用于金属离子电池的电活性材料
1.本发明总体上涉及适合在可充电金属离子电池的电极中使用的电活性材料,并且更具体地涉及适合作为可充电金属离子电池中的阳极活性材料使用的具有高电化学容量的颗粒材料。
2.可充电金属离子电池广泛用于便携式电子设备,比如手机和笔记本电脑,并且越来越多地应用于电动汽车或混合动力汽车。可充电金属离子电池通常包括阳极层、阴极层、用于在阳极层和阴极层之间传输金属离子的电解质以及设置在阳极和阴极之间的电绝缘多孔隔膜。阴极一般包括具有含金属离子的金属氧化物系复合材料层的金属集流体,并且阳极一般包括具有电活性材料层的金属集流体,所述电活性材料在本文中被定义为在电池的充电和放电期间能够嵌入和释放金属离子的材料。为了避免疑义,术语“阴极”和“阳极”在本文中按以下意义使用:将电池接入负载中,使得阴极为正极,并且阳极为负极。当对金属离子电池进行充电时,金属离子从含金属离子的阴极层经由电解质传输到阳极,并且嵌入到阳极材料中。术语“电池”在本文中既指含有单个阳极和单个阴极的装置又指含有多个阳极和/或多个阴极的装置。
3.感兴趣的是,提高可充电金属离子电池的重量容量和/或体积容量。锂离子电池的使用在与其他电池技术相比时已经提供了实质性的改善,但是仍有进一步发展的空间。迄今为止,商业锂离子电池很大程度上受限于使用石墨作为阳极活性材料。当对石墨阳极进行充电时,锂嵌入石墨层之间形成具有经验式li
x
c6(其中x大于0且小于或等于1)的材料。因此,在锂离子电池中石墨的最大理论容量为372mah/g,实际容量稍低(大约340至360mah/g)。其他材料比如硅、锡和锗与石墨相比能够以明显更高的容量嵌入锂,但是由于在多次充电/放电循环时难以保持足够的容量而尚未得到广泛的商业应用。
4.特别地,硅由于其非常高的锂容量(参见例如用于可充电锂电池的插入电极材料(insertion electrode materials for rechargeable lithium batteries),winter,m等,adv.mater.1998,10,第10期)而被认为是有前景的用于制造具有高重量容量和体积容量的可充电金属离子电池的石墨替代物。在室温,在锂离子电池中硅的理论最大比容量为约3,600mah/g(基于li
15
si4计)。然而,硅作为阳极材料的使用由于在充电和放电时的大体积变化而是复杂的。
5.锂嵌入到本体硅(bulk silicon)中导致硅材料体积大幅增加,在硅被锂化至其最大容量时高达其原始体积的400%,并且反复的充电
‑
放电循环在硅材料中引起明显的机械应力,导致硅阳极材料断裂和脱层。硅粒子在脱锂时的体积收缩可能导致阳极材料和集流体之间的电接触损失。另一个困难是在硅表面上形成的固体电解质界面(sei)层没有足够的机械容差来适应硅的膨胀和收缩。结果,新暴露的硅表面导致进一步的电解质分解和sei层厚度增大以及不可逆的锂消耗。这些破坏机制在连续的充电和放电循环时共同导致不可接受的电化学容量损失。
6.已经提出了多种方法来克服与在对含硅阳极进行充电时观察到的体积变化相关的问题。最普遍的解决含硅阳极的不可逆容量损失的方法是使用一些形式的精细结构化硅作为电活性材料。已经报道了与在微米尺寸范围内的硅粒子相比,横截面低于大约150nm的
微细硅结构比如硅膜和硅纳米粒子更能容忍在充电和放电时的体积变化。然而,它们均不是特别适合以它们的未改进形式用于商业规模的应用;纳米尺寸的粒子难以制备和处理,并且硅膜无法提供足够的体容量(bulk capacity)。例如,纳米尺寸的粒子倾向于形成团聚体,使得难以得到可用的在阳极材料基体内的粒子分散体。另外,纳米尺寸粒子的团聚体的形成在反复的充电
‑
放电循环时导致不可接受的容量损失。
7.ohara等(journal of power sources 136(2004)303
‑
306)已经描述了将硅作为薄膜蒸镀到镍箔集流体上以及使用此结构作为锂离子电池的阳极。尽管此方法得到了良好的容量保持率,但是薄膜结构无法给出每单位面积的容量的可用量,并且当膜厚度增加时,消除了任何改善。
8.wo 2007/083155公开了可以通过使用具有高长径比(即粒子的最大尺寸与最小尺寸的比率)的硅粒子来获得改善的容量保持率。这样的粒子的小横截面减小了由于充电和放电时的体积变化引起的材料上的结构应力。然而,这样的粒子的制造可能困难且昂贵,并且可能是易碎的。另外,高表面积可能导致过多的sei形成,在首次充电
‑
放电循环时造成过多的容量损失。
9.一般还已知的是,可以将电活性材料比如硅沉积在多孔载体材料比如活性炭材料的孔隙内。这些复合材料提供纳米尺寸硅粒子的一些有益的充电
‑
放电性质,同时避免纳米粒子的处理困难。例如,guo等(journal of materials chemistry a,2013,14075
‑
14079页)公开了一种硅
‑
碳复合材料,其中多孔碳基底提供导电骨架,硅纳米粒子沉积在该基底的均匀分布的孔隙结构中。在初始充电循环时的sei形成受限于剩余的孔体积,使得剩余的硅在随后的充电循环中不暴露于电解质。已知复合材料在多次充电循环时具有改善的容量保持率,但是复合材料的初始容量(以mah/g计)明显低于硅纳米粒子的初始容量。
10.jp 2003100284公开了一种活性材料,其包括具有从一些较大孔隙分支的小孔隙的碳基支架。电活性材料(例如硅)任意地位于大孔隙和小孔隙二者的壁上,以及位于碳基支架的外表面上。
11.硅低氧化物材料(例如sio
x
,其中0<x<2)一直用于主要包含石墨作为活性材料的“混合型”电极。然而,由于sio
x
在锂化时的膨胀和在首次充电循环期间的相对高的不可逆锂损失,sio
x
的最大负载量一般为电极中总电活性材料的约10重量%。更高的sio
x
负载量导致过多的电极膨胀和对电极的不可逆损坏。因此,需要具有与硅氧化物相当的锂化容量但在首次充电循环期间具有减少的膨胀和减少的容量损失的高容量电极材料。这将会使得能够以比sio
x
更高的负载量使用该高容量材料,而不将总电极膨胀增加到超过可能发生结构损坏的极限。
12.电极材料的理想膨胀性质必须与其他重要性质一起获得。特别地,可商购得到的替代电极材料需要提供高锂化容量以及随着大量充电
‑
放电循环的高容量保持率的益处。另外,重要的是,任何新的电活性材料都应该可容易替代为常规电极制作过程中的已知材料。这些过程一般依赖于将电极材料压延到集流体上以使电极层致密化并且提高在电池设计内的空间利用率。多孔材料在电极制作期间易断裂,导致电化学性能受损。因此,特别要求新的电化学材料应具有足够的结构强度,以及增加的电化学储存容量和可逆的容量保持率。
13.本发明人已经确定,包含多孔碳骨架和位于多孔碳骨架内的电活性材料的复合材
料的机械性能可以通过具有特定的孔隙结构、仔细控制的孔隙大小分布和电活性材料在多孔碳骨架内的受控负载的多孔碳骨架来改善。通过将电活性材料定位在碳骨架内,还减少了与电池中的电解质溶剂的接触,使在首次和后续充电循环期间导致不可逆锂损失的化学副反应最小化。
14.在第一方面,本发明提供了一种由多个复合粒子组成的颗粒材料,其中所述复合粒子包含:
15.(a)包括微孔和介孔的多孔碳骨架,
16.其中所述微孔和介孔的通过气体吸附测量的总孔体积为p1cm3/g,其中p1表示值为0.4至0.75的自然数字,并且
17.其中基于微孔和介孔的总体积,微孔体积分数在0.5至0.85的范围内;以及
18.(b)位于所述多孔碳骨架的孔隙内的多个纳米尺寸元素硅结构域;
19.其中所述复合粒子中的硅与所述多孔碳骨架的重量比在[0.9
×
p1至1.9
×
p1]:1的范围内。
[0020]
本发明特别依赖于具有相对低孔隙率的多孔碳骨架的使用,从而提供用于容纳电活性硅结构域的高强度骨架。特别地,0.75cm3/g的孔隙率上限确保了多孔碳骨架在压缩应力下的耐断裂性非常高。
[0021]
多孔碳骨架包括三维互连的开放孔隙网络,其包括微孔和介孔。多孔碳骨架可以任选地还包括小体积的大孔。根据常规iupac术语学,术语“微孔”在本文中用来指直径小于2nm的孔隙,术语“介孔”在本文中用来指直径为2
‑
50nm的孔隙,并且术语“大孔”用来指直径大于50nm的孔隙。
[0022]
多孔碳骨架中的微孔和介孔的总体积为0.4至0.75cm3/g。为了避免疑义,本文中提到多孔碳骨架的孔体积(在不存在任何相反指示的情况下)涉及单独的多孔碳骨架的孔体积,即,在不存在占据多孔碳骨架的孔隙的任何电活性材料(或任何其他材料)的情况下测量的多孔碳骨架的孔体积。
[0023]
微孔和介孔的总体积(即直径在0至50nm范围内的孔隙的总孔体积)在本文中被称为p1cm3/g,其中p1表示值为0.4至0.75的无量纲自然数字。p1的值用于将多孔碳骨架中的可用孔体积和如以下所述的硅与多孔碳骨架的重量比相关联。
[0024]
元素硅位于多孔碳骨架的孔隙网络内。因此,硅采取多个纳米尺寸硅结构域的形式。如本文中使用的,术语“纳米尺寸硅结构域”是指具有受硅布置在多孔碳骨架的微孔和/或介孔内确定的尺寸的纳米尺寸硅体。
[0025]
多微孔碳骨架提供如下益处:电活性材料以尺寸为大约几纳米以下的小结构域的形式位于微孔网络内。这些精细电活性结构与更大的电活性结构相比具有更低的对弹性形变的耐性和更高的耐断裂性,因此能够锂化和脱锂而没有过度的结构应力。因此,多孔碳骨架的微孔隙率不仅提供了改善的粒子机械强度,而且确保了电活性材料自身具有足够的弹性以禁受在多次充放电循环时的反复体积变化而没有显著的容量损失。
[0026]
低孔体积的多孔碳骨架需要硅对孔体积的相对高的占据率,从而提供具有足够高的重量容量和体积容量的材料。硅与多孔碳骨架的重量比通过比率[0.9
×
p1至1.9
×
p1]:1与总微孔/介孔体积相关联。通过限定基于p1的值的硅的重量比,硅对孔体积的体积占据百分比被控制在特定界限内。作为参考,[0.9
×
p1至1.9
×
p1]:1的硅与多孔碳骨架的重量比对
应于硅体积为微孔和介孔的总体积的大约40至82%。
[0027]
已经发现,仅在多孔碳骨架的孔隙大小分布被控制在特定的界限内的情况下,才可以得到所要求的硅与多孔碳骨架的重量比。由本技术人为其他申请研发的高孔隙率骨架具有高度互连的孔隙网络,具有多条进入到最内部孔隙中的路径,而低孔隙率骨架具有少得多的在孔隙网络内的互连。为了使硅优先沉积在多孔碳骨架的孔隙内而不是沉积在外表面上,多孔碳骨架中必须具有相对高的微孔分数,因为硅到微孔中的沉积是动力学有利的。然而,如果微孔隙率过高,则发现硅基本上沉积在多孔碳骨架的外表面上。据认为这是由于硅的初始沉积物在孔隙网络中形成堵塞。通过孔隙网络的替代路径的可利用性是总孔体积的函数。在低孔隙率材料中,相对缺乏通过孔隙网络的替代路径意味着此类型的堵塞可能使一部分孔体积对硅前体来说是不可接近的。因此,确保硅在低孔隙率碳骨架中的内部沉积是本发明人已经解决的一个特定挑战。概括地说,已经发现,得到具有高抗压强度、有限的膨胀、高电化学容量和高容量保持率的所需性能的电活性材料依赖于总孔体积、孔隙大小分布和硅对孔隙的占据程度的组合。
[0028]
与低孔隙率骨架中的孔隙网络的互连相关的另一种因素是粒度。较大的粒子必然具有较长的从孔隙网络到最内部孔隙的路径,因此,粒子越大,越难以获得所需重量比的硅的所需内部沉积。因此,在本发明的优选实施方案中,还仔细地控制多孔碳骨架的中值粒度(d
50
)以及粒度分布。受控的粒度分布额外地有助于粒子强度,以及实现改善的复合粒子在电极活性层中的紧密填充,从而减少了对过度压延压力的需求。
[0029]
本发明的复合材料的高度微孔碳骨架具有高拉伸断裂强度,因此可以容纳电活性材料在孔体积内的显著量的膨胀而不发生断裂。尽管电活性材料的完全锂化可能导致整个复合材料的一些外部膨胀,但是由于受控的总孔体积以及因此的硅与多孔碳骨架的最大重量比,外部膨胀的量是有限的。尽管与更高孔隙率的骨架相比,高强度多孔碳骨架不太能够在应力下发生弹性形变,但是硅与多孔碳骨架的受控的比率确保了在最大锂化时的多孔碳骨架上的膨胀应力保持低于引起破裂所需要的水平。
[0030]
在本发明的复合材料的超常性能中的又一个因素是使sei形成最小化。通过将电活性材料定位在孔隙网络,仅小面积的电活性材料表面对于电解质来说是可接近的,因此在电活性材料表面上的sei形成是有限的,从而使得在首次充电循环期间的不可逆锂损失最小化。还在很大程度上防止了在后续充电
‑
放电循环中的电活性材料的额外暴露,使得sei形成不是一种导致容量损失的明显的破坏机制。这与作为例如guo所公开材料(参见上文)的特征的过量sei形成形成了鲜明的对比。
[0031]
由于本发明的颗粒材料的独特粒子构造,复合粒子具有改善现有sio
x
技术的电化学性能。特别地,本发明的颗粒材料具有更大的电化学容量、更低的总体膨胀和相当的可逆容量保持率,导致高容量电活性材料的负载量比先前已经实现的更高的可能性。
[0032]
本文中提到多孔碳骨架中的微孔、介孔和大孔的体积以及任何提到孔体积在多孔碳骨架内的分布是指单独(即在不存在占据孔体积中的一些或全部的任何电活性材料或其他材料的情况下)的多孔碳骨架的内部孔体积。
[0033]
p1的值优选为至少0.42,更优选至少0.45,更优选至少0.47,更优选至少0.5。更高孔隙率的骨架是有利的,因为其使得能够在孔隙结构内容纳更大量的硅,而不破坏在电极制造期间的压缩应力下或在由于硅的锂化引起的膨胀应力下多孔碳骨架对破裂的耐性。如
上所述,p1的值上限是确保维持粒子的高抗压强度的值。因此,p1的值可以是至多0.75。然而,更优选地,p1的值可以是至多0.72,更优选地至多0.7,更优选地至多0.67,更优选地至多0.65,更优选地至多0.62,更优选地至多0.6。任选地,p1的值可以小于0.6。
[0034]
p1可以在0.42至0.75的范围内,或在0.42至0.72的范围内,或在0.42至0.7的范围内,或在0.42至0.67的范围内,或在0.42至0.65的范围内,或在0.42至0.62的范围内,或在0.42至0.6的范围内,或在0.42至小于0.6的范围内,或在0.45至0.75的范围内,或在0.45至0.72的范围内,或在0.45至0.7的范围内,或在0.45至0.67的范围内,或在0.45至0.65的范围内,或在0.45至0.62的范围内,或在0.45至0.6的范围内,或在0.45至小于0.6的范围内,或在0.47至0.75的范围内,或在0.47至0.72的范围内,或在0.47至0.7的范围内,或在0.47至0.67的范围内,或在0.47至0.65的范围内,或在0.47至0.62的范围内,或在0.47至0.6的范围内,或在0.47至小于0.6的范围内,或在0.5至0.75的范围内,或在0.5至0.72的范围内,或在0.5至0.7的范围内,或在0.5至0.67的范围内,或在0.5至0.65的范围内,或在0.5至0.62的范围内,或在0.5至0.6的范围内,或在0.5至小于0.6的范围内。
[0035]
如本文中使用的,微孔体积分数是指表示为微孔和介孔的总体积的分数的微孔的体积。换句话说,微孔体积分数是直径为2nm以下的孔隙相对于直径为至多50nm的孔隙的总体积的体积分数。如以上所讨论的,基于微孔和介孔的总体积,微孔体积分数需要被仔细地控制在0.5至0.85的范围内。优选地,基于微孔和介孔的总体积,微孔体积分数为至少0.55,或至少0.56,或至少0.58,或至少0.6,或至少0.62,或至少0.64,或至少0.65。优选地,基于微孔和介孔的总体积,微孔体积分数为至多0.84,或至多0.82,或至多0.8,或至多0.78,或至多0.76,或至多0.75。
[0036]
基于微孔和介孔的总体积,微孔体积分数可以任选地在0.5至0.84的范围内,或在0.5至0.82的范围内,或在0.5至0.8的范围内,或在0.5至0.78的范围内,或在0.5至0.76的范围内,或在0.5至0.75的范围内,或在0.55至0.84的范围内,或在0.55至0.82的范围内,或在0.55至0.8的范围内,或在0.55至0.78的范围内,或在0.55至0.76的范围内,或在0.55至0.75的范围内,或在0.6至0.84的范围内,或在0.6至0.82的范围内,或在0.6至0.8的范围内,或在0.6至0.78的范围内,或在0.6至0.76的范围内,或在0.6至0.75的范围内,或在0.65至0.84的范围内,或在0.65至0.82的范围内,或在0.65至0.8的范围内,或在0.65至0.78的范围内,或在0.65至0.76的范围内,或在0.65至0.75的范围内。
[0037]
在这些范围内的微孔分数确保了硅纳米结构具有足够小的尺寸以防止在锂化期间的过多应力,同时还确保了孔隙尺寸足够大以使得能够在可接受的加工时间下在多孔碳骨架的孔隙结构内沉积(例如,通过化学气相侵入技术)高的硅水平。如果微孔分数比规定的高,则硅可能在其被沉积时形成堵塞物和/或封盖物,特别是以较高的沉积速率被沉积时,导致不可接近并且因此不能被硅填充的孔体积口袋,从而导致硅在外部粒子表面上的过多沉积。
[0038]
多孔碳骨架的pd
90
孔径优选为至多20nm,更优选地至多15nm,更优选地至多12nm,更优选地至多10nm,更优选地至多8nm,更优选地至多6nm,更优选地至多5nm。
[0039]
如本文中使用的术语“pd
90
孔径”是指基于微孔和介孔的总体积的体积第90百分位的孔径。换言之,如本文中使用的术语“d
90
孔径”是指测得总微孔和介孔体积(由p1表示)的90%比某一孔径低时的该孔径。为了避免疑义,为了确定pd
90
值,不考虑任何大孔体积(孔径
大于50nm)。
[0040]
优选地,pd
95
孔径为至多20nm,或至多15nm,或至多12nm,或至多10nm,或至多8nm。如本文中使用的“pd
95
孔径”是指体积第95百分位孔径。
[0041]
小体积分数的直径在较大介孔范围内的孔隙可以有利地促进电解质接近硅。因此,直径在10至50nm范围内的孔隙(即较大的介孔)可以任选地占多孔碳骨架的总微孔和介孔体积的至少1%、至少2%、至少5%或至少10%。
[0042]
多孔碳骨架的孔隙大小分布优选为双峰或多峰的。如本文中使用的,术语“孔隙大小分布”涉及多孔碳骨架的孔隙大小相对于累积总内部孔体积的分布。双峰或多峰的孔隙大小分布可以是优选的,因为微孔和具有更大直径的孔隙之间的紧密接近提供了从多孔网络到硅的高效离子传输的优点。因此,颗粒材料具有高的离子扩散性以及因此改善的倍率性能。
[0043]
按照iso 15901
‑
2和iso 15901
‑
3中所述的方法,使用氮气吸附在77k下利用淬火固体密度泛函理论(qsdft)来确定微孔和介孔的总体积以及微孔和介孔的孔隙大小分布。氮气吸附是一种通过使气体在固体的孔隙中冷凝来表征材料的孔隙率和孔径分布的技术。随着压力升高,气体首先在具有最小直径的孔隙中冷凝,并且压力升高直至达到饱和点,在该饱和点所有孔隙都被液体填充。然后氮气压力逐渐降低,以使液体从体系中蒸发。对吸附和脱附等温线以及它们之间的滞后的分析使得能够确定孔体积和孔隙大小分布。合适的用于通过氮气吸附测量孔体积和孔隙大小分布的仪器包括tristar ii和tristar ii plus孔隙率分析仪(它们可由美国的micromeritics instrument corporation获得)。
[0044]
氮气吸附对于测量直径最大为50nm的孔隙的孔体积和孔隙大小分布是有效的,但是对于具有大得多的直径的孔隙较不可靠。为了本发明的目的,因此,仅对直径最大为50nm(包括50nm)的孔隙使用氮气吸附来确定孔体积和孔隙大小分布。如上所述,通过仅考虑直径最大为50nm(包括50nm)的孔隙(即仅微孔和介孔)来确定p1的值,并且同样相对于仅微孔和介孔的总体积来确定pd
90
和pd
95
的值以及微孔分数。
[0045]
鉴于可用分析技术的限制,无法使用单一技术测量在微孔、介孔和大孔的整个范围上的孔体积和孔隙大小分布。在多孔碳骨架包括大孔的情况下,在大于50nm到最大100nm范围内的孔隙的体积在本文中被认为具有p2cm3/g的值,并且通过压汞法进行测量。如上所述,p2的值涉及单独测量时的多孔碳骨架的孔体积,即,在不存在占据多孔碳骨架的孔隙的硅或任何其他材料的情况下的多孔碳骨架的孔体积。
[0046]
为了避免疑义,p2的值仅考虑直径从大于50nm到最大100nm(包括100nm)的孔隙,即其仅包括直径最大为100nm的大孔的体积。为了确定p2的值,不考虑通过压汞法测量的在50nm以下的孔隙大小的任何孔体积(如上所述,使用氮气吸附来表征介孔和微孔)。为了本发明的目的,通过压汞法测量的在高于100nm的情况下的孔体积被假定为粒子间孔隙率,并且在确定p2的值时也不考虑。
[0047]
压汞法是一种通过对浸入汞中的材料样品施加不同水平的压力来表征该材料的孔隙率和孔径分布的技术。使汞侵入到样品的孔隙中所需的压力与孔隙的大小成反比。本文所报道的通过压汞法获得的值根据astm uop578
‑
11获得,其中对于在室温的汞,表面张力γ为480mn/m,并且接触角为140
°
。汞在室温的密度为13.5462g/cm3。可商购获得多种高精度压汞法仪器,比如autopore iv系列的自动化汞孔隙率计,其可由美国的
micromeritics instrument corporation获得。关于压汞法的完整概述,可以参考p.a.webb和c.orr,“微细粒子技术中的分析方法(analytical methods in fine particle technology)”,1997,micromeritics instrument corporation,isbn 0
‑
9656783
‑
0。
[0048]
与微孔和介孔的体积(以及因此p1的值)相比,大孔的体积(以及因此p2的值)优选较小。尽管一小部分的大孔可以对促进电解质进入到孔隙网络中是有用的,但是本发明的优点基本上通过在微孔和较小的介孔中容纳硅来获得。
[0049]
因此,根据本发明,通过水银孔率法(也称作压汞法)测量的多孔碳骨架中的大孔的总体积为p2cm3/g,其中p2的值优选为至多0.2
×
p1,或至多0.1
×
p1,或至多0.05
×
p1,或至多0.02
×
p1,或至多0.01
×
p1,或至多0.005
×
p1。
[0050]
应理解,侵入技术比如气体吸附和压汞法仅对确定氮气或汞从多孔碳骨架外部可及的孔隙的孔体积有效。如本文中所指明的孔隙率值(p1和p2)应被理解为是指开孔(流体从多孔碳骨架外部可及的孔隙)的体积。在本文中,当指明孔隙率值时,不应考虑无法通过氮气吸附或压汞法鉴定的完全被包封的孔隙。同样,对于确定p1的值,也不考虑位于小到低于氮气吸附的检测极限的孔隙中的任何孔体积。
[0051]
多孔碳骨架可以包括结晶碳或无定形碳,或者无定形和结晶碳的混合物。多孔碳骨架可以是硬碳或软碳骨架,并且可以适宜地通过涉及含碳材料(包括有机材料、树脂和聚合物)热解的已知工序获得。多孔碳材料也可以通过其他方法获得,例如由含碳化物的前体获得。
[0052]
多孔碳骨架优选地具有包含至少90重量%的碳、更优选至少95重量%的碳、更优选至少98重量%的碳的元素组成。多孔碳骨架可以任选地包含少量的其他元素,比如氧、氮、硫和氢。多孔碳骨架的元素组成可以通过在不存在硅的情况下进行的常规元素分析技术进行测定。
[0053]
如本文中使用的,术语“硬碳”是指其中发现碳原子主要为在纳米尺度聚芳烃结构域中的sp2杂化状态(三键)的无序碳基质。聚芳烃结构域利用化学键例如c
‑
o
‑
c键交联。
[0054]
由于聚芳烃结构域之间的化学交联,硬碳不能在高温转化为石墨。硬碳具有石墨状特性,这通过在拉曼光谱中大的g带(~1600cm
‑1)所证明的。然而,该碳不完全是石墨性的,这通过拉曼光谱中明显的d带(~1350cm
‑1)所证明的。
[0055]
如本文中使用的,术语“软碳”也是指其中发现碳原子主要在尺寸在5
‑
200nm范围内的聚芳烃结构域中的sp2杂化状态(三键)的无序碳基质。与硬碳相比,软碳中的聚芳烃结构域通过分子间力相缔合,而不是利用化学键交联。这意味着它们将会在高温石墨化。多孔碳骨架优选包含至少50%的sp2杂化碳(通过xps测量)。例如,多孔碳骨架可以适宜地包含50%至98%的sp2杂化碳,55%至95%的sp2杂化碳,60%至90%的sp2杂化碳,或70%至85%的sp2杂化碳。
[0056]
可以使用各种各样的不同材料来制备合适的多孔碳骨架。可以使用的有机材料的实例包括:植物生物质,其包括可木质纤维素材料(比如椰子壳、稻壳、木材等),和化石碳源,比如煤。在热解时形成多孔碳骨架的树脂和聚合物材料的实例包括:酚醛树脂,酚醛清漆树脂,沥青,三聚氰胺,聚丙烯酸酯,聚苯乙烯,聚乙烯醇(pva),聚乙烯吡咯烷酮(pvp),以及包含丙烯酸酯、苯乙烯、α
‑
烯烃、乙烯吡咯烷酮和其他烯属不饱和单体的单体单元的各种共聚物。根据热解过程的原料和条件,在本领域中可获得各种不同的硬碳材料。
[0057]
多孔碳骨架可以进行化学或气体活化过程以增加介孔和微孔的体积。合适的活化过程包括使热解碳与氧气、水蒸气、co、co2和koh中的一种或多种在600至1000℃范围内的温度接触。
[0058]
也可以通过已知的模板化过程,使用可取出的造孔剂比如mgo和其他胶体或聚合物模板(其可以通过在热解或活化后的热或化学手段移除)来获得介孔。
[0059]
本发明的复合粒子中的硅的量被选择为使得多孔碳骨架的至少约40%且至多约78%的内部孔体积被硅占据(在未充电的状态下)。优选地,硅占据多孔碳骨架的约50%至约75%的内部孔体积,更优选多孔碳骨架的约55%至70%的内部孔体积。在这些优选范围内,多孔碳骨架的孔体积有效地容纳在充电和放电期间的硅的膨胀,但是避免了过量的孔体积,这种过量的孔体积不利于颗粒材料的体积容量。然而,由于金属离子扩散速率不足或膨胀体积不足导致对锂化的机械阻力,硅的量也并非高到阻碍有效锂化。
[0060]
如上所述,通过要求硅与多孔碳骨架的重量比在[0.9
×
p1至1.9
×
p1]:1的范围内,可以使多孔碳骨架中的硅的量与可获得的孔体积相关联。此关系考虑了硅的密度和多孔碳骨架的孔体积来限定硅的重量比,估计在所述重量比下被占据的孔体积为大约40%至82%。优选地,硅与多孔碳骨架的重量比在[0.9
×
p1至1.8
×
p1]:1的范围内,这表明被占据的孔体积为大约40%至78%。
[0061]
优选地,硅与多孔碳骨架的重量比为至少0.95
×
p1,或至少1
×
p1,或至少1.05
×
p1,或至少1.1
×
p1,或至少1.15
×
p1,或至少1.2
×
p1。
[0062]
优选地,硅与多孔碳骨架的重量比为至多1.85
×
p1,或至多1.8
×
p1,或至多1.75
×
p1,或至多1.7
×
p1,或至多1.65
×
p1,或至多1.6
×
p1。
[0063]
例如,硅与多孔碳骨架的重量比可以在[0.95
×
p1至1.85
×
p1]:1的范围内,或在[0.95
×
p1至1.8
×
p1]:1的范围内,或在[1
×
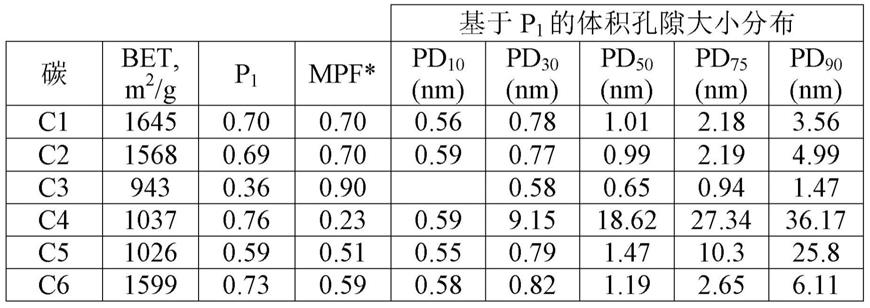
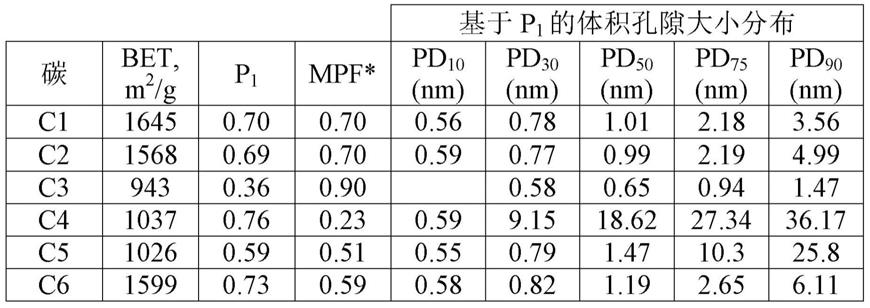
p1至1.8
×
p1]:1的范围内,或在[1.05
×
p1至1.75
×
p1]:1的范围内,或在[1.1
×
p1至1.7
×
p1]:1的范围内,或在[1.15
×
p1至1.7
×
p1]:1的范围内,或在[1.2
×
p1至1.65
×
p1]:1的范围内,或在[1.25
×
p1至1.65
×
p1]:1的范围内,或在[1.3
×
p1至1.6
×
p1]:1的范围内,或在[1.35
×
p1至1.6
×
p1]:1的范围内,或在[1.4
×
p1至1.55
×
p1]:1的范围内。
[0064]
基于复合粒子的总重量,复合粒子优选地包含小于10重量%的氧,更优选小于5重量%的氧,更优选小于2重量%的氧。优选地,硅和碳一起占复合粒子的至少90重量%,更优选地占复合粒子的至少95重量%。
[0065]
复合粒子可以任选地包括其中完全包封的空隙空间被硅封盖的孔隙,因而防止了电解质进入到空隙空间中。
[0066]
复合粒子中的硅的优选至少80重量%、更优选至少85重量%、更优选至少90重量%、更优选至少95重量%、甚至更优选至少98重量%位于多孔碳骨架的内部孔体积内,使得没有或有非常少的硅位于复合粒子的外表面上。
[0067]
本发明的颗粒材料的特征可以进一步在于它们在热重分析(tga)下在空气中的性能。优选地,当通过tga在空气中以10℃/min的升温速率分析所述颗粒材料时,所述颗粒材料中的硅含量的不超过10%、更优选不超过5%、更优选不超过2%在800℃未被氧化。
[0068]
未被氧化的硅的量的测定得自这些材料的特征tga追踪。在大约300
‑
500℃的质量增大对应于硅最初氧化为sio2,然后是在大约500
‑
600℃当碳被氧化为co2气体时的质量损
失。在高于大约600℃时,存在对应于硅继续转化为sio2的进一步质量增大,随着硅氧化完成而在高于1000℃增加至渐近值。
[0069]
为了此分析的目的,假设高于800℃的任何质量增大对应于硅氧化为sio2,并且在氧化完成时的总质量为sio2。这使得能够根据下式确定在800℃未被氧化的硅的百分比(作为硅总量的比例):
[0070]
z=1.875
×
[(m
f
‑
m
800
)/m
f
]
×
100%
[0071]
其中z是在800℃未被氧化的硅的百分比,m
f
是在氧化完成时的样品的质量,并且m
800
是在800℃的样品的质量。
[0072]
不受理论束缚,应理解,在tga下硅被氧化的温度大致对应于在硅上的氧化物涂层的长度尺度,这是因为氧原子扩散通过氧化物层而被热活化。硅纳米结构的尺寸及其位置限制了氧化物涂层厚度的长度尺度。因此,应理解,与硅在粒子表面上的沉积物相比,沉积在微孔和较小的介孔中的硅将会在更低的温度氧化,因为在这些结构上存在必然更薄的氧化物涂层。因此,根据本发明的优选材料表现出硅在低温的基本上完全氧化,这与位于微孔和较小的介孔中的硅纳米结构的小长度尺度相符。
[0073]
优选地,通过氮气吸附测量的复合粒子中的微孔和任选的介孔的总体积(即,在硅的存在下)为至多0.15
×
p1,或至多0.10
×
p1,或至多0.05
×
p1,或至多0.02
×
p1。
[0074]
可以通过元素分析来确定硅与多孔碳骨架的重量比。使用元素分析确定复合粒子中的硅和碳两者的重量百分比。任选地,氢、氮和氧的量也可以通过元素分析来确定。优选地,还使用元素分析确定在单独的多孔碳骨架中的碳(和任选的氢、氮和氧)的重量百分比。确定在单独的多孔碳骨架中的碳的重量百分比考虑了以下可能性:多孔碳骨架在其分子骨架内含有少量的杂原子。一起进行的两种测量使得能够可靠地确定相对于整个多孔碳骨架的硅的重量百分比。
[0075]
优选地通过icp
‑
oes(诱导耦合等离子体
‑
光学发射光谱法)来确定硅含量。可商购获得多种icp
‑
oes仪器,比如7000系列的icp
‑
oes分析仪(可由thermofisher scientific获得)。优选地通过ir吸收来确定复合粒子和单独的多孔碳骨架的碳含量(以及如果需要的话,氢含量、氮含量和氧含量)。合适的用于确定碳含量、氢含量、氮含量和氧含量的仪器是micro元素分析仪(可由leco corporation获得)。
[0076]
复合粒子优选具有低的总氧含量。氧可以例如作为多孔碳骨架的一部分或者作为在任何暴露的硅表面上的氧化物层存在于复合粒子中。优选地,复合粒子的总氧含量低于15重量%,更优选低于10重量%,更优选低于5重量%,例如低于2重量%,或低于1重量%,或低于0.5重量%。
[0077]
硅可以任选地包含少量的一种或多种掺杂剂。合适的掺杂剂包括硼和磷、其他n型或p型掺杂剂、氮或锗。优选地,掺杂剂以基于硅和一种或多种掺杂剂的总量计不超过2重量%的总量存在。
[0078]
为了避免疑义,如本文中使用的术语“粒径”是指等效球径(esd),即与给定粒子具有相同体积的球体的直径,其中粒子体积应理解为包括任何粒子内孔隙的体积。如本文中使用的术语“d
50”和“d
50
粒径”是指体积中值粒径,即,测得50%体积的粒子群体低于某一直径时的该直径。如本文中使用的术语“d
10”和“d
10
粒径”是指第10百分位的体积中值粒径,即,测得10%体积的粒子群体低于某一直径时的该直径。如本文中使用的术语“d
90”和“d
90
粒径”是指第90百分位的体积中值粒径,即,测得90%体积的粒子群体低于某一直径时的该直径。
[0079]
应将在本文中用来限定粒径分布的术语“dn”与如上所述在本文中用来限定孔径分布的术语“pd
n”区分开。
[0080]
可以根据iso 13320:2009通过常规激光衍射技术来确定粒径和粒度分布。激光衍射依赖于以下原理:粒子将以一定角度散射光,所述角度根据粒子的大小而变化,并且多个粒子将会产生由可以与粒度分布相关的强度和角度限定的散射光图案。可商购获得多种激光衍射仪器用于粒度分布的快速且可靠的确定。除非另有说明,否则本文中指明或报告的粒度分布测量是通过来自malvern instruments的常规malvern mastersizer
tm 3000粒度分析仪测量的。malvern mastersizer
tm 3000粒度分析仪通过将氦
‑
氖气体激光束投射通过含有悬浮于水溶液中的所关注粒子的透明池来运行。撞击到粒子的光线散射通过与粒度成反比的角度,并且光电探测器阵列以多个预定角度测量光的强度,并且使用标准理论原理通过计算机处理在不同角度测得的强度以确定粒度分布。本文中报告的激光衍射值使用粒子在蒸馏水中的湿分散体获得。粒子折射率为3.50,并且分散剂指数为1.330。使用米氏散射模型(mie scattering model)来计算粒度分布。
[0081]
复合粒子的d
50
粒径可以在1至30μm的范围内。任选地,d
50
粒径可以是至少1μm,或至少2μm,或至少3μm,或至少4μm,或至少5μm。任选地,d
50
粒径可以是至多20μm,或至多18μm,或至多16μm,或至多14μm,或至多12μm,或至多10μm。
[0082]
例如,复合粒子的d
50
粒径可以在1至20μm的范围内,或在1至18μm的范围内,或在1至16μm的范围内,或在2至16μm的范围内,或在2至14μm的范围内,或在3至12μm的范围内,或在4至10μm的范围内。在这些尺寸范围内且具有本文所述的孔隙率和孔径分布的粒子理想地适用于金属离子电池的阳极,原因在于其在浆料中的分散性、其结构稳健性、其随着反复充电
‑
放电循环的容量保持率以及其对形成具有在20至50μm常规范围内的均匀厚度的致密电极层的适用性。
[0083]
复合粒子的d10粒径优选为至少0.5μm,或至少0.8μm,或至少1μm,或至少1.5μm,或至少2μm。通过将d
10
粒径保持在0.5μm以上,减少了不期望的亚微米尺寸粒子的团聚的可能性,得到改善的颗粒材料分散性和改善的容量保持率。
[0084]
复合粒子的d
90
粒径优选为至多50μm,或至多40μm,或至多30μm,或至多25μm,或至多20μm。非常大的粒子的存在导致粒子在电极活性层中的不均匀地形成填充物,由此破坏致密电极层、特别是厚度在20至50μm范围内的电极层的形成。因此,优选的是,d
90
粒径为至多40μm,并且更优选甚至更小。
[0085]
复合粒子优选具有窄的尺寸分布跨度。例如,粒度分布跨度(定义为(d
90
‑
d
10
)/d
50
)优选为5以下,更优选4以下,更优选3以下,更优选2以下,并且最优选1.5以下。通过保持窄的尺寸分布跨度,能够更容易地实现粒子到致密电极层中的高效填充。
[0086]
复合粒子的形状可以是类球形。如本文中所定义的类球形粒子可以包括球形粒子和椭球形粒子两者,并且本发明的复合粒子的形状可以适宜地参照本发明的粒子的球形度和长径比进行限定。发现类球形粒子特别好地适合于在浆料中的分散体,而不形成团聚体。另外,出人意料地发现,当与不规则形貌的多孔粒子和多孔粒子片段相比时,使用多孔类球形粒子提供了强度的进一步改善。
[0087]
物体的球形度常规地定义为球体的表面积与物体的表面积的比率,其中所述物体和所述球体具有相同的体积。然而,实际上难以测量在微米尺度的个体粒子的表面积和体积。然而,可以通过扫描电子显微镜(sem)和通过动态图像分析来获得微米尺度粒子的高度精确的二维投影,其中使用数字摄像机来记录粒子投射的阴影。如本文中使用的术语“球形度”应被理解为粒子投影的面积与圆形的面积的比率,其中所述粒子投影和圆形具有相同的周长。因此,对于个体粒子来说,球形度s可以定义为:
[0088][0089]
其中a
m
是测得的粒子投影的面积,并且c
m
是测得的粒子投影的周长。如本文中使用的多个粒子的平均球形度s
av
被定义为:
[0090][0091]
其中n表示群体中的粒子的数量。
[0092]
如本文中使用的,应用于本发明的复合粒子的术语“类球形”应被理解为是指平均球形度为至少0.70的材料。优选地,本发明的多孔类球形粒子的平均球形度为至少0.85,更优选至少0.90,更优选至少0.92,更优选至少0.93,优选至少0.94,更优选至少0.95。任选地,多孔类球形粒子的平均球形度可以是至少0.96,或至少0.97,或至少0.98,或至少0.99。
[0093]
要理解的是,在不是完美类球形的任何粒子的情况下,二维粒子投影的周长和面积将会取决于粒子的取向。然而,可以通过报告有具有随机取向的多个粒子获得的作为平均值的球形度和长径比来抵消粒子取向的影响。可商购获得多种sem和动态图像分析仪器,使得能够快速且可靠地确定颗粒材料的球形度和长径比。除非另外说明,否则如本文中规定或报告的球形度值通过来自retsch technology gmbh的camsizer xt粒子分析仪测量。camsizer xt是一种动态图像分析仪器,其能够以100mg至100g的样品体积获得高度精确的颗粒材料的尺寸和形状的分布,使得能够直接通过该仪器计算出诸如平均球形度和长径比之类的性能。
[0094]
本发明的复合粒子的bet表面积优选为至多100m2/g,或至多80m2/g,或至多60m2/g,或至多50m2/g,或至多40m2/g,或至多30m2/g,或至多25m2/g,或至多20m2/g,或至多15m2/g,或至多10m2/g。如本文中使用的术语“bet表面积”应被认为是指根据iso 9277,使用brunauer
‑
emmett
‑
teller原理,由气体分子在固体表面上的物理吸附的测量结果计算出的每单位质量的表面积。通常,低bet表面积是优选的,以使在包含本发明的颗粒材料的阳极的首次充电
‑
放电循环期间在复合粒子表面处的固体电解质界面(sei)层的形成最小化。然而,过低的bet表面积由于电活性材料本体对周围电解质中的金属离子的不可接近性而导致不可接受地低的充电率和容量限制。例如,bet表面积优选为至少0.1m2/g,或至少1m2/g,或至少2m2/g,或至少5m2/g。例如,bet表面积可以在1m2/g至25m2/g的范围内,更优选在2至15m2/g的范围内。
[0095]
本发明的颗粒材料在首次锂化的充电比容量一般为1200至2000mah/g。优选地,本发明的颗粒材料在首次锂化的充电比容量为至少1400mah/g。
[0096]
本发明的复合粒子适宜地通过将含硅前体化学气相渗透(cvi)到多孔碳骨架的孔隙结构中来制备。如本文中使用的,cvi是指其中将气态含硅气体在表面上热分解以形成在表面处的元素硅和气态副产物的过程。
[0097]
合适的气态含硅前体包括硅烷(sih4)、硅烷衍生物(例如,二硅烷、三硅烷和四硅烷)和三氯硅烷(sihcl3)。含硅前体可以以纯净的形式使用,或者更通常地作为用惰性载气比如氮气或氩气稀释的混合物使用。例如,含硅前体可以以基于含硅前体和惰性载气的总体积计在0.5
‑
20体积%、或1
‑
10体积%或1
‑
5体积%范围内的量使用。cvi过程适宜地在总压为101.3kpa(即1atm)的情况下以低的硅前体分压进行,剩余分压使用惰性填充气体比如氢气、氮气或氩气补足到大气压。使用在400
‑
700℃范围内的沉积温度,例如450
‑
550℃或450
‑
500℃。cvi过程可以适宜地在固定床反应器、回转窑或流化床反应器(包括喷动床反应器)中进行。
[0098]
作为固定床反应器法的一个实例,在不锈钢板上沿其长度以1mm的恒定厚度放置1.8g的颗粒多孔骨架。然后将该板放置到位于甑式炉的热区中的具有气体入口和出口管线的外径60mm的不锈钢管中。将炉管在室温用氮气吹扫30分钟,然后将样品温度升高至450
‑
500℃。调整氮气流速以确保在炉管中的至少90秒的气体停留时间,并且将氮气流速在该速率保持30分钟。然后,将气体供应从氮气切换为1.25体积%浓度的甲硅烷在氮气中的混合物。甲硅烷的进料在反应器压力保持在101.3kpa(1atm)的情况下在5小时的时段内进行。在进料完成后,将气体流速保持不变,同时使用氮气从炉中吹扫硅烷。将炉在氮气下吹扫30分钟,然后在数小时内冷却至室温。然后在两小时的时间内通过将气流从氮气切换为来自压缩空气供应的空气而将气氛逐渐切换为空气。
[0099]
本发明的颗粒材料可以任选地包含导电碳涂层。适宜地,可以通过化学气相沉积(cvd)法获得导电碳涂层。cvd是一种本领域中众所周知的方法,并且包括将挥发性含碳气体(例如乙烯)热分解到颗粒材料的表面上。备选地,可以通过将含碳化合物的溶液沉积到颗粒材料的表面上然后进行热解来形成碳涂层。导电碳涂层有足够的渗透性,以使得锂能够接近复合粒子的内部,而没有过度的阻力,从而不降低复合粒子的倍率性能。例如,碳涂层的厚度可以适宜地在2至30nm的范围内。任选地,碳涂层可以是多孔的,和/或可以仅部分覆盖复合粒子的表面。
[0100]
碳涂层具有以下优点:其通过使任何表面缺陷平滑并且通过填充任何残留的表面微孔而进一步减小颗粒材料的bet表面积,从而进一步减少首次循环损失。另外,碳涂层提高了复合粒子的表面的导电性,从而减少了对电极组合物中的导电添加剂的需求,并且还产生了用于形成稳定sei层的改善表面,从而得到改善的在循环时的容量保持率。
[0101]
根据本发明的第一方面,还提供了根据以下方面1
‑
1至1
‑
25的颗粒材料。
[0102]
方面1
‑
1:根据本发明第一方面所述的颗粒材料,其中:
[0103]
(i)微孔体积分数在0.55至0.85的范围内;
[0104]
(ii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.8
×
p1]:1的范围内。
[0105]
方面1
‑
2:根据本发明第一方面所述的颗粒材料,其中:
[0106]
(i)p1在0.45至0.75的范围内;
[0107]
(ii)微孔体积分数在0.55至0.8的范围内;
[0108]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0109]
方面1
‑
3:根据本发明第一方面所述的颗粒材料,其中:
[0110]
(i)p1在0.45至0.75的范围内;
[0111]
(ii)微孔体积分数在0.55至0.75的范围内;
[0112]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0113]
方面1
‑
4:根据本发明第一方面所述的颗粒材料,其中:
[0114]
(i)p1在0.45至0.75的范围内;
[0115]
(ii)微孔体积分数在0.6至0.75的范围内;
[0116]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0117]
方面1
‑
5:根据本发明第一方面所述的颗粒材料,其中:
[0118]
(i)p1在0.5至0.75的范围内;
[0119]
(ii)微孔体积分数在0.55至0.8的范围内;
[0120]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0121]
方面1
‑
6:根据本发明第一方面所述的颗粒材料,其中:
[0122]
(i)p1在0.5至0.75的范围内;
[0123]
(ii)微孔体积分数在0.55至0.75的范围内;
[0124]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0125]
方面1
‑
7:根据本发明第一方面所述的颗粒材料,其中:
[0126]
(i)p1在0.5至0.75的范围内;
[0127]
(ii)微孔体积分数在0.6至0.75的范围内;
[0128]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0129]
方面1
‑
8:根据本发明第一方面所述的颗粒材料,其中:
[0130]
(i)p1在0.5至0.7的范围内;
[0131]
(ii)微孔体积分数在0.55至0.8的范围内;
[0132]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0133]
方面1
‑
9:根据本发明第一方面所述的颗粒材料,其中:
[0134]
(i)p1在0.5至0.7的范围内;
[0135]
(ii)微孔体积分数在0.55至0.75的范围内;
[0136]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0137]
方面1
‑
10:根据本发明第一方面所述的颗粒材料,其中:
[0138]
(i)p1在0.5至0.7的范围内;
[0139]
(ii)微孔体积分数在0.6至0.75的范围内;
[0140]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0141]
方面1
‑
11:根据本发明第一方面所述的颗粒材料,其中:
[0142]
(i)p1在0.45至小于0.6的范围内;
[0143]
(ii)微孔体积分数在0.55至0.8的范围内;
[0144]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0145]
方面1
‑
12:根据本发明第一方面所述的颗粒材料,其中:
[0146]
(i)p1在0.45至小于0.6的范围内;
[0147]
(ii)微孔体积分数在0.55至0.75的范围内;
[0148]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0149]
方面1
‑
13:根据本发明第一方面所述的颗粒材料,其中:
[0150]
(i)p1在0.45至小于0.6的范围内;
[0151]
(ii)微孔体积分数在0.6至0.75的范围内;
[0152]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[0.9
×
p1至1.6
×
p1]:1的范围内。
[0153]
方面1
‑
14:根据本发明第一方面所述的颗粒材料,其中:
[0154]
(i)p1在0.45至0.75的范围内;
[0155]
(ii)微孔体积分数在0.55至0.8的范围内;
[0156]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0157]
(iv)pd
90
孔径为至多10nm。
[0158]
方面1
‑
15:根据本发明第一方面所述的颗粒材料,其中:
[0159]
(i)p1在0.45至0.75的范围内;
[0160]
(ii)微孔体积分数在0.55至0.75的范围内;
[0161]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0162]
(iv)pd
90
孔径为至多10nm。
[0163]
方面1
‑
16:根据本发明第一方面所述的颗粒材料,其中:
[0164]
(i)p1在0.45至0.75的范围内;
[0165]
(ii)微孔体积分数在0.6至0.75的范围内;
[0166]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0167]
(iv)pd
90
孔径为至多10nm。
[0168]
方面1
‑
17:根据本发明第一方面所述的颗粒材料,其中:
[0169]
(i)p1在0.5至0.75的范围内;
[0170]
(ii)微孔体积分数在0.55至0.8的范围内;
[0171]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0172]
(iv)pd
90
孔径为至多10nm。
[0173]
方面1
‑
18:根据本发明第一方面所述的颗粒材料,其中:
[0174]
(i)p1在0.5至0.75的范围内;
[0175]
(ii)微孔体积分数在0.55至0.75的范围内;
[0176]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0177]
(iv)pd
90
孔径为至多10nm。
[0178]
方面1
‑
19:根据本发明第一方面所述的颗粒材料,其中:
[0179]
(i)p1在0.5至0.75的范围内;
[0180]
(ii)微孔体积分数在0.6至0.75的范围内;
[0181]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0182]
(iv)pd
90
孔径为至多10nm。
[0183]
方面1
‑
20:根据本发明第一方面所述的颗粒材料,其中:
[0184]
(i)p1在0.5至0.7的范围内;
[0185]
(ii)微孔体积分数在0.55至0.8的范围内;
[0186]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0187]
(iv)pd
90
孔径为至多10nm。
[0188]
方面1
‑
21:根据本发明第一方面所述的颗粒材料,其中:
[0189]
(i)p1在0.5至0.7的范围内;
[0190]
(ii)微孔体积分数在0.55至0.75的范围内;
[0191]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0192]
(iv)pd
90
孔径为至多10nm。
[0193]
方面1
‑
22:根据本发明第一方面所述的颗粒材料,其中:
[0194]
(i)p1在0.5至0.7的范围内;
[0195]
(ii)微孔体积分数在0.6至0.75的范围内;
[0196]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0197]
(iv)pd
90
孔径为至多10nm。
[0198]
方面1
‑
23:根据本发明第一方面所述的颗粒材料,其中:
[0199]
(i)p1在0.45至小于0.6的范围内;
[0200]
(ii)微孔体积分数在0.55至0.8的范围内;
[0201]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0202]
(iv)pd
90
孔径为至多10nm。
[0203]
方面1
‑
24:根据本发明第一方面所述的颗粒材料,其中:
[0204]
(i)p1在0.45至小于0.6的范围内;
[0205]
(ii)微孔体积分数在0.55至0.75的范围内;
[0206]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0207]
(iv)pd
90
孔径为至多10nm。
[0208]
方面1
‑
25:根据本发明第一方面所述的颗粒材料,其中:
[0209]
(i)p1在0.45至小于0.6的范围内;
[0210]
(ii)微孔体积分数在0.6至0.75的范围内;
[0211]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.5
×
p1]:1的范围内;
[0212]
(iv)pd
90
孔径为至多10nm。
[0213]
方面1
‑
26:根据本发明第一方面所述的颗粒材料,其中:
[0214]
(i)p1在0.45至0.75的范围内;
[0215]
(ii)微孔体积分数在0.55至0.8的范围内;
[0216]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.8
×
p1]:1的范围内;
[0217]
(iv)pd
90
孔径为至多8nm。
[0218]
方面1
‑
27:根据本发明第一方面所述的颗粒材料,其中:
[0219]
(i)p1在0.45至0.75的范围内;
[0220]
(ii)微孔体积分数在0.55至0.8的范围内;
[0221]
(iii)复合粒子中的硅与多孔碳骨架的重量比在[1
×
p1至1.8
×
p1]:1的范围内;
[0222]
(iv)pd
90
孔径为至多6nm。
[0223]
根据本发明,应理解,在上述方面1
‑
1至1
‑
27范围内的与本发明第一方面相关的本文所公开的优选的/任选的特征也被认为是方面1
‑
1至1
‑
27的优选的/任选的特征。同样,在上述方面1
‑
1至1
‑
27范围内的从属权利要求的任何特征也应被解释为如同那些权利要求也
从属于方面1
‑
1至1
‑
27。
[0224]
在本发明的第二方面,提供了一种包含根据本发明第一方面的颗粒材料和至少一种其他组分的组合物。特别地,提供了一种包含根据本发明第一方面的颗粒材料和选自以下各项的至少一种其他组分的组合物:(i)粘结剂;(ii)导电添加剂;和(iii)另外的颗粒电活性材料。根据本发明的第二方面的组合物可用作电极组合物,因此可以用于形成电极的活性层。
[0225]
用于制备本发明第二方面的组合物的颗粒材料可以具有关于本发明第一方面被描述为优选的或任选的特征中的任一个。
[0226]
组合物可以是混合型电极组合物,其包含根据本发明第一方面的颗粒材料和至少一种另外的颗粒电活性材料。另外的颗粒电活性材料的实例包括石墨、硬碳、硅、锡、锗、铝和铅。至少一种另外的颗粒电活性材料优选地选自石墨和硬碳,并且最优选地,至少一种另外的颗粒电活性材料是石墨。
[0227]
在混合型电极组合物的情况下,基于组合物的总干重,组合物优选地包含15至60重量%、或20至50重量%、或30至50重量%的根据本发明第一方面的颗粒材料。
[0228]
至少一种另外的颗粒电活性材料适宜地以20至70重量%、或25至65重量%、或30至60重量%的量的至少一种另外的颗粒电活性材料存在。
[0229]
至少一种另外的颗粒电活性材料的d50粒径优选在10至50μm的范围内,优选在10至40μm的范围内,更优选在10至30μm的范围内,并且最优选在10至25μm的范围内,例如在15至25μm的范围内。
[0230]
至少一种另外的颗粒电活性材料的d
10
粒径优选为至少5μm,更优选至少6μm,更优选至少7μm,更优选至少8μm,更优选至少9μm,并且仍更优选至少10μm。
[0231]
至少一种另外的颗粒电活性材料的d
90
粒径优选为至多100μm,更优选地至多80μm,更优选地至多60μm,更优选地至多50μm,并且最优选至多40μm。
[0232]
至少一种另外的颗粒电活性材料优选地选自包含碳的粒子、石墨粒子和/或硬碳粒子,其中石墨粒子和硬碳粒子的d
50
粒径在10至50μm的范围内。仍更优选地,所述至少一种另外的颗粒电活性材料选自石墨粒子,其中石墨粒子的d
50
粒径在10至50μm的范围内。
[0233]
组合物也可以是非混合型(或“高负载”)的电极组合物,其基本上不含另外的颗粒电活性材料。在此情况下,术语“基本上不含另外的颗粒电活性材料”应被解释为意指基于组合物的总干重,组合物包含小于15重量%、优选地小于10重量%、优选地小于5重量%、优选地小于2重量%、更优选地小于1重量%、更优选地小于0.5重量%的任何另外的电活性材料(即,另外的在电池的充电和放电期间能够嵌入和释放金属离子的材料)。
[0234]
基于组合物的总干重,此类型的“高负载”电极组合物优选地包含至少50重量%、或至少60重量%、或至少70重量%、或至少80重量%、或至少90重量%的根据本发明第一方面的颗粒材料。
[0235]
组合物可以任选地包含粘结剂。粘结剂起将组合物粘附至集流体并且保持组合物的完整性的作用。根据本发明可以使用的粘结剂的实例包括聚偏二氟乙烯(pvdf)、聚丙烯酸(paa)及其碱金属盐、改性聚丙烯酸(mpaa)及其碱金属盐、羧甲基纤维素(cmc)、改性羧甲基纤维素(mcmc)、羧甲基纤维素钠(na
‑
cmc)、聚乙烯醇(pva)、藻酸盐及其碱金属盐、丁苯橡胶(sbr)和聚酰亚胺。组合物可以包含多种粘结剂的混合物。优选地,粘结剂包括选自以下
各项的聚合物:聚丙烯酸(paa)及其碱金属盐,和改性聚丙烯酸(mpaa)及其碱金属盐,sbr和cmc。
[0236]
粘结剂可以适宜地以基于组合物的总干重计0.5至20重量%、优选1至15重量%、优选2至10重量%并且最优选5至10重量%的量存在。
[0237]
粘结剂可以任选地与一种或多种改变粘结剂的性质的添加剂组合存在,所述添加剂比如是交联促进剂、偶联剂和/或促粘剂。
[0238]
组合物可以任选地包含一种或多种导电添加剂。优选的导电添加剂是非电活性材料,其被包含是为了提高组合物的电活性组分之间的导电性以及组合物的电活性组分与集流体之间的导电性。导电添加剂可以适宜地选自炭黑、碳纤维、碳纳米管、石墨烯、乙炔黑、科琴黑、金属纤维、金属粉末和导电金属氧化物。优选的导电添加剂包括炭黑和碳纳米管。
[0239]
一种或多种导电添加剂可以适宜地以基于组合物的总干重计0.5至20重量%、优选1至15重量%、优选2至10重量%并且最优选5至10重量%的总量存在。
[0240]
在第三方面,本发明提供了一种电极,所述电极包含与集流体电接触的根据本发明第一方面限定的颗粒材料。用于制备本发明第三方面的电极的颗粒材料可以具有关于本发明第一方面被描述为优选的或任选的特征中的任一个。
[0241]
如本文中使用的,术语“集流体”是指能够将电流携带至组合物中的电活性粒子以及从组合物中的电活性粒子携带出电流的任何导电基底。可以用作集流体的材料的实例包括铜、铝、不锈钢、镍、钛和烧结碳。铜是优选的材料。集流体一般是厚度为3至500μm的箔或网状物的形式。可以将本发明的颗粒材料施用至集流体的一个表面或两个表面,达到优选在10μm至1mm、例如20至500μm、或50至200μm范围内的厚度。
[0242]
优选地,电极包含与集流体电接触的根据本发明第二方面限定的组合物。组合物可以具有关于本发明第二方面被描述为优选的或任选的特征中的任一个。
[0243]
本发明第三方面的电极可以适宜地通过以下方式制备:将本发明的颗粒材料(任选地是本发明的组合物的形式)与溶剂和任选的一种或多种粘度改性添加剂组合而形成浆料。然后将浆料流延到集流体的表面上,并且去除溶剂,从而在集流体的表面上形成电极层。可以视情况进行另外的步骤,比如用于使任何粘结剂固化的热处理和/或电极层的压延。电极层的厚度适宜地在20μm至2mm、优选20μm至1mm、优选20μm至500μm、优选20μm至200μm、优选20μm至100μm、优选20μm至50μm的范围内。
[0244]
备选地,可以例如通过以下方式将浆料形成为包含本发明的颗粒材料的自立式膜或垫:将浆料流延到合适的流延模板上,去除溶剂以及然后去除流延模板。所得膜或垫是有粘性的自立物体的形式,然后可以通过已知方法将其与集流体结合。
[0245]
可以使用本发明第三方面的电极作为金属离子电池的阳极。因此,在第四方面,本发明提供了一种可充电金属离子电池,其包括:阳极,该阳极包括如上所述的电极;阴极,该阴极包含能够释放和再吸收金属离子的阴极活性材料;以及在阳极和阴极之间的电解质。
[0246]
金属离子优选为锂离子。更优选地,本发明的可充电金属离子电池是锂离子电池,并且阴极活性材料能够释放和接受锂离子。
[0247]
阴极活性材料优选为基于金属氧化物的复合材料。合适的阴极活性材料的实例包括licoo2、lico
0.99
al
0.01
o2、linio2、limno2、lico
0.5
ni
0.5
o2、lico
0.7
ni
0.3
o2、lico
0.8
ni
0.2
o2、lico
0.82
ni
0.18
o2、lico
0.8
ni
0.15
al
0.05
o2、lini
0.4
co
0.3
mn
0.3
o2和lini
0.33
co
0.33
mn
0.34
o2。阴极集流
体通常具有3至500μm的厚度。可以用作阴极集流体的材料的实例包括铝、不锈钢、镍、钛和烧结碳。
[0248]
电解质适宜地是含有金属盐(例如锂盐)的非水性电解质,并且可以包括但不限于非水性电解液、固体电解质和无机固体电解质。可以使用的非水性电解质溶液的实例包括非质子有机溶剂,比如碳酸亚丙酯、碳酸亚乙酯、碳酸亚丁酯、碳酸二甲酯、碳酸二乙酯、γ
‑
丁内酯、1,2
‑
二甲氧基乙烷、2
‑
甲基四氢呋喃、二甲亚砜、1,3
‑
二氧戊环、甲酰胺、二甲基甲酰胺、乙腈、硝基甲烷、甲酸甲酯、乙酸甲酯、磷酸三酯、三甲氧基甲烷、环丁砜、甲基环丁砜和1,3
‑
二甲基
‑2‑
咪唑烷酮。
[0249]
有机固体电解质的实例包括聚乙烯衍生物、聚氧化乙烯衍生物、聚氧化丙烯衍生物、磷酸酯聚合物、聚酯硫化物、聚乙烯醇、聚偏二氟乙烯和含有离子解离基团的聚合物。
[0250]
无机固体电解质的实例包括锂盐的氮化物、卤化物和硫化物(比如li5ni2、li3n、lii、lisio4、li2sis3、li4sio4、lioh和li3po4)。
[0251]
锂盐适宜地可溶于所选择的溶剂或溶剂混合物中。合适的锂盐的实例包括licl、libr、lii、liclo4、libf4、libc4o8、lipf6、licf3so3、liasf6、lisbf6、lialcl4、ch3so3li和cf3so3li。
[0252]
在电解质是非水性有机溶液的情况下,金属离子电池优选地具有介于阳极和阴极之间的隔膜。隔膜一般由具有高离子渗透率和高机械强度的绝缘材料形成。隔膜一般具有0.01至100μm的孔径和5至300μm的厚度。合适的电极隔膜的实例包括微孔聚乙烯膜。
[0253]
可以用聚合物电解质材料代替隔膜,并且在这样的情况下,聚合物电解质材料存在于复合阳极层和复合阴极层两者内。聚合物电解质材料可以是固体聚合物电解质或凝胶型聚合物电解质。
[0254]
在第五方面,本发明提供了根据本发明第一方面限定的颗粒材料作为阳极活性材料的用途。优选地,颗粒材料是根据本发明第二方面限定的组合物的形式,并且最优选地,组合物包含如上所限定的一种或多种另外的颗粒电活性材料。
实施例
[0255]
在以下实施例中使用的多孔碳骨架c1至c5具有表1中所列的特性。
[0256]
表1
[0257][0258]
*mpf=微孔分数
[0259]
实施例1
‑
在固定床反应器中的复合粒子的制各
[0260]
通过将1.8g的具有表1中所列性质的颗粒多孔骨架以1mm的恒定厚度沿不锈钢板的长度放置在不锈钢板上来制备硅
‑
碳复合粒子。然后将该板放置到位于甑式炉的热区中的具有气体入口和出口管线的外径60mm的不锈钢管中。将炉管在室温用氮气吹扫30分钟,然后将样品温度升高至450
‑
500℃。调整氮气流速以确保在炉管中的至少90秒的气体停留时间,并且将氮气流速在该速率保持30分钟。然后,将气体供应从氮气切换为1.25体积%浓度的甲硅烷在氮气中的混合物。甲硅烷的进料在反应器压力保持在101.3kpa(1atm)的情况下在5小时的时段内进行。在进料完成后,将气体流速保持不变,同时使用氮气从炉中吹扫硅烷。将炉在氮气下吹扫30分钟,然后在数小时内冷却至室温。然后在两小时的时间内通过将气流从氮气切换为来自压缩空气供应的空气而将气氛逐渐切换为空气。
[0261]
根据实施例1制备的复合材料具有以下表2中所列的特性。
[0262]
表2
[0263][0264]
*粗si=在800℃氧化的硅的重量%,通过上述tga法测定并且使用关于z的公式计算
[0265]
**比较例
[0266]
数据显示,多孔碳骨架必须具有仔细控制的粒度分布以使得硅能够高效地渗透到微孔中。
[0267]
对样品s3的tga分析证明,如果微孔分数过高,则不能将硅渗透到多孔碳骨架的孔隙中,并且代替地,硅沉积在多孔碳骨架的表面上。这由通过tga分析测得的高含量的粗硅证实。认为这是因为初始硅沉积快速地阻塞孔隙开口,阻止硅烷气体接近内部孔隙空间。因此,需要最低量的介孔以确保硅烷气体到微孔的足够输送。然而,对样品s4的tga分析证明,如果微孔分数过低,则再次得到过量的粗硅。认为微孔体积快速地耗尽,导致在介孔中和在多孔碳骨架表面上均形成较大的硅沉积物。
[0268]
与此相比,对样品s1、s2、s5和s6的tga分析显示出非常低含量的粗硅,这表明硅基本上全部以位于微孔和较小的介孔中的硅纳米结构的形式存在。与粗硅结构相比,这些微细硅纳米结构具有低的对形变的耐性和更高的耐断裂性,因此更好地适合作为锂离子电池中的电活性材料使用。
[0269]
实施例2
‑
在旋转管式炉反应器中的复合粒子的制备
[0270]
通过将5g的具有表1中所列性质的颗粒多孔骨架放置到具有泡形部分(11.4em长)的石英管中来制备硅
‑
碳复合粒子。然后将石英管放置到旋转反应器管式炉内部,所述旋转反应器管式炉具有大约15
×
20cm(l
×
d)的加热区,以及位于与炉的热区相距大约29em处的气体入口和出口管线。在炉内的石英管顺时针旋转大约315
°
,然后逆时针旋转,从而连续移
动/旋转多孔碳。将炉管在室温用氮气吹扫30分钟,然后将样品温度升高至450
‑
500℃。调整氮气流速以确保在炉管中的至少90秒的气体停留时间,并且将氮气流速在该速率保持30分钟。然后,将气体供应从氮气切换为1.25体积%浓度的甲硅烷在氮气中的混合物。甲硅烷的进料在反应器压力保持在101.3kpa(1atm)的情况下在5小时的时段内进行。在进料完成后,将气体流速保持不变,同时使用氮气从炉中吹扫硅烷。将炉在氮气下吹扫30分钟,然后在数小时内冷却至室温。然后在两小时的时间内通过将气流从氮气切换为来自压缩空气供应的空气而将气氛逐渐切换为空气。
[0271]
根据实施例2制备的复合材料具有以下表3中所列的特性。
[0272]
表3
[0273][0274]
实施例3
‑
电极的制备
[0275]
使用以下方法制备包含表1的颗粒si
‑
c复合材料的阳极和测试电池:
[0276]
用如上所述制备的包含硅系材料的负极制备测试纽扣电池。将carbon super p(导电性碳)在cmc粘结剂中的分散体在thinky
tm
混合器中混合。将硅系材料加入到混合物中并且在thinky
tm
混合器中混合30min。然后加入sbr粘结剂以提供1∶1的cmc∶sbr比,得到硅系材料:cmc/sbr:导电碳的重量比为70%∶16%∶14%的浆料。将浆料在thinky
tm
混合器中再混合30min,然后涂覆到10μm厚的铜基底(集流体)上并在50℃干燥10分钟,随后再在110℃干燥12小时,由此形成包括在铜基底上的活性层的电极。
[0277]
实施例4
‑
全电池制造和循环
[0278]
使用从实施例3的电极切下的0.8cm半径的圆形负极与多孔聚乙烯隔膜和镍锰钴(nmc532)正极来制备全纽扣电池。正极和负极被设计为形成平衡对,使得电极的设计容量比为大约0.9。然后在密封前,将电解质加入到电池中,所述电解质包含处于含有3重量%碳酸亚乙酯的emc/fec(碳酸甲乙酯/氟代碳酸亚乙酯)的7:3溶液中的1m lipf6。
[0279]
对于每种复合材料,制备3个纽扣电池。
[0280]
如下对全纽扣电池进行循环:以c/25的倍率施加恒电流以将阳极锂化,截止电压为4.3v。当达到截止时,施加4.3v的恒电压,直到达到c/100的截止电流。然后将电池在锂化状态下静置10分钟。然后将阳极以c/25的恒电流脱锂,截止电压为2.75v。然后将电池静置10分钟。在此初始循环后,施加c/2的恒电流以将阳极锂化,截止电压为4.3v,然后施加4.3v恒电压,截止电流为c/40,静置时间为5分钟。然后将阳极以c/2的恒电流脱锂,截止为2.75v。然后将此过程重复所需数量的循环。继续充电和放电容量直到1000次循环,并且测定各样品的在100次和300次循环时的容量保持率(cr100、cr300和cr500)。在s2的情况下,还测定了cr1000。此数据连同各样品的第1次锂化容量、第1次脱锂容量和首次循环损失(fcl)一起在表4中提供。
[0281]
计算每次循环的每单位质量硅
‑
碳复合材料的充电(锂化)和放电(脱锂)容量,并且作为第二次循环的放电容量的%,计算每次放电容量的容量保持率值。首次循环损失(fcl)是(1
‑
(第1次脱锂容量/第1次锂化容量))
×
100%。表4中的值是每个样品的3个纽扣
电池的平均值。
[0282]
通过以下表4可以证实,用根据本发明的材料制成的电池具有高的初始容量和一致的循环性能以及高的容量保持率。与粗硅含量较高(10.1重量%)的s7材料相比,粗硅含量较低(4.8重量%)的s2材料在高循环次数时显示出更好的容量保持率。
[0283]
表4
[0284][0285]
实施例5
‑
半电池制造
[0286]
使用从实施例3的电极切割的0.8cm半径的圆形电极以及多孔聚乙烯隔膜、作为对电极的锂箔和电解质来制作纽扣半电池,所述电解质包含处于含有3重量%碳酸亚乙酯的fec∶ec∶emc 1∶5∶14(氟代碳酸亚乙酯/碳酸亚乙酯/碳酸甲乙酯,v/v/v)溶液中的1m lipf6。
[0287]
使用这些半电池来测量活性层的初始体积能量密度(ved1)、首次循环损失(fcl)和首次脱锂容量。相关值在表4中列出。通过以下方式对半电池进行测试:施加c/25的恒电流(其中“c”表示电极的以mah计的比容量,并且“25”是指25小时),以将包括多孔粒子的电极锂化,截止电压为10mv。当达到截止时,施加10mv的恒电压,截止电流为c/100。然后将电池在锂化状态下静置1小时。然后将电极以c/25的恒电流脱锂,截止电压为1v,然后将电池静置1小时。然后施加c/25的恒电流以再次将电池锂化,截止电压为10mv,然后施加10mv恒电压,截止电流为c/100。表5中的值是每个样品的3个电池的平均值。
[0288]
表5
[0289]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。