员可以彼此具有序列同源性。这样的成员可以与模板核酸分子具有序列同源性。在
一些情况下,这样的成员可与模板核酸分子(例如,如果是单链)的互补序列具有序列同源性。克隆群体的成员可以是双链或单链的。群体的成员可以不是100%同一或互补的,因为,例如,在合成过程中可能会发生“误差”,使得给定群体的少数可以与该群体的大多数不具有序列同源性。例如,群体成员的至少50%可以彼此或与参考核酸分子(即,用作序列比较基础的限定序列的分子)基本相同。群体成员的至少60%、至少70%、至少80%、至少90%、至少95%、至少99%或更多可以与参考核酸分子基本相同。如果两个分子之间的同一性百分比为至少60%、70%、75%、80%、85%、90%、95%、98%、99%、99.9%或更高,则可以认为这两个分子基本相同(或同源)。如果两个分子之间的百分比互补性为至少60%、70%、75%、80%、85%、90%、95%、98%、99%、99.9%或更高,则可以认为这两个分子基本互补。可能会发生非同源核酸的低水平或非实质性水平混合,因此克隆群体可以包含少数不同的核酸(例如,少于30%,例如,少于10%)。
[0091]
用于从单分子进行克隆扩增的有用方法包括滚环扩增(rca)(lizardi等人,nat.genet.19:225
‑
232(1998),其通过引用并入本文)、桥式pcr(adams和kron,method for performing amplification of nucleic acid with two primers bound to a single solid support,mosaic technologies,inc.(winter hill,mass.);whitehead institute for biomedical research,cambridge,mass.,(1997);adessi等人,nucl.acids res.28:e87(2000);pemov等人,nucl.acids res.33:e11(2005);或美国专利号5,641,658,其各自通过引用并入本文)、聚合酶克隆产生(mitra等人,proc.natl.acad.sci.usa 100:5926
‑
5931(2003);mitra等人,anal.biochem.320:55
‑
65(2003),其各自通过引用并入本文)以及使用乳剂(dressman等人,proc.natl.acad.sci.usa 100:8817
‑
8822(2003),其通过引用并入本文)或连接到基于珠的衔接子文库(brenner等人,nat.biotechnol.18:630
‑
634(2000);brenner等人,proc.natl.acad.sci.usa 97:1665
‑
1670(2000);reinartz等人,brief funct.genomic proteomic 1:95
‑
104(2002),其各自通过引用并入本文)在珠上的克隆扩增。克隆扩增提供的增强的信噪比超过了循环测序要求的缺点。
[0092]
如本文所用,术语“邻近序列依赖性”或“邻近序列依赖关系”通常是指与局部序列、相对核苷酸表示或基因组位点的信号相关性。给定序列的信号可因邻近序列依赖性而变化,邻近序列依赖性可取决于局部序列、序列的相对核苷酸表示或序列的基因组位点。
[0093]
流式合成测序(sbs)可以包括执行重复的dna延伸循环,其中核苷酸和/或标记的类似物的单个种类呈递给引物
‑
模板
‑
聚合酶复合物,然后在互补的情况下掺入该核苷酸。可针对模板的每个克隆群体(例如,珠或集落)测量每个流的产物。所得核苷酸掺入物可通过对应于或零、一、二、三、四、五、六、七、八、九、十或多于十个顺序的掺入物或者与之相关的明确区分性信号来检测和量化。对这样的多个顺序的掺入物的准确量化包括对每个流中的集落上掺入的0,1,2,
…
,n个顺序的核苷酸的每种可能的同聚物的特征性信号进行量化。例如,含有顺序的a核苷酸的同聚物可表示为a、aa、aaa,
…
,最多n个顺序的a核苷酸。同聚物长度的准确量化(例如,序列中顺序的相同核苷酸的数目)可能会由于信号水平的随机和不可预测的系统变化而遇到挑战,其可导致在同聚物长度量化中的错误。在一些情况下,仪器和检测系统可通过监测仪器诊断和大量集落之间的共模行为来校准和移除。同聚物长度的准确量化(例如,序列中顺序的相同核苷酸的数目)还可能由于对于每个序列可能不同的邻近序列依赖性信号而遇到挑战。例如,在稀释标记的核苷酸的荧光测量的情况下,邻近序列
既可影响标记类似物的数目(用于并入标记类似物的可变耐受性),也可影响单个标记类似物的荧光(例如,受
±
5碱基的局部邻近序列影响的染料的量子产率,如[kretschy,等人,sequence
‑
dependent fluorescence of cy3
‑
and cy5
‑
labeled double
‑
stranded dna,bioconjugate chem.,27(3),840
‑
848页]所述,其通过引用整体并入本文)。实际上,通过染料终止剂桑格循环测序,已经鉴别出对于3碱基邻近序列的信号的实质性系统变化(例如,如[zakeri,等人,peak height pattern in dichloro
‑
rhodamine and energy transfer dye terminator sequencing,biotechniques,25(3),406
‑
10页]所述,其通过引用整体并入本文)。
[0094]
一般而言,本文使用的命名法和本公开的方法和系统中使用的实验室程序可包括分子学、生物化学、微生物学和重组dna技术。此类技术的细节可以见于例如"molecular cloning:a laboratory manual"sambrook等人,(1989);"current protocols in molecular biology"volumes i
‑
iii ausubel,r.m.,编著(1994);ausubel等人,"current protocols in molecular biology",john wiley and sons,baltimore,maryland(1989);perbal,"a practical guide to molecular cloning",john wiley&sons,new york(1988);watson等人,"recombinant dna",scientific american books,new york;birren等人(编著)"genome analysis:a laboratory manual series",vols.1
‑
4,cold spring harbor laboratory press,new york(1998);美国专利号4,666,828、4,683,202、4,801,531、5,192,659和5,272,057中所述的方法论;"cell biology:alaboratory handbook",volumes i
‑
iii cellis,j.e.,编著(1994);freshney的"culture of animal cells
‑
a manual of basic technique",wiley
‑
liss,n.y.(1994),第三版;"current protocols in immunology"volumes i
‑
iii coligan j.e.,编著(1994);stites等人(eds),"basic and clinical immunology"(第八版),appleton&lange,norwalk,ct(1994);mishell和shiigi(编著),"strategies for protein purification and characterization
‑
a laboratory course manual"cshl press(1996),所有这些都通过引用并入本文。
[0095]
如本文中所使用的,术语“可信信号”或“可信测序信号”通常是指为理想信号的测序信号,其是无误差的或至少是足够准确到可信的信号。可以以各种方式确定准确度水平。在一些情况下,可信信号可以是满足准确度水平的预定阈值的信号。可信测序信号可用作生成训练集或训练算法(例如,分类器,如机器学习分类器)的参考。例如,可信测序信号可对应于已知核苷酸序列(例如,已知碱基的序列),使得可信测序信号集和已知核苷酸序列集可用于构建训练集。
[0096]
本公开可以参考(为便于解释)大肠杆菌基因组、人类基因组、神经网络和鸟枪法测序。这些分别是不同大小的基因组、机器学习过程和某种类型的测序的示例。
[0097]
检测器可以输出受不准确度和噪声影响的实际人类片段测序信号。由于其随机性,这些不准确度和噪声可能很难或不可能提前被分析计算。本公开提供了方法和系统,其应用机器学习来帮助在包括实际人类片段测序信号(其可以是有噪声且不准确的)的输入数据集与包括准确人类片段测序信号的输出数据集之间生成映射或分类。准确人类片段测序信号可以进一步被处理—例如,与准确人类基因组比对,以用于下游应用,例如诊断和其他精准健康应用。
[0098]
人类基因组的长度超过三十亿个碱基对。因此,人类基因组的大小可能会给仅使
用人类基因组来生成实际人类片段测序信号集(可以是有噪声且不准确的)与准确人类片段测序信号集之间的直接映射带来挑战或困难。
[0099]
本公开提供了将机器学习过程应用于小得多的基因组的方法和系统—例如应用在约几千个基因长的大肠杆菌基因组上—以便提供实际人类片段测序信号集与准确人类片段测序信号集之间的此类直接映射。尽管大肠杆菌基因组不同于人类基因组,但它可以在包括以下一项或多项的多阶段过程中使用:(a)获得包括在实际参考测序信号与可信参考测序信号之间的第一映射(例如,分类或回归)的第一经训练的算法(例如,机器学习过程);(b)获得对应于第二基因组的实际测序信号;以及(c)生成用于训练包括在对应于第二基因组的实际测序信号与对应于第二基因组的可信测序信号之间的第二映射(例如,分类或回归)的第二经训练的算法(例如,机器学习过程)的训练集。在一些实施方案中,实际参考测序信号和可信参考测序信号代表与第二属的第二基因组不同的第一属的参考基因组的部分。在一些实施方案中,参考基因组小于第二基因组。在一些实施方案中,训练集是基于第一映射采用对应于第二基因组的实际测序信号生成的。
[0100]
该多阶段过程使用一个或多个具有合理复杂性和成本的机器学习过程生成第二映射。
[0101]
应当理解,虽然本公开是相对于将例如人类基因组和大肠杆菌基因组用各种训练算法关联和/或映射来说明的,但本公开的方法和系统可以适用于任何两个基因组,例如其中一个基因组比另一个基因组更大和/或更复杂。例如,可以接收或生成非人类样品的实际测序信号。
[0102]
本公开提供了基于与具有小于人类基因组的基因组的属对应的第一映射来生成第二映射的系统、方法和计算机可读介质。第二映射可用于处理实际人类片段测序信号以产生准确人类片段测序信号,其可与参考人类基因组比对以提供受试者基因组的评估。
[0103]
该方法可以包括获得或生成第一经训练的算法,该算法包括参考实际测序信号与参考可信测序信号之间(例如,实际大肠杆菌片段测序信号与准确大肠杆菌片段测序信号之间)的第一映射。可以使用机器学习过程训练被配置为应用第二映射的第二经训练的算法。
[0104]
机器学习过程可以包括(i)使用经训练以应用第一映射处理实际大肠杆菌片段测序信号的第一经训练的算法(例如,第一神经网络)以产生准确大肠杆菌片段测序信号,以及(ii)使用经训练以应用第二映射处理实际人类片段测序信号的第二经训练的算法(例如,第二神经网络)以生成准确人类片段测序信号。然后准确人类片段测序信号可以与参考人类基因组进行比对(例如,用于进一步的基因组分析)。
[0105]
第一经训练的算法可以生成训练集(例如训练数据集),该训练集可以用于训练第二经训练的算法(例如第二神经网络)以应用在对应于人类基因组的实际测序信号与准确测序信号之间(例如,实际人类片段测序信号与准确人类片段测序信号之间)的第二映射。
[0106]
系统、方法和计算机可读介质在存储器和/或计算资源方面可以是高效的,因为它们被配置为在比人类基因组小得多的大肠杆菌基因组上应用机器学习算法。因此,这样的系统、方法和计算机可读介质可以有利地以更高的准确度和效率执行序列判定(sequence calling)或碱基判定,同时使用更少的存储器和/或计算资源。
[0107]
图1示出了用于训练神经网络的方法100的示例,该神经网络被配置为应用大肠杆
菌的实际片段测序信号与大肠杆菌的可信片段测序信号之间的第一映射。在一些实施方案中,方法100可以包括操作110、112、120、122、124、130、134和136中的一个或多个。
[0108]
方法100可以包括接收对应于不同于人类基因组的属或种的基因组(例如,大肠杆菌基因组)(如在操作110中)。例如,大肠杆菌基因组可包含约460万个碱基对,这明显小于人类基因组(可包含约30亿个碱基对)。使用较小的基因组可以有利于降低计算复杂性(从而以较少的计算资源实现更快的运行时间),该复杂性可以与基因组的大小成线性比例。
[0109]
接下来,方法100可以包括模拟检测器(例如,特别是模拟检测器对大肠杆菌基因组的响应)—假设为基本无误差的过程(如在操作112中)。
[0110]
方法100可以包括模拟由检测器执行的化学和/或光学过程(如在操作112中)。操作112的结果可以是大肠杆菌密钥(115),其包括可预期从检测器(在基本无误差的检测过程下)获得的针对整个大肠杆菌基因组的可信测序信号。大肠杆菌密钥115可以包括针对整个大肠杆菌基因组的a、c、t、g元件的强度值。
[0111]
接下来,方法100可以包括使用检测器处理一组大肠杆菌样品片段(如在操作120中)。
[0112]
接下来,方法100可以包括获得针对每个区段的实际片段测序信号(如在操作122中)。
[0113]
接下来,方法100可以包括选择新的片段组(如在操作124中)并行进到操作120。可以重复或迭代操作120、122和124的集合,直到接收到针对整个大肠杆菌基因组的实际片段测序信号,或直到接收到充分量的实际片段测序信号。
[0114]
在一些实施方案中,操作122可以包括(或可以随后是)拒绝可能有缺陷的实际片段测序信号。
[0115]
例如,虽然无噪声片段测序信号可被预期代表整数个同聚物,但实际片段测序信号可提供非整数个同聚物。与预期的整数个同聚物的偏差可以表示实际片段测序信号中的误差,并且一旦误差超过预定阈值,则实际片段测序信号可以被忽略并且不能在后续操作例如操作130和136中被处理。可以以各种方式计算误差,例如均方误差等。可以以任何方式设置预定阈值。
[0116]
接下来,方法100可以包括将实际片段测序信号与大肠杆菌密钥115比对(如在操作130中)。操作130可以包括将实际片段测序信号与整个大肠杆菌密钥关联,以在大肠杆菌密钥中找到最佳匹配的可信片段测序信号的位置。
[0117]
接下来,方法100可以包括选择一组新的片段(如在操作134中)并行进到操作130。可以重复或迭代操作130和134的集合,直到对于实际片段测序信号中的每一个,在大肠杆菌密钥中找到最佳匹配的可信片段测序信号。在一些情况下,基本上所有的实际片段测序信号都可以与可信片段测序信号相匹配。在一些情况下,所有实际片段测序信号都可以与可信片段测序信号相匹配。在一些情况下,实际片段测序信号集中的任何百分比,例如至少10%、20%、30%、40%、50%、60%、70%、80%、90%、95%、96%、97%、98%、99%或更多可以与可信片段测序信号相匹配。
[0118]
在一些实施方案中,实际片段测序信号与大肠杆菌密钥中(针对实际片段测序信号)最匹配的可信片段测序信号的配对、或阵列或配对可以形成第一训练集。
[0119]
接下来,方法100可以包括使用包括{大肠杆菌的实际片段测序信号和大肠杆菌的
可信片段测序信号}配对的第一训练集来训练神经网络,以执行大肠杆菌的实际片段测序信号与大肠杆菌的可信片段测序信号之间的第一映射(例如,分类或回归)(如在操作136中)。
[0120]
图2示出了使用神经网络(经训练以应用第一映射)生成第二训练集的方法200的示例,该第二训练集可用于将某个人的实际片段测序信号映射到参考人类基因组的可信片段测序信号。
[0121]
方法200可以包括使用检测器处理一组人类dna片段(如在操作210中)。例如,操作210可以包括使用已知变体的已知人类dna并且忽略变体或补偿变体。
[0122]
接下来,方法200可以包括获得针对每个区段的实际片段测序信号(如在操作212中)。这些实际片段测序信号可以是检测器的输出。
[0123]
接下来,方法200可以包括选择新的片段组(如在操作214中)并行进到操作210。可以重复或迭代操作210、212和214的集合,直到接收到针对整个人类基因组的实际片段测序信号,或直到接收到充分量的实际片段测序信号。
[0124]
在一些实施方案中,操作212可以包括(或可以随后是)拒绝可能有缺陷的实际片段测序信号。例如,虽然无噪声片段测序信号可预期代表整数个同聚物,但实际片段测序信号可提供非整数个同聚物。与预期的整数个同聚物的偏差可以指示实际片段测序信号中的误差,并且一旦误差超过预定阈值,实际片段测序信号可以被忽略并且不能在操作218和220中被处理。可以以各种方式计算误差,例如均方误差等。可以以任何方式设置预定阈值。
[0125]
接下来,方法200可以包括使用经训练以输出第一映射的神经网络来处理每个片段的实际片段测序信号,从而提供第一映射测序信号(如在操作218中)。
[0126]
接下来,方法200可以包括将第一映射测序信号与参考人类基因组比对,以确定最匹配第一映射测序信号的可信片段测序信号(如在操作220中)。这些可信片段测序信号可视为与实际片段测序信号最匹配。方法200可以包括针对操作212中提供的每个实际片段测序信号重复操作218和220。在一些情况下,基本上所有的第一映射测序信号可以与可信片段测序信号相匹配。在一些情况下,所有第一映射片段测序信号可与可信片段测序信号相匹配。在一些情况下,第一映射片段测序信号集中的任何百分比,例如至少10%、20%、30%、40%、50%、60%、70%、80%、90%、95%、96%、97%、98%、99%或更多可以与可信片段测序信号相匹配。
[0127]
接下来,方法200可以包括生成“人类”训练集(如在操作230中),该训练集包括对应于人类基因组的{实际片段测序信号和可信片段测序信号}配对。
[0128]
接下来,方法200可以包括使用“人类”训练集训练神经网络(如在操作232中)。在训练之后,神经网络被配置为应用对应于人类基因组的实际片段测序信号和对应于人类基因组的可信片段测序信号之间的第二映射(例如分类或回归)。
[0129]
使用本公开的系统、方法和介质,当使用截短的实际人类测序信号和截短的可信参考测序信号时,可以提供更稳健的方法。将这些信号截短成例如单比特实际人类测序信号和单比特可信参考测序,可以提供对测量误差具有鲁棒性的方法,同时使在比对过程中为每个散列值寻找更多候选物的成本可容忍。
[0130]
在方法100和200完成之后,可以生成受试者基因组的评估。
[0131]
图3示出了用于评估受试者的基因组的方法300的示例。
[0132]
方法300可以包括使用检测器处理受试者的人类dna的一组片段。
[0133]
接下来,方法300可以包括获得针对每个区段的实际片段测序信号(如在操作312中)。
[0134]
在一些实施方案中,操作312可以包括(或可以随后是)将置信水平分配给实际片段测序信号。例如,虽然无噪声片段测序信号可预期代表整数个同聚物,但实际片段测序信号可提供非整数个同聚物。与预期的整数个同聚物的偏差可以表示实际片段测序信号中的误差,该误差可以影响分配给实际片段测序信号的置信水平。
[0135]
接下来,方法300可以包括选择新的片段组(如在操作314中)并行进到操作310。可以重复或迭代操作310、312和314的集合,直到接收到针对受试者的整个基因组的实际片段测序信号,或直到接收到充分量的实际片段测序信号。
[0136]
方法300可以包括针对操作312中提供的每个实际片段测序信号重复操作320和322。
[0137]
在一些实施方案中,操作320可以包括使用神经网络处理实际片段测序信号,该神经网络被使用“人类”训练集训练以提供第二映射测序信号。
[0138]
接下来,方法300可以包括将第二映射片段测序信号与人工密钥比对(如在操作322中)。例如,比对可以是基于散列的。
[0139]
接下来,操作322的一次或多次迭代之后可以是提供受试者的基因组的评估(如在操作324中)。
[0140]
图4示出了用于基于散列的比对的方法400的示例(例如,根据操作322)。
[0141]
方法400可以包括将实际片段测序信号412划分成较小的部分重叠部分414,以便简化操作322的执行。例如,约一百个值的实际片段测序信号412可以划分为每个约二十个值的部分。
[0142]
接下来,方法400可以包括对每个部分应用散列函数(416)以提供散列值418。
[0143]
在一些实施方案中,散列值418用作对应于参考人类基因组420的散列表的索引。
[0144]
由某个散列值访问的散列表420的条目可以将候选物(具有相同散列值)的位置存储在数据结构中,该数据结构存储在处理参考人类基因组时通过模拟检测器的输出而生成的参考数据库(430)。模拟可以假设为基本无误差的过程。
[0145]
接下来,方法400可以包括使用散列值418访问条目422,该条目存储参考数据库430中的候选物的位置(432)。
[0146]
在一些实施方案中,不同的参考与参考人类基因组中的不同位置相关。为了选择选定的候选物,确定实际片段测序信号(412)与位于每个不同位置的参考的部分(430)之间的相关性(434)。该选择可以包括选择具有最高相关性的位置。
[0147]
图5示出了可以在方法100和/或方法200期间训练的神经网络500的示例,其可以用于执行方法300。
[0148]
神经网络可以包括输入层510、多个中间层520和输出层530。
[0149]
在一些实施方案中,神经网络500是回归网络,例如全连接回归网络。
[0150]
输入层可以包括每个实际片段测序信号的一个神经元。例如,如果输入层由一百个值的实际片段测序信号馈送,则输入层510可以包括一百个神经元。类似的示例可以适用于输出层。每个中间层可以比输入层大得多。例如,中间层可以比输入层大约1.5x、2x、3x、
4x、5x、6x、7x、8x、9x、10x或超过10x。可以使用其他比率。
[0151]
图6示出了用于生成训练集的方法600的示例。
[0152]
方法600可以包括使用第一经训练的算法(例如,机器学习过程)生成实际参考测序信号到可信参考测序信号之间的第一映射(例如,分类或回归)。实际参考测序信号和可信参考测序信号可以代表第一属(例如,人类基因组)的参考基因组的部分。
[0153]
接下来,方法600可以包括将方法100的操作应用于可不同于大肠杆菌的第一属的第一基因组(例如,人类基因组)。
[0154]
接下来,方法600可以包括接收或生成对应于第二属的第二基因组的实际测序信号(如在操作620中)。第一个属可以与第二属不同。第一基因组可以比第二基因组小例如至少约2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、15倍、20倍、25倍、30倍、35倍、40倍、45倍、50倍、55倍、60倍、65倍、70倍、75倍、80倍、85倍、90倍、95倍、100倍、200倍、300倍、400倍、500倍、600倍、700倍、800倍、900倍、1000倍、2000倍、3000倍、4000倍、5000倍、6000倍、7000倍、8000倍、9000倍或10000倍。可以应用其他倍数。
[0155]
接下来,方法600可以包括生成用于训练第二经训练的算法(例如,机器学习过程)的第二基因组训练集,以提供对应于第二基因组的实际测序信号到对应于第二基因组的可信测序信号之间的第二映射(例如,分类或回归)(如在操作630中)。
[0156]
操作630可以基于第一映射执行,并且可以包括使用第二经训练的算法(例如,机器学习过程)以处理对应于第二基因组的实际测序信号。
[0157]
操作630可以将方法200的操作应用于可不用于人类(例如,大肠杆菌)的第二属的第二基因组。
[0158]
操作630之后可以是使用第二基因组训练集来训练经训练的算法(例如,机器学习过程)。
[0159]
在一些实施方案中,第一经训练的算法(例如,机器学习过程)可以不同于第二经训练的算法(例如,机器学习过程)或可以与第二经训练的算法(例如,机器学习过程)相同。
[0160]
图7示出了用于评估第二属的某个实体(例如,受试者)的基因组的方法700的示例。评估可以基于第一属进行,并且方法700可以被称为用于基于第一属评估第二属的基因组的方法。
[0161]
方法700可以包括针对在第二属的基因组的多个部分中的第二属的某个实体(例如,受试者)的基因组的每个部分执行操作710和720。方法700可以包括执行操作710和720的集合的一个或多个重复或迭代以提供第二属的某个实体(例如,受试者)的基因组的评估。
[0162]
操作710可以包括接收或生成代表第二属的基因组的部分的实际测序信号。
[0163]
操作720可以包括评估第二属的某个实体(例如,受试者)的基因组的部分。
[0164]
操作720可以包括将第二经训练的算法(例如,机器学习过程)应用于实际测序信号。第二经训练的算法(例如,机器学习过程)可被训练以提供在对应于第二基因组的实际测序信号与对应于第二基因组的可信测序信号之间的第二映射(例如,分类或回归)。可以基于在实际参考测序信号与可信参考测序信号之间的第一映射来生成第二映射。实际参考测序信号和可信参考测序信号可以代表与第二属的第二基因组不同的第一属的参考基因组的部分。参考基因组可以小于第二基因组。
[0165]
操作710和720可以包括将方法300的操作应用于可以不同于人类的第二属,其中第一映射可以涉及除大肠杆菌之外的第一属。经训练的算法
1.在处理生物样品以生成核酸的测序信号之后,可以使用经训练的算法处理测序信号以执行测序判定(例如,基于序列信号确定碱基判定)。例如,经训练的算法可用于确定核酸的多个核苷酸位置中每处的序列信号的定量测量。经训练的算法可以被配置为以至少约50%、至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%、至少约95%、至少约96%、至少约97%、至少约98%、至少约99%、或超过99%的准确度确定序列信号的定量测量。
2.经训练的算法可以包括有监督机器学习算法。经训练的算法可以包括分类和回归树(cart)算法。有监督机器学习算法可以包括例如随机森林、支持向量机(svm)、神经网络或深度学习算法。经训练的算法可以包括无监督机器学习算法。
3.经训练的算法可以被配置为接受多个输入变量并且基于多个输入变量产生一个或多个输出值。多个输入变量可以基于处理核酸的测序信号生成。例如,输入变量可以包括与参考基因组或参考基因组的基因组位点对应或比对的多个序列。作为另一个示例,输入变量可以包括由测序仪产生的测序信号的模拟值。
4.经训练的算法可以包括分类器,使得一个或多个输出值中的每一个包括固定数目的可能值(例如,线性分类器、逻辑回归分类器等)中的一个,其指示该分类器对测序信号的分类。经训练的算法可以包括二元分类器,使得一个或多个输出值中的每一个包括两个值(例如,{0、1}、{阳性、阴性}或{存在、不存在})中的一个,其指示该分类器对测序信号的分类。经训练的算法可以是另一种类型的分类器,使得一个或多个输出值中的每一个包括多于两个值(例如,{0、1、2}、{阳性、阴性或不确定}、{存在、不存在或不确定}、{a、c、g、t}或{a、c、g、u})中的一个,其指示该分类器对测序信号的分类。输出值可以包括描述性标记、数值或其组合。一些输出值可以包括描述性标记。这种描述性标记可以提供序列信号的碱基判定的标识,并且可以包括例如{a、c、g、t}或{a、c、g、u}。这种描述性标记可以提供碱基判定的邻近序列指示,或碱基判定的置信度或准确度。作为另一个示例,这种描述性标记可以提供被判定为测序信号的不同碱基的可能性的相对评估。一些描述性标记可以映射到数值,例如,通过将“阳性”或“存在”映射为1并将“阴性”或“不存在”映射为0。
5.一些输出值可以包括数值,例如二进制、整数值或连续值。这样的二进制输出值可以包括例如{0、1}、{阳性、阴性}或{存在、不存在}。这种整数输出值可以包括例如{0、1、2}。这样的连续输出值可以包括例如至少0且不超过1的概率值(例如,指示测序信号的碱基判定的可能性)。这样的连续输出值可以包括例如至少为0的非归一化概率值。一些数值可以映射到描述性标记,例如,通过将1映射到“阳性”或“存在”并将0映射到“阴性”或“不存在”。
6.一些输出值可以基于一个或多个截止值进行分配。例如,如果特定核苷酸位置处的测序信号有至少50%的概率被判定为给定碱基(例如,a、c、g、t或u),则测序信号的二进制分类可以分配“阳性”或1的输出值。例如,如果特定核苷酸位置处的测序信号有至少50%的概率被判定为给定碱基(例如,a、c、g、t或u),则样品的二进制分类可以分配“阴性”或0的输出值。在这种情况下,使用50%的单个截止值将测序信号的碱基分类为两个可能的二进制输出值之一。单个截止值的示例可包括约1%、约2%、约5%、约10%、约15%、约20%、约
25%、约30%、约35%、约40%、约45%、约50%、约55%、约60%、约65%、约70%、约75%、约80%、约85%、约90%、约91%、约92%、约93%、约94%、约95%、约96%、约97%、约98%和约99%。
7.作为另一个示例,如果特定核苷酸位置处的测序信号有至少约50%、至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%、至少约99%或更多的概率被判定为给定碱基(例如,a、c、g、t或u),则测序信号的分类可以分配“阳性”或1的输出值。如果特定核苷酸位置的测序信号有超过约50%、超过约55%、超过约60%、超过约65%、超过约70%、超过约75%、超过约80%、超过约85%、超过约90%、超过约91%、超过约92%、超过约93%、超过约94%、超过约95%、超过约96%、超过约97%、超过约98%,或超过约99%的概率被判定为给定碱基(例如,a、c、g、t或u),则测序信号的分类可以分配“阳性”或1的输出值。
8.如果特定核苷酸位置处的测序信号有小于约50%、小于约45%、小于约40%、小于约35%、小于约30%、小于约25%、小于约20%、小于约15%、小于约10%、小于约9%、小于约8%、小于约7%、小于约6%、小于约5%、小于约4%、小于约3%、小于约2%或小于约1%的概率被判定为给定碱基(例如,a、c、g、t或u),则测序信号的分类可以分配“阴性”或0的输出值。如果特定核苷酸位置的测序信号有不超过约50%、不超过约45%、不超过约40%、不超过约35%、不超过约30%、不超过约25%、不超过约20%、不超过约15%、不超过约10%、不超过约9%、不超过约8%、不超过约7%、不超过约6%、不超过约5%、不超过约4%、不超过约3%、不超过约2%、或不超过约1%的概率被判定为给定碱基(例如,a、c、g、t或u),则测序信号的分类可以分配“阴性”或0的输出值。
9.如果样品未被分类为“阳性”、“阴性”、1或0,则测序信号的分类可以分配“不确定”或2的输出值。在这种情况下,使用两个截止值的集合将测序信号分类为三个可能的输出值之一。截止值集合的示例可以包括{1%、99%}、{2%、98%}、{5%、95%}、{10%、90%}、{15%、85%}、{20%、80%}、{25%、75%}、{30%、70%}、{35%、65%}、{40%、60%}和{45%、55%}。类似地,n个截止值的集合可用于将测序信号分类为n 1个可能的输出值之一,其中n是任意正整数。
10.经训练的算法可以用多个独立的训练样品进行训练。每个独立的训练样品可包含从核酸(例如,来自受试者的生物样品)生成的测序信号集和对应于测序信号的一个或多个已知输出值(例如,对应于测序信号的一组碱基判定或核苷酸序列)。独立的训练样品可以从多个不同的受试者获得或来源于多个不同的受试者。独立的训练样品可以包含从核酸(例如,来自受试者的生物样品)生成的测序信号集和对应于在多个不同时间点(例如,定期,例如每周、每两周或每月)从同一受试者获得的测序信号的一个或多个已知输出值(例如,对应于测序信号的一组碱基判定或核苷酸序列)。
11.可以用至少约5、至少约10、至少约15、至少约20、至少约25、至少约30、至少约35、至少约40、至少约45、至少约50、至少约100、至少约150、至少约200、至少约250、至少约300、至少约350、至少约400、至少约450、或至少约500个独立训练样品来训练经训练的算法。可以用不超过约500、不超过约450、不超过约400、不超过约350、不超过约300、不超过约250、不超过约200、不超过约150、不超过约100或不超过约50个独立训练样品来训练经训练的算
法。
12.经训练的算法可以被配置为以至少约50%、至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约81%、至少约82%、至少约83%、至少约84%、至少约85%、至少约86%、至少约87%、至少约88%、至少约89%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%、至少约99%或至少约99%的准确度鉴别测序信号的碱基判定。通过经训练的算法鉴别测序信号的碱基判定的准确度可以被计算为正确鉴别或分类(例如,特定碱基的存在或不存在)的碱基判定的百分比。
13.经训练的算法可以被配置为以至少约5%、至少约10%、至少约15%、至少约20%、至少约25%、至少约30%、至少约35%、至少约40%、至少约50%、至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约81%、至少约82%、至少约83%、至少约84%、至少约85%、至少约86%、至少约87%、至少约88%、至少约89%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%、至少约99%或更高的阳性预测值(ppv)鉴别测序信号的碱基判定。使用经训练的算法鉴别测序信号的碱基判定的ppv可以被计算为对应于真正存在的碱基的被鉴别或分类为存在的碱基判定的百分比。
14.经训练的算法可以被配置为以至少约5%、至少约10%、至少约15%、至少约20%、至少约25%、至少约30%、至少约35%、至少约40%、至少约50%、至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约81%、至少约82%、至少约83%、至少约84%、至少约85%、至少约86%、至少约87%、至少约88%、至少约89%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%、至少约99%或更高的阴性预测值(npv)鉴别测序信号的碱基判定。使用经训练的算法鉴别测序信号的碱基判定的npv可以被计算为对应于真正不存在(例如未存在)的碱基的被鉴别或分类为不存在的碱基判定的百分比。
15.可以调整或调准经训练的算法以改进鉴别测序信号的碱基判定的性能、准确度、ppv或npv中的一个或多个。可以通过调整经训练的算法的参数(例如,如本文别处所述的用于鉴别测序信号的碱基判定的截止值集,或神经网络的权重)来调整或调准经训练的算法。经训练的算法可以在训练过程中或在训练过程完成后不断地进行调整或调准。
16.在最初训练经训练的算法之后,输入的子集可以被鉴别为最有影响力或最重要的,以被包括用于进行高质量分类。多个输入变量或其子集可以基于分类度量来排序,该分类度量指示每个输入变量对进行高质量分类或测序信号的碱基判定的鉴别的重要性。在一些情况下,此类度量可用于显著降低可用于将经训练的算法训练到所需性能水平(例如,基于所需最小准确度、ppv、或npv或其组合)的输入变量(例如预测变量)的数目。例如,如果在经训练的算法中有多个、包括几十个或数百个的输入变量的情况下训练经训练的算法导致超过99%的分类准确度,则替代地仅使用所述多个中的不超过约5、不超过约10、不超过约15、不超过约20、不超过约25、不超过约30、不超过约35、不超过约40、不超过约45、不超过约50或不超过约100个这种最有影响力或最重要的输入变量的选定子集来练经训练的算法可产生降低但仍可接受的分类准确度(例如,至少约50%、至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约81%、至少约82%、至少约83%、至少约
84%、至少约85%、至少约86%、至少约87%、至少约88%、至少约89%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%或至少约99%)。可以通过对全部多个输入变量进行排序并选择预定数目(例如,不超过约5个、不超过约10个、不超过约15个、不超过约20个、不超过约25个、不超过约30个、不超过约35个、不超过约40个、不超过约45个、不超过约50个或不超过约100个)的具有最佳分类度量的输入变量来选择该子集。
[0166]
本公开提供了被编程用于实现本公开的方法的计算机系统。在一些实施方案中,用于实现方法100和/或方法200的神经网络可以是u
‑
net。
[0167]
u
‑
net是一种卷积神经网络,在德国弗莱堡大学计算机科学系开发用于生物医学图像分割。该网络可以基于全卷积神经网络,其架构经过修改和扩展,以使用更少的训练图像并产生更精确的分割。例如,可以在现代gpu上使用u
‑
net在不到一秒的时间内执行512
×
512图像的分割。
[0168]
u
‑
net可以是两种深度学习方法的组合:卷积神经网络(cnn)和编码器
‑
解码器。cnn可以配置为在网络中使用相对少量的权重处理大输入图像。这是可能的,因为输入图像通常是位置不变的—在输入图像的一个部分中操作的过滤器与输入图像的其他部分中的过滤器相同。因此,cnn在输入图像的所有部分应用相同的过滤器,从而允许使用合理数目的参数进行优化,并实现在合理的时间内用可管理的样品数目进行机器学习过程。编码器
‑
解码器是一种用于在机器学习过程中执行降维的方法。它可以包括具有将所有输入变量映射到少量权重,并将权重解码回输入图像的网络。这种技术能够实现以少量参数使用来自整个输入图像的信息。
[0169]
u
‑
net可以并行使用cnn和编码器
‑
解码器技术,从而允许在输入图像中重复重用相同的过滤器并考虑到图像的大规模效应。
[0170]
本公开的方法、系统和介质可以通过利用一些并行元件以与用于语义分割的方式类似的方式执行实际人类片段测序信号的处理。
[0171]
在一些实施方案中,实际人类片段测序信号可被视为一维(1d)图像。输入图像和实际人类片段测序信号都可以表现出具有大部分信息流动不变的特性—因为实际人类片段测序信号的序列判定或碱基判定可以包括实际人类片段测序信号的值的分析以及对实际人类片段测序信号的邻近周围值的分析。然而,实际人类片段测序信号的处理也可以使用来自整个读数的信息,因此使用网络的编码器部分可以是有益的。
[0172]
u
‑
net可以由各种类型的信息馈送。不同类型的信息可以看作是不同的信息通道。例如,不同的信息类型可以包括实际人类片段测序信号并且还可以包括一种或多种其他附加类型的信息。作为示例,附加类型的信息可以包括测光背景噪声的计算,这被发现是有益的信息。
[0173]
作为另一示例,附加类型的信息可以包括从前导码获得的测序信号。前导码可以附加到测试的人类基因组片段上,并且可以是预先已知的。可以预期从前导码获得的测序信号对于所有读数基本上相同。从前导码获得的测序信号的强度可以指示珠中的链数目的近似值。该强度可用于对从前导码获得的测序信号进行归一化。作为另一示例,附加类型的信息可以包括对应于读数附近的本地信息。例如,本地信息可以用图块(tile)表示读数,例如每个流的读数。支持样品的基底可以虚拟地分割成
图块(例如,数十直到数千个图块),并且本地信息可以反映对应于给定图块的读数。例如,读数可以被计算为测光图像图块中所有珠和每个流的平均信号。可以使用其他函数(例如加权和、线性或非线性函数)。该本地信息可用于补偿基底上的不均匀性(例如,与另一图块相比,某些图块可以被用更强的辐射照射)。
[0174]
作为另一示例,附加类型的信息可以包括指示流基准(在流期间使用的基准)和/或流位置的信息。这种附加信息可以包括流基准合成整数向量和流位置合成整数向量。可以提供第四附加类型信息的任何其他表示。
[0175]
本公开的系统、方法和介质的u
‑
net可以是例如并行连接到编码器
‑
解码器的6层cnn模型。模型可以包含约1000、5000、10000、50000、100000、200000、300000、400000、500000、600000、700000、800000、900000、100万或超过100万个参数数目。进一步地,可以使用约100万、500万、1000万、1500万、2000万、2500万、3000万、3500万、4000万、4500万、5000万、5500万、6000万、6500万、7000万、7500万、8000万、8500万、9000万、9500万、1亿、1.5亿、2亿、2.5亿、3亿、3.5亿、4亿、4.5亿、5亿、6亿、7亿、8亿、9亿或10亿个读数来训练模型。可以通过比对来创建表示基本事实(ground truth)的读数,并且可以基于比对的高置信度来选择训练中使用的读数。具有可疑方差的读数和其中信息在序列结束之前结束的读数可以从训练中丢弃。
[0176]
图8示出了经训练以评估第二属的某个实体的基因组的u
‑
net900的示例。可以根据方法100和/或200的一个或多个操作训练和/或使用u
‑
net 900。可以用输入901(其可以包括实际人类片段测序信号和可选的一个或多个其他附加类型的信息)和输出902(其可以包括例如准确人类片段测序信号)馈送u
‑
net。
[0177]
u
‑
net 900包括第一到第四下卷积单元(“downconv”)921、923、925和927,第一到第三最大池单元922、924和926,第一到第三上采样单元934、931和928,第一至第三连接单元935、932和929,以及第一至第三上卷积单元933、933和930。
[0178]
图10和图11示出了馈送到神经网络的输入信号和由神经网络生成的输出的示例。输入信号包括代表每个同聚物测得的核苷酸数目的实际测序信号(例如,具有不准确度和噪声),并且输出信号包括代表每个同聚物评估的核苷酸数目的无噪声(或噪声降低的)信号。
[0179]
图10示出了图示输入信号1001和输出信号1002的图1000的示例。输出信号1002集中在每个同聚物约0、1、2和3个核苷酸附近,而输入数1001覆盖更大范围的值。
[0180]
图11示出了输入信号直方图1010和输出信号直方图1020的示例。
[0181]
输入值的第一分布1011由神经网络映射到值0附近的第一窄分布1021(其可以为近似δ函数)。
[0182]
输入值的第二分布1012由神经网络映射到值1附近的第二窄分布1022(其可以为近似δ函数)。
[0183]
输入值的第三分布1013由神经网络映射到值2附近的第三窄分布1023(其可以为近似δ函数)。
[0184]
输入值的第四分布1014由神经网络映射到值3附近的第四窄分布1024(其可以为近似δ函数)。
[0185]
在一些实施方案中,计算机系统可用于随着时间的推移执行本公开的方法的操作
并生成一种或多种生物体的基因组的一种或多种评估。
[0186]
在一些实施方案中,机械条件、检查条件、收集条件和化学条件中的至少一个可以随时间改变,从而导致曾经准确的一个或多个模型变得不准确。因此,可以根据需要随时间替换、调整或修正模型。例如,修正可以包括最初使用在计算机系统的初始设置时产生的初始模型。可以使用本文公开的任何方法来生成初始模型。
[0187]
在一些实施方案中,初始模型随着时间被修正和/或替换。例如,可以通过使用新的实际测序信号再训练经训练的算法(例如,机器学习过程)来修正或替换初始模型。新的实际测序信号可包括在一个或多个完成的评估期间获取的信息或先前未处理的信息。
[0188]
在一些实施方案中,响应于某些事件、在运行每个评估之后和/或在运行多个(n)个评估之后,模型替换或修正以周期性方式发生。在其他情况下,可以根据手动校准程序触发模型替换或更改。
[0189]
模型替换可以基于当前模型的评价而发生,例如使用在先前评估中所用的模型推断新的实际测序信号的样品。从样品中,可以使用比对程序创建基本事实。可以比较推断的结果和新的基本事实,并且可以计算错误率或任何其他可靠性或准确度分数。如果结果足够准确,则可以保持当前模型。如果结果不够准确,则样品数据可用于训练经训练的算法(例如,机器学习过程)以提供用于新的实际测序信号的新模型。
[0190]
经训练的算法(例如,机器学习过程)的再训练可以包括训练机器学习过程从开始(例如,从头)生成新模型,或获得先前使用的模型并运行一代或多代以更新模型。
[0191]
可以以各种方式执行再训练,例如应用迁移学习和调整模型的仅一部分(例如,调整模型中的几个初始输入层)。由于训练时间限制可以变得至关重要,因此可能需要这种高效的再训练。
[0192]
图12图示了用于评估属的基因组的方法1200的示例。
[0193]
方法1200可以包括(a)接收或生成代表属的基因组的第一部分的实际测序信号;(b)对至少部分实际测序信号应用当前模型以提供部分当前结果;其中当前模型是由经训练的算法(例如,机器学习过程)生成的;(c)评价部分当前结果的准确度;以及(d)基于部分当前结果的准确度,确定是否继续使用当前模型完成基因组的评估(例如,使用当前基因组)(如操作1210)。部分当前结果的准确度可以使用本文所述的任何方法(例如,相对于基本事实进行处理)来评价。
[0194]
如果方法1200已经确定继续使用当前模型,则操作1210之后可以是使用当前模型完成基因组的评估(如在操作1220中)。
[0195]
如果方法1200已经确定不继续使用当前模型,则操作1210之后可以是获得具有足够评估准确度的第二模型,并且使用第二模型评估(例如,第二属的)基因组(如在操作1230中)。在一些情况下,可以再训练或修正当前模型并且重复操作1210直到确定所评价的模型具有足够的准确度。
[0196]
在一些实施方案中,当前模型是基于与小于(例如,显著小于)该属的基因组的参考基因组对应的信息生成的。例如,如本文公开的任何方法中所述,可以使用比第二基因组(基因组)短的第一基因组(参考基因组)。
[0197]
评估可以由计算机系统执行。在一些实施方案中,在使用当前模型之前由计算机系统使用的至少一个模型是基于与小于(例如,显著小于)该属的基因组的参考基因组对应
的信息生成的。该至少一个模型可以是初始模型或任何其他模型。
[0198]
在一些实施方案中,方法1200可以包括执行操作1210、1220和1230的集合的多次迭代。
[0199]
图13图示了用于评估属的多个生物体的基因组的方法1300的示例。
[0200]
方法1300可以包括执行多个不同的评估过程以评估多个生物体的基因组(如在操作1310中)。
[0201]
在一些实施方案中,执行多个评估过程包括使用多个不同的评估模型。
[0202]
在一些实施方案中,多个不同模型中的至少一个是通过再训练经训练的算法(例如,机器学习过程)以提供新的和/或修正的模型生成的(如在操作1320中)。
[0203]
在一些实施方案中,再训练至少部分地基于与小于(例如,显著小于)该属的基因组(例如,第二基因组)的参考基因组对应的信息进行。
[0204]
在一些实施方案中,多个不同模型中的至少一个是基于与小于(例如,显著小于)该属的基因组的参考基因组对应的信息生成的。
[0205]
在一些实施方案中,方法1300可以包括在多个预定持续时间中的每一个期间用第二模型替换多个不同模型中的模型(如在操作1330中)。
[0206]
在一些实施方案中,方法1300可以包括在多个预定数目的评估过程的每一个期间用第二模型替换多个不同模型中的模型。
[0207]
在一些实施方案中,方法1300可以包括基于模型准确度的评价,用第二模型替换多个不同模型中的模型。
[0208]
图14图示了用于评估属的基因组的方法1400的示例。
[0209]
方法1400可以包括评估该属的基因组。评估可以包括提供多个模型(如在操作1410中);从多个模型中选择待在评估过程中使用的模型(如在操作1430中);以及使用选定的模型评估基因组(如在操作1440中)。
[0210]
可以基于与对应于多个模型的评估准确度相关的评估来执行选择(如在操作1420中)。
[0211]
可以基于对基因组的部分进行的测试来执行评估(如在操作1425中)。可以使用本文描述的任何方法(例如,相对于基本事实进行处理)评价模型的准确度。例如,可以使用误差的统计量度来评价模型的准确度,例如r平方值、均方误差(mse)、均方根误差(rmse)、误差平方和(sse)、平均绝对误差(mae)、平均绝对百分比误差(mape)等(例如,其中较低的误差度量表示模型的较高准确度)。在一些情况下,可以在基因组的单个部分或基因组的多个部分上测试每个模型。在一些情况下,可以通过测试参考基因组来评价模型。在一些情况下,可以通过测试另一基因组来评价模型。例如,可以将基因组的一个或多个部分与参考基因组或另一基因组进行比较以评价模型的准确度。
[0212]
在替代实施方案中,方法1400可以包括从多个模型中选择一个或多个模型,并且使用所选择的一个或多个模型评估基因组。例如,相同的基因组可以基于多个模型进行评估以生成多个评估。多个评估可以进一步被处理以例如生成综合评估。多个评估可用于评价所选模型(如在操作1425中),例如以确定是否必须再训练和/或修正此类所选模型中的一个或多个。例如,与评估的其余部分显著偏离的评估可以指示不准确的模型。
[0213]
提供了一种用于评估属的基因组的方法。该方法可以包括执行多个不同的评估过
程以评估多个多种生物的基因组;其中,多个不同评估过程中的评估过程包括从有待在评估过程中使用的多个不同模型中选择模型。
[0214]
在一些实施方案中,选择是基于与对应于多个模型的评估准确度相关的评估。
[0215]
在一些实施方案中,评估是基于对基因组的部分进行的测试。
[0216]
在一些实施方案中,评估由计算机系统执行。
[0217]
图15图示了用于评估属的基因组的方法1500的示例。
[0218]
方法1500可以包括接收或生成代表属的基因组的至少部分的实际测序信号。可以通过对可包括多个基底区段的基底进行成像来生成实际测序信号(如在操作1510中)。图16示出了基底(例如,晶片)及其区段的两个示例—具有其区段的晶片1610(例如,以网格状图案排列)和具有其区段的晶片1620(例如,以同心圆图案排列)。应当理解,基底可以以任何排列、图案或构造被分割成任何数目的区段。
[0219]
方法1500可以包括鉴别不同的基底区段(如在操作1520中)。在一些情况下,可以在成像之前、成像期间或成像之后鉴别不同的基底区段。例如,在成像之前,基底可以被分割成可以被定界也可以不被定界的不同区段。在另一示例中,在成像之后,可以从来自成像的一个或多个图像鉴别不同的基底区段。可以鉴别任意数目的基底区段。
[0220]
接下来,方法1500可以包括通过将第一模块应用于与多个基底区段的第一基底区段相关的信号(例如,来自实际测序信号)并将不同于第一模块的第二模块应用于与多个基底区段中的第二基底区段相关的信号(例如,来自实际测序信号),来评估该属的基因组。不同的模块可以应用于不同基底区段中的每一个。模块可以应用于多个不同的基底区段。在一些情况下,经鉴别的基底区段集合可被分组成多个组,并且不同的模块可应用于每个组,使得相同的模块应用于组的每个成员。模块可包括如本文别处所述的模型。
[0221]
在一些实施方案中,基于多个基底区段的照度之间的预期或实际差异来确定多个基底区段。
[0222]
在一些实施方案中,基于来自多个基底区段的辐射的收集或测量之间的预期或实际差异来确定多个基底区段。
[0223]
在一些实施方案中,基于化学材料在多个基底区段上的预期或实际的分布来确定多个基底区段。
[0224]
在一些实施方案中,基于样品或样品源在多个基底区段上的预期或实际分布来确定多个基底区段。例如,此类样品(例如,包含多个珠,每个珠包含扩增产物的克隆群)可以固定在不同的基底区段上。
[0225]
在一些实施方案中,多个基底区段包括相同的形状和/或大小。
[0226]
在一些实施方案中,多个基底区段中的至少两个在形状和大小中的至少一个上有所不同。
[0227]
提供了一种用于评估属的基因组的方法。
[0228]
该方法可以包括接收或生成代表属的基因组的至少部分的实际测序信号;其中实际测序信号属于与多个dna珠连接的基底的至少一部分的至少一幅图像。
[0229]
接下来,该方法可以包括通过将至少一个模型应用于实际测序信号来评估该属的基因组。
[0230]
计算机系统
17.本公开提供了被编程用于实现本公开的方法的计算机系统。图9示出了计算机系统901,其被编程或以其他方式配置用于例如执行方法100、200、300、600和700的一个或多个操作。
18.计算机系统901可以调节本公开的分析、计算和生成的各个方面,例如执行方法100、200、300、600和700的一个或多个操作。计算机系统901可以是用户的电子设备或相对于电子设备远程定位的计算机系统。该电子设备可以是移动电子设备。
19.计算机系统901包括中央处理单元(cpu,本文也称为“处理器”和“计算机处理器”)905,其可以是单核或多核处理器,或者是用于并行处理的多个处理器。计算机系统901还包括存储器或存储器位置910(例如,随机存取存储器、只读存储器、闪存)、电子存储单元915(例如,硬盘)、用于与一个或多个其他系统通信的通信接口920(例如,网络适配器)以及外围设备925,诸如高速缓存、其他存储器、数据存储和/或电子显示适配器。存储器910、存储单元915、接口920和外围设备925通过诸如主板等通信总线(实线)与cpu 905通信。存储单元915可以是用于存储数据的数据存储单元(或数据存储库)。计算机系统901借助于通信接口920可操作地耦合到计算机网络(“网络”)930。网络930可以是因特网、互联网和/或外联网,或者与因特网通信的内联网和/或外联网。
20.在一些情况下,网络930是电信和/或数据网络。网络930可以包括一个或多个计算机服务器,其可以实现分布式计算,诸如云计算。例如,一个或多个计算机服务器可以通过网络930(“云”)实现云计算以执行本公开的分析、计算和生成的各个方面,例如执行方法100、200、300、600和700的一个或多个操作。这样的云计算可以由云计算平台提供,例如亚马逊网络服务(aws)、微软azure、谷歌云平台和ibm云。在一些情况下,网络930可以借助于计算机系统901实现对等网络,这可以使得耦合到计算机系统901的设备能够起到客户端或服务器的作用。
21.cpu 905可以包括一个或多个计算机处理器和/或一个或多个图形处理单元(gpu)。cpu 905可以执行一系列机器可读指令,该机器可读指令可以体现在程序或软件中。指令可以存储在存储位置如存储器910中。指令可以针对cpu 905,该指令随后可以编程或以其他方式配置cpu 905以实现本公开的方法。由cpu 905执行的操作的示例可以包括提取、解码、执行和回写。
22.cpu 905可以是电路如集成电路的一部分。电路中可以包括系统901的一个或多个其他组件。在一些情况下,该电路是专用集成电路(asic)。
23.存储单元915可以存储文件,诸如驱动程序、库和保存的程序。存储单元915可以存储用户数据,例如用户偏好和用户程序。在一些情况下,计算机系统901可以包括一个或多个附加数据存储单元,所述附加数据存储单元位于计算机系统901外部,诸如位于通过内联网或因特网与计算机系统901通信的远程服务器上。
24.计算机系统901可通过网络930与一个或多个远程计算机系统通信。例如,计算机系统901可以与用户的远程计算机系统通信。远程计算机系统的示例包括个人计算机(例如,便携式pc)、平板或平板型pc(例如,ipad、galaxy tab)、电话、智能手机(例如,iphone、支持android的设备、)或个人数字助理。用户可以经由网络930访问计算机系统901。
25.本文所述的方法可通过机器(例如,计算机处理器)可执行代码的方式来实现,该机器可执行代码存储在计算机系统901的电子存储位置上,例如存储器910或电子存储单元915上。机器可执行代码或机器可读代码可以以软件的形式提供。在使用期间,该代码可由处理器905执行。在一些情况下,可从存储单元915检索代码并将其存储在存储器910上,以供处理器905迅速存取。在一些情况下,可排除电子存储单元915,并且将机器可执行指令存储在存储器910上。
26.该代码可以被预编译并配置用于由具有适于执行代码的处理器的机器使用,或者可以在运行期间被编译。代码可以用编程语言提供,可以选择编程语言以使代码能够以预编译或即时编译(as
‑
compiled)的方式执行。
27.本文提供的系统和方法的各个方面,诸如计算机系统901,可以在编程中体现。该技术的各个方面可以被认为是“产品”或“制品”,其一般为在一种类型的机器可读介质上携带或体现的机器(或处理器)可执行代码和/或相关数据的形式。机器可执行代码可以存储在电子存储单元如存储器(例如,只读存储器、随机存取存储器、闪存)或硬盘上。“存储”型介质可以包括计算机的任何或全部有形存储器、处理器等,或其相关模块,诸如各种半导体存储器、磁带驱动器、磁盘驱动器等,其可以在任何时间为软件编程提供非暂时性存储。软件的全部或部分有时可以通过因特网或各种其他电信网络进行通信。例如,这样的通信可以使软件从能够一台计算机或处理器加载到另一台计算机或处理器中,例如从管理服务器或主机加载到应用服务器的计算机平台中。因此,可以承载软件元件的另一类型的介质包括光波、电波和电磁波,诸如跨本地设备之间的物理接口、通过有线和光学陆线网络以及各种空中链路而使用的。携载此类波的物理元件,诸如有线或无线链路、光学链路等,也可以被视为承载软件的介质。如本文所用,除非仅限于非暂时性有形的“存储”介质,否则计算机或机器“可读介质”等术语是指参与向处理器提供指令以供执行的任何介质。
28.因此,机器可读介质如计算机可执行代码可采取多种形式,包括但不限于有形存储介质、载波介质或物理传输介质。非易失性存储介质包括例如光盘或磁盘,诸如任何计算机中的任何存储设备等,诸如可用于实现如附图中所示的数据库等。易失性存储介质包括动态存储器,诸如这样的计算机平台的主存储器。有形传输介质包括同轴缆线、铜线和光纤,包括构成计算机系统内的总线的线。载波传输介质可以采取电信号或电磁信号或者声波或光波的形式,诸如在射频(rf)和红外(ir)数据通信期间产生的那些。因此,计算机可读介质的常见形式包括例如:软盘、柔性盘、硬盘、磁带、任何其他磁性介质、cd
‑
rom、dvd或dvd
‑
rom、任何其他光学介质、穿孔卡片纸带、任何其他具有孔洞图案的物理存储介质、ram、rom、prom和eprom、flash
‑
eprom、任何其他存储器芯片或匣盒、传送数据或指令的载波、传送此类载波的电缆或链路,或者计算机可以从中读取编程代码和/或数据的任何其他介质。这些计算机可读介质形式中的许多可涉及将一个或多个指令的一个或多个序列携带到处理器以供执行。
29.计算机系统901可以包括电子显示器935,或者与电子显示器935通信,电子显示器935包括用于提供例如指示测序信号、实际测序信号、准确测序信号等的视觉显示的用户界面(ui)940。ui的示例包括但不限于图形用户界面(gui)和基于网络的用户界面。
30.本公开的方法和系统可通过一种或多种算法来实现。算法可以在由中央处理单元905执行时通过软件的方式来实现。算法可以例如执行方法100、200、300、600和700的一个
或多个操作。实施例
[0231]
实施例1
[0232]
使用本公开的系统、方法和介质,从多个核酸生成原始测序信号。如图17所示,绘制了具有给定幅度的每个原始测序信号的碱基数的直方图。将经训练的神经网络应用于原始测序信号,以鉴别和解卷积原始测序信号的系统学(例如定相、信号衰减和邻近序列),如子图a所示,从而生成经处理的测序信号(例如,校正或准确的测序信号),如子图b所示。经处理信号的直方图(图17)显示了具有约0、1、2和3的幅度的经处理序列的多个碱基的窄分布。经处理的测序信号在未使用参考的情况下产生,由此提高序列判定(例如,含有同聚物的序列)的准确度。
[0233]
实施例2
[0234]
使用本公开的系统、方法和介质,训练神经网络以产生在人类或其他大基因组的多个输入测序信号(例如,从多个核酸生成)与多个输出序列(例如,包含多个碱基判定)之间的“基本事实”映射。首先,对多个输入测序信号执行碱基判定,从而产生多个初始序列。这可以使用全碱基判定模型(例如,基于大基因组,例如人类基因组)来执行。多个初始序列可以任选地被hpn截短,使得初始序列中的所有同聚物(例如,长度为2、3、4
……
)被截短至长度为1(例如,由单个碱基表示)或另一较小数字n,以保证比对的低错误率。接下来,将hpn截短的序列与匹配的hpn截短的人类参考(例如,hpn截短的人类基因组)进行比对。接下来,使用hpn比对的序列中的一些或全部(作为输出)和相关的测序信号(作为输入)构建训练集。接下来,使用该训练集训练神经网络,从而产生经训练的神经网络。
[0235]
替代地或组合地,可以将hpn截短的序列的至少部分与匹配的大肠杆菌(或其他较小的基因组)参考进行比对。可以使用hpn比对的序列中的一些或全部(作为输出)和相关的测序信号(作为输入)构建训练集。可以使用该训练集训练神经网络,从而产生经训练的神经网络。可以相对于该训练集测试现有模型,以便基于准确度选择模型(例如,最小化碱基判定错误的模型)。
[0236]
虽然本文已经示出和描述了本公开的优选实施方案,但是对于本领域技术人员而言显而易见的是,这些实施方案仅以示例的方式提供。本发明不意在受说明书中提供的具体示例的限制。虽然已经参考上述说明书描述了本发明,但是本文实施方案的描述和说明并不意味着以限制性意义进行解释。在不偏离本公开的情况下,本领域技术人员现将想到许多变化、改变和替代。此外,应当理解,本发明的所有方面不限于本文阐述的特定描述、配置或相对比例,而是取决于各种条件和变量。应当理解,本文所述的本发明实施方案的各种替代方案可用于实践本发明。因此,考虑到本发明还应涵盖任何此类替代、修改、变化或等同物。以下权利要求旨在限定本发明的范围,并由此涵盖这些权利要求范围内的方法和结构及其等同物。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。