一种用于nox和cvocs的协同脱除的催化剂及其制备方法和用途
技术领域
1.本发明涉及一种用于nox和cvocs协同脱除的催化剂及其制备方法和用途,属于环境保护中的大气环境污染治理领域。
背景技术:
2.氮氧化物(nox)和含氯挥发性有机物(cvocs,chlorinated volatile organic compounds)是造成大气中雾霾、臭氧超标的主要污染物,对人体健康和生态环境会造成极大的危害。我国垃圾焚烧、钢铁烧结以及有色冶炼等行业所产生的烟气中均含有大量的nox和二噁英,垃圾焚烧炉中的二噁英通常是由cvocs(氯苯、二氯苯等)通过自由基缩合等反应生成,并因其高毒性和持久性,成为目前国内外的关注点。随着国家环保政策的逐渐严格,更多垃圾焚烧、钢铁烧结以及有色冶炼等相关企业将开始实施烟气超低排放控制工程。
3.目前我国垃圾焚烧、钢铁烧结以及有色冶炼等行业一般采用选择性催化还原技术(nh3‑
scr)作为烟气脱硝技术,采用吸附剂喷射结合布袋除尘器作为烟气中的cvocs等物质脱除的主要方法。然而,吸附法一方面会显著增加除尘设备运行负荷,提高体系的运行成本,并且该方法仅仅把cvocs由烟气转移至吸附剂中,并没有真正实现cvocs的完全消除。因此针对垃圾焚烧烟气、钢铁烧结以及有色冶炼等行业的烟气特征,利用现有催化反应装置,开发nox和cvocs协同脱除的双功能催化体系,把nox和含氯挥发性有机物催化分解为n2、co2、h2o和hcl(cl2)等成分,在降低运行成本的同时真正实现nox和cvocs的减量化和无害化,具有重要的经济和社会价值。
4.在目前的nh3‑
scr催化领域中,v2o5‑
wo3/tio2是应用最为广泛的商业scr催化剂,其中钒的含量一般不超过1wt.%,较多用于燃煤发电厂。该类催化剂的反应温度窗口较窄,在300
‑
400℃之间。而相应的垃圾焚烧、钢铁烧结以及有色冶炼等行业所排放得烟气温度较低(<300℃)。传统的v2o5‑
wo3/tio2催化剂不适合垃圾焚烧、钢铁烧结以及有色冶炼等行业的复杂烟气。与此同时,传统的scr催化剂对于cvocs的催化氧化脱除能力较为有限,且反应过程中会产生大量的一氧化碳和多氯有机副产物,因此需要开发新型高效的协同反应催化剂。
5.专利文献1中公开了一种低温耐硫钒钛系脱硝催化剂,其为采用浸渍法制备的在催化剂载体表面负载v2o5、wo3以及ru的催化剂,催化剂的载体为tio2,活性成分为v2o5,助剂为wo3和ru。该催化剂在150~400℃温度下的脱硝效率为90%以上,但是其协同脱除nox和cvocs的效率不能说是令人满意的。
6.引用文献
7.专利文献1:cn106111135a
技术实现要素:
8.发明要解决的问题
9.为了克服上述现有技术的缺点,本发明的目的在于提供一种能够适用于垃圾焚烧、钢铁烧结和有色冶炼等行业烟气的氮氧化物和含氯挥发性有机物的协同脱除的催化剂。
10.用于解决问题的方案
11.为了实现上述目的,本发明采用二氧化铈作为基体,使活性组分掺杂于二氧化铈的晶格中,由此获得了能够以良好的效率协同脱除氮氧化物和含氯挥发性有机物的催化剂。
12.1、本发明提供一种用于氮氧化物nox和含氯挥发性有机物cvocs协同脱除的催化剂,所述催化剂包括基体和活性组分,其特征在于,
13.所述活性组分掺杂于的所述基体的晶格中,
14.其中所述活性组分包括钒,所述基体为二氧化铈。
15.2、根据上述1所述的催化剂,其中钒在所述催化剂中的含量为1
‑
10wt.%、优选3~8wt.%。
16.3、根据上述1或2所述的催化剂,其中所述活性组分还包括贵金属;和/或,贵金属在所述催化剂中的含量为0.1
‑
3wt.%、优选0.5~2wt.%。
17.4、根据上述3所述的催化剂,其中所述贵金属为钌、铂、钯中的一种或多种。
18.5、一种上述1
‑
4任一项所述的催化剂的制备方法,其特征在于,其包括以下步骤:
19.将钒前驱体加入至草酸水溶液中,以得到混合溶液a;
20.将铈前驱体和任选的贵金属前驱体加入所述混合溶液a中,以得到混合溶液b;
21.将柠檬酸加入至所述混合溶液b中;和
22.将所得产物进行干燥、焙烧。
23.6、根据上述5所述的制备方法,其中所述钒前驱体为偏钒酸铵、草酸氧钒、乙酰丙酮氧钒中的一种或多种;和/或,所述贵金属前驱体为亚硝酰硝酸钌、三氯化钌、硝酸铂、硝酸钯中的一种或多种;和/或,所述铈前驱体为硝酸铈、硝酸铈铵中的一种或多种。
24.7、根据上述5或6所述的制备方法,其中在将所得产物进行干燥之前的各步骤中,反应温度为60
‑
100℃、优选70
‑
90℃。
25.8、根据上述4
‑
6任一项所述的制备方法,其中干燥温度为120
‑
200℃;和/或,焙烧温度为400
‑
600℃,焙烧时的升温速度为1
‑
3℃/min。
26.9、根据上述1
‑
4任一项所述的催化剂或者根据上述5
‑
8任一项所述的制备方法获得的催化剂在氮氧化物nox和含氯挥发性有机物cvocs的协同脱除中的用途。
27.10、根据上述9所述的用途,其中所述氮氧化物nox为一氧化氮和二氧化氮中的一种或多种,所述含氯挥发性有机物cvocs为氯苯、二氯苯、二氯甲烷和二氯乙烯中的一种或多种。
28.发明的效果
29.与现有技术相比,本发明的优点在于:
30.1)本发明的催化剂中,活性组分掺杂于二氧化铈晶格中,提供了丰富的酸性位点和氧化还原位点,并且可避免催化剂因积氯导致的中毒失活,因此使得本发明催化剂对氮氧化物nox和含氯挥发性有机物cvocs的协同脱除具有较高活性,具有较宽的反应温度窗口150~400℃,同时在300
‑
400℃的工作范围内具有较高的消除活性、较高的氮气选择性、较
高的二氧化碳选择性以及较高的氯化氢选择性;
31.2)本发明的催化剂具有较高的反应空速,有效地减少了催化剂的用量,极大地降低了成本;
32.3)本发明采用溶胶凝胶法制备,制备方法简单,方便蜂窝式/条状催化剂成型工艺探索。
附图说明
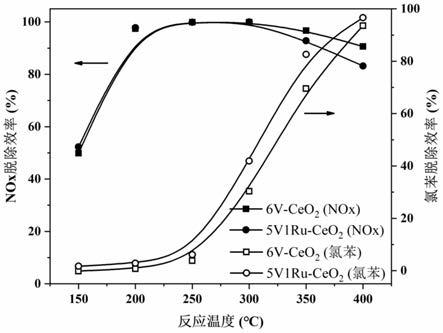
33.图1为6v
‑
ceo2和5v1ru
‑
ceo2在协同脱除过程中氮氧化物和氯苯的脱除效率。
34.图2为6v
‑
ceo2和5v1ru
‑
ceo2在协同脱除过程中氮气的选择性。
35.图3为6v
‑
ceo2和5v1ru
‑
ceo2在协同脱除过程中二氧化碳的选择性。
36.图4为6v
‑
ceo2和5v1ru
‑
ceo2在协同脱除过程中出口的氯化氢浓度。
37.图5为1ru
‑
ceo2在协同脱除过程中氮氧化物和氯苯的脱除效率。
具体实施方式
38.以下,针对本发明的内容进行详细说明。以下所记载的技术特征的说明基于本发明的代表性的实施方案、具体例子而进行,但本发明不限定于这些实施方案、具体例子。需要说明的是:
39.本说明书中,使用“数值a~数值b”表示的数值范围是指包含端点数值a、b的范围。
40.本说明书中,使用“以上”或“以下”表示的数值范围是指包含本数的数值范围。
41.本说明书中,使用“可以”表示的含义包括了进行某种处理以及不进行某种处理两方面的含义。
42.本说明书中,使用“任选”或“任选的”表示某些物质、组分、执行步骤、施加条件等因素使用或者不使用。
43.本说明书中,所使用的单位名称均为国际标准单位名称。
44.本说明书中,如没有特别声明,则“多(个/种)”指的是具有两个/种或两个/种以上的情况。
45.本说明书中,所提及的“一些具体/优选的实施方案”、“另一些具体/优选的实施方案”、“实施方案”等是指所描述的与该实施方案有关的特定要素(例如,特征、结构、性质和/或特性)包括在此处所述的至少一种实施方案中,并且可存在于其它实施方案中或者可不存在于其它实施方案中。另外,应理解,所述要素可以任何合适的方式组合在各种实施方案中。
46.本说明书中,如有出现“室温”、“常温”等,其温度一般可以是15
‑
35℃。
47.<第一方面>
48.本发明的第一方面提供一种催化剂。本发明的催化剂具有活性组分和基体,且活性组分掺杂于基体的晶格中,由此使得本发明的催化剂具有优异的对nox和cvocs的协同脱除活性。
49.本发明的实施方案中,催化剂的活性组分包括钒,催化剂的基体为二氧化铈,并且钒掺杂于二氧化铈的晶格中。其中,钒在催化剂中的含量为1
‑
10wt.%、优选3~8wt.%、更优选4~6wt.%。
50.在本发明的优选实施方案中,催化剂的活性组分还进一步包括贵金属。当存在贵金属时,贵金属在催化剂中的含量为0.1
‑
3wt.%、优选0.5~2wt.%。并且,在此情况下,钒和贵金属均掺杂于二氧化铈的晶格中。
51.在本发明的具体实施方案中,贵金属的实例可包括钌、铂、钯中的一种或多种。
52.本发明的催化剂中,活性组分中的钒掺杂于二氧化铈晶格中后,为催化剂提供了丰富的酸性位点,促进nh3的活化,有利于nox还原的进行。活性组分中的贵金属掺杂于二氧化铈晶格中后,为催化剂提供了丰富的氧化还原位点,促进氧空位的形成,有利于cvocs的氧化分解。钒和贵金属协同促进cvocs分解后残留于催化剂表面的cl物种的脱附释放,避免催化剂因积氯导致的中毒失活。因此,本发明的催化剂具有优异的对nox和cvocs的协同脱除活性和特定选择性。
53.<第二方面>
54.本发明的第二方面提供催化剂的制备方法,所述催化剂与上述<第一方面>所描述或定义的催化剂相同。
55.本发明的催化剂可采用溶胶凝胶法制备。具体地,本发明催化剂的制备方法可包括以下步骤:
56.将钒前驱体加入至草酸水溶液中,以得到混合溶液a;
57.将铈前驱体和任选的贵金属前驱体加入所述混合溶液a中,以得到混合溶液b;
58.将柠檬酸加入至所述混合溶液b中;和
59.将所得产物进行干燥、焙烧。
60.本发明的具体实施方案中,钒前驱体的实例可包括偏钒酸铵、草酸氧钒、乙酰丙酮氧钒中的一种或几种。贵金属前驱体的实例可包括亚硝酰硝酸钌、三氯化钌、硝酸铂、硝酸钯中的一种或多种。铈前驱体的实例可包括硝酸铈、硝酸铈铵中的一种或多种。
61.更具体地,在混合溶液a的制备步骤中,先将草酸加入去离子水中,在转速为400
‑
600转/min的条件下磁力搅拌5
‑
10分钟使其充分溶解以得到草酸水溶液,并使用水浴将溶液加热至60
‑
100℃、优选70
‑
90℃。然后,在保持水浴温度恒定为60
‑
100℃、优选70
‑
90℃的条件下,将钒前驱体加入至草酸水溶液中,并在转速为400
‑
600转/min的条件下磁力搅拌5
‑
10分钟,使钒前驱体充分溶解,从而得到混合溶液a。
62.在混合溶液b的制备步骤中,在保持水浴温度恒定为60
‑
100℃、优选70
‑
90℃的条件下,将任选的贵金属前驱体、铈前驱体加入至混合溶液a中,并在转速为400
‑
600转/min的条件下磁力搅拌5
‑
10分钟使其充分混合。
63.接下来,将柠檬酸加入至混合溶液b中,并在保持水浴温度恒定为60
‑
100℃、优选70
‑
90℃的情况下,在转速为400
‑
600转/min的条件下持续磁力搅拌至溶液成胶状物质。
64.然后,将胶状产物置于鼓风干燥箱中在120
‑
200℃的温度下处理10
‑
14小时,从而获得固体产物。
65.之后,将所得的固体产物置于马弗炉中从室温以1
‑
3℃/min的升温速度慢速升温至400
‑
600℃来进行焙烧2
‑
5小时,并最终得到催化剂成品。
66.在本发明的催化剂的制备方法中,钒前驱体的用量以最终产生的催化剂中的钒计,保证钒在催化剂中的含量为1
‑
10wt.%优选3~8wt.%、更优选4~6wt.%。
67.另外,当催化剂中含有贵金属时,贵金属前驱体的用量以最终产生的催化剂中的
贵金属计,保证贵金属在催化剂中的含量为0.1
‑
3wt.%、优选0.5~2wt.%。
68.在本发明的制备方法中,由于用作基体的氧化铈的前驱体与活性组分的前驱体混合在一起,因此使得在最终的产物中活性组分可掺杂于基体的晶格中,有利于活性组分的分散,使催化剂具有良好的酸性和氧化还原性。由此,使得本发明的催化剂具有高效的nox催化还原和cvocs(如氯苯、二氯苯、二氯甲烷等)催化氧化的性能。
69.<第三方面>
70.本发明的第三方面中,提供将上述催化剂用于处理固定源烟气中的氮氧化物nox和含氯挥发性有机物cvocs脱除的用途。
71.本发明中所述的烟气为垃圾焚烧、钢铁烧结和有色冶炼等行业的烟气。烟气中的氮氧化物nox主要包括一氧化氮和二氧化氮中的一种或两种。烟气中的含氯挥发性有机物可包括氯苯、二氯苯、二氯甲烷和二氯乙烯中的一种或多种,优选包括氯苯与二氯苯。
72.对于催化性能测试的条件没有特别限制,可使用本领域通常的测试条件。在一些具体的实施方案中,测试条件如下:
73.反应温度范围为150
‑
450℃,催化剂的用量为200mg,模拟气体总流量为100ml/min,质量空速whsv为30000ml/(g
·
h),一氧化氮的浓度为500ppm,氨气的浓度为500ppm,氯苯的浓度为50ppm,氧气的浓度为10%,氮气平衡,反应压力为0.1mpa。
74.在上述测试条件下,氮氧化物脱除效率随着反应温度的升高先升高后下降,在200
‑
350℃范围内可保持在85%以上;氯苯氧化脱除效率随着反应温度的升高逐渐升高,在反应温度为350℃时,氯苯脱除效率可达到69%以上,在反应温度为400℃时,氯苯脱除效率可达到93%以上;氮气选择性在反应温度350℃时达到70%以上;二氧化碳选择性在反应温度350℃时达到70%以上;氯化氢浓度在反应温度为350℃时达到16ppm以上。
75.实施例
76.下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售获得的常规产品。
77.实施例1:
78.一种用于nox和氯苯协同脱除催化剂6v
‑
ceo2,其中钒质量分数为6%,二氧化铈为基体,其制备方法包括以下步骤:
79.步骤一:将0.4740g草酸加入100ml去离子水中,磁力搅拌5分钟使其充分溶解,随后使用水浴将其加热至80℃;
80.步骤二:将0.4740g偏钒酸铵加入至草酸溶液中,磁力搅拌5分钟使其充分溶解,该过程保持水浴加热温度为80℃恒定;
81.步骤三:将10.96g硝酸铈铵加入至上一步骤的混合溶液中,磁力搅拌10分钟使其充分混合,该过程保持水浴加热温度为80℃恒定;
82.步骤四:将3.84g柠檬酸加入至上一步骤的混合溶液中,持续磁力搅拌至溶液成胶状物质,该过程保持水浴加热温度为80℃恒定;
83.步骤五:将上一步骤蒸干所得的固体产物置于鼓风干燥箱中干燥12小时,鼓风干燥箱温度为180℃;
84.步骤六:将上一步骤所得的固体产物置于马弗炉中在室温下以2℃/min慢速升温至500℃焙烧3小时,最终得到所需催化剂,标记为6v
‑
ceo2。
85.将制备所得的6v
‑
ceo2催化剂研磨、压片、破碎,最终筛分为40
‑
60目,并在固定床微反应器(内径6mm)中进行氮氧化物和氯苯协同脱除的活性评价。反应温度范围为150
‑
450℃,温度采用k型热电偶自动控制。催化剂的用量为200mg,模拟气体总流量为100ml/min,质量空速whsv为30000ml/(g
·
h),一氧化氮的浓度为500ppm,氨气的浓度为500ppm,氯苯的浓度为50ppm,氧气的浓度为10%,氮气平衡,反应压力为0.1mpa。
86.协同反应过程中氮氧化物脱除效率测试结果和氯苯氧化脱除效率测试结果见图1,氮气选择性测试结果见图2,二氧化碳选择性测试结果见3,出口氯化氢浓度测试结果见图4。
87.可以看出,氮氧化物脱除效率随着反应温度的升高先升高后下降,在200
‑
350℃范围内可保持在85%以上;氯苯氧化脱除效率随着反应温度的升高逐渐升高,在反应温度为350℃时,氯苯脱除效率达到69.56%,在反应温度为400℃时,氯苯脱除效率达到93.56%;氮气选择性在150
‑
400℃范围内可保持在85%以上,其中150
‑
300℃范围内接近100%;二氧化碳选择性随着反应温度的升高逐渐降低,在反应温度为350℃时,二氧化碳选择性达到70.50%,在反应温度为400℃时,二氧化碳选择性达到68.68%;氯化氢浓度随着反应温度的升高逐渐升高,在反应温度为350℃时,氯化氢浓度达到16.24ppm,在反应温度为400℃时,氯化氢浓度达到37.43ppm。
88.实施例2:
89.一种用于nox和氯苯协同脱除催化剂5v1ru
‑
ceo2,其中钌质量分数为1%,钒质量分数为5%,二氧化铈为基体,其制备方法包括以下步骤:
90.步骤一:将0.3950g草酸加入100ml去离子水中,磁力搅拌5分钟使其充分溶解,随后使用水浴将其加热至80℃;
91.步骤二:将0.3950g偏钒酸铵加入至草酸溶液中,磁力搅拌5分钟使其充分溶解,该过程保持水浴加热温度为80℃恒定;
92.步骤三:将0.0706g三氯化钌三水合物和10.96g硝酸铈铵加入至上一步骤的混合溶液中,磁力搅拌10分钟使其充分混合,该过程保持水浴加热温度为80℃恒定;
93.步骤四:将3.84g柠檬酸加入至上一步骤的混合溶液中,持续磁力搅拌至溶液成胶状物质,该过程保持水浴加热温度为80℃恒定;
94.步骤五:将上一步骤蒸干所得的固体产物置于鼓风干燥箱中干燥12小时,鼓风干燥箱温度为180℃;
95.步骤六:将上一步骤所得的固体产物置于马弗炉中在室温下以2℃/min慢速升温至500℃焙烧3小时,最终得到所需催化剂,标记为5v1ru
‑
ceo2。
96.将制备所得的5v1ru
‑
ceo2催化剂研磨、压片、破碎,最终筛分为40
‑
60目,并在固定床微反应器(内径6mm)中进行氮氧化物和氯苯协同脱除的活性评价。反应温度范围为150
‑
450℃,温度采用k型热电偶自动控制。催化剂的用量为200mg,模拟气体总流量为100ml/min,质量空速whsv为30000ml/(g
·
h),一氧化氮的浓度为500ppm,氨气的浓度为500ppm,氯苯的浓度为50ppm,氧气的浓度为10%,氮气平衡,反应压力为0.1mpa。
97.协同反应过程中氮氧化物脱除效率测试结果和氯苯氧化脱除效率测试结果见图
1,氮气选择性测试结果见图2,二氧化碳选择性测试结果见图3,出口氯化氢浓度测试结果见图4。
98.可以看出,氮氧化物脱除效率随着反应温度的升高先升高后下降,在200
‑
350℃范围内可保持在85%以上;氯苯氧化脱除效率随着反应温度的升高逐渐升高,在反应温度为350℃时,氯苯脱除效率达到82.62%,在反应温度为400℃时,氯苯脱除效率达到96.60%;氮气选择性在150
‑
400℃范围内可保持在85%以上,其中150
‑
300℃范围内接近100%;二氧化碳选择性随着反应温度的升高逐渐升高,在反应温度为350℃时,二氧化碳选择性达到78.84%,在反应温度为400℃时,二氧化碳选择性达到90.76%;氯化氢浓度随着反应温度的升高逐渐升高,在反应温度为350℃时,氯化氢浓度达到32.49ppm,在反应温度为400℃时,氯化氢浓度达到41.75ppm。
99.对比例1:
100.一种用于nox和氯苯协同脱除催化剂1ru
‑
ceo2,其中钌质量分数为1%,二氧化铈为基体,其制备方法包括以下步骤:
101.步骤一:将0.0706g三氯化钌三水合物加入100ml去离子水中,磁力搅拌5分钟使其充分溶解,并使用水浴将其加热至80℃;
102.步骤二:将10.96g硝酸铈铵加入至上一步骤的混合溶液中,磁力搅拌10分钟使其充分混合,该过程保持水浴加热温度为80℃恒定;
103.步骤三:将3.84g柠檬酸加入至上一步骤的混合溶液中,持续磁力搅拌至溶液成胶状物质,该过程保持水浴加热温度为80℃恒定;
104.步骤四:将上一步骤蒸干所得的固体产物置于鼓风干燥箱中干燥12小时,鼓风干燥箱温度为180℃;
105.步骤五:将上一步骤所得的固体产物置于马弗炉中在室温下以2℃/min慢速升温至500℃焙烧3小时,最终得到所需催化剂,标记为1ru
‑
ceo2。
106.将制备所得的1ru
‑
ceo2催化剂研磨、压片、破碎,最终筛分为40
‑
60目,并在固定床微反应器(内径6mm)中进行氮氧化物和氯苯协同脱除的活性评价。反应温度范围为150
‑
450℃,温度采用k型热电偶自动控制。催化剂的用量为200mg,模拟气体总流量为100ml/min,质量空速whsv为30000ml/(g
·
h),一氧化氮的浓度为500ppm,氨气的浓度为500ppm,氯苯的浓度为50ppm,氧气的浓度为10%,氮气平衡,反应压力为0.1mpa。
107.协同反应过程中氮氧化物脱除效率测试结果和氯苯氧化脱除效率测试结果见图5。可以看出,氮氧化物脱除效率随着反应温度的升高逐渐下降,在250
‑
400℃范围内为负值;氯苯氧化脱除效率随着反应温度的升高逐渐升高,在反应温度为350℃时,氯苯脱除效率达到38.09%,在反应温度为400℃时,氯苯脱除效率达到84.91%。
108.以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变形,这些改进和变形也应视为本发明的保护范围。
109.产业上的可利用性
110.本发明的催化剂具有高效的nox催化还原和cvocs(如氯苯、二氯苯、二氯甲烷等)催化氧化、具有较宽的反应温度窗口且制备方法简便、成本较低,因此便于工业上推广利用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。