1.本发明涉及中药质量分析检测技术领域,尤其涉及一种小承气汤指纹图谱的构建方法。
背景技术:
2.小承气汤来源于东汉张仲景的《伤寒论》,由大黄、厚朴、枳实三味药组成。主治阳明腑实轻症而偏于痞满者,具有攻下通腑、轻下热结之良效。方中大黄荡涤肠胃,推陈致新;枳实破气消痞;厚朴下气除满。三者合用具有泻热通便、除满消滞的作用,在临床中得到广泛应用,常用于肠梗阻、术后胃肠功能障碍、便秘、慢性胃炎等的治疗。
3.小承气汤组方明确,疗效确切,现已被收录于国家中医药管理局发布的《古代经典名方目录(第一批)》中。近年来,为阐明小承气汤治疗疾病的物质基准,保障临床用药的有效性和安全性,许多学者对小承气汤中的化学成分和有效组分进行药理学、药效学和药动学等方面的研究,并且不断完善其质量标准,为经典名方的开发奠定了基础。
4.目前,对小承气汤的研究主要集中在药理学的研究方面,对其汤剂的物质基础、提取工艺、多指标成分含量测定及指纹图谱研究较少,缺少系统性。且对于如何衡量大生产制剂与传统汤剂质量的一致性也研究较少。
技术实现要素:
5.本发明所要解决的技术问题在于,提供一种小承气汤指纹图谱的构建方法,该方法可为小承气汤大生产质量控制提供数据基础,保证小承气汤产品质量的稳定性和可控性。
6.为了解决上述技术问题,本发明提供了一种小承气汤指纹图谱的构建方法,所述小承气汤由以下组分组成:大黄、厚朴和枳实;所述构建方法包括:
7.(1)分别取芦荟大黄素对照品、大黄素对照品、大黄素甲醚对照品、大黄酸对照品、没食子酸对照品、大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、大黄酚对照品、大黄酸
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、大黄酚
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、芦荟大黄素
‑8‑
o
‑
葡萄糖苷对照品、儿茶素对照品、川陈皮素对照品、橙皮苷对照品、新橙皮苷对照品、柚皮苷对照品、厚朴酚对照品、和厚朴酚对照品,加入溶剂溶解或提取,制得对照品溶液;
8.(2)取小承气汤制剂,加提取溶剂提取,制得供试品溶液;
9.(3)取预设量的对照品溶液和供试品溶液,注入液相色谱仪,所述液相色谱仪以十八烷基硅烷键合硅胶为填充剂,以甲醇为流动相a,磷酸水溶液为流动相b进行梯度洗脱,构建小承气汤的特征图谱。
10.作为上述技术方案的改进,所述梯度洗脱按下述程序进行:
11.0~5min,流动相a从3%
→
21%,流动相b从97%
→
79%;
12.5~20min,流动相a从21%
→
36%,流动相b从79%
→
64%;
13.20~32min,流动相a从36%
→
50%,流动相b从64%
→
50%;
14.32~42min,流动相a从50%
→
62%,流动相b从50%
→
38%;
15.42~50min,流动相a从62%
→
85%,流动相b从38%
→
15%;
16.50~60min,流动相a从85%
→
95%,流动相b从15%
→
5%。
17.作为上述技术方案的改进,步骤(3)中,分别吸取对照品溶液和供试品溶液各1~3μl,注入液相色谱仪进行检测,所述液相色谱仪的色谱柱以十八烷基硅烷键合硅胶为填充剂,所述液相色谱仪以甲醇为流动相a,以0.1%~0.2vol%的磷酸水溶液为流动相b;流速为0.18~0.22ml/min;柱温为28~32℃,检测波长为220~290nm。
18.作为上述技术方案的改进,步骤(3)中,分别吸取对照品溶液和供试品溶液各1μl,注入液相色谱仪进行检测,所述液相色谱仪的色谱柱以十八烷基硅烷键合硅胶为填充剂,其柱长为150mm,内径为2.1mm,粒径为1.6μm;所述液相色谱仪以甲醇为流动相a,以0.1vol%的磷酸溶液为流动相b;流速为0.2ml/min;柱温为30℃,检测波长为260nm。
19.作为上述技术方案的改进,步骤(1)中,分别取芦荟大黄素对照品、大黄素对照品、大黄素甲醚对照品、大黄酸对照品、没食子酸对照品、大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、大黄酚对照品、大黄酸
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、大黄酚
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、芦荟大黄素
‑8‑
o
‑
葡萄糖苷对照品、儿茶素对照品、川陈皮素对照品、橙皮苷对照品、新橙皮苷对照品、柚皮苷对照品、厚朴酚对照品、和厚朴酚对照品,加入甲醇制成每1ml含芦荟大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品15μg、柚皮苷对照品150μg、新橙皮苷对照品200μg、大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷对照品20μg、大黄酚
‑8‑
o
‑
葡萄糖苷对照品30μg、大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品20μg、芦荟大黄素对照品10μg、川陈皮素对照品10μg、大黄酸对照品30μg、和厚朴酚对照品15μg、厚朴酚对照品15μg、大黄素对照品10μg、大黄酚对照品15μg、大黄素甲醚对照品5μg的混合溶液,得到对照品溶液。
20.作为上述技术方案的改进,步骤(2)中,所述提取溶剂为50~100%甲醇,提取时间为15~30min,提取方式为超声提取或回流提取。
21.作为上述技术方案的改进,步骤(2)包括:
22.取小承气汤制剂0.2g,置于锥形瓶中,精密加入80%甲醇10ml,密塞,称定重量,超声处理30min,放冷,再次称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;或取小承气汤煎液2ml,精密加入甲醇8ml,超声处理30分钟,用80%甲醇补足减失的重量,取续滤液,即得供试品溶液。
23.作为上述技术方案的改进,所述小承气汤的特征图谱包括18个特征峰;其中,峰1为没食子酸,峰2为儿茶素,峰3为芦荟大黄素
‑8‑
o
‑
葡萄糖苷,峰4为柚皮苷,峰5为橙皮苷,峰6为大黄酸
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰7为新橙皮苷,峰8为大黄酚
‑1‑
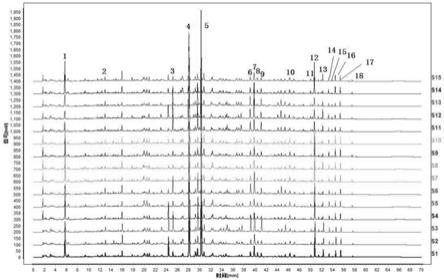
o
‑
β
‑
d
‑
葡萄糖苷,峰9为大黄酚
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰10为大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰11为芦荟大黄素,峰12为川陈皮素,峰13为大黄酸,峰14为和厚朴酚,峰15为厚朴酚,峰16为大黄素,峰17为大黄酚,峰18为大黄素甲醚。
24.作为上述技术方案的改进,所述小承气汤由以下重量份的组分组成:生大黄55.2份,姜厚朴27.6份,麸炒枳实36份。
25.作为上述技术方案的改进,所述小承气汤的制备方法为:取大黄、厚朴和枳实,加500~1200ml水浸泡,武火煮沸,文火沸腾至药液为200~300ml,滤过即得。
26.作为上述技术方案的改进,所述小承气汤的制备方法为:取生大黄55.2g、姜厚朴27.6g和麸炒枳实27.6g,加800ml水浸泡,武火煮沸,文火沸腾至药液为240ml,滤过即得。
27.实施本发明,具有如下有益效果:
28.本发明对小承气汤建立了uplc
‑
uv特征图谱。采用uplc
‑
ms进行相关物质基础研究的确认,标定了共18个特征峰,可充分展示小承气汤的化学成分的特征,特征峰信息量丰富,该方法方法稳定,准确可靠,实现对小承气汤中多个药味的特征成分的质量监控。
附图说明
29.图1是本发明小承气汤采用不同色谱柱测定时的特征图谱;
30.图2是本发明小承气汤采用不同波长测定时的特征图谱;
31.图3是本发明小承气汤采用不同流动相测定时的特征图谱;
32.图4是本发明小承气汤采用不同浓度磷酸的流动相测定时的特征图谱;
33.图5是本发明小承气汤采用洗脱梯度1测定时的特征图谱;
34.图6是本发明小承气汤采用洗脱梯度2测定时的特征图谱;
35.图7是本发明小承气汤采用洗脱梯度3测定时的特征图谱;
36.图8是本发明小承气汤采用洗脱梯度4测定时的特征图谱;
37.图9是本发明小承气汤采用不同流速测定时的特征图谱;
38.图10是本发明小承气汤采用不同柱温测定时的特征图谱;
39.图11是本发明小承气汤特征图谱专属性考察中小承气汤、大黄对照药材、缺大黄阴性样品的特征图谱;其中,峰1为没食子酸,峰2为儿茶素,峰3为芦荟大黄素
‑8‑
o
‑
葡萄糖苷,峰4为柚皮苷,峰5为橙皮苷,峰6为大黄酸
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰7为新橙皮苷,峰8为大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷,峰9为大黄酚
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰10为大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰11为芦荟大黄素,峰12为川陈皮素,峰13为大黄酸,峰14为和厚朴酚,峰15为厚朴酚,峰16为大黄素,峰17为大黄酚,峰18为大黄素甲醚;
40.图12是本发明小承气汤特征图谱专属性考察中小承气汤、厚朴对照药材、缺厚朴阴性样品的特征图谱;其中,峰1为没食子酸,峰2为儿茶素,峰3为芦荟大黄素
‑8‑
o
‑
葡萄糖苷,峰4为柚皮苷,峰5为橙皮苷,峰6为大黄酸
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰7为新橙皮苷,峰8为大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷,峰9为大黄酚
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰10为大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰11为芦荟大黄素,峰12为川陈皮素,峰13为大黄酸,峰14为和厚朴酚,峰15为厚朴酚,峰16为大黄素,峰17为大黄酚,峰18为大黄素甲醚;
41.图13是本发明小承气汤特征图谱专属性考察中小承气汤、枳实对照药材、缺枳实阴性样品的特征图谱;其中,峰1为没食子酸,峰2为儿茶素,峰3为芦荟大黄素
‑8‑
o
‑
葡萄糖苷,峰4为柚皮苷,峰5为橙皮苷,峰6为大黄酸
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰7为新橙皮苷,峰8为大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷,峰9为大黄酚
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰10为大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰11为芦荟大黄素,峰12为川陈皮素,峰13为大黄酸,峰14为和厚朴酚,峰15为厚朴酚,峰16为大黄素,峰17为大黄酚,峰18为大黄素甲醚;
42.图14是本发明小承气汤对照指纹图谱;其中,峰1为没食子酸,峰2为儿茶素,峰3为芦荟大黄素
‑8‑
o
‑
葡萄糖苷,峰4为柚皮苷,峰5为橙皮苷,峰6为大黄酸
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰7为新橙皮苷,峰8为大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷,峰9为大黄酚
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰10
为大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰11为芦荟大黄素,峰12为川陈皮素,峰13为大黄酸,峰14为和厚朴酚,峰15为厚朴酚,峰16为大黄素,峰17为大黄酚,峰18为大黄素甲醚;
43.图15是15批次小承气汤样品的特征图谱叠加图;其中,峰1为没食子酸,峰2为儿茶素,峰3为芦荟大黄素
‑8‑
o
‑
葡萄糖苷,峰4为柚皮苷,峰5为橙皮苷,峰6为大黄酸
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰7为新橙皮苷,峰8为大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷,峰9为大黄酚
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰10为大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷,峰11为芦荟大黄素,峰12为川陈皮素,峰13为大黄酸,峰14为和厚朴酚,峰15为厚朴酚,峰16为大黄素,峰17为大黄酚,峰18为大黄素甲醚。
具体实施方式
44.为使本发明的目的、技术方案和优点更加清楚,下面将结合附图及具体实施方式对本发明作进一步地详细描述。
45.本发明的小承气汤出自伤寒论,现有记载中一般认为其由大黄(酒洗)、厚朴(去皮,炙)、枳实(炙)组成。基于古籍文献进行多方面循古考证,所确定的小承气汤方剂如下:生大黄55.2g,姜厚朴27.6g,麸炒枳实36.0g。其中,各药材经鉴定符合2020年版《中国药典》一部相关项下规定。需要说明的是,张仲景对《伤寒论》中药物下方标注的炙,与现代药典中的清炒法最为接近,古代不加辅料炒制分为炒黄和炒炭两种方法,结合2020年版《中国药典》并参照了其他地方的炮制规范,确定了枳实是麸炒枳实、厚朴是姜厚朴。药物大黄结合临床药效及应用,采用生大黄。
46.进一步的,基于古籍文献考证确定小承气汤传统汤剂的制法为:取生大黄55.2g,姜厚朴27.6g,麸炒枳实36.0g,加水800ml,浸泡,武火(500w)煮沸,文火(300w)保持沸腾至药液约240ml,筛网(350目)滤过,得小承气汤汤剂;
47.本发明中所采用的小承气汤样品均为小承气汤冻干粉,具体的,汤剂减压低温浓缩至约120ml的浸膏,搅匀分装棕色西林瓶中,转移至真空循环泵真空冷冻干燥机中冷冻干燥,取出,得冻干粉,即得小承气汤冻干粉。此外,为了全面地反映小承气汤的质量信息,发明人针对各药材收集了不少于3产地15个批次,并制备样品进行研究。
48.为了全面地反映小承气汤的质量信息,实现全面、有效地控制小承气汤产品质量,本发明提供了一种小承气汤指纹图谱的构建方法,以下对其进行详细说明:
49.1仪器与试药
50.本发明所用到的仪器、试剂、试药以及组方所采用药物的信息如表1~表7所示:
51.表1仪器信息汇总表
[0052][0053][0054]
表2试剂信息汇总表
[0055][0056]
表3对照品信息
[0057][0058][0059]
表4大黄药材信息表
[0060][0061]
表5厚朴药材信息表
[0062][0063][0064]
表6枳实药材信息表
[0065][0066]
表7小承气汤组方表
[0067]
[0068][0069]
2色谱条件与参照物溶液、供试品溶液的制备
[0070]
2.1色谱条件
[0071]
色谱条件:以十八烷基硅烷键合硅胶为填充剂(柱长150mm,内径为2.1mm,粒径为1.6μm,色谱柱:waters cortecs t3柱);以甲醇为流动相a,以0.1vol%磷酸水溶液为流动相b,按表8中的规定进行梯度洗脱;柱温为30℃,流速为每分钟0.2ml,检测波长为260nm。理论板数按儿茶素峰计算应不低于10000。
[0072]
表8小承气汤指纹图谱用梯度洗脱表
[0073][0074]
2.2对照品溶液的制备
[0075]
取芦荟大黄素对照品、大黄素对照品、大黄素甲醚对照品、大黄酸对照品、没食子酸对照品、大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、大黄酚对照品、大黄酸
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、大黄酚8
‑
o
‑
葡萄糖苷对照品、芦荟大黄素
‑8‑
o
‑
葡萄糖苷对照品、儿茶素对照品、川陈皮素对照品、橙皮苷对照品、新橙皮苷对照品、柚皮苷对照品、橙皮内酯水合物对照品、厚朴酚对照品、和厚朴酚对照品,精密称定,分别加甲醇制成每1ml含芦荟大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品15μg、柚皮苷对照品150μg、新橙皮苷对照品200μg、大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷对照品20μg、大黄酚
‑8‑
o
‑
葡萄糖苷对照品30μg、大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品20μg、芦荟大黄素对照品10μg、川陈皮素对照品10μg、大黄酸对照品30μg、和厚朴酚对照品15μg、厚朴酚对照品15μg、大黄素对照品10μg、大黄酚对照品15μg、大黄素甲醚对照品5μg的混合溶液,得到对照品溶液。
[0076]
2.3供试品溶液的制备
[0077]
取小承气汤冻干粉0.2g,置于锥形瓶中,精密加入80%甲醇10ml,密塞,称定重量,超声处理30min,放冷,再次称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;或取小承气汤煎液2ml,精密加入甲醇8ml,超声处理30分钟,用80%甲醇补足减失的重量,取续滤液,即得供试品溶液。
[0078]
2.4测定法
[0079]
分别精密吸取对照品溶液与供试品溶液各1μl,注入液相色谱仪,测定,即得指纹图谱。供试品指纹图谱中应分别呈现与参照物色谱峰保留时间相对应的色谱峰。按中药色谱指纹图谱相似度评价系统计算,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90。
[0080]
3色谱条件的确定
[0081]
3.1色谱柱
[0082]
分别比较了waters cortecs t3柱(1.6μm,150mm
×
2.1mm)、waters beh c18柱(1.7μm,150mm
×
2.1mm)、waters acquity uplc hss t3柱(1.8μm,150mm
×
2.1mm)三根色谱柱对小承气汤指纹图谱色谱行为的影响。除色谱柱外,其他测试条件如2.1节。结果见图1。
[0083]
实验结果表明,不同色谱柱对小承气汤指纹图谱色谱峰峰数、峰形、峰分离度存在较大影响,以waters cortecs t3柱(1.6μm,150mm
×
2.1mm)为色谱柱,色谱峰峰数稍多,且峰形较好,基线较平稳,故选择waters cortecs t3柱为小承气汤指纹图谱研究色谱柱。
[0084]
3.2最佳吸收波长
[0085]
对不同吸收波长进行考察;吸收波长分别为220nm、260nm、280nm、290nm。采用waters cortecs t3柱(1.6μm,150mm
×
2.1mm)为色谱柱;以甲醇为流动相a,以0.1%磷酸为流动相b,按表8中的规定进行梯度洗脱;流速为每分钟0.2ml;进样量为1μl;结果见图2。
[0086]
通过对比220nm、260nm、280nm、290nm的图谱可知,当检测波长为260nm时,图谱中主要色谱峰峰面积较大,峰数目较多,满足信息量最大的原则,因此,将小承气汤指纹图谱的检测波长确定为260nm。
[0087]
3.3流动相
[0088]
对流动相类型进行考察,分别选用乙腈
‑
磷酸、甲醇
‑
水、甲醇
‑
0.1vol%磷酸、甲醇
‑
0.2vol%磷酸水溶液作为流动相,并采用如表8的梯度洗脱程序进行考察。采用waters cortecs t3柱(1.6μm,150mm
×
2.1mm)为色谱柱;以甲醇为流动相a,以0.1%磷酸为流动相b,按表8中的规定进行梯度洗脱;流速为每分钟0.2ml;进样量为1μl;结果见图3、图4。
[0089]
结果显示,若以乙腈
‑
磷酸为流动相,供试品色谱较多成分出峰较早,色谱峰分离差;若以甲醇
‑
水为流动相,色谱峰较少,且分离度较差;若以甲醇
‑
磷酸作为流动相,色谱图信息较为丰富,色谱峰分离度较好,基线较平稳,出峰时间适宜。流动相以浓度为0.1%磷酸为佳,若采用0.2%磷酸,个别色谱峰分离度稍差。最终确定以甲醇为流动相a,以0.1%磷酸水溶液为流动相b,按表8的程序进行梯度洗脱。
[0090]
3.4不同梯度的考察
[0091]
梯度1:以waters cortecs t3 c18柱(1.6μm,150mm
×
2.1mm)为色谱柱,以甲醇为流动相a,以0.1%磷酸为流动相b,按表9中的规定进行梯度洗脱;流速为每分钟0.3ml;进样量为1μl;结果见图5。
[0092]
表9洗脱梯度1
[0093][0094]
梯度2:以waters cortecs t3 c18柱(2.1mm
×
150mm,1.6μm)为色谱柱;以甲醇为流动相a,以0.1%磷酸为流动相b,按照表10进行梯度洗脱;检测波长为260nm,柱温为30℃,流速为每分钟0.3ml,进样量为1μl,结果见图6。
[0095]
表10洗脱梯度2
[0096][0097]
梯度3:以waters cortecs t3 c18柱(2.1mm
×
150mm,1.6μm)为色谱柱;以甲醇为流动相a,以0.1%磷酸为流动相b,按照表11进行梯度洗脱;检测波长为260nm,柱温为30℃,流速为每分钟0.2ml,进样量为1μl,结果见图7。
[0098]
表11洗脱梯度3
[0099][0100]
梯度4:以waters cortecs t3 c18柱(2.1mm
×
150mm,1.6μm)为色谱柱;以甲醇为流动相a,以0.1%磷酸为流动相b,按照表8进行梯度洗脱;检测波长为260nm,柱温为30℃,
流速为每分钟0.2ml,进样量为1μl,结果见图8。
[0101]
对比梯度1和梯度2,梯度1各色谱峰出峰位置均靠后,且各色谱峰分离较差,梯度2所得指纹图谱的后半部分色谱峰分离仍不佳;对比梯度2与梯度3,梯度3的色谱峰分离效果大大提高,但仍有个别峰分离较差;而梯度4所得的指纹图谱中的各色谱峰均达到了较好的分离效果,因此选择梯度4为洗脱梯度。
[0102]
3.5流速的考察
[0103]
分别考察了流速为每分钟0.18ml、每分钟0.2ml、每分钟0.22ml对小承气汤指纹图谱色谱行为的影响。除流速外,其他测试条件如2.1节。结果如图9所示。
[0104]
结果显示,不同流速对小承气汤指纹图谱色谱行为存在一定的影响,综合考虑色谱峰分离度、柱压等情况,研究确定小承气汤指纹图谱流动相流速为每分钟0.2ml。
[0105]
3.6柱温的考察
[0106]
分别考察了柱温为28℃、30℃、32℃对小承气汤指纹图谱色谱行为的影响。除柱温外,其他测试条件如2.1节。结果如图10所示。
[0107]
实验结果表明,不同色谱柱温度对小承气汤指纹图谱的无明显影响,表明该方法能适应一定的柱温变动,但综合考虑色谱峰峰形、峰分离度、基线等情况,本实验优选小承气汤指纹图谱研究的色谱柱温为30℃。
[0108]
3.7色谱条件的确定
[0109]
根据以上实验,确定色谱条件如下:以十八烷基硅烷键合硅胶为填充剂(柱长150mm,内径为2.1mm,粒径为1.6μm,色谱柱:waters cortecs t3柱);以甲醇为流动相a,以0.1vol%磷酸水溶液为流动相b,按表8中的规定进行梯度洗脱;柱温为30℃,流速为每分钟0.2ml,检测波长为260nm。
[0110]
4供试品溶液制备方法考察
[0111]
4.1提取溶剂考察
[0112]
分别考察了不同提取溶剂对小承气汤指纹图谱的影响,选取50%甲醇、80%甲醇、100%甲醇、稀乙醇(参中国药典第三部通则和指导原则试液部分)、100%乙醇作为提取溶剂,并比较不同提取溶剂指纹图谱结果。
[0113]
其中,供试品溶液制备方法为:取小承气汤冻干粉0.2g,置于锥形瓶中,精密加入上述提取溶剂10ml,密塞,称定重量,超声处理30min,放冷,再次称定重量,用相应的提取溶剂补足减失的重量,摇匀,滤过,取续滤液,即得。
[0114]
按照2.1节的条件进行测试,测试结果如表12所示。
[0115]
表12小承气汤指纹图谱提取溶剂考察结果表
[0116]
[0117][0118]
实验结果显示,不同提取溶剂的图谱中色谱峰的数目差异并不显著,以80%甲醇的“总峰面积/称样量”值最大,因此,选择以80%甲醇作为提取溶剂。
[0119]
4.2提取方式考察
[0120]
取小承气汤冻干粉0.2g,置于锥形瓶中,精密加入80%甲醇10ml,密塞,称定重量,分别超声处理、加热回流30min,放冷,再次称定重量,用相应浓度的80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
[0121]
按照2.1节的条件进行测试,测试结果如表13所示。
[0122]
表13小承气汤指纹图谱提取方式考察结果表
[0123][0124]
实验结果显示,不同提取方式对小承气汤指纹图谱的“总峰面积/称样量”的值影响并不显著,因此,选择操作简便的超声作为提取方式。
[0125]
4.3提取时间考察
[0126]
考察不同提取时间对小承气汤指纹图谱的影响,选取考察15分钟、30分钟、45分钟三个不同提取时间。
[0127]
其中,供试品溶液制备方法为:取小承气汤冻干粉0.2g,置于锥形瓶中,精密加入80%甲醇10ml,密塞,称定重量,各超声处理15min、30min、45min,放冷,再次称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
[0128]
按照2.1节的条件进行测试,测试结果如表14所示。
[0129]
表14小承气汤指纹图谱提取时间考察结果表
[0130][0131]
实验结果显示,不同提取时间的“总峰面积/称样量”值不存在明显的差异,为保证提取完全,选择30min作为提取时间。
[0132]
4.4提取溶剂用量考察
[0133]
考察不同提取溶剂用量对小承气汤指纹图谱的影响,选取10ml、25ml两个不同提取溶剂用量。
[0134]
其中,供试品溶液制备方法为:取小承气汤冻干粉0.2g,置于锥形瓶中,精密加入
80%甲醇10ml、25ml,密塞,称定重量,超声处理30min,放冷,再次称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
[0135]
按照2.1节的条件进行测试,测试结果如表15所示。
[0136]
表15小承气汤指纹图谱提取时间考察结果表
[0137][0138]
实验结果显示,增加溶剂用量,指纹图谱的“总峰面积
×
稀释倍数/称样量”值并无显著增大,表明10ml溶剂可以完全提取小承气汤中的化学成分,因此,为节省溶剂,选择10ml作为提取溶剂用量。
[0139]
4.5供试品溶液制备方法的确定
[0140]
根据上述实验结果,小承气汤指纹图谱样品前处理方法确定为:
[0141]
取小承气汤冻干粉0.2g,置于锥形瓶中,精密加入80%甲醇10ml,密塞,称定重量,超声处理30min,放冷,再次称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
[0142]
5方法学验证
[0143]
5.1专属性考察
[0144]
分别取缺每味药物的小承气汤,按照供试品溶液的制备方法制备,得到缺每味药材的阴性样品溶液。
[0145]
分别取大黄、姜厚朴、麸炒枳实饮片,按照小承气汤冻干粉样品的制备方法制备每味药材的冻干粉,再按照供试品溶液的制备方法制备每味药材的指纹图谱对照药材溶液。
[0146]
取芦荟大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、柚皮苷对照品、新橙皮苷对照品、大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、大黄酚
‑8‑
o
‑
葡萄糖苷对照品、大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品、芦荟大黄素对照品、川陈皮素对照品、大黄酸对照品、和厚朴酚对照品、厚朴酚对照品、大黄素对照品、大黄酚对照品、大黄素甲醚对照品适量,精密称定,加甲醇制成每1ml含芦荟大黄素
‑8‑0‑
β
‑
d
‑
葡萄糖苷对照品15μg、柚皮苷对照品150μg、新橙皮苷对照品200μg、大黄酚
‑1‑
o
‑
β
‑
d
‑
葡萄糖苷对照品20μg、大黄酚
‑8‑
o
‑
葡萄糖苷对照品30μg、大黄素
‑8‑
o
‑
β
‑
d
‑
葡萄糖苷对照品20μg、芦荟大黄素对照品10μg、川陈皮素对照品10μg、大黄酸对照品30μg、和厚朴酚对照品15μg、厚朴酚对照品15μg、大黄素对照品10μg、大黄酚对照品15μg、大黄素甲醚对照品5μg的溶液,即得每种对照品的对照品溶液。
[0147]
将供试品溶液、缺每味药材的阴性样品溶液、每味药材的指纹图谱对照药材溶液、每种对照品的对照品溶液各1μl注入液相色谱仪,按2.1节的色谱条件进样分析,结果如图11~图13所示。
[0148]
由图11~图13可知:检测出小承气汤冻干粉指纹图谱18个共有色谱峰,其中13个峰(1、2、3、6、8、9、10、11、13、16、17、18号峰)来源于大黄,2个峰(14、15号峰)来源于姜厚朴,4个峰(4、5、7、12号峰)来源于麸炒枳实(参表16);建立的指纹图谱能反映来源于处方中所有药味成分。
[0149]
供试品色谱在与对照品色谱相应保留时间处有相同的色谱峰,阴性无干扰,综上
所述,说明该方法专属性良好。
[0150]
表16小气汤特征图谱峰归属
[0151][0152][0153]
5.2精密度考察
[0154]
取同一批小承气汤冻干粉0.2g,按2.3节的方法制备供试品溶液,按2.1节的色谱条件重复进样6次,计算指纹图谱的相对保留时间、相对峰面积与相似度。结果显示:保留时间rsd在0.02%~0.08%范围内,相对峰面积在0.03%~1.63%范围内,rsd值均<3%,相似度均为1.0000;表明该方法精密度良好。
[0155]
5.3重复性考察
[0156]
取同一批小承气汤冻干粉0.2g,平行6份,按2.3节的方法制备供试品溶液,按2.1节的色谱条件进样测定,并将指纹图谱导入《中药色谱指纹图谱相似度评价系统》(2012年版)计算相似度、相对保留时间与相对峰面积。结果显示:保留时间rsd在0.02%~0.06%范围内,相对峰面积在0.02%~1.91%范围内,rsd值均<3%,相似度均为1.0000;表明该方法重复性良好。
[0157]
5.4稳定性考察
[0158]
取同一批小承气汤冻干粉0.2g,平行6份,按2.3节的方法制备供试品溶液,按2.1节的色谱条件,分别在0,3,6,9,12,18,24h进样测定,计算指纹图谱的相对保留时间、相对
峰面积与相似度。结果显示:保留时间rsd在0.02%~0.10%范围内,相对峰面积在0.08%~2.41%范围内,rsd值均<3%,相似度均为1.0000;表明该方法稳定性良好。
[0159]
5.5中间精密度考察
[0160]
由另外一名分析人员在不同日期取同一批小承气汤冻干粉,按2.3节的方法制备供试品溶液,按2.1节的色谱条件,使用不同的仪器进行测定,计算指纹图谱的相对保留时间、相对峰面积与相似度。结果显示:指纹图谱相对保留时间rsd在0.19%~0.82%范围内,相对峰面积在0.26%~8.30%范围内,相似度均为1.0000;表明该方法中间精密度良好。
[0161]
6不同批次样品的测定及共有峰的确定
[0162]
取15批次小承气汤对应实物,按确定的供试品溶液制备方法(2.3节)制得供试品溶液,再按确定的色谱条件(2.1节),分别进样1μl,测定。以儿茶素为参照峰,计算各共有特征峰的相对保留时间及相对峰面积,并计算rsd值,同时采用中药色谱指纹图谱相似度(2012年版)评价软件计算各个指纹图谱的相似度。结果见表17,色谱图见图14、图15。
[0163]
结果表明,按平均数法生成小承气汤冻干粉对照指纹图谱,得到各批次小承气汤指纹图谱相似度,15批小承气汤指纹图谱相似度均大于0.90,相似度较高。
[0164]
表17 15批次小承气汤冻干粉制剂指纹图谱相似度表
[0165][0166]
7指纹图谱的化学成分试验研究
[0167]
根据小承气汤方中各药味所含化学成分,采用高分辨质谱,对小承气汤所含化学成分进行指认。
[0168]
质谱条件:thermo vanquish型超高效液相色谱仪;uplc
‑
q
‑
exactive focus
‑
ms/ms型高分辨质谱仪;waters cortecs t3(柱长150mm,内径2.1mm,粒径1.6μm)色谱柱。离子源hesi,正负离子模式分别检测,鞘气流速35μl/min,辅助气流速10μl/min,喷雾电压分别为3.8kv、3.2kv,毛细管温度350℃,离子源温度350℃,s
‑
lens rf level 50;扫描模式为full ms/dd
‑
ms2,扫描范围m/z 100~1000。碰撞能量20、40ev。
[0169]
色谱条件:以十八烷基硅烷键合硅胶为填充剂(柱长150mm,内径为2.1mm,粒径为1.6μm,色谱柱:waters cortecs t3柱);以甲醇为流动相a,以0.1%甲酸水溶液为流动相b,按表8的规定进行梯度洗脱;柱温为30℃;流速为每分钟0.2ml;检测波长为260nm。
[0170]
鉴定结果详见表18。
[0171]
表18小承气汤指纹图谱成分指认结果
[0172][0173]
综上,本发明建立了小承气汤的指纹图谱,该指纹图谱可充分展示小承气汤的化学成分特征,特征峰信息量丰富,可全面地反映小承气汤的质量信息,从而能够达到全面、有效地控制小承气汤产品质量的目的。同时,本发明中的特征图谱构建方法重现性良好,准确可靠,稳定性好。
[0174]
以上所述是发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。