包含皂苷的抗体药物缀合物(adc)
技术领域
1.本发明涉及抗体
‑
药物缀合物(adc),其通过共施用adc与包含共价连接的皂苷的部分而加强。本发明还涉及抗体
‑
寡核苷酸缀合物 (aoc),其通过共施用aoc与包含共价连接的皂苷的部分而加强。本发明还涉及经由共价接头与皂苷缀合的adc和aoc。本发明还涉及经由共价键与皂苷缀合的效应子部分(如毒素)或反义寡核苷酸诸如例如bna。本发明还涉及与靶向部分(如抗体)共价缀合的bna。本发明还涉及治疗性组合,其包含第一药物组合物以及包含第二药物组合物,所述第一药物组合物包含细胞靶向部分(如抗体)和与其共价结合的反义寡核苷酸(如bna)的缀合物,所述第二药物组合物包含游离皂苷或细胞靶向部分(如抗体)和与其共价连接的皂苷的缀合物。此外,本发明涉及这些缀合物或治疗性组合物或治疗性组合中的任一种,其用作药物。本发明还涉及这些缀合物或治疗性组合物或治疗性组合中的任一种,其用于治疗或预防癌症的方法中。最后,本发明涉及用于产生本发明的这些缀合物中的任一种的方法。
2.背景
3.在理论上,具有治疗性生物活性的分子在许多情况下适合应用为有效治疗药物以用于治疗有需要的人类患者中的疾病,如癌症。一个典型的实例是小分子生物活性部分。然而,目前临床中使用的许多(如果不是全部)潜在的类药(drug
‑
like)分子和治疗剂至少存在众多缺点和缺陷之一。当施用至人体时,除了针对待治疗疾病或健康问题的潜在方面的生物学活性之外,治疗活性分子还可发挥脱靶效应。此类脱靶效应是不希望的,并且存在所施用分子诱发健康或甚至危及生命的副作用的风险。正是此类不良事件的发生导致许多类药化合物和治疗性部分未能通过iii期临床试验或甚至iv期临床试验(进入市场后随访)。因此,强烈希望提供药物分子,如小分子治疗剂,其中药物分子的治疗性效应应该,如,(1)对驱动疾病的生物因素或生物过程具有高度特异性,(2)足够安全,(3)足够有效,(4)充分针对患病细胞且对非患病细胞几乎没有脱靶活性,(5)具有足够及时的作用模式(如施用的药物分子应在一定的时间范围内到达人类患者中的靶向部位,并应在靶向部位处保持一定的时间范围),和/或(6)在患者体内具有足够长的持续治疗活性,等等。不幸的是,迄今为止,尽管已经进行了长期和深入的研究并且尽管在个别遇到困难和缺陷的几个领域取得的令人印象深刻的进展,但具有许多或甚至所有上述有益特征的
‘
理想’治疗剂对于患者来说仍是不可用的。
4.化疗是癌症治疗中最重要的治疗选项之一。然而,它通常与低治疗窗有关,因为与健康组织中的分裂细胞相比,它对癌细胞没有特异性。单克隆抗体的发明提供了利用其特异性结合特性作为将细胞毒性剂靶向递送至癌细胞同时保留正常细胞的机制的可能性。这可以通过细胞毒性效应子(也称为有效载荷或弹头)化学缀合至抗体以产生抗体
‑ꢀ
药物缀合物(adc)来实现。通常,使用非常有效的有效载荷(如美坦新 (dm1)),它们的未缀合形式具有有限的治疗指数(比较毒性剂量与有效剂量的比率)。dm1与曲妥珠单抗(ado
‑
曲妥珠单抗美坦新)的缀合,也称为kadcycla,将猴子中的dm1耐受剂量提高至少两倍。在过去的几十年中,已经为开发治疗性adc做出了巨大的努力和投资。然而,尽管临床前数据很有希望,但
将adc引入临床仍然具有挑战性。2000 年,第一个获批用于临床使用的adc是吉妥珠单抗奥佐米星(麦罗塔 (mylotarg),cd33靶向的,pfizer/wyeth)用于复发性急性髓系白血病 (aml)。然而,由于包括其安全性概况在内的诸多问题,麦罗塔应美国联邦药品管理局(federal drug administration,fda)的要求从市场上撤出。与用常规化疗治疗的患者相比,用麦罗塔治疗的患者更常被发现死亡。麦罗塔在2017年再次以较低的推荐剂量、不同的时间表联合化疗或其自身以及新的患者群体进入市场。迄今为止,只有5种adc被批准用于临床使用,同时大约55种adc的临床开发已停止。然而,人们的兴趣仍然很高,并且目前大约有80种adc在近600项临床试验中仍处于临床开发。
5.尽管有可能使用患者通常不能耐受的毒性有效载荷,但低治疗指数(比较毒性剂量与有效剂量的比率)是导致许多adc在临床开发中停止的主要问题,这可能由若干种机制导致,如对正常细胞的脱靶毒性、对细胞毒性剂产生抗性以及药物在循环中过早释放。fda对adc的系统审查发现,大多数adc的毒性特征可以根据所使用的有效载荷进行分类,但不能根据所使用的抗体进行分类,这表明毒性主要是由有效载荷的过早释放决定的。在停止使用的大约55种adc中,估计至少有23种是由于治疗指数不佳导致。例如,曲妥珠单抗特司林 (trastuzumab tesirine)缀合物(adct
‑
502,her
‑
2靶向的,adc治疗剂) 的开发最近因治疗指数窄而停止,这可能是由于在表达相当水平的 her2的肺组织中的中靶、脱组织效应。此外,由于缺少主要终点,3 期试验中的几种adc已停止。例如,在新诊断的成胶质细胞瘤患者中测试的德帕妥昔组单抗莫福汀(depatuxizumab mafodotin)缀合物(abt
‑ꢀ
414,egfr靶向的,abbvie)和在患有铂抗性卵巢癌的患者中测试的索星
‑
米妥昔单抗(mirvetuximab soravtansine)缀合物(imgn853,叶酸受体α(frα)靶向的,immunogen)的3期试验最近停止了,显示没有存活益处。重要的是注意一些adc的临床使用剂量可能不足以发挥其全部抗癌活性。例如,ado
‑
曲妥珠单抗美坦新在人体内的mtd为3.6mg/kg。在乳腺癌的临床前模型中,ado
‑
曲妥珠单抗美坦新在3mg/kg以上的剂量水平下诱导肿瘤消退,但在15mg/kg下观察到更有效的功效。这表明,在临床施用剂量下,ado
‑
曲妥珠单抗美坦新可能不会发挥其最大的潜在抗肿瘤效应。
6.adc主要由抗体、细胞毒性部分(如有效载荷)和接头组成。在新的adc的设计和开发中已经提出并实施了几种新型策略,以克服靶向adc的每种组分的现有问题。例如,通过鉴定和验证针对抗体组分的足够抗原靶标,通过选择在肿瘤中具有高表达水平而在正常组织中几乎不表达的抗原,存在于细胞表面上以易于接近循环adc的抗原以及在结合后允许adc内化到细胞中的抗原;以及活性的替代机制;设计和优化提高adc的溶解度和药物抗体比(dar)并克服可将化疗剂转运出细胞的蛋白质诱导的抗性的接头;通过包含更多有效载荷来提高dar比率,选择和优化抗体以提高抗体的同质性和可开发性。除了adc的技术开发外,还正在部署新的临床和转化策略以最大化治疗指数,如通过分次给药改变给药时间表;进行生物分布研究;包括生物标志物以优化患者选择,及早捕获响应信号并监测响应的持续时间和深度,以及告知组合研究。
7.具有临床潜力的adc的实例是那些adc,如本妥昔单抗维多汀 (brentuximab vedotin)、英妥珠单抗奥佐米星(inotuzumab ozogamicin)、 moxetumomab pasudotox和泊洛妥珠单抗维多汀(polatuzumab vedotin),它们被评价为针对淋巴恶性肿瘤和多发性骨髓瘤的治疗选项。结合(恶性)b细胞上的cd79b的泊洛妥珠单抗维多汀和结合cd22的匹那妥
珠单抗维多汀(pinatuzumab vedotin)在临床试验中进行测试,其中adc 各自与共施用的利妥昔单抗(一种结合cd20但不提供有效载荷的单克隆抗体)组合[b.yu和d.liu,antibody
‑
drug conjugates in clinical trialsfor lymphoid malignancies and multiple myeloma;journal of hematology &oncology(2019)12:94]。单克隆抗体的组合(如这些实例)是进一步的方法,并试图获得
‘
魔弹(magic bullet)’,其组合adc的许多或甚至所有上述所需特征。
[0008]
与此同时,在过去的几十年里,基于核酸的治疗剂正在开发中。治疗性核酸可基于脱氧核糖核酸(dna)或核糖核酸(rna)、反义寡核苷酸(aso,aon)和短干扰rna(sirna)、微小rna以及dna和rna 适配体,用于诸如基因疗法、rna干扰(rnai)的方法中。它们中的许多通过抑制dna或rna表达而共有相同的基本作用基础,从而防止疾病相关的异常蛋白质的表达。基因疗法领域中正在进行最大数量的临床试验,全球有近2600项正在进行或已完成的临床试验,但只有约 4%进入3期。随后是aso的临床试验。与adc类似,尽管探索了大量技术,但治疗性核酸在临床开发过程中共有两个主要问题:递送到细胞中和脱靶效应。例如,正在研究aso,如肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)和桥接核酸(bna)作为特异性抑制靶基因,尤其是那些难以用小分子抑制剂或中和抗体靶向的基因的有吸引力的策略。目前,正在研究不同aso在许多神经退行性疾病 (如亨廷顿氏病(huntington’s disease)、帕金森氏病(parkinson’s disease)、阿尔茨海默氏病(alzheimer’s disease)和肌萎缩侧索硬化症)以及还有几个癌症阶段中的功效。aso作为潜在治疗剂的应用需要安全且有效的方法将其递送至靶细胞和组织的细胞质和/或细胞核。尽管已经证明了 aso的临床相关性,但体外和体内低效的细胞摄取限制了aso的功效,并且已经成为治疗剂开发的障碍。细胞摄取可小于剂量的2%,导致活性位点处的aso浓度过低,无法获得有效和持续的结果。因此,这需要增加施用剂量,这会诱导脱靶效应。最常见的副作用是补体级联的激活、凝血级联的抑制和toll样受体介导的免疫系统的刺激。
[0009]
化疗剂是最常见的小分子,然而,它们的功效受到严重的脱靶副毒性以及差的溶解性、快速清除和有限的肿瘤暴露的阻碍。支架
‑
小分子药物缀合物如聚合物
‑
药物缀合物(pdc)是具有药理学活性的大分子构建体,其包含结合载体支架(如聚乙二醇(peg))的一个或多个分子的小分子药物。
[0010]
此类缀合物原理引起了广泛的关注,并且已经被研究了几十年。处于临床前或临床开发中的大多数小分子药物的缀合物用于肿瘤适应症。然而,最近仅一种与癌症无关的药物(movantik,一种阿片类药物拮抗剂纳洛酮的peg寡聚物缀合物,astrazeneca)被批准用于2014年慢性疼痛患者中阿片类药物引起的便秘,这是一种非肿瘤学适应症。到目前为止,将药物
‑
支架缀合物的应用转化为人类对象的治疗几乎没有取得临床成功。例如,pk1(n
‑
(2
‑
羟丙基)甲基丙烯酰胺(hpma)共聚物多柔比星;由pharmacia,pfizer开发)在鼠模型的实体瘤和白血病中均显示出极大的抗癌活性,并且处于肿瘤适应症的临床研究中。尽管它在人体中证明了非特异性毒性的显著降低和改善的药代动力学,但在患者中抗癌功效的改善却是微不足道的,并且因此停止了对pk1的进一步开发。
[0011]
支架
‑
小分子药物缀合物的失败至少部分归因于其在肿瘤部位处的不良累积。例如,虽然在鼠模型中,pk1显示了在肿瘤中的累积比健康组织(肝、肾、肺、脾和心脏)中高45
‑
250倍,但在临床试验中仅在一小亚组的患者中观察到肿瘤中的累积。
[0012]
上述问题的一个潜在解决方案是将纳米颗粒系统应用于药物递送,如脂质体。脂质体是由一个或多个磷脂双层组成的球形囊泡,其当磷脂分散在水中时自发形成。磷脂的两亲性特征为其提供了自组装特性、乳化和润湿特征,并且这些特性可用于新药和新药递送系统的设计。与直接施用药物相比,封装在脂质体递送系统中的药物可具有多种优势,如改善和控制药代动力学和药效学、组织靶向特性、降低的毒性和增强的药物活性。此类成功的一个实例是脂质体封装形式的小分子化疗剂多柔比星(doxil:一种聚乙二醇化脂质体封装形式的多柔比星; myocet:一种非聚乙二醇化脂质体多柔比星),其已被批准用于临床使用。
[0013]
因此,仍然需要找到一种解决方案,其允许药物疗法,如抗肿瘤疗法,在需要时适用于非全身性使用,其中药物具有例如可接受的安全性特性、几乎没有脱靶活性、足够的功效,从患者体内清除率足够低等。
[0014]
概述
[0015]
对于本发明的一个实施方案,第一目标是提供改善的生物活性化合物或包含此类改善的生物活性化合物的组合物。
[0016]
本发明的实施方案的几个目标之一是提供在向有需要的人类患者施用小分子治疗活性化合物时遇到的非特异性问题的解决方案。本发明的实施方案的几个目标之一是提供对驱动疾病的生物因素或生物过程具有非最佳特异性的药物的问题的解决方案。本发明的实施方案的几个目标之一是提供当施用至有需要的人类患者时当前药物的安全特性不足的问题的解决方案。本发明的实施方案的几个目标之一是提供当施用至有需要的人类患者时当前药物不如所需有效的问题的解决方案。本发明的实施方案的几个目标之一是提供当施用至有需要的人类患者时当前药物未充分针对患病细胞且对非患病细胞几乎没有脱靶活性的问题的解决方案。本发明的实施方案的几个目标之一是提供当施用至有需要的人类患者时当前药物没有足够及时的作用模式的问题(如,施用的药物分子应在一定的时间范围内到达人类患者的靶向部位,并应在靶向部位处保持一定的时间范围)的解决方案。本发明的实施方案的几个目标之一是提供当施用至有需要的人类患者时当前药物在患者体内没有足够长的持久治疗活性的问题的解决方案。
[0017]
本发明的实施方案的上述目标中的至少一个是通过提供优选包含细胞靶向部分和至少一个皂苷的本发明的缀合物来实现的,根据本发明,缀合物也适合用作药物或者适用于参与根据本发明的药物组合,以及适合用作制备本发明的adc或抗体
‑
寡核苷酸缀合物(aoc)的半成品。治疗性组合包含例如含有共价结合的皂苷的本发明的缀合物并且例如包含含有效应分子(也称为效应子部分或有效载荷)的第二缀合物,其中缀合物包含针对暴露在靶细胞的不同细胞表面分子上的不同表位的不同结合位点,其中不同细胞表面分子由相同靶细胞表达并且暴露于相同靶细胞的表面上,或者其中缀合物包含针对所述靶细胞的相同细胞表面分子的相同表位的相同结合位点。
[0018]
将关于特定实施方案描述本发明,但本发明不限于此而仅由权利要求书限制。除非另有说明,否则本文描述的本发明的实施方案可以组合和协作操作。
[0019]
本发明的一方面涉及包括共价连接在一起的抗体和反义寡核苷酸,如反义bna,或者由其组成的缀合物。在图1
‑
5中,描绘了此类缀合物的基因沉默活性(在动物肿瘤模型中的体内测试)。还参考实施例部分。
[0020]
本发明的一方面涉及第一组合物和第二组合物的组合,所述第一组合物包含缀合物,所述缀合物包含共价连接在一起的抗体和反义寡核苷酸,如反义bna,或者由其组成,所述第二组合物包含本发明的游离皂苷(参见表a1,方案i)。在图1
‑
7a和图1
‑
7c中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0021]
本发明的一方面涉及药物组合,其包含第一组合物和第二组合物,或者由其组成,所述第一组合物包含第一缀合物,所述第一缀合物包含抗体和反义寡核苷酸,如反义bna,或者由其组成,所述第二组合物包含第一缀合物,所述第一缀合物包含相同抗体和本发明的至少一个皂苷,或者由其组成。在图1
‑
5中,描绘了此类缀合物的基因沉默活性(在动物肿瘤模型中的体内测试)。在图8
‑
5中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0022]
本发明的一方面涉及药物组合,其包含第四组合物和第五组合物,或者由其组成,所述第四组合物包含第四缀合物,所述第四缀合物包含抗体和反义寡核苷酸,如反义bna,或者由其组成,所述第五组合物包含第一缀合物,所述第一缀合物包含不同抗体和本发明的至少一个皂苷,或者由其组成。在图10
‑
6a和图10
‑
6c中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0023]
本发明的一方面涉及缀合物,其包含共价连接至本发明的至少一个皂苷的反义寡核苷酸,如反义bna,或者由其组成。在图1
‑
3中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0024]
本发明的一方面涉及缀合物,其包含共价偶联至聚合支架,如树枝化基元(如g4
‑
树枝化基元)的反义寡核苷酸(如反义bna),或者由其组成,其中聚合支架与本发明的一个或多个皂苷分子(如四个皂苷分子) 共价缀合。在图1
‑
3中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0025]
本发明的一方面涉及缀合物,其包含以下或由以下组成:共价连接至至少一个反义寡核苷酸分子(如反义bna)以及共价连接至本发明的至少一个皂苷分子的对肿瘤标志物或肿瘤细胞受体具有特异性的抗体(如单克隆抗体)。在图2
‑
4中,描绘了此类缀合物的基因沉默活性(在动物肿瘤模型中的体内测试)。还参考实施例部分。
[0026]
本发明的一方面涉及缀合物,其包含以下或由以下组成:经由三官能接头(如方案ii的接头)共价连接至至少一个反义寡核苷酸分子(如反义bna)以及经由相同的三官能接头共价连接至本发明的至少一个皂苷分子的对肿瘤标志物或肿瘤细胞受体具有特异性的抗体(如单克隆抗体)。在图1
‑
1中,描绘了此类缀合物的基因沉默活性(在动物肿瘤模型中的体内测试)。还参考实施例部分。
[0027]
本发明的一方面涉及治疗性组合,其包含以下或由以下组成:第八组合物,所述第八组合物包含缀合物,所述缀合物包含以下或由以下组成:经由至少一个接头,优选在生理酸性条件下可裂解的至少一个可裂解接头共价连接至本发明的至少一个皂苷分子的对肿瘤标志物或肿瘤细胞受体具有特异性的抗体,优选单克隆抗体;以及还包含第九组合物,所述第九组合物包含反义寡核苷酸(如反义bna分子)。在图6
‑
2中,描绘了此类缀合物的基因沉默活性(在动物肿瘤模型中的体内测试)。在图5
‑
2a和图5
‑
2c中,描绘了此类缀合物的基因沉默活性 (用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0028]
本发明的一方面涉及上述缀合物或组合物或治疗性组合中的任一种,其用作药物。
[0029]
本发明的一方面涉及上述缀合物或组合物或治疗性组合中的任一种,其用于治疗或预防癌症。
[0030]
本发明的一方面涉及具有化合物i的化学结构的治疗性分子:
[0031]
a1
m
((
‑
l9
w
)((
‑
l1
q
‑
b1
n
)
u
((
‑
l2
r
‑
l3
s
)(
‑
l4
v
‑
c)
p
)
t
))
x
[0032]
(化合物i),
[0033]
其中
[0034]
如果b1是第一效应子部分,则a1是第一配体,或者如果b1是第一配体,则a1是第一效应子部分;
[0035]
c是皂苷;
[0036]
如果a1是第一配体并且b1是第一效应子部分,则m=0或1;
[0037]
如果a1是第一效应子部分并且b1是第一配体,则m=0
‑
32;
[0038]
如果b1是第一配体并且a1是第一效应子部分,或者如果a1是第一配体并且b1是第一效应子部分,则n=0或1;
[0039]
p=1
‑
128中的任一个;
[0040]
l1是用于共价偶联两个化学基团的至少一个接头;
[0041]
l2是用于共价偶联两个化学基团的至少一个接头;
[0042]
l3是用于共价偶联两个化学基团的至少一个寡聚支架或多聚支架;
[0043]
l4是用于共价偶联两个化学基团的至少一个接头;
[0044]
l9是用于共价偶联三个化学基团的三官能接头;
[0045]
q=0或1;
[0046]
r=0或1;
[0047]
s=0或1;
[0048]
如果s=0时,则t=0、1或2,以及如果s=1,则t=0
‑
16中的任一个;
[0049]
如果a1是第一配体并且b1是第一效应子部分,则u=0
‑
32中的任一个,或者如果a1是第一效应子部分并且b1是第一配体,则u=1;
[0050]
v=0或1;
[0051]
w=1或0;以及
[0052]
x=1
‑
16。
[0053]
一个实施方案涉及治疗性组合,其包含根据本发明的治疗性分子和具有化合物ii的化学结构的第二治疗性分子:
[0054]
a2
a
((
‑
l10
j
)((
‑
l5
d
‑
b2
b
)
h
((
‑
l6
e
‑
l7
f
)(
‑
l8
i
‑
c)
c
)
g
))
k
[0055]
(化合物ii),
[0056]
其中
[0057]
如果b2是第二效应子部分,则a2是第二配体,或者如果b2是第二配体,则a2是第二效应子部分;
[0058]
c是皂苷;
[0059]
如果a2是第二配体并且b2是第二效应子部分,则a=0或1,或者如果a2是第二效应
子部分并且b2是第二配体,则a=0
‑
32;
[0060]
如果b2是第二配体并且a2是第二效应子部分,或者如果a2是第二配体并且b2是第二效应子部分,则b=0或1;
[0061]
c=1
‑
128中的任一个;
[0062]
l5是用于共价偶联两个化学基团的至少一个接头;
[0063]
l6是用于共价偶联两个化学基团的至少一个接头;
[0064]
l7是用于共价偶联两个化学基团的至少一个寡聚支架或多聚支架;
[0065]
l8是用于共价偶联两个化学基团的至少一个接头;
[0066]
l10是用于共价偶联三个化学基团的三官能接头;
[0067]
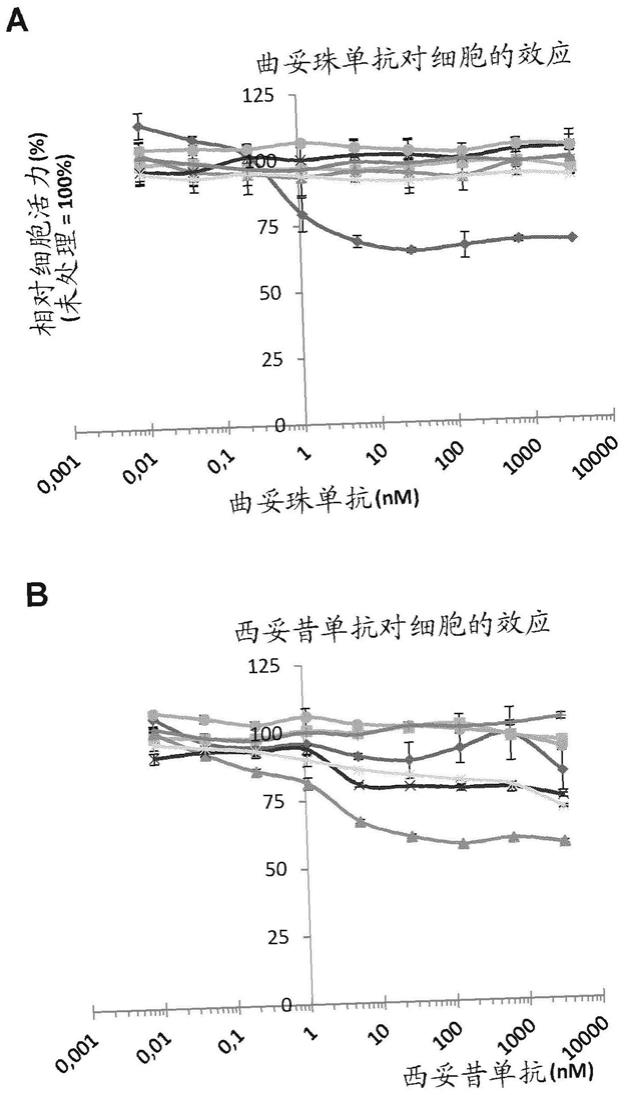
d=0或1;
[0068]
e=0或1;
[0069]
f=0或1;
[0070]
如果f=0,则g=0、1或2,并且如果f=1,则g=0
‑
16中的任一个;
[0071]
如果a2是第二配体并且b2是第二效应子部分,则h=0
‑
32中的任一个,或者如果a2是第二效应子部分并且b2是第二配体,则h=1;
[0072]
i=0或1;
[0073]
j=1或0,以及;
[0074]
k=1
‑
16。
[0075]
一个实施方案涉及本发明的治疗性分子,其中r=0,s=0,v=0,s=0, v=0,p=0,t=0,配体a1是权利要求3
‑
7中任一项所述的单克隆抗体或其至少一个结合片段或结合结构域,m=1,效应子部分b1是权利要求8所述的效应子部分,优选bna,q=0,n=1并且u=2
‑
4,或者q=1, n=2
‑
4,u=2
‑
4以及接头l1是权利要求21
‑
30中任一项所述的寡聚支架或多聚支架l3。
[0076]
一个实施方案是本发明的治疗性组合,其中治疗性分子是之前实施方案的治疗性分子,并且其中第二配体a2是根据本发明的单克隆抗体或其至少一个结合片段或结合结构域,a=1,d=0,b=0,h=0,e=0, l7是根据本发明的支架并且f=1,或者l7不存在并且f=0,l8是根据本发明的接头或可裂解接头,i=1,c=2
‑
4并且如果f=1,则g=2
‑
4以及如果f=0,则g=0,以及皂苷c是根据本发明的皂苷,优选皂苷c是 so1861和/或qs
‑
21,条件是配体a1和配体a2是相同的或不同的。
[0077]
本发明的一方面涉及治疗性组合,其中治疗性组合包含:(a)第一药物组合物,其包含具有根据本发明的化合物i的化学结构的治疗性分子,第一药物组合物任选地还包含药学上可接受的赋形剂;以及(b) 第二药物组合物,其包含具有根据本发明的化合物ii的化学结构的第二治疗性分子,第二药物组合物任选地还包含药学上可接受的赋形剂。
[0078]
本发明的一方面涉及本发明的第一药物组合物,其用作药物。
[0079]
本发明的一方面涉及治疗性组合,其用于治疗或预防人类对象中的癌症,其中治疗性组合包含:(a)本发明的第一药物组合物;以及(b) 本发明的第二药物组合物,其中配体a1或b1和配体a2或b2能够结合肿瘤细胞表面分子,优选肿瘤细胞特异性表面分子上的肿瘤细胞表位,优选肿瘤细胞特异性表位,条件是配体a1或b1能够结合的肿瘤细胞表位或肿瘤细胞特异性表位与配体a2或b2能够结合的肿瘤细胞表位或肿瘤细胞特异性表位是相同
的或不同的。
[0080]
本发明的一方面涉及本发明的第一药物组合物,其还包含本发明的第二治疗性分子。
[0081]
本发明的一方面涉及还包含本发明的第二治疗性分子的本发明的第一药物组合物,其用作药物。
[0082]
本发明的一方面涉及还包含本发明的第二治疗性分子的本发明的第一药物组合物,其用于治疗或预防人类对象中的癌症。
[0083]
本发明的一方面涉及以下adc和aoc中的任一种,及其半成品缀合物,其包含含有本发明的共价连接的皂苷的缀合物和/或包含含有连接至如本发明的抗体的有效载荷或效应子部分并且分别任选地还包含本发明的至少一个效应分子或任选地还包含本发明的至少一个皂苷或两者的缀合物;
[0084]
抗egfr抗体
‑
皂苷;
[0085]
抗egfr抗体
‑
三萜皂苷和/或双糖链三萜皂苷,其属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团;
[0086]
抗egfr抗体
‑
so1861;
[0087]
抗egfr抗体
‑
ge1741;
[0088]
抗egfr抗体
‑
sa1641;
[0089]
抗egfr抗体
‑
quil
‑
a;
[0090]
抗egfr抗体
‑
qs
‑
21;
[0091]
抗egfr抗体
‑
皂皮树(quillaja saponaria)的水溶性皂苷级分中的皂苷;
[0092]
西妥昔单抗
‑
皂苷;
[0093]
西妥昔单抗
‑
三萜皂苷和/或双糖链三萜皂苷,其属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团;
[0094]
西妥昔单抗
‑
so1861;
[0095]
西妥昔单抗
‑
ge1741;
[0096]
西妥昔单抗
‑
sa1641;
[0097]
西妥昔单抗
‑
quil
‑
a;
[0098]
西妥昔单抗
‑
qs
‑
21;
[0099]
西妥昔单抗
‑
皂皮树的水溶性皂苷级分中的皂苷;
[0100]
抗her2抗体
‑
皂苷;
[0101]
抗her2抗体
‑
三萜皂苷和/或双糖链三萜皂苷,其属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团;
[0102]
抗her2抗体
‑
so1861;
[0103]
抗her2抗体
‑
ge1741;
[0104]
抗her2抗体
‑
sa1641;
[0105]
抗her2抗体
‑
quil
‑
a;
[0106]
抗her2抗体
‑
qs
‑
21;
[0107]
抗her2抗体
‑
皂皮树的水溶性皂苷级分中的皂苷;
[0108]
曲妥珠单抗
‑
皂苷;
[0109]
曲妥珠单抗
‑
三萜皂苷和/或双糖链三萜皂苷,属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团;
[0110]
曲妥珠单抗
‑
so1861;
[0111]
曲妥珠单抗
‑
ge1741;
[0112]
曲妥珠单抗
‑
sa1641;
[0113]
曲妥珠单抗
‑
quil
‑
a;
[0114]
曲妥珠单抗
‑
qs
‑
21;
[0115]
曲妥珠单抗
‑
皂皮树的水溶性皂苷级分中的皂苷;
[0116]
抗cd71抗体
‑
皂苷;
[0117]
抗cd71抗体
‑
三萜皂苷和/或双糖链三萜皂苷,其属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团;
[0118]
抗cd71抗体
‑
so1861;
[0119]
抗cd71抗体
‑
ge1741;
[0120]
抗cd71抗体
‑
sa1641;
[0121]
抗cd71抗体
‑
quil
‑
a;
[0122]
抗cd71抗体
‑
qs
‑
21;
[0123]
抗cd71抗体
‑
皂皮树的水溶性皂苷级分中的皂苷;
[0124]
okt
‑9‑
皂苷;
[0125]
okt
‑9‑
三萜皂苷和/或双糖链三萜皂苷,其属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团;
[0126]
okt
‑9‑
so1861;
[0127]
okt
‑9‑
ge1741;
[0128]
okt
‑9‑
sa1641;
[0129]
okt
‑9‑
quil
‑
a;
[0130]
okt
‑9‑
qs
‑
21;
[0131]
okt
‑9‑
皂皮树的水溶性皂苷级分中的皂苷;
[0132]
抗egfr抗体
‑
寡核苷酸;
[0133]
抗egfr抗体
‑
反义寡核苷酸;
[0134]
抗egfr抗体
‑
sirna;
[0135]
抗egfr抗体
‑
反义bna;
[0136]
抗egfr抗体
‑
反义bna(hsp27);
[0137]
抗egfr抗体
‑
蛋白质毒素;
[0138]
抗egfr抗体
‑
核糖体失活蛋白;
[0139]
抗egfr抗体
‑
石竹素;
[0140]
抗egfr抗体
‑
皂草素;
[0141]
西妥昔单抗
‑
寡核苷酸;
[0142]
西妥昔单抗
‑
反义寡核苷酸;
[0143]
西妥昔单抗
‑
sirna;
[0144]
西妥昔单抗
‑
反义bna;
[0145]
西妥昔单抗
‑
反义bna(hsp27);
[0146]
西妥昔单抗
‑
蛋白质毒素;
[0147]
西妥昔单抗
‑
核糖体失活蛋白;
[0148]
西妥昔单抗
‑
石竹素;
[0149]
西妥昔单抗
‑
皂草素;
[0150]
抗her2抗体
‑
寡核苷酸;
[0151]
抗her2抗体
‑
反义寡核苷酸;
[0152]
抗her2抗体
‑
sirna;
[0153]
抗her2抗体
‑
反义bna;
[0154]
抗her2抗体
‑
反义bna(hsp27);
[0155]
抗her2抗体
‑
蛋白质毒素;
[0156]
抗her2抗体
‑
核糖体失活蛋白;
[0157]
抗her2抗体
‑
石竹素;
[0158]
抗her2抗体
‑
皂草素;
[0159]
曲妥珠单抗
‑
寡核苷酸;
[0160]
曲妥珠单抗
‑
反义寡核苷酸;
[0161]
曲妥珠单抗
‑
sirna;
[0162]
曲妥珠单抗
‑
反义bna;
[0163]
曲妥珠单抗
‑
反义bna(hsp27);
[0164]
曲妥珠单抗
‑
蛋白质毒素;
[0165]
曲妥珠单抗
‑
核糖体失活蛋白;
[0166]
曲妥珠单抗
‑
石竹素;
[0167]
曲妥珠单抗
‑
皂草素;
[0168]
抗cd71抗体
‑
寡核苷酸;
[0169]
抗cd71抗体
‑
反义寡核苷酸;
[0170]
抗cd71抗体
‑
sirna;
[0171]
抗cd71抗体
‑
反义bna;
[0172]
抗cd71抗体
‑
反义bna(hsp27);
[0173]
抗cd71抗体
‑
蛋白质毒素;
[0174]
抗cd71抗体
‑
核糖体失活蛋白;
[0175]
抗cd71抗体
‑
石竹素;
[0176]
抗cd71抗体
‑
皂草素;
[0177]
okt
‑9‑
寡核苷酸;
[0178]
okt
‑9‑
反义寡核苷酸;
[0179]
okt
‑9‑
sirna;
[0180]
okt
‑9‑
反义bna;
[0181]
okt
‑9‑
反义bna(hsp27);
[0182]
okt
‑9‑
蛋白质毒素;
[0183]
okt
‑9‑
核糖体失活蛋白;
[0184]
okt
‑9‑
石竹素;
[0185]
okt
‑9‑
皂草素;
[0186]
抗egfr抗体(
‑
寡核苷酸)皂苷),其中寡核苷酸是以下中的任何一种或多种:反义寡核苷酸、sirna、反义bna和反义bna(hsp27),并且其中皂苷是以下中的任何一种或多种:属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷、so1861、ge1741、sa1641、quil
‑
a、qs
‑
21和皂皮树的水溶性皂苷级分中的皂苷,其中抗egfr抗体优选是西妥昔单抗;
[0187]
抗egfr抗体(
‑
蛋白质毒素)(
‑
皂苷),其中蛋白质毒素是以下中的任何一种或多种:核糖体失活蛋白、石竹素和皂草素,并且其中皂苷是以下中的任何一种或多种:属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷、so1861、 ge1741、sa1641、quil
‑
a、qs
‑
21和皂皮树的水溶性皂苷级分中的皂苷,其中抗egfr抗体优选是西妥昔单抗;
[0188]
抗her2抗体(
‑
寡核苷酸)(
‑
皂苷),其中寡核苷酸是以下中的任何一种或多种:反义寡核苷酸、sirna、反义bna和反义bna(hsp27),并且其中皂苷是以下中的任何一种或多种:属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷、so1861、ge1741、sa1641、quil
‑
a、qs
‑
21和皂皮树的水溶性皂苷级分中的皂苷,其中抗her2抗体优选是曲妥珠单抗;
[0189]
抗her2抗体(
‑
蛋白质毒素)(
‑
皂苷),其中蛋白质毒素是以下中的任何一种或多种:核糖体失活蛋白、石竹素和皂草素,并且其中皂苷是以下中的任何一种或多种:属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷、so1861、 ge1741、sa1641、quil
‑
a、qs
‑
21和皂皮树的水溶性皂苷级分中的皂苷,其中抗her2抗体优选是曲妥珠单抗;
[0190]
抗cd71抗体(
‑
寡核苷酸)(
‑
皂苷),其中寡核苷酸是以下中的任何一种或多种:反义寡核苷酸、sirna、反义bna和反义bna(hsp27),并且其中皂苷是以下中的任何一种或多种:属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷、so1861、ge1741、sa1641、quil
‑
a、qs
‑
21和皂皮树的水溶性皂苷级分中的皂苷,其中抗cd71抗体优选是okt
‑
9;和
[0191]
抗cd71抗体(
‑
蛋白质毒素)(
‑
皂苷),其中蛋白质毒素是以下中的任何一种或多
种:核糖体失活蛋白、石竹素和皂草素,并且其中皂苷是以下中的任何一种或多种:属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷、so1861、 ge1741、sa1641、quil
‑
a、qs
‑
21和皂皮树的水溶性皂苷级分中的皂苷,其中抗cd71抗体优选是okt
‑
9。
[0192]
一个实施方案是本发明的第一蛋白质分子、本发明的半成品缀合物或本发明的缀合物,其中第一结合位点选自西妥昔单抗、曲妥珠单抗、okt
‑
9,和/或其中效应分子选自石竹素、皂草素和反义 bna(hsp27),和/或其中皂苷选自so1861、ge1741、sa1641、quil
‑ꢀ
a、qs
‑
21和皂皮树的水溶性皂苷级分中的皂苷。
[0193]
一个实施方案是根据本发明的缀合物,其中第一蛋白质分子选自西妥昔单抗、曲妥珠单抗、okt
‑
9,和/或其中效应分子选自石竹素、皂草素和反义bna(hsp27),和/或其中皂苷选自so1861、ge1741、 sa1641、quil
‑
a、qs
‑
21和皂皮树的水溶性皂苷级分中的皂苷。
[0194]
本发明的一方面涉及结构c的adc或aoc或半成品adc缀合物或半成品aoc缀合物,其包含本发明的抗体
‑
皂苷缀合物并且包含本发明的至少一个效应分子和/或包含本发明的至少一个皂苷:
[0195]
a(
‑
s)b(
‑
e)c
[0196]
结构c,
[0197]
其中a是第一结合位点;
[0198]
s是皂苷;
[0199]
e是效应分子;
[0200]
b=0
‑
64,优选0、1、2、3、4、8、16、32、64或者其间的任何整数或分数;
[0201]
c=0
‑
8,优选0、1、2、3、4、6、8或者其间的任何整数或分数,
[0202]
其中s偶联至a和/或e,e偶联至a和/或s,优选s偶联至a 并且e偶联至a。
[0203]
一个实施方案是本发明的结构c,其中a是抗egfr抗体如西妥昔单抗、抗her2抗体如曲妥珠单抗、抗cd71抗体如okt
‑
9,和/或其中s是以下中的任何一种或多种:皂苷、属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷、so1861、ge1741、sa1641、quil
‑
a、qs
‑
21和皂皮树的水溶性皂苷级分中的皂苷,和/或其中e是以下中的任何一种或多种:寡核苷酸、反义寡核苷酸、sirna、反义bna和反义bna(hsp27),和/或以下中的任何一种或多种:蛋白质毒素、核糖体失活蛋白、石竹素和皂草素。
[0204]
一个实施方案是本发明的结构c、本发明的缀合物或本发明的半成品缀合物或本发明的第一蛋白质分子,其中皂苷(如果存在)和/或效应分子(如果存在)经由至少一个接头(如可裂解接头)和/或经由至少一个寡聚支架或多聚支架,如基于n
‑
ε
‑
马来酰亚胺基己酸酰肼(emch)、 3
‑
(2
‑
吡啶基二硫代)丙酸琥珀酰亚胺酯或3
‑
(2
‑
吡啶基二硫代)丙酸n
‑
羟基琥珀酰亚胺酯(spdp)和1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并 [4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐(hatu)的接头以及如基于树枝化基元(如g4
‑
树枝化基元)的支架,或三官能接头(如方案ii的三官能接头) 共价偶联,和/或其中抗体(优选单克隆抗体或其片段或结构域)的第一结合位点的至少一条赖氨酸侧链和/或半胱氨酸侧链参与同皂苷和/或效应分子和/或接头和/或可裂解接头和/或支架的共价键,其中优选地,皂苷和/或效应分
子共价连接至抗体(优选单克隆抗体)的第一结合位点,其中共价连接包含以下或由以下组成:酰胺键、腙键、二硫键。
[0205]
本发明的一方面涉及本发明的上述缀合物中的任一种或者本发明的半成品缀合物或本发明的抗体
‑
皂苷缀合物中的任一种作为药物的用途。
[0206]
本发明的一方面涉及本发明的缀合物或本发明的半成品缀合物或本发明的抗体
‑
皂苷缀合物中的任一种用于治疗或预防癌症或自身免疫疾病的用途。
[0207]
图25和图26显示了具有共价偶联的皂苷的本发明的adc和具有共价偶联的皂苷的本发明的oac的实例。
[0208]
当然,a、b、c、d、e、f、g、h、i、j、k、m、n、p、q、r、s、t、 u、v、w和/或x中的任一个和全部对于根据本发明的任何和全部上述方面和实施方案,具有根据本发明的每个单独实施方案和方面的值。此外,(三官能)接头l1、l2、l4、l5、l6、l8、l9和/或l10,如果存在于本发明的分子或缀合物或部分中,则为本发明的上述方面和实施方案中的每一个和任一个所示的(三官能)接头,如技术人员容易理解的。寡聚支架或多聚支架l3和/或l7,如果存在于本发明的分子或缀合物或部分中,则为本发明的上述方面和实施方案中的每一个和任一个所示的寡聚支架或多聚支架,也如技术人员容易理解的。此外,第一配体a1和第一效应子部分b1(如果存在),以及第二配体a2和第二效应子部分b2(如果存在),以及第一效应子部分a1和第一配体b1(如果存在),以及第二效应子部分a2和第二配体b2(如果存在)是选定和指定的配体和效应子部分,如针对本发明的第一、第二、第三、第四、第五和第六系列的实施方案和方面以及上文列出的本发明的所有另外的实施方案和方面所公开的。皂苷c是本发明的上述方面和实施方案的任一个中提及和列出的皂苷中的任何一种或多种,特别是选自方案i和/或表a1的一种或多种皂苷。
[0209]
定义
[0210]
术语“接头”具有其常规的科学含义,并且在本文是指通过肽键复合的氨基酸残基的化学部分或线性区段,其将分子或原子连接至另一个分子,如连接至配体或效应分子或支架。通常,接头包含通过化学键连接的原子链。本领域已知的任何接头分子或接头技术均可用于本公开内容中。在指示的情况下,接头是用于通过适合与接头形成共价连键或共价键的此类分子上的化学基团共价结合分子的接头。接头可以是不可裂解接头,如,接头在生理条件下是稳定的。接头可以是可裂解接头,如在酶的存在下或在特定的ph范围或值下或在生理条件下(如在内体(如晚期内体)和哺乳动物细胞(如人类细胞)的溶酶体中的胞内条件)可裂解的接头。可用于本公开内容上下文中的示例性接头包括但不限于n
‑
ε
‑
马来酰亚胺基己酸酰肼(emch)、3
‑
(2
‑
吡啶基二硫代) 丙酸琥珀酰亚胺酯或3
‑
(2
‑
吡啶基二硫代)丙酸n
‑
羟基琥珀酰亚胺酯 (spdp)和1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑ꢀ
氧化物六氟磷酸盐(hatu)。
[0211]
术语“三官能接头”具有其常规的科学含义,并且在本文是指经由三个分子中的每一个上的化学基团连接三个分子的接头。技术人员能够基于本公开内容和公知常识设计此类三官能接头。此类三官能接头可以表现出例如马来酰亚胺基基团,马来酰亚胺基基团可用于缀合至表现出硫醇基团的靶向配体以进行硫醇
‑
烯反应。此外,三官能接头可以表现出二苯并环辛炔(dbco)基团,以与具有叠氮基的皂苷进行所谓的应变促进的炔烃
‑
叠氮化物环加成(spaac,点击化学)。最后,三官能接头可以获得第三个官能团,如反式环辛烯(tco)
基团,以与具有四嗪(tz)的效应分子进行所谓的逆电子需求狄尔斯
‑
阿尔德(inverseelectron demand diels
‑
alder,iedda)反应。技术人员将理解,三官能接头的化学基团可以是所有三个相同或不同的,或者接头可以包含用于将分子连接到三官能接头的两个相同的化学基团。三官能接头之间形成的键可以是共价的或非共价的,并且优选共价键。三官能接头与一个或两个或三个结合的分子之间经由各自的化学基团形成的键,可以是可裂解的(不稳定的)键,如在酸性条件下在细胞内(如哺乳动物细胞(如人类细胞)的内体和溶酶体)裂解,或者可以是不可裂解的。当然,三官能接头可以涵盖一个或两个用于形成共价键的化学基团,而另外的两个或一个化学基团分别用于形成非共价键。当然,三官能接头可以涵盖一个或两个用于形成可裂解键的化学基团,而另外的两个或一个化学基团分别用于形成不可裂解的键。
[0212]
诸如在术语“可裂解接头”或“可裂解键”中使用的术语“可裂解的”具有其常规的科学含义,并且在本文是指在酸性条件、还原条件、酶促条件或光诱导条件下进行裂解。例如,可裂解接头可以在酸性条件下进行裂解,优选地所述可裂解接头在如存在于哺乳动物细胞,优选人类细胞的内体和/或溶酶体的酸性条件下,优选在ph 4.0
‑
6.5下,并且更优选在ph≤5.5下在体内进行裂解。作为另一个实例,可裂解接头可以通过酶,如通过组织蛋白酶进行裂解。此外,在还原条件下可裂解的共价键的一个实例是二硫键。
[0213]
在寡聚支架或多聚支架的上下文中的术语“寡聚物”和“多聚物”具有其常规的科学含义。本文的多聚物是指分子结构主要或完全由大量相同或相似的单元键合在一起构成的物质;本文的寡聚物是指其分子由相对较少的重复单元组成的聚合物。例如,包含5
‑
10个或更少相同或相似单元的结构可以被称为寡聚结构,而包含10
‑
50个单体单元或更多的结构可以被称为多聚结构,而10个单体单元的结构可以被称为寡聚的或多聚的。
[0214]
术语“结合位点”具有其常规的科学含义,并且在本文是指分子(如,蛋白质、dna或rna)上的另一分子能够结合的区域或表位。
[0215]
术语“支架”具有其常规的科学含义,并且在本文是指一个或多个分子(如配体分子、效应分子)可以直接或经由接头(如可裂解接头)共价结合的寡聚或多聚模板或者载体或者基础(base)(基础分子或基础结构)。支架可具有结构上有序的形成,如多聚物、寡聚物、树状物、树枝化基元化多聚物或树枝化基元化寡聚物或者具有组装的聚合结构,如水凝胶、微凝胶、纳米凝胶、稳定的聚合胶束或脂质体,但不包括由单体的非共价组装体(如胆固醇/磷脂混合物)组成的结构。支架可包含多聚结构或寡聚结构,如聚
‑
或寡聚(胺),如聚乙烯亚胺和聚(酰氨基胺);或者结构,如聚乙二醇,聚或寡聚(酯),如聚(丙交酯)、聚(内酰胺)、聚丙交酯
‑
共
‑
乙交酯共聚物;或聚(糊精)、聚糖或寡糖,如环糊精或聚葡萄糖;或者结构,如天然和/或人工聚氨基酸或寡氨基酸,如聚赖氨酸或肽或蛋白质、dna寡聚物或多聚物、稳定的rna聚合物或 pna(肽核酸)聚合物。优选地,多聚结构或寡聚结构是生物相容的,其中生物相容意指多聚结构或寡聚结构在有机体内不显示出显著的急性或慢性毒性,并且可以原样排泄或通过身体的代谢完全降解为可排泄的化合物和/或生理化合物。
[0216]
术语“配体”具有其常规的科学含义,并且在本文是指可以选择性地结合靶细胞(如靶标癌细胞或靶标自身免疫细胞)处表达的靶细胞表面分子或靶细胞表面受体的任何一种或多种分子。配体可以结合靶细胞上的受体或其它抗原所包含的表位。优选地,细胞结合配体是抗体。
[0217]
如本文所用的术语“抗体”以最广泛的含义使用,可以指被定义为属于igg类、igm类、ige类、iga类或igd类(或其任何亚类)的蛋白质的免疫球蛋白(ig)或者免疫球蛋白的功能结合片段或结合结构域。在本发明的上下文中,免疫球蛋白的“结合片段”或“结合结构域”被定义为亲本免疫球蛋白的抗原结合片段或抗原结合结构域或其它衍生物,其基本上保持了此类亲本免疫球蛋白的抗原结合活性。功能片段和功能结构域是本发明意义上的抗体,即使它们对抗原的亲和力低于亲本免疫球蛋白。根据本发明的“功能片段和功能结构域”包括但不限于 f(ab')2片段、fab'片段、fab片段、scfv、dsfv、单域抗体(sdab)、单价igg、scfv
‑
fc、还原的igg(rigg)、微抗体、双链抗体、三链抗体、四链抗体、fc融合蛋白、纳米抗体、可变v结构域,如vhh、vh和其它类型的抗原识别免疫球蛋白片段和结构域。可以工程化片段和结构域以最小化或完全去除发生在ch1和cl结构域之间的分子间二硫化物相互作用。功能片段和功能结构域因其较小的尺寸而提供了更大的肿瘤渗透优势。此外,与整个免疫球蛋白相比,功能片段或功能结构域可以更均匀地分布在整个肿瘤团块中。
[0218]
本发明的抗体(免疫球蛋白)可以是双功能的或多功能的。例如,双功能抗体的一个臂对一种受体或抗原具有特异性,而另一臂识别不同的受体或抗原。可替代地,双功能抗体的每个臂可以对靶细胞的相同受体或抗原的不同表位具有特异性。
[0219]
本发明的抗体(免疫球蛋白)可以是但不限于多克隆抗体、单克隆抗体、人抗体、人源化抗体、嵌合抗体、重新表面化抗体、抗独特型抗体、小鼠抗体、大鼠抗体、大鼠/小鼠杂合抗体、美洲驼抗体、仅美洲驼重链抗体、仅重链抗体和兽用抗体。优选地,本发明的抗体(免疫球蛋白)是单克隆抗体。重新表面化的、嵌合的、人源化的和全人抗体也是更优选的,因为它们不太可能在人类中引起免疫原性。本发明的 adc的抗体优选特异性结合在癌细胞、自身免疫细胞、患病细胞、异常细胞的表面上表达的抗原,同时使任何健康细胞基本上未改变(如通过不结合此类正常细胞,或者通过在数量上和/或亲和力上以较小程度结合此类健康细胞)。
[0220]
可用于本发明的adc的特异性抗体包括但不限于抗her2单克隆抗体如曲妥珠单抗和帕妥珠单抗,抗cd20单克隆抗体如利妥昔单抗、奥法木单抗、托西莫单抗和异贝莫单抗(ibritumomab),抗ca125 单克隆抗体如奥瑞戈单抗(oregovomab),抗epcam(17
‑
1a)单克隆抗体如依决洛单抗,抗egfr单克隆抗体如西妥昔单抗、帕尼单抗和尼妥珠单抗,抗cd30单克隆抗体如本妥昔单抗,抗cd33单克隆抗体如吉妥珠单抗和humy9
‑
6,抗血管整合素α
‑
vβ
‑
3单克隆抗体如埃达珠单抗(etaracizumab),抗cd52单克隆抗体如阿仑单抗,抗cd22单克隆抗体如依帕珠单抗,抗cea单克隆抗体如拉贝珠单抗(labetuzumab),抗cd44v6单克隆抗体如比伐珠单抗(bivatuzumab),抗fap单克隆抗体如西罗珠单抗(sibrotuzumab),抗cd19单克隆抗体如hub4,抗canag 单克隆抗体如huc242,抗cd56单克隆抗体如hun901,抗cd38单克隆抗体如达雷木单抗,抗ca6单克隆抗体如ds6,抗igf
‑
ir单克隆抗体如西妥木单抗和3b7,抗整合素单克隆抗体如cnto 95以及抗多配体蛋白聚糖
‑
1单克隆抗体如b
‑
b4。
[0221]
除了结合靶细胞的细胞受体或抗原的抗体以外的任何其它分子也可用作本发明的配体
‑
药物缀合物的细胞结合配体以及提供有根据本发明的共价结合的皂苷的配体。这些配体包括但不限于蛋白质、多肽、肽、小分子。这些非抗体配体的实例是干扰素(如ifn
‑
α、ifn
‑
β和ifn
‑ꢀ
γ)、转铁蛋白、凝集素、表皮生长因子(egf)和egf样结构域、胃泌素释放肽(grp)、血小板衍生生长因子(pdgf)、转化生长因子(tgf)、痘苗生长因子(vgf)、胰岛素和胰
岛素样生长因子(igf,如igf
‑
1和igf
‑ꢀ
2)、其它适合的激素如促甲状腺素释放激素(trh)、黑素细胞刺激激素 (msh)、类固醇激素(如雌激素和雄激素)、生长激素抑制素、淋巴因子 (如il
‑
2、il
‑
3、il
‑
4和il
‑
6)、集落刺激因子(csf,如g
‑
csf、m
‑
csf 和gm
‑
csf)、蛙皮素、胃泌素、arg
‑
gly
‑
asp或rgd、适配体(如as
‑ꢀ
1411、gbi
‑
10、针对hiv糖蛋白的rna适配体)、小分子(如叶酸、茴香酰胺苯基硼酸)、维生素(如维生素d)、碳水化合物(如透明质酸、半乳糖)。
[0222]“效应分子”或“效应子部分”或“有效载荷”具有其常规的科学含义,并且在本发明的上下文中是通过与胞内效应分子靶标相互作用而影响细胞代谢的任何物质,其中该效应分子靶标是除了内吞和循环途径的区室和囊泡的腔之外但包括这些区室和囊泡的膜的细胞内的任何分子或结构。因此,细胞内的所述结构包括细胞核、线粒体、叶绿体、内质网、高尔基体、其它转运囊泡、质膜的内部和胞质。
[0223]
效应分子或效应子部分是药学活性物质,如毒素,如蛋白质毒素、药物、多肽或多核苷酸。本发明中的药学活性物质是用于在有机体内,优选脊椎动物,更优选哺乳动物,如非人类对象或人类/对象中实现有益结果的效应分子或效应子部分。益处包括疾病和/或症状和/或健康问题的诊断、预后、治疗、治愈和防止(预防)。药学活性物质也可能导致不希望的并且有时甚至是有害的副作用(如临床试验期间观察到的不良事件)。在这种情况下,必须权衡利弊,以决定药学活性物质是否适用于特定情况。如果药学活性物质在细胞内的效应主要对整个有机体有益,则该细胞被称为靶细胞。如果细胞内的效应主要对整个有机体有害,则该细胞被称为脱靶细胞。在人工系统如细胞培养和生物反应器中,靶细胞和脱靶细胞取决于目的并由用户定义。效应分子和效应子部分的实例是药物、毒素、多肽(如酶)、多核苷酸(包括包含非天然氨基酸或核酸的多肽和多核苷酸)及其任何组合。
[0224]
作为药物的效应分子或效应子部分可以包括但不限于抗癌剂、抗炎剂和抗感染(如,抗真菌、抗细菌、抗寄生虫、抗病毒)剂。优选地,本发明的药物分子是抗癌剂或抗自身免疫剂。适合的抗癌剂包括但不限于烷化剂、抗代谢物、纺锤体毒植物生物碱、细胞毒性/抗肿瘤抗生素、拓扑异构酶抑制剂、光敏剂和激酶抑制剂。“抗癌剂”的定义中还包括:如,(i)抗激素剂,其用于调控或抑制激素对肿瘤的作用,如抗雌激素和选择性雌激素受体调节剂;(ii)抑制芳香酶的芳香酶抑制剂,其调控肾上腺中的雌激素产生;(iii)抗雄激素;(iv)蛋白激酶抑制剂; (v)脂质激酶抑制剂;(vi)反义寡核苷酸,特别是抑制参与异常细胞增殖的信号传导途径中的基因表达的那些;(vii)核酶,如vegf表达抑制剂和her2表达抑制剂;(viii)疫苗,如基因疗法疫苗;拓扑异构酶1 抑制剂;(ix)抗血管生成剂;以及上述中任一种的药学上可接受的盐、酸、溶剂化物和衍生物。
[0225]
作为毒素的效应分子或效应子部分可以包括但不限于:蛋白质毒性(如细菌源性毒素和植物源性毒素)、靶向微管蛋白丝的毒素、靶向 dna的毒素、靶向rna的毒素。蛋白质毒素的实例是皂草素、石竹素、蓖麻毒素、蒴莲根毒素、相思子毒素、蒴莲毒素、槲寄生素、志贺毒素、志贺样毒素、假单胞菌外毒素(pe,也称为外毒素a)、白喉毒素(dt)和霍乱毒素。靶向微管蛋白细丝的毒素的实例是美登素(如dm1 和dm4)、澳瑞他汀(如单甲基澳瑞他汀e(mmae)和单甲基澳瑞他汀 f(mmaf))、类毒素、微管菌素、念珠藻素、根瘤菌素。dna靶向毒素的实例是卡奇霉素:n
‑
乙酰基
‑
γ
‑
卡奇霉素、cc
‑
1065类似物、倍癌霉素、多柔比星、甲氨蝶呤、苯并二氮杂喜树碱类似物和蒽环类药物。dna靶向毒性的实例是鹅膏蕈碱、斯考他汀(spliceostatins)和泰兰斯他汀(thailanstatins)。如本发明中使用的毒素被定义为能够
杀伤或灭活细胞的药学活性物质。优选地,靶向毒素是仅或至少主要对靶细胞有毒但对脱靶细胞无毒的毒素。靶向毒素的净效应优选对整个有机体是有益的。
[0226]
作为多肽的效应分子或效应子部分可以是如恢复失去的功能、诸如例如酶替代、基因调控功能的多肽,或毒素。作为效应分子的多肽的实例是如cas9;毒素(如皂草素、石竹素、白树毒素、(去)三角梅核糖体失活蛋白((de)bouganin)、麦仙翁毒素(agrostin)、蓖麻毒素(毒素a 链);商陆抗病毒蛋白、凋亡蛋白、白喉毒素、假单胞菌外毒素);代谢酶(如精氨基琥珀酸裂解酶、精氨基琥珀酸合成酶)、凝血级联酶、修复酶;用于细胞信号传导的酶;细胞周期调控因子;基因调控因子(转录因子如nf
‑
κb或基因阻遏子如甲硫氨酸阻遏子)。
[0227]
作为多核苷酸的效应分子或效应子部分可以是如包含编码信息如编码蛋白质的基因或开放阅读框的多核苷酸。它还可以包含调控信息,如启动子或调控元件结合区,或编码微小rna的序列。此类多核苷酸可以包括天然和人工核酸。人工核酸包括如肽核酸(pna)、吗啉代和锁核酸(lna)以及二醇核酸(gna)和苏糖核酸(tna)。这些中的每一个都通过分子主链的变化与天然存在的dna或rna区分开来。作为效应分子的核苷酸的实例是但不限于,如,dna:单链dna(如,腺嘌呤磷酸核糖转移酶的dna);线性双链dna(如凝血因子ix基因);环状双链dna(如质粒);rna:mrna(如tal效应分子核酸酶)、trna、 rrna、sirna、mirna、反义rna;反义寡核苷酸(aso、aon,如 pna、pmo、lna和bna)。
[0228]
在如“蛋白质分子”和“蛋白质毒素”中使用的术语“蛋白质”是包含至少一串氨基酸残基的分子和毒素,其可以作为表达产物从单一 mrna获得。此类分子或毒素还可包含任何翻译后修饰、碳水化合物如n
‑
或o
‑
连接的碳水化合物、二硫键、磷酸化、硫酸化等作为任何翻译后修饰的结果,和/或还可包含任何其它修饰如由化学修饰产生的那些(如,连接效应子部分、皂苷、支架、配体等,直接连接至如氨基酸侧链,或经由至少一个接头(共价)结合用于化学修饰蛋白质分子的分子以及化学结合(共价)蛋白质分子)。术语“蛋白质”还涵盖并包括此类分子的组装体,如同源二聚体、异源三聚体、异源六聚体或复杂的组装体如核糖体。
[0229]
在例如“特异性结合”和“特异性地存在或表达于肿瘤细胞表面上的受体或分子靶标”等的上下文中的术语“特异性的(specific)”和“特异性地(specifically)”具有本领域已知的其正常的科学含义,并且在本文是指如相对于第一分子与不同于第二分子的另一分子的任何推定结合以更高的亲和力发生的第一分子与第二分子的结合相互作用,或者是指如当例如考虑受体或分子靶标的数量时,相对于在第二类型细胞(如健康细胞等)处相同受体或分子靶标的表达程度,第一类型细胞(如肿瘤细胞、自身免疫细胞、患病细胞、异常细胞)的表面上细胞表面受体或分子靶标的表达或表达至更高程度,其中在第二类型细胞处的表达相对于肿瘤细胞等上的任何表达程度可完全不存在或非常低。此外,例如在“特异性结合”中的术语“特异性(specificity)”具有本领域已知的其正常科学含义,并且在本文具有表示可以具有以比分子之间的本底相互作用更高的结合亲和力与另一分子相互作用的分子的含义。类似地,术语“特异性(specificity)”是指,例如,两个分子之间或细胞与分子之间的相互作用,其具有比分子之间的本底相互作用更高的结合亲和力。结合分子如免疫球蛋白经由其结合位点(如免疫球蛋白的免疫球蛋白可变区)以比分子之间的本底相互作用更高的结合亲和力结合分子上的结合位点,如表位、细胞表面受体等。在本发明的上下文中,本底相互作用通常是具有低于10e
‑
4m的k
d
的亲和力的相互作用。类似地,“特异性结合
结构域”是以比分子之间的本底相互作用更高的结合亲和力优先结合分子上的结合位点,如表位、细胞表面受体等的结构域。在本发明的上下文中,“本底相互作用”通常是具有低于10e
‑
4m 的k
d
的亲和力的相互作用。优选地,特异性结合结构域以高于约10e
‑ꢀ
5m的k
d
的亲和力结合。
[0230]
术语“结合”被定义为可以与本底相互作用区分开来的分子之间的相互作用。
[0231]
在整个说明书中,术语“片段”是指作为蛋白质结构域的一部分或构建完整蛋白质结构域的氨基酸序列。根据本发明的结合片段必须对各自的靶标,如例如诸如肿瘤细胞的患病细胞的表面上的细胞表面受体具有结合特异性。
[0232]
术语“adc”或“抗体
‑
药物缀合物”具有技术人员已知的其常规的科学含义,并且在本文是指被设计为用于治疗如癌症的靶向疗法的一类生物制药药物。与化疗不同,adc旨在靶向并杀伤肿瘤细胞,同时保留健康细胞。adc由与具有生物活性的细胞毒性(抗癌)有效载荷或药物连接的抗体组成。adc将单克隆抗体的靶向能力与细胞毒性药物的杀癌能力组合在一起。它们被设计用于区分健康细胞和患病组织(如肿瘤中的肿瘤细胞)。
[0233]
术语“saponinum album”具有其正常含义,并且在本文是指由 merck kgaa(darmstadt,germany)生产的皂苷混合物,其含有来自圆锥石头花(gypsophila paniculata)和gypsophila arostii的皂苷,含有 sa1657并且主要是sa1641。
[0234]
术语“皂皮树皂苷(quillajasaponin)”具有其正常含义,并且在本文是指皂皮树的皂苷级分并因此是所有其它qs皂苷的来源,主要含有 qs
‑
18和qs
‑
21。
[0235]“qs
‑
21”或“qs21”具有其常规的科学含义,并且在本文是指qs
‑
21 a
‑
apio(~63%)、qs
‑
21a
‑
xylo(~32%),qs
‑
21b
‑
apio(~3.3%)和qs
‑
21b
‑ꢀ
xylo(~1.7%)的混合物。
[0236]
类似地,“qs
‑
21a”具有其常规的科学含义,并且在本文是指qs
‑ꢀ
21a
‑
apio(~65%)和qs
‑
21a
‑
xylo(~35%)的混合物。
[0237]
类似地,“qs
‑
21b”具有其常规的科学含义,并且在本文是指qs
‑ꢀ
21b
‑
apio(~65%)和qs
‑
21b
‑
xylo(~35%)的混合物。
[0238]
术语“quil
‑
a”是指可商购获得的来自皂皮树的半纯化提取物,并且含有可变数量的50多种不同的皂苷,其中许多在qs
‑
7、qs
‑
17、 qs18和qs
‑
21中存在的c
‑
3β
‑
oh基团处掺入三萜
‑
三糖亚结构gal
‑ꢀ
(1
→
2)
‑
[xyl
‑
(1
→
3)]
‑
glca
‑
。quil
‑
a中发现的皂苷列于van setten(1995),表2中[dirk c.van setten、gerrit van de werken、gijsbert zomer和gideon f.a.kersten,glycosyl compositions and structuralcharacteristics of the potential immuno
‑
adjuvant active saponins in thequillaja saponaria molina extract quil a,rapid communicationsin mass spectrometry,第9卷,660
‑
666(1995)]。quil
‑
a和皂皮树皂苷是来自皂皮树的皂苷的级分,并且两者都含有各种各样不同的皂苷,其含量大部分重叠。这两种级分的具体组成不同,因为两种级分是通过不同的纯化程序获得的。
[0239]
术语“qs1861”和术语“qs1862”是指qs
‑
7和qs
‑
7api。qs1861的分子质量是1861道尔顿,qs1862的分子质量是1862道尔顿。qs1862 描述于fleck等人(2019)的表1,第28行[juliane deise fleck,andresaheemann betti,francini pereira da silva,eduardo artur troian,cristinaolivaro,fernando ferreira和simone gasparin verza,
saponins fromquillaja saponaria and quillaja brasiliensis:particular chemicalcharacteristics and biological activities,molecules 2019,24,171; doi:10.3390/molecules24010171]。所述的结构是qs
‑
7的api
‑
变体 qs1862。分子质量是1862道尔顿,因为该质量是包括葡糖醛酸处的质子在内的正式质量。在中性ph下,分子被去质子化。当以负离子模式在质谱法中测量时,测量的质量是1861道尔顿。
[0240]
说明书和权利要求中的术语第一、第二、第三等用于区分相似的要素,并不一定用于描述依序或时间顺序。在适当的情况下,术语可以互换。本发明的实施方案可以以不同于本文描述或例示的其它顺序操作。
[0241]
此外,各个实施方案,虽然被称为“优选的”或“如”或“例如”或“特别地”,但应被解释为可以实施本发明的示例性方式而不是限制本发明的范围。
[0242]
权利要求中使用的术语“包括/包含”不应被解释为限于其后列出的要素或步骤;它不排除其它要素或步骤。需要将其解释为指定所提及的所述特征、整数、步骤或组分的存在,但不排除一个或多个其它特征、整数、步骤或组分或其群组的存在或添加。因此,表述“包含a 和b的药物组合物”的范围不应限于仅由组分a和b组成的药物组合物,而是就本发明而言,药物组合物的唯一列举的组分是a和b,而且权利要求应被解释为包括那些组分的等效物。类似地,表述“包括步骤a和步骤b的方法”的范围不应限于仅由步骤a和b组成的方法,而就本发明而言,该方法仅列举的步骤为a和b,而且权利要求应被解释为包括那些步骤的等效物。
[0243]
此外,不定冠词“一个/一种(a)”或“一个/一种(an)”对特征的提及不排除存在多于一个特征(如例如组分、赋形剂、皂苷等)的可能性,除非上下文清楚地要求有一个且只有一个特征。因此,不定冠词“一个/ 一种(a)”或“一个/一种(an)”通常意指“至少一个/种”。
[0244]
附图简述
[0245]
图1
‑
1:用30mg/kg西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑ꢀ
hsp27bna)、25mg/kg西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4或25mg/kg 西妥昔单抗
‑
(cys
‑
l
‑
so1861)处理的a431异种移植
‘
裸’鼠肿瘤模型中的体内hsp27表达。
[0246]
图2
‑
1:通过用西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑ꢀ
hsp27bna)、西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4或西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)处理,表达egfr的a431细胞中的体外增强的hsp27基因沉默。
[0247]
图3
‑
1:图a、b、c和d的图例和坐标轴是相同的。a.西妥昔单抗、西妥昔单抗 10pm西妥昔单抗
‑
皂草素、西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
和西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑ꢀ
so1861)4)
3,9
10pm西妥昔单抗
‑
皂草素在表达egfr的细胞(mda
‑ꢀ
mb
‑
468)中的细胞杀伤活性。b.曲妥珠单抗、曲妥珠单抗 50pm曲妥珠单抗
‑
皂草素、曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4和曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4 50pm曲妥珠单抗
‑
皂草素在表达her2的细胞(sk
‑
br
‑
3)中的细胞杀伤活性。c.西妥昔单抗、西妥昔单抗 10pm西妥昔单抗
‑
皂草素、西妥昔单抗
‑
cys
‑
(树枝化基元(
‑ꢀ
l
‑
so1861)4)
3,9
和西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
10pm 西妥昔单抗
‑
皂草素在表达egfr的细胞(hela)中的细胞杀伤活性。d. 曲妥珠单抗、曲妥珠单抗 50pm曲妥珠单抗
‑
皂草素、曲妥珠单抗
‑
cys
‑ꢀ
(树枝化基元(
‑
l
‑
so1861)4)4和曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑ꢀ
so1861)4)4 50pm曲妥珠单抗
‑
皂草素在表达her2的细胞(jimt
‑
1)中的细胞杀伤活性。
[0248]
图4
‑
1:图a、b、c和d的图例和坐标轴是相同的。a.西妥昔单抗、西妥昔单抗 10pm cd71mab
‑
皂草素、西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
、西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑ꢀ
so1861)4)
3,9
10pm cd71mab
‑
皂草素、西妥昔单抗
‑
lys
‑
(树枝化基元(
‑ꢀ
l
‑
so1861)4)
4,4
或西妥昔单抗
‑
lys
‑
(树枝化基元(
‑
l
‑
so1861)4)
4,4
10pmcd71mab
‑
皂草素在egfr
/cd71
细胞(mda
‑
mb
‑
468)中的细胞杀伤活性。b.曲妥珠单抗、曲妥珠单抗 10pm cd71mab
‑
皂草素、曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4、曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑ꢀ
l
‑
so1861)4)4 10pm cd71mab
‑
皂草素、曲妥珠单抗
‑
lys
‑
(树枝化基元 (
‑
l
‑
so1861)4)
4,7
或曲妥珠单抗
‑
lys
‑
(树枝化基元(
‑
l
‑
so1861)4)
4,7
10 pm cd71mab
‑
皂草素在her2
/cd71
(sk
‑
br
‑
3)细胞系中的细胞杀伤活性。c.西妥昔单抗、西妥昔单抗 10pm cd71mab
‑
皂草素、西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
、西妥昔单抗
‑
cys
‑
(树枝化基元 (
‑
l
‑
so1861)4)
3,9
10pm cd71mab
‑
皂草素、西妥昔单抗
‑
lys
‑
(树枝化基元(
‑
l
‑
so1861)4)
4,4
或西妥昔单抗
‑
lys
‑
(树枝化基元(
‑
l
‑
so1861)4)
4,4
10 pm cd71mab
‑
皂草素在egfr
/cd71
细胞(caski)中的细胞杀伤活性。 d.曲妥珠单抗、曲妥珠单抗 10pm cd71mab
‑
皂草素、曲妥珠单抗
‑ꢀ
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4、曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑ꢀ
so1861)4)4 10pm cd71mab
‑
皂草素、曲妥珠单抗
‑
lys
‑
(树枝化基元(
‑
l
‑ꢀ
so1861)4)
4,7
或曲妥珠单抗
‑
lys
‑
(树枝化基元(
‑
l
‑
so1861)4)
4,7
10pmcd71mab
‑
皂草素在her2
/
‑
/cd71
细胞(jimt
‑
1)中的细胞杀伤活性。
[0249]
图5
‑
1:t
‑
dm1、t
‑
dm1 25.6nm曲妥珠单抗或t
‑
dm1 5.3nm曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4在表达her2的细胞(sk
‑ꢀ
br
‑
3)中的细胞杀伤活性。
[0250]
图6
‑
1:与单独的hsp27bna相比,hsp27bna
‑
树枝化基元(
‑
l
‑ꢀ
so1861)4的hsp27基因沉默活性。
[0251]
图7
‑
1:在酸性条件下由树枝化基元(
‑
l
‑
so1861)4释放so1861的示意图。
[0252]
图8
‑
1:图a和b的图例和坐标轴是相同的。a.
‘
裸’树枝化基元 (树枝化基元(nem)4)、树枝化基元(nem)4 10pm egf石竹素、树枝化基元(
‑
l
‑
so1861)4或树枝化基元(l
‑
so1861)4 10pm egf石竹素在表达egfr的a431细胞中的细胞杀伤活性。b.
‘
裸’树枝化基元(树枝化基元(nem)4)、树枝化基元(nem)4 10pm egf石竹素、树枝化基元(
‑ꢀ
l
‑
so1861)4或树枝化基元(l
‑
so1861)4 10pm egf石竹素在表达 egfr的hela细胞中的细胞杀伤活性。
[0253]
图9
‑
1:图a、b和c的图例和坐标轴是相同的。a.曲妥珠单抗对一系列癌细胞的细胞活力的效应。b.西妥昔单抗对一系列癌细胞的细胞活力的效应。c.t
‑
dm1对一系列癌细胞的细胞活力的效应。
[0254]
图10
‑
1:单克隆抗体
‑
(so1861
‑
支架
‑
反义bna寡核苷酸)缀合物的示意图。
[0255]
图11
‑
1:使用结合毒素的单克隆抗体和结合包含皂苷的支架的相同单克隆抗体的1
‑
靶标2
‑
组分系统的示意图。
[0256]
图12
‑
1:使用结合毒素的单克隆抗体和具有结合包含皂苷的支架的不同靶标的不同单克隆抗体的2
‑
靶标2
‑
组分系统的示意图。
[0257]
图13
‑
1:树枝化基元(
‑
l
‑
so1861)4的示意图。
[0258]
图14
‑
1:树枝化基元(
‑
l
‑
so1861)8的示意图。
[0259]
图15
‑
1:so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna的示意图。
[0260]
图16
‑
1:树枝化基元(so1861)4‑
hsp27bna寡核苷酸缀合物的合成的反应方案。
[0261]
图17
‑
1:由用于经由希夫碱(schiff base)(亚胺)结合皂苷的四个分子臂和用于点击化学的一个臂组成的模型支架。聚合结构是第一代的基于五价聚乙二醇的树状物。
[0262]
图18
‑
1:在用五聚物的树状物(pentrimer)、sa1641存在下的 pentrimer、石竹素
‑
egf、sa1641存在下的石竹素
‑
efg、石竹素
‑
egf 存在下的pentrimer及石竹素
‑
egf和sa1641存在下的pentrimer处理之后her14细胞的细胞活力。
[0263]
图19
‑
1:用于将增强内体逃逸的皂苷缀合到聚合结构的具有突出显示的化学基团的so1861结构。突出显示的基团是醛(黑色圆圈)、羧酸(虚线圆圈)、烯烃(虚线五边形)和醇(虚线框)。
[0264]
图20
‑
1:a.so
‑
1861
‑
emch缀合物的标准分子结构。马来酰亚胺基团用圆圈标记。b.so1861
‑
emch缀合物的3d模型。马来酰亚胺基团用圆圈标记。
[0265]
图21
‑
1:使用so1861
‑
emch作为单体、aps/tmeda系统作为聚合引发剂和氨基丙硫醇作为自由基淬灭剂生成聚(so1861)的反应方案。
[0266]
图22
‑
1:dna方法的示意图。利用dna
‑
origami的原理,产生了能够缀合并释放糖苷分子的基于dna的支架。此外,一条dna链获得了点击化学部分,其可用于与靶向毒素缀合以形成官能化支架。bp:碱基对。
[0267]
图23
‑
1:聚(肽
‑
so1861)方法的示意图。利用肽序列,其可以缀合并释放糖苷分子并且可以与自身反应而形成聚(肽
‑
so1861)构建体。多 (肽)链末端还可以用点击化学部分(如,bcn
‑
nhs接头)修饰,所述点击化学部分可用于与毒素缀合。
[0268]
图24
‑
1:具有保护的氨基基团的g4
‑
树枝化基元的分子结构。
[0269]
图25
‑
1:具有连接任何所需效应分子的点击化学官能团的基本支架的示意图。
[0270]
图26
‑
1:具有预结合的效应分子和连接任何所需配体的点击化学官能团的官能化支架的示意图。任选地,可以提供ph敏感连键以在到达内体之后从支架释放效应分子。
[0271]
图1
‑
2.抗体
‑
蛋白质毒素 未缀合的so1861体内研究。用各种浓度的曲妥珠单抗
‑
皂草素(i.v.) 1.5mg/kg未缀合的so1861(曲妥珠单抗
ꢀ‑
皂草素处理之前1小时subq注射)处理bt474荷瘤小鼠。
[0272]
图2
‑
2.未缀合的皂苷介导的内体逃逸和靶细胞杀伤增强。a)在具有或没有1.5pm egf石竹素的情况下,用so1861、so1832、 so1862(so1861的异构体)或so1904处理的hela细胞(egfr
)的细胞活力分析。b)用egf石竹素和固定浓度的so1861、so1832、so1862(so1861的异构体)或so1904处理的hela细胞(egfr
)的细胞活力分析。c)在具有或没有1.5pm egf石竹素的情况下,用so1861 或ge1741处理的hela细胞(egfr
)的细胞活力分析。d)在具有或没有1.5pm egf石竹素的情况下用各种qsmix(来自皂皮树(quillaiasaponaria)的皂苷混合物)处理的hela细胞(egfr
)的细胞活力分析。
[0273]
图3
‑
2.未缀合的so1861对比so1861
‑
emch活性。根据本发明,癌细胞中egfr靶向的反义bna寡核苷酸递送和基因沉默。a,b, c)在具有或没有1.5pm eg石竹素的情况下,用so1861或so1861
‑ꢀ
emch处理的a431(egfr
)、hela(egfr
)或a2058(egfr
‑
)细胞的细胞活力分析。d,e)在具有或没有1.5pm egf石竹素的情况下,用 so1861或so1861
‑
n3处理的a431(egfr
)或hela(egfr
)细胞的细胞活力分析。
[0274]
图4
‑
2.未缀合的so1861对比so1861
‑
emch(不稳定的)对比 so1861
‑
s(稳定的)。在具有或没有egf石竹素的情况下用so1861、 so1861
‑
s(s=hatu,稳定接头)和so1861
‑
emch
(不稳定接头)处理的 hela细胞(egfr
)的细胞活力分析。
[0275]
图5
‑
2.egfr靶向的反义bna寡核苷酸递送和基因沉默。用西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3.9
或西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3.9
100nmhsp27bna处理的a431(egfr
)和a2058(egfr
‑
)细胞的hsp27 mrna表达分析。
[0276]
图6
‑
2.荷瘤小鼠中肿瘤靶向的反义bna寡核苷酸递送和基因沉默。与对照相比,在a431荷瘤小鼠中用hsp27bna 西妥昔单抗
‑
(cys
‑ꢀ
l
‑
so1861)
3,9
处理的小鼠揭示了有效的肿瘤靶向基因沉默。
[0277]
图1
‑
3:a431癌细胞系中hsp27bna、hsp27bna
‑
so1861和 hsp27bna
‑
树枝化基元
‑
(so1861)4的hsp27bna基因沉默活性。
[0278]
图1
‑
4:在荷瘤小鼠中,肿瘤靶向蛋白质毒素递送导致肿瘤体积减小和肿瘤生长抑制。a.与对照相比,a431荷瘤小鼠中西妥昔单抗
‑
(cys
‑ꢀ
l
‑
so1861)
3,9
(lys
‑
s
‑
石竹素)2的剂量递增(腹膜内,i.p.)揭示了肿瘤体积减小。b.与对照相比,a431荷瘤小鼠中西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,9
(lys
‑
l
‑
石竹素)2的剂量递增(i.p)揭示了肿瘤生长减小。c.与对照相比,a431荷瘤小鼠中西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
l
‑
石竹素)2的静脉内剂量递增(i.v.)揭示了生长减小。
[0279]
图2
‑
4:荷瘤小鼠中肿瘤靶向的反义bna寡核苷酸递送和基因沉默。与对照相比,a431荷瘤小鼠中30mg/kg西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,9
(lys
‑
l
‑
hsp27bna)
1,8
揭示了诱导的有效肿瘤靶向的基因沉默。
[0280]
图3
‑
4:荷瘤小鼠中肿瘤靶向的反义bna寡核苷酸递送和基因沉默。与对照相比,a431荷瘤小鼠中30mg/kg西妥昔单抗
‑
cys
‑
(so1861
‑ꢀ
l
‑
三官能接头
‑
l
‑
hsp27bna)
3,7
揭示了诱导的有效肿瘤靶向的基因沉默。
[0281]
图4
‑
4:根据本发明,癌细胞中her2或egfr靶向的蛋白质毒素递送和细胞杀伤。a.sk
‑
br
‑
3细胞(her2
)上曲妥珠单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
(lys
‑
l
‑
石竹素)
1,7
或曲妥珠单抗
‑
(cys
‑
l
‑
so1861)
3,8
(lys
‑
s
‑
石竹素)
1,7
处理和对照。b.mda
‑
mb
‑
468细胞(her2
‑
)上的曲妥珠单抗
‑ꢀ
(cys
‑
l
‑
so1861)
3,8
(lys
‑
l
‑
石竹素)
1,7
或曲妥珠单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
(lys
‑
s
‑
石竹素)
1,7
处理和对照。c.a431细胞(egfr
)上的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
(lys
‑
l
‑
石竹素)
1,7
或西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
(lys
‑
s
‑
石竹素)
1,7
处理和对照。d.a2058细胞(egfr
‑
)上的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
(lys
‑
l
‑
石竹素)
1,7
或西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
(lys
‑
s
‑
石竹素)
1,7
处理和对照。
[0282]
图5
‑
4:根据本发明,癌细胞中egfr靶向的反义bna寡核苷酸递送和基因沉默。a.a431细胞(egfr
)上的西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
(lys
‑
l
‑
hsp27bna)
1,7
处理和对照。b.a2058细胞(egfr
‑
)上的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
(lys
‑
l
‑
hsp27bna)
1,7
处理和对照。
[0283]
图6
‑
4:根据本发明,癌细胞中her2靶向的反义bna寡核苷酸递送和基因沉默。sk
‑
br
‑
3细胞(her2
)上曲妥珠单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
(lys
‑
l
‑
hsp27bna)
3,5
处理和对照。
[0284]
图7
‑
4:根据本发明,癌细胞中egfr靶向的反义bna寡核苷酸递送和基因沉默。a.a431细胞(egfr
)上的西妥昔单抗
‑
cys
‑
(so1861
‑ꢀ
l
‑
三官能接头
‑
l
‑
hsp27bna)
3,7
处理和对照。b.a2058细胞(egfr
‑
)上的西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna)
3,7
处理和对照。
[0285]
图8
‑
4:对所有细胞系的对照处理。当曲妥珠单抗(a)、西妥昔单抗(b)、t
‑
dm1、(c)游离毒素:皂草素和石竹素(d)或偶联至非细胞结合igg的皂草素(d)用作对指定细胞系sk
‑
br
‑
3、jimt
‑
1、mda
‑
mb
‑ꢀ
468、a431、caski、hela、a2058、bt
‑
474的处理时的细胞活力。
[0286]
图9
‑
4:(s)n
‑
(l)(e)概念:mab
‑
(so1861)
n
(蛋白质毒素)
n
。半胱氨酸残基(cys)处的so1861和赖氨酸残基处的蛋白质毒素(核糖体失活蛋白)两者均缀合至相同的抗体(mab),以递送和内化到靶细胞中。1) mab
‑
(cys
‑
l
‑
so1861)4(lys
‑
蛋白质毒素)2结合其相应的细胞表面受体, 2)发生受体介导的缀合物的内吞作用,3)在低内溶酶体ph值和适当浓度下,so1861变得有活性,以使内溶酶体逃逸,4)发生毒素释放到细胞质中,以及5)毒素诱导细胞死亡。
[0287]
图10
‑
4:(s)n
‑
(l)(e)概念:mab
‑
(so1861)
n
(反义bna寡核苷酸)
n
. 半胱氨酸残基(cys)处的so1861和赖氨酸残基处的反义bna寡核苷酸两者均缀合至相同的抗体(mab),以递送和内化到靶细胞中。1)mab
‑ꢀ
(cys
‑
so1861)4(lys
‑
bna寡核苷酸)2结合其相应的细胞表面受体,2)发生受体介导的两种缀合物的内吞作用,3)在低内溶酶体ph和适当浓度下,so1861变得有活性,以使内溶酶体逃逸,4)发生bna寡核苷酸释放到细胞质中,以及5)诱导靶基因沉默。
[0288]
图11
‑
4:(s)n
‑
(l)(e)概念:mab
‑
(so1861
‑
支架
‑
反义bna寡核苷酸)
n
.(so1861
‑
三官能接头
‑
bna寡核苷酸)
n
缀合至抗体(mab),以用于递送并内化至靶细胞中。1)mab
‑
(so1861
‑
三官能接头
‑
bnaoligo)4结合其相应的细胞表面受体,2)发生受体介导的两种缀合物的内吞作用, 3)在低内溶酶体ph和适当浓度下,so1861变得有活性,以使内溶酶体逃逸,4)发生bna寡核苷酸释放到细胞质中,以及5)诱导靶基因沉默。
[0289]
图12
‑
4:抗体
‑
so1861缀合程序。显示了植物来源的皂苷so1861 的四个部分与抗体轻链中的四个半胱氨酸连接的偶联反应。首先,igg 中的二硫键在暴露于tcep(三(2
‑
羧乙基)膦)的影响下被破坏;其次,包含与其结合的化学接头的皂苷so1861与三氟乙酸一起添加,并且四个皂苷部分连接至igg。为了产生可裂解的
‘
准备缀合(ready toconjugate)’皂苷,将so1861的醛基团与emch(ε
‑
马来酰亚胺基己酸酰肼)接头反应。emch的酰肼基团与so1861的醛形成酸可裂解的腙键。同时,emch接头呈现具有硫醇(巯基基团)反应性的马来酰亚胺基团,因此可缀合至igg的硫醇(即配体部分)。在此,提供了本发明的内体逃逸增强缀合物,和/或提供了本发明的第一结合分子。
[0290]
图1
‑
5:1t2c体内活性。与对照相比,a431荷瘤小鼠中50mg/kg 西妥昔单抗
‑
(cys
‑
l
‑
so1861)
4
25mg/kg西妥昔单抗
‑
(
‑
l
‑
hsp27bna)4的1t2c组合揭示了强烈的肿瘤靶向基因沉默。
[0291]
图2
‑
5:1t2c体内活性。pdx肿瘤小鼠模型(高her2表达)中40 mg/kg曲妥珠单抗
‑
(cys
‑
l
‑
so1861)
4
0.02/0.03mg/kg曲妥珠单抗
‑
皂草素的1t2c组合显示了有效的肿瘤生长抑制。
[0292]
图3
‑
5:1
‑
靶标2
‑
组分(1
‑
target 2
‑
component)。根据本发明的治疗性组合对a431细胞(egfr
)(a,c)和caski细胞(egfr
)(b,d)中的 egfr靶向的细胞杀伤。a,b)a431(a)和caski(b)细胞上的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
滴定 固定浓度的10pm西妥昔单抗
‑
皂草素和对照。c,d)a431(c)和caski(d)细胞上的西妥昔单抗
‑
皂草素滴定 固定浓度的75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
和对照。备注:对于每个细胞系的靶受体表达数据(通过facs
分析确定),参见表19。
[0293]
图4
‑
5:1
‑
靶标2
‑
组分。根据本发明的治疗性组合对hela细胞 (egfr
/
‑
)(a,c)和a2058细胞(egfr
‑
)(b,d)中的egfr靶向的细胞杀伤。a,b)hela(a)和caski(b)细胞上的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
滴定 固定浓度的10pm西妥昔单抗
‑
皂草素和对照。c,d)hela(c)和 a2058(d)细胞上的西妥昔单抗
‑
皂草素滴定 固定浓度的75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
和对照。备注:对于每个细胞系的靶受体表达数据(通过facs分析确定),参见表19。
[0294]
图5
‑
5:1
‑
靶标2
‑
组分。根据本发明的治疗性组合对skbr3细胞 (her2
)(a,b)中的her2靶向的细胞杀伤。a)skbr3细胞上的曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4滴定 固定浓度的50pm曲妥珠单抗
‑
皂草素和对照。b)skbr3细胞上的曲妥珠单抗
‑
皂草素滴定 固定浓度的 2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4和对照。备注:对于每个细胞系的靶受体表达数据(通过facs分析确定),参见表19。
[0295]
图6
‑
5:1
‑
靶标2
‑
组分。根据本发明的治疗性组合对jimt
‑
1细胞 (her2
/
‑
)(a,c)和mda
‑
mb
‑
468细胞(her2
‑
)(b,d)中的her2靶向的细胞杀伤。a,b)jimt
‑
1(a)和mda
‑
mb
‑
468(b)细胞上的曲妥珠单抗
‑ꢀ
(cys
‑
l
‑
so1861)4滴定 固定浓度的50pm曲妥珠单抗
‑
皂草素和对照。 c,d)jimt
‑
1(c)和mda
‑
mb
‑
468(d)细胞上的曲妥珠单抗
‑
皂草素滴定 固定浓度的2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4和对照。备注:对于每个细胞系的靶受体表达数据(通过facs分析确定),参见表19。
[0296]
图7
‑
5:氯喹抑制1
‑
靶标2
‑
组分。根据本发明的治疗性组合 氯喹在sk
‑
br
‑
3(her2
)和a431细胞(egfr
)中的her2和egfr靶向的细胞杀伤。a)sk
‑
br
‑
3细胞上的曲妥珠单抗
‑
皂草素滴定 固定浓度的5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4 0.5μm氯喹和对照。b)a431细胞上的西妥昔单抗
‑
皂草素滴定 固定浓度的5nm西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
0.5μm氯喹和对照。备注:对于每个细胞系的靶受体表达数据(通过facs分析确定),参见表19。
[0297]
图8
‑
5:1
‑
靶标2
‑
组分。根据本发明的治疗性组合对a431细胞 (egfr
)和a2058细胞(egfr
‑
)中的egfr靶向的基因沉默。a,b) a431细胞(a)和a2058细胞(b)上的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
滴定 固定浓度的100nm西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4和对照。c,d) a431细胞(c)和a2058细胞(d)上的西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4滴定 固定浓度的77nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
和对照。备注:对于每个细胞系的靶受体表达数据(通过facs分析确定),参见表19。
[0298]
图9
‑
5:2
‑
靶标2
‑
组分。a)根据本发明的治疗性组合对mda
‑
mb
‑ꢀ
468细胞(egfr
)和hela细胞(egfr
/
‑
)中的egfr和her2靶向的细胞杀伤,以及在sk
‑
br
‑
3细胞(her2
)和jimt
‑
1细胞(her2
/
‑
)中的her2靶向的细胞杀伤。a)mda
‑
mb
‑
468细胞(a)和hela细胞(b) 上的西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
滴定 固定浓度的10 pm西妥昔单抗
‑
皂草素和对照。c,d)sk
‑
br
‑
3细胞(c)和jimt
‑
1细胞(d)上的曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4滴定 固定浓度的 50pm曲妥珠单抗
‑
皂草素和对照。备注:对于每个细胞系的靶受体表达数据(通过facs分析确定),参见表19。
[0299]
图10
‑
5:1
‑
靶标2
‑
组分。sk
‑
br
‑
3细胞(her2
/
‑
)可被根据本发明的治疗性组合、曲妥珠单抗
‑
皂草素 2.5nm曲妥珠单抗
‑
(cys
‑
l
‑ꢀ
so1861)4有效杀伤,然而滴定t
‑
dm1 2.5nm曲妥珠单抗
‑
(cys
‑
l
‑ꢀ
so1861)4在如此低的毒素浓度下是无效的。t
‑
dm1是曲妥珠单抗
‑
美
坦新每抗体携带~3.5个美坦新(dm1)毒素分子(dar3.5)。备注:对于每个细胞系的靶受体表达数据(通过facs分析确定),参见表19。
[0300]
图11
‑
5:对所有细胞系的对照处理。a
‑
d)当用曲妥珠单抗(a)、西妥昔单抗(b)、t
‑
dm1(c)、游离毒素:皂草素和石竹素(d)或偶联至非细胞结合igg的皂草素(d)处理指定细胞系sk
‑
br
‑
3、jimt
‑
1、mda
‑ꢀ
mb
‑
468、a431、caski、hela、a2058、bt
‑
474时的细胞活力。备注:对于每个细胞系的靶受体表达数据(通过facs分析确定),参见表19。
[0301]
图12
‑
5:1
‑
靶标2
‑
组分。根据本发明的治疗性组合对a431细胞 (egfr
)(a)和caski细胞(egfr
)(b)和a2058细胞(egfr
‑
)中的 egfr靶向的细胞杀伤。a,b,c)a431细胞(a)、caski细胞(b)和 a2058细胞(c)中的西妥昔单抗
‑
(cys
‑
l
‑
qsmix)
4,1
滴定 固定浓度的10 pm西妥昔单抗
‑
皂草素或10pm西妥昔单抗
‑
石竹素和对照。qsmix是来自皂皮树提取物的皂苷的混合物。备注:对于每个细胞系的靶受体表达数据(通过facs分析确定),参见表19。
[0302]
图13
‑
5:1
‑
靶标2
‑
组分概念:mab1
‑
so1861 mab1
‑
蛋白质毒素。 so1861和毒素(核糖体失活蛋白)各自独立地缀合至抗体(mab1),以递送和内化到靶细胞中。1)mab1
‑
so1861和mab1
‑
蛋白质毒素结合细胞表面受体,2)发生受体介导的两种缀合物的内吞作用,3)在低内溶酶体 ph值和适当浓度下,so1861变得有活性,以使内溶酶体逃逸,4)发生毒素释放到细胞质中,以及5)毒素诱导细胞死亡
[0303]
图14
‑
5:1
‑
靶标2
‑
组分概念:mab1
‑
so1861 mab2
‑
bna寡核苷酸。 so1861和反义bna寡核苷酸各自独立地缀合至抗体(mab1),以递送和内化到靶细胞中。1)mab1
‑
so1861和mab1
‑
bna寡核苷酸结合细胞表面受体,2)发生受体介导的两种缀合物的内吞作用,3)在低内溶酶体ph和适当浓度下,so1861变得有活性,以使内溶酶体逃逸,4)发生bna寡核苷酸释放到细胞质中,以及5)靶基因沉默。
[0304]
图15
‑
5:1
‑
靶标2
‑
组分概念:mab1
‑
(支架(
‑
so1861)
n
)
n
mab1
‑
蛋白质毒素。树枝化基元(
‑
so1861)
n
和蛋白质毒素(核糖体失活蛋白)各自独立地缀合至抗体(mab1),以递送和内化到靶细胞中。1)mab1
‑
树枝化基元(
‑
so1861)4和mab1
‑
蛋白质毒素结合细胞表面受体,2)发生受体介导的两种缀合物的内吞作用,3)在低内溶酶体ph值和适当浓度下,so1861变得有活性,以使内溶酶体逃逸,4)发生毒素释放到细胞质中,以及5)毒素诱导细胞死亡。
[0305]
图1
‑
6:在a431荷瘤小鼠模型中测试的2t2组分系统揭示了肿瘤消退。
[0306]
图2
‑
6:在a431荷瘤小鼠模型中测试的2t2组分系统揭示了肿瘤消退和根除。
[0307]
图3
‑
6:2
‑
靶标2
‑
组分。根据本发明的治疗性组合对a431细胞 (egfr
/her2
/
‑
)(a,c)和caski细胞(egfr
/her2
/
‑
)(b,d)中的 egfr/her2靶向的细胞杀伤。a,b)a431细胞上的西妥昔单抗
‑
(cys
‑ꢀ
l
‑
so1861)
3,7
滴定 固定浓度的50pm曲妥珠单抗
‑
皂草素和对照。c, d)caski细胞上的曲妥珠单抗
‑
皂草素滴定 固定浓度的75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
和对照。对于所有a、b、c或d,图例和/或坐标轴是相同的。
[0308]
图4
‑
6:2
‑
靶标2
‑
组分。根据本发明的治疗性组合对hela细胞 (egfr
/
‑
/her2
/
‑
)(a,c)和a2058细胞(egfr
‑
/her2
/
‑
)(b,d)中的 egfr/her2靶向的细胞杀伤。a,b)hela细胞上的西妥昔单抗
‑
(cys
‑ꢀ
l
‑
so1861)
3,7
滴定 固定浓度的50pm曲妥珠单抗
‑
皂草素和对照。c, d)a2058细胞上的曲妥珠单抗
‑
皂草素滴定 固定浓度的75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
和对照。对于所有a、b、c或d,图例和/或坐标轴是相同的。
[0309]
图5
‑
6:2
‑
靶标2
‑
组分。根据本发明的治疗性组合对skbr3细胞(her2
/egfr
/
‑
)(a,
b)中的her2/egfr靶向的细胞杀伤。a)skbr3 细胞上的曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4滴定 固定浓度的1.5pm egf 石竹素和对照。b)skbr3细胞上的egf石竹素滴定 固定浓度的 2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4和对照。
[0310]
图6
‑
6:2
‑
靶标2
‑
组分。根据本发明的治疗性组合对jimt
‑
1细胞 (her2
/
‑
egfr
/
‑
)(a,c)和mda
‑
mb
‑
468细胞(her2
‑
/egfr
)(b,d)中的her2/egfr靶向的细胞杀伤。a,b)jimt
‑
1细胞上的曲妥珠单抗
‑ꢀ
(cys
‑
l
‑
so1861)4滴定 固定浓度的1.5pm egf石竹素和对照。c,d) mda
‑
mb
‑
468细胞上的egf石竹素滴定 固定浓度的2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4和对照。对于所有a、b、c或d,图例和/或坐标轴是相同的。
[0311]
图7
‑
6:2
‑
靶标2
‑
组分。根据本发明的治疗性组合对skbr3细胞 (her2
/egfr
/
‑
)(a,b)中的her2/egfr靶向的细胞杀伤。a)skbr3 细胞上的曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4滴定 固定浓度的10pm西妥昔单抗
‑
皂草素和对照。b)skbr3细胞上的西妥昔单抗
‑
皂草素滴定 固定浓度的2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4和对照。
[0312]
图8
‑
6:2
‑
靶标2
‑
组分。根据本发明的治疗性组合对jimt
‑
1细胞 (her2
/
‑
egfr
/
‑
)(a,c)和mda
‑
mb
‑
468细胞(her2
‑
/egfr
)(b,d)中的her2/egfr靶向的细胞杀伤。a,b)jimt
‑
1细胞上的曲妥珠单抗
‑ꢀ
(cys
‑
l
‑
so1861)4滴定 固定浓度10pm西妥昔单抗
‑
皂草素和对照。c, d)mda
‑
mb
‑
468细胞上的西妥昔单抗
‑
皂草素滴定 固定浓度的2.5nm 曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4和对照。对于所有a、b、c或d,图例和/或坐标轴是相同的。
[0313]
图9
‑
6:氯喹抑制2
‑
靶标2
‑
组分。根据本发明的治疗性组合 氯喹对a431细胞(egfr
/her2
/
‑
/cd71
)(a,b)、mda
‑
mb
‑
468细胞 (egfr
/her2
‑
/cd71
)(c)或sk
‑
br
‑
3(her2
/egfr
/
‑
/cd71
)(d)中的egfr/her2、egfr/cd71或her2/cd71靶向的细胞杀伤。a)a431 细胞上的曲妥珠单抗
‑
石竹素或曲妥珠单抗
‑
皂草素滴定 固定浓度的 75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
800nm氯喹和对照。b)a431细胞上的cd71mab
‑
皂草素滴定 固定浓度的10.5nm西妥昔单抗
‑
(cys
‑ꢀ
l
‑
so1861)
3,9
500nm氯喹和对照。c)mda
‑
mb
‑
468细胞上的 cd71mab
‑
皂草素滴定 固定浓度的10.5nm西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,9
500nm氯喹和对照。d)sk
‑
br
‑
3细胞上的cd71mab
‑
皂草素滴定 固定浓度的5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)
3,9
500nm氯喹和对照。
[0314]
图10
‑
6:2
‑
靶标2
‑
组分。根据本发明的治疗性组合对a431细胞 (egfr
/her2
/
‑
)(a)和a2058细胞(egfr
‑
/her2
/
‑
)(b)中的 egfr/her2靶向的基因沉默。a)a431细胞(a)和a2058细胞(b)上的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
滴定 固定浓度的100nm曲妥珠单抗
‑ꢀ
(lys
‑
l
‑
hsp27bna)
4,4
和对照。c,d)a431细胞(a)和a2058细胞(b)上的曲妥珠单抗
‑
(lys
‑
l
‑
hsp27bna)
4,4
滴定 固定浓度的77nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
和对照。对于所有a、b、c或d,图例和/或坐标轴是相同的。
[0315]
图11
‑
6:2
‑
靶标2
‑
组分。根据本发明的治疗性组合对a)mda
‑
mb
‑ꢀ
468细胞(egfr
/cd71
)(a)hela细胞(egfr
/
‑
/cd71
)、sk
‑
br
‑
3细胞(her2
/cd71
)(b)和jimt
‑
1细胞(her2
/
‑
/cd71
)中的egfr/cd71 或her2/cd71靶向的细胞杀伤。a)mda
‑
mb
‑
468细胞上的西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
滴定 固定浓度的10pm cd71mab
‑ꢀ
皂草素和对照。b)a)hela细胞上的西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑ꢀ
so1861)4)
3,9
滴定 固定浓度的10pm cd71mab
‑
皂草素和对照。c)sk
‑ꢀ
br
‑
3细胞上的曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4滴定 固定浓度的10pm cd71mab
‑
皂草素和对照。d)jimt
‑
1细胞上的曲妥珠单抗
‑
cys
‑
(树枝
化基元(
‑
l
‑
so1861)4)4滴定 固定浓度的10pm cd71mab
‑ꢀ
皂草素和对照。
[0316]
图12
‑
6:2
‑
靶标2
‑
组分对比t
‑
dm1。a431细胞(egfr
/her2
/
‑
) 可被根据本发明的治疗性组合、曲妥珠单抗
‑
皂草素 75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
有效杀伤,然而滴定t
‑
dm1 75nm西妥昔单抗
‑ꢀ
(cys
‑
l
‑
so1861)
3,9
在如此低的毒素浓度下是无效的。t
‑
dm1是曲妥珠单抗
‑
美坦新每抗体携带~3.5个美坦新(dm1)毒素分子。
[0317]
图13
‑
6:2
‑
靶标2
‑
组分。根据本发明的治疗性组合对a431细胞(egfr
/her2
/
‑
)(a)和caski细胞(egfr
/her2
/
‑
)(b)和a2058细胞(egfr
‑
/her2
/
‑
)中的egfr/cd71和egfr/her2靶向的细胞杀伤。 a,b,c)a431细胞(a)、caski细胞(b)和a2058细胞(c)上的西妥昔单抗
‑
(cys
‑
l
‑
qsmix)
4,1
滴定 固定浓度的10pm曲妥珠单抗
‑
皂草素或 10pm cd71mab
‑
皂草素和对照。qsmix是来自皂皮树提取物的皂苷的混合物。
[0318]
图14
‑
6:对所有细胞系的对照处理。a
‑
d)当用曲妥珠单抗(a)、西妥昔单抗(b)、t
‑
dm1(c)、游离毒素:皂草素和石竹素(d)或偶联至非细胞结合igg的皂草素(d)处理指定细胞系sk
‑
br
‑
3、jimt
‑
1、mda
‑ꢀ
mb
‑
468、a431、caski、hela、a2058、bt
‑
474时的细胞活力。对于所有a、b、c或d,图例和/或坐标轴是相同的。
[0319]
图15
‑
6:2
‑
靶标2
‑
组分概念:mab1
‑
so1861 mab2
‑
蛋白质毒素。 so1861和毒素(核糖体失活蛋白)各自分别缀合至抗体(mab),以递送和内化到靶细胞中。1)mab1
‑
so1861和mab2
‑
蛋白质毒素结合其相应的细胞表面受体,2)发生受体介导的两种缀合物的内吞作用,3)在低内溶酶体ph值和适当浓度下,so1861变得有活性,以使内溶酶体逃逸, 4)发生毒素释放到细胞质中,以及5)毒素诱导细胞死亡。
[0320]
图16
‑
6:2
‑
靶标2
‑
组分概念:mab1
‑
so1861 mab2
‑
bna寡核苷酸。 so1861和反义bna寡核苷酸各自分别缀合至抗体(mab),以递送和内化到靶细胞中。1)mab1
‑
so1861和mab2
‑
bna寡核苷酸结合其相应的细胞表面受体,2)发生受体介导的两种缀合物的内吞作用,3)在低内溶酶体ph和适当浓度下,so1861变得有活性,以使内溶酶体逃逸, 4)发生bna寡核苷酸释放到细胞质中,以及5)靶基因沉默。
[0321]
图1
‑
7:未缀合的so1861
‑
emch和hsp27bna的组合对a431 细胞(egfr
)(a)和a2058细胞(egfr
‑
)(b)中的egfr靶向的基因沉默,以及a431细胞(a)和a2058细胞(b)上的so1861
‑
emch和西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)滴定 固定浓度的4000nm so1861
‑
emch 和对照。(a)和(b)是基于hsp27bna或西妥昔单抗
‑
hsp27bna缀合物(nm)的图。未缀合的so1861
‑
emch与游离的hsp27bna的组合对 a431细胞(egfr
)(a)和a2058细胞(egfr
‑
)(b)中的egfr靶向的基因沉默,以及a431细胞(c)和a2058细胞(d)上的so1861
‑
emch和缀合物西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)滴定 固定浓度的4000nmso1861
‑
emch和对照。(c)和(d)是基于hsp27bna(nm)的图。图1
‑ꢀ
7b和d的图例也应用于图1
‑
7a和c。
[0322]
详述
[0323]
为了使生物活性分子发挥作用,该分子必须能够与其如在血清中、在细胞表面的外部或者细胞或细胞器的内部的靶标结合。例如,几乎所有基于蛋白质的靶向毒素的活性部分都必须进入靶细胞的胞质以介导其靶标调节作用。在许多配合(constellation)中,毒素保持无效,因为 (1)靶向部分内化较差并保持与细胞外部结合,(2)内化后循环回到细胞表面或(3)运输到内溶酶体中,在那,其被降解。尽管这些基本问题几十年来已为人所知并且在过去几十年中已经研究了500多种靶向毒素,但所述问题仍未得到解决,并且迄今为止
只有一种抗体靶向蛋白质毒素moxetumomab pasudotox
‑
tdfk(astrazenecapharmaceuticals lp)已被fda批准用于复发性或难治性毛细胞白血病。
[0324]
为了克服这些问题,已经描述了许多策略,包括将毒素重定向到内质网中的生物合成途径的内源性细胞膜转运复合物的方法,以及破坏或削弱内体(即细胞中内吞途径的区室)的膜完整性,从而促进内体逃逸的技术。这包括使用亲溶酶体胺,羧酸离子载体,钙通道拮抗剂,病毒、细菌、植物、动物、人类和合成来源的各种细胞穿透肽,其它有机分子和光诱导技术。尽管靶向毒素的功效通常在细胞培养中增强了数百或数千倍,在特殊情况下超过百万倍,但需要将内体逃逸增强剂与其它物质共施用会带来新的问题,包括另外的副作用、损失靶标特异性、难以确定治疗窗和细胞类型依赖性变化。
[0325]
所有策略(包括物理化学技术)都需要增强剂分子,其或多或少直接与膜相互作用,并且基本上包括小化学分子、次级代谢物、肽和蛋白质。所有这些物质的一个共同特征是它们本身不是靶细胞特异性的,并且以不同于靶向毒素的其它动力学分布。这是当前方法的一个主要缺点。
[0326]
将关于特定实施方案描述本发明,但本发明不限于此而仅由权利要求书限制。除非另有说明,否则本文描述的本发明的实施方案可以组合和协作操作。
[0327]
虽然本发明已经根据几个实施方案进行了描述,但预期在阅读说明书和研究附图和图表后,本领域的普通技术人员将明白其替代、修改、置换和等价物。本发明不以任何方式限于例示的实施方案。在不脱离由所附权利要求限定的范围的情况下可以进行改变。
[0328]
本发明的一方面涉及具有化合物i的化学结构的治疗性分子:
[0329]
a1
m
((
‑
l9
w
)((
‑
l1
q
‑
b1
n
)
u
((
‑
l2
r
‑
l3
s
)(
‑
l4
v
‑
c)
p
)
t
))
x
[0330]
(化合物i),
[0331]
其中
[0332]
如果b1是第一效应子部分,则a1是第一配体,或者如果b1是第一配体,则a1是第一效应子部分;
[0333]
c是皂苷;
[0334]
如果a1是第一配体并且b1是第一效应子部分,则m=0或1;
[0335]
如果a1是第一效应子部分并且b1是第一配体,则m=0
‑
32;
[0336]
如果b1是第一配体并且a1是第一效应子部分,或者如果a1是第一配体并且b1是第一效应子部分,则n=0或1;
[0337]
p=1
‑
128中的任一个;
[0338]
l1是用于共价偶联两个化学基团的至少一个接头;
[0339]
l2是用于共价偶联两个化学基团的至少一个接头;
[0340]
l3是用于共价偶联两个化学基团的至少一个寡聚支架或多聚支架;
[0341]
l4是用于共价偶联两个化学基团的至少一个接头;
[0342]
l9是用于共价偶联三个化学基团的三官能接头;
[0343]
q=0或1;
[0344]
r=0或1;
[0345]
s=0或1;
[0346]
如果s=0时,则t=0、1或2,以及如果s=1,则t=0
‑
16中的任一个;
[0347]
如果a1是所述第一配体并且b1是所述第一效应子部分,则u=0
‑ꢀ
32中的任一个,或者如果a1是所述第一效应子部分并且b1是所述第一配体,则u=1;
[0348]
v=0或1;
[0349]
w=1或0;以及
[0350]
x=1
‑
16。
[0351]
本发明的一方面涉及治疗性组合,其包含根据本发明的治疗性分子和具有化合物ii的化学结构的第二治疗性分子:
[0352]
a2
a
((
‑
l10
j
)((
‑
l5
d
‑
b2
b
)
h
((
‑
l6
e
‑
l7
f
)(
‑
l8
i
‑
c)
c
)
g
))
k
[0353]
(化合物ii),
[0354]
其中
[0355]
如果b2是第二效应子部分,则a2是第二配体,或者如果b2是第二配体,则a2是第二效应子部分;
[0356]
c是皂苷;
[0357]
如果a2是第二配体并且b2是第二效应子部分,则a=0或1,或者如果a2是第二效应子部分并且b2是第二配体,则a=0
‑
32;
[0358]
如果b2是第二配体并且a2是第二效应子部分,或者如果a2是第二配体并且b2是第二效应子部分,则b=0或1;
[0359]
c=1
‑
128中的任一个;
[0360]
l5是用于共价偶联两个化学基团的至少一个接头;
[0361]
l6是用于共价偶联两个化学基团的至少一个接头;
[0362]
l7是用于共价偶联两个化学基团的至少一个寡聚支架或多聚支架;
[0363]
l8是用于共价偶联两个化学基团的至少一个接头;
[0364]
l10是用于共价偶联三个化学基团的三官能接头;
[0365]
d=0或1;
[0366]
e=0或1;
[0367]
f=0或1;
[0368]
如果f=0,则g=0、1或2,并且如果f=1,则g=0
‑
16中的任一个;
[0369]
如果a2是第二配体并且b2是第二效应子部分,则h=0
‑
32中的任一个,或者如果a2是第二效应子部分并且b2是第二配体,则h=1;
[0370]
i=0或1;
[0371]
j=1或0,以及;
[0372]
k=1
‑
16。
[0373]
一个实施方案是本发明的第二治疗性分子,其中如果f=0并且t> 0,则g=0、1或2,如果f=0并且t=0,则g=1或2,如果f=1并且t> 0,则g=0
‑
16中任一个,以及如果f=1并且t=0,则g=1
‑
16中任一个。
[0374]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第一配体a1或b1和/或第二配体a2或b2包含以下或由以下组成:免疫球蛋白、免疫球蛋白的结合结构域或免疫球蛋白的结合片段,如抗体、igg、包含vhh结构域或vh结构域或者由其组成的分子、 fab、scfv、fv、dab、f(ab)2、fcab片段;或者包含以下或由以下组成:至少一种非蛋白质
配体和/或至少一种蛋白质配体,所述配体如 egf或细胞因子用于结合细胞表面分子,条件是第一配体和第二配体是相同的或不同的。
[0375]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第一配体a1或b1和/或第二配体a2或b2结合肿瘤细胞受体,优选肿瘤细胞特异性受体的肿瘤细胞表位,优选肿瘤细胞特异性表位,所述肿瘤细胞特异性受体优选选自cd71、ca125、epcam(17
‑
1a)、 cd52、cea、cd44v6、fap、egf
‑
ir、整合素、多配体蛋白聚糖
‑
1、血管整合素α
‑
vβ
‑
3、her2、egfr、cd20、cd22、叶酸受体1、cd146、 cd56、cd19、cd138、cd27l受体、psma、canag、整合素
‑
αv、 ca6、cd33、间皮素、cripto、cd3、cd30、cd239、cd70、cd123、 cd352、dll3、cd25、肝配蛋白a4、muc1、trop2、ceacam5、 ceacam6、her3、cd74、ptk7、notch3、fgf2、c4.4a、flt3、 cd38、fgfr3、cd7、pd
‑
l1、ctla4、cd52、pdgfra、vegfr1、 vegfr2,更优选选自cd71、egfr和her2,条件是第一配体和第二配体结合相同或不同的肿瘤细胞表位,优选肿瘤细胞特异性表位,和/或其中第一配体能够结合的肿瘤细胞受体,优选肿瘤细胞特异性受体与第二配体能够结合的肿瘤细胞受体,优选地,肿瘤细胞特异性受体是相同的或不同的。
[0376]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第一配体a1或b1和/或第二配体a2或b2包含以下或由以下组成:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗(inotuzumab)、莫西莫单抗(moxetumomab)、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型okt
‑
9抗cd71单克隆抗体、帕妥珠单抗、利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、okt
‑
10、抗cd38单克隆抗体、表a2或表a3或表a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑
9,或者其至少一个肿瘤细胞受体结合结构域和/或其至少一个肿瘤细胞受体结合片段,其优选是肿瘤细胞特异性受体结合结构域和/或肿瘤细胞特异性受体结合片段,条件是第一配体与第二配体是相同的或不同的。
[0377]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第一配体a1或b1在所述第一配体与其在肿瘤细胞上的结合伴侣结合之后由所述肿瘤细胞内化,并且其中优选地,第一配体与肿瘤细胞结合之后是例如经由第一配体和肿瘤细胞上第一配体的结合伴侣的复合物的内吞作用进行的肿瘤细胞受体介导的内化。
[0378]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第二配体a2或b2在第二配体与其在肿瘤细胞上的结合伴侣结合之后由所述肿瘤细胞内化,并且其中优选地,第二配体与肿瘤细胞结合之后是例如经由第二配体和肿瘤细胞上的第二配体的结合伴侣的复合物的内吞作用进行的肿瘤细胞受体介导的内化。
[0379]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第一效应子部分a1或b1和/或所述第二效应子部分a2或b2包含寡核苷酸、核酸和异源核酸中的任何一种或多种的至少一种或者由其组成,所述寡核苷酸、核酸和异源核酸优选选自以下中的任何一种或多种:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸(rna)、反义寡核苷酸(aso,aon)、短干扰rna(sirna)、微小rna(mirna)、dna适配体、rna适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)、桥接核酸(bna)、2
’‑
脱氧
‑2’‑
氟阿拉伯糖核酸(fana)、2
’‑
o
‑
甲氧基乙基
‑ꢀ
rna(moe)、2
’‑
o,4
’‑
氨基亚乙基桥接核酸、3
’‑
氟己糖醇核酸(fhna)、质粒、二醇核酸(gna)和苏糖核酸(tna)、或它们的衍生物,更优选 bna,例如用于沉默hsp27蛋白表达的bna,条件是所述第一效应子部分和
所述第二效应子部分是相同的或不同的。
[0380]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中其中第一效应子部分a1或b1和/或第二效应子部分a2或b2包含至少一种蛋白质分子或由其组成,所述蛋白质分子优选选自以下中的任何一种或多种:肽、蛋白质、酶如脲酶和cre重组酶、蛋白质毒素、核糖体失活蛋白、选自表a5的至少一种蛋白质毒素和/或细菌毒素、植物毒素,更优选选自以下中的任何一种或多种:病毒毒素,如凋亡蛋白;细菌毒素,如志贺毒素、志贺样毒素、铜绿假单胞菌外毒素(pe)或pe的外毒素a、全长或截短的白喉毒素(dt)、霍乱毒素;真菌毒素,如α
‑
帚曲菌素;植物毒素,包括核糖体失活蛋白和2型核糖体失活蛋白的a链,如石竹素例如石竹素
‑
30或石竹素
‑
32、皂草素例如皂草素
‑
s3或皂草素
‑
s6、三角梅核糖体失活蛋白或三角梅核糖体失活蛋白(bouganin)的去免疫化衍生物去三角梅核糖体失活蛋白 (debouganin)、志贺样毒素a、商陆抗病毒蛋白、蓖麻毒素、蓖麻毒素 a链、蒴莲根毒素、蒴莲根毒素a链、相思子毒素、相思子毒素a链、蒴莲毒素、蒴莲毒素a链、槲寄生素、槲寄生素a链;或者动物或人类毒素,如蛙rna酶或者来自人类的颗粒酶b或血管生长素,或者它们的任何片段或衍生物;优选地,所述蛋白质毒素是石竹素和/或皂草素,条件是第一效应子部分和第二效应子部分是相同的或不同的。
[0381]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第一效应子部分a1或b1和/或第二效应子部分a2或b2包含至少一种有效载荷或由其组成,所述有效载荷优选选自以下中的任何一种或多种:靶向核糖体的毒素、靶向延长因子的毒素、靶向微管蛋白的毒素、靶向dna的毒素和靶向rna的毒素,更优选选自以下中的任何一种或多种:美坦新、pasudotox、美登素衍生物dm1、美登素衍生物dm4、单甲基澳瑞他汀e(mmae,维多汀)、单甲基澳瑞他汀f(mmaf,莫福汀)、卡奇霉素、n
‑
乙酰基
‑
γ
‑
卡奇霉素、吡咯并苯并二氮杂(pbd)二聚体、苯并二氮杂cc
‑
1065类似物、倍癌霉素、多柔比星、紫杉醇、多西他赛、顺铂、环磷酰胺、依托泊苷、多西他赛、 5
‑
氟尿嘧啶(5
‑
fu)、米托蒽醌、微管菌素、吲哚啉苯并二氮杂 az13599185、念珠藻素、根瘤菌素、甲氨蝶呤、蒽环类药物、喜树碱类似物、sn
‑
38、dx
‑
8951f、甲磺酸依喜替康、截短形式的铜绿假单胞菌外毒素(pe38)、倍癌霉素衍生物、鹅膏蕈碱、α
‑
鹅膏蕈碱、斯考他汀、泰兰斯他汀、奥佐米星、特司林、amberstatin269和soravtansine或它们的衍生物。
[0382]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中治疗性分子和/或第二治疗性分子包含以下中的任一种或由其组成:吉妥珠单抗奥佐米星、本妥昔单抗维多汀、曲妥珠单抗美坦新、英妥珠单抗奥佐米星、moxetumomab pasudotox和泊洛妥珠单抗维多汀以及表a2和表a3的抗体
‑
药物缀合物,或者其至少一个肿瘤细胞特异性受体结合结构域和/或其至少一个肿瘤细胞特异性受体结合片段,其优选是肿瘤细胞特异性受体结合结构域和/或肿瘤细胞特异性受体结合片段,条件是治疗性分子和第二治疗性分子是相同的或不同的。
[0383]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中皂苷c是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且任选地在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中包含葡糖醛酸官能团的三萜皂苷或双糖链三萜皂苷,和/或是从石头花属物种和/或肥皂草属物种和/或麦仙翁属物种和/或皂皮树属物种如皂皮树分离的
皂苷。
[0384]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中皂苷c是单一特异性皂苷或者是两个或更多个不同皂苷的混合物,如表a1或方案i中的皂苷中的一种或多种、so1861、sa1657、ge1741、 sa1641、qs
‑
21、qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21a
‑
xyl、qs
‑
21b、qs
‑ꢀ
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、qs
‑
17
‑
xyl、 qs1861、qs1862、皂皮树皂苷、saponinum album、qs
‑
18、quil
‑
a、 gyp1、丝石竹皂苷a、ag1、ag2、so1542、so1584、so1658、so1674、so1832,或者它们的立体异构体中的任一种和/或它们的任何组合,优选地,所述皂苷是so1861和/或ge1741和/或sa1641和/或qs
‑
21和 /或具有皂皮酸苷元核心、在c
‑
3β
‑
oh基团处具有gal
‑
(1
→
2)
‑
[xyl
‑ꢀ
(1
→
3)]
‑
glca碳水化合物取代基以及在c
‑
28
‑
oh基团处具有glc
‑ꢀ
(1
→
3)
‑
xyl
‑
(1
→
4)
‑
rha
‑
(1
→
2)
‑
[xyl
‑
(1
→
3)
‑4‑
oac
‑
qui
‑
(1
→
4)]
‑
fuc碳水化合物取代基的皂苷,和/或是3
‑
o
‑
β
‑
d
‑
半乳吡喃糖基
‑
(1
→
2)
‑
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)]
‑
β
‑
d
‑
葡糖醛酸吡喃糖基皂皮酸28
‑
o
‑
β
‑
d
‑
葡吡喃糖基
ꢀ‑
(1
→
3)
‑
β
‑
d
‑
木吡喃糖基
‑
(1
→
4)
‑
α
‑
l
‑
鼠李吡喃糖基
‑
(1
→
2)
‑
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)
‑4‑
oac
‑
β
‑
d
‑
奎诺吡喃糖基
‑
(1
→
4)]
‑
β
‑
d
‑
岩藻吡喃糖苷,更优选地,所述皂苷是so1861和/或qs
‑
21。
[0385]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中所述皂苷c是具有至少1.500道尔顿的分子质量并且包含在c
‑
23 位含有醛基团并且任选地在c
‑
16位含有羟基基团,以及在c
‑
3位具有第一支链碳水化合物侧链的齐墩果烷型三萜的双糖链皂苷,所述第一支链碳水化合物侧链任选地含有葡糖醛酸,其中所述皂苷含有在c
‑
28 位具有第二支链碳水化合物侧链的酯基团,所述第二支链碳水化合物链优选包含至少四个碳水化合物单元,任选地含有至少一个乙酰基残基如两个乙酰基残基和/或至少一个脱氧碳水化合物和/或奎诺糖和/或葡萄糖和/或4
‑
甲氧基肉桂酸和/或任选地包含5
‑
o
‑
[5
‑
o
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸和/或任选地包含经由酯键与碳水化合物结合的5
‑
o
‑
[5
‑
o
‑
rha
‑
(1
→
2)
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸,或者其中至少一个皂苷是qs
‑
21 或者以下中的任何一种或多种:qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21a
‑
xyl、 qs
‑
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、 qs
‑
17
‑
xyl、qs
‑
18、qs1861、质子化的qs1861(qs1862)、quil
‑
a。
[0386]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中所述皂苷c是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型的双糖链三萜皂苷,其中所述皂苷c经由皂苷c中的醛官能团,优选c
‑
23位的所述醛官能团,优选经由接头l2、l4、l6、l8、 l9和/或l10,更优选经由可裂解接头l2、l4、l6、l8、l9和/或l10 共价偶联至所述第一配体a1或b1和/或所述第一效应子部分b1或 a1和/或所述第二配体a2或b2和/或所述第二效应子部分b2或a2 的氨基酸残基,其中所述氨基酸残基优选选自半胱氨酸和赖氨酸。
[0387]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中所述皂苷c是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型的双糖链三萜皂苷,其中所述至少一个皂苷的c
‑
23位的醛官能团共价偶联至接头n
‑
ε
‑
马来酰亚胺基己酸酰肼,所述接头经由硫醚键共价偶联至所述第一配体a1或b1和/或所述第一效应子部分b1或 a1中和/或所述第二配体a2或b2和/或所述第二效应子部分b2或a2 中的巯基基团,如半胱氨酸的巯基基团。
[0388]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中所述皂苷
c是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在所述皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中包含葡糖醛酸官能团的双糖链三萜皂苷,其中所述皂苷c经由所述皂苷c 中的葡糖醛酸官能团(如果存在时),优选经由接头l2、l4、l6、l8、 l9和/或l10共价偶联至所述第一配体a1或b1和/或所述第一效应子部分b1或a1和/或所述第二配体a2或b2和/或所述第二效应子部分b2或a2的氨基酸残基,其中所述氨基酸残基优选选自半胱氨酸和赖氨酸,更优选地,所述氨基酸残基是赖氨酸。
[0389]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中所述皂苷c是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在所述皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中包含葡糖醛酸官能团的双糖链三萜皂苷,其中所述至少一个皂苷的c
‑
3β
‑ꢀ
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头 1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至所述第一配体a1或b1中和/或所述第一效应子部分b1或a1中和/或所述第二配体a2或b2中和/或所述第二效应子部分b2或a2中的胺基团,如所述第一配体a1 或b1和/或所述第一效应子部分b1或a1和/或所述第二配体a2或 b2和/或所述第二效应子部分b2或a2的赖氨酸或n
‑
末端的胺基团。
[0390]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第一配体a1或b1和/或第一效应子部分b1或a1和/或第二配体 a2或b2和/或第二效应子部分b2或a2包含一个或多于一个共价结合的皂苷c,优选2、3、4、5、6、8、10、16、32、64、128或1
‑
100 个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷。
[0391]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第一配体a1或b1和/或第一效应子部分b1或a1和/或第二配体 a2或b2和/或第二效应子部分b2或a2包含一种或多于一种共价结合的皂苷c,其中当r、s、v、e、f和i是0时,所述皂苷c直接共价结合第一配体a1或b1和/或第一效应子部分b1或a1和/或第二配体 a2或b2和/或第二效应子部分b2或a2的氨基酸残基,优选半胱氨酸和/或赖氨酸,和/或所述皂苷c经由至少一个接头l2、l4、l6、l8、 l9和/或l10或经由至少一个可裂解接头l2、l4、l6、l8、l9和/或 l10和/或经由至少一个寡聚支架或多聚支架l3和/或l7,优选1
‑
8个此类支架或2
‑
4个此类支架共价结合,其中所述至少一个支架任选地基于树枝化基元,其中1
‑
32个皂苷,优选2、3、4、5、6、8、10、16、 32个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷共价结合至少一个支架。
[0392]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中所述皂苷c是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型的双糖链三萜皂苷,其中所述皂苷c经由皂苷c中的醛官能团,优选c
‑
23位的所述醛官能团,优选经由接头l2、l4、l6、l8、 l9和/或l10,更优选经由可裂解接头l2、l4、l6、l8、l9和/或l10 共价偶联至第一配体a1或b1和/或第一效应子部分b1或a1和/或第二配体a2或b2和/或第二效应子部分b2或a2的氨基酸残基,其中氨基酸残基优选选自半胱氨酸和赖氨酸,并且其中可裂解接头l2、l4、 l6、l8、l9和/或l10在酸性条件、还原条件、酶促条件或光诱导条件下进行裂解,并且优选地可裂解接头包含在酸性条件下进行裂解的腙键或酰肼键(当结合皂苷时),和/或包含易于蛋白质水解(例如由组织蛋白酶b进行蛋白质水解)的键(当结合皂苷时),和/或可裂解接头包含易于在还原条件下裂解的二硫键。
[0393]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中所述皂苷
c是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型的双糖链三萜皂苷,其中所述皂苷c经由皂苷c中的醛官能团,优选c
‑
23位的所述醛官能团,优选经由接头l2、l4、l6、l8、 l9和/或l10,更优选经由可裂解接头l2、l4、l6、l8、l9和/或l10 共价偶联至第一配体a1或b1和/或第一效应子部分b1或a1和/或第二配体a2或b2和/或第二效应子部分b2或a2的氨基酸残基,其中氨基酸残基优选选自半胱氨酸和赖氨酸,并且其中所述可裂解接头l2、 l4、l6、l8、l9和/或l10在如存在于哺乳动物细胞,优选人类细胞的内体和/或溶酶体的酸性条件下,优选在ph 4.0
‑
6.5下并且更优选在 ph≤5.5下在体内进行裂解。
[0394]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中多聚支架或寡聚支架l3和/或l7包含多聚结构或寡聚结构并且包含化学基团,所述化学基团用于所述多聚支架或寡聚支架l3和/或 l7与第一配体和/或第一效应子部分和/或第二配体和/或第二效应子部分的氨基酸残基的共价偶联。
[0395]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中所述皂苷c是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中包含葡糖醛酸官能团的双糖链三萜皂苷,其中在至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至第一配体a1或b1中和/或第一效应子部分b1或a1中和/或第二配体a2或b2中和/或第二效应子部分 b2或a2中的氨基团,如第一配体a1或b1和/或第一效应子部分b1 或a1和/或第二配体a2或b2和/或第二效应子部分b2或a2的赖氨酸或n
‑
末端的胺基团,并且其中至少一个皂苷经由根据本发明的可裂解接头l4和/或l8共价结合支架l3和/或l7的多聚结构或寡聚结构。
[0396]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中多聚支架或寡聚支架l3和/或l7的用于支架与第一配体和/或第一效应子部分和/或第二配体和/或第二效应子部分的氨基酸残基的共价偶联的化学基团是点击化学基团,其优选选自四嗪、叠氮化物、烯烃或炔烃或者这些基团的环状衍生物,更优选地,所述点击化学基团是叠氮化物。
[0397]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第一配体a1或b1和/或第一效应子部分b1或a1和/或第二配体 a2或b2和/或第二效应子部分b2或a2包含一个或多于一个共价结合的皂苷c,优选2、3、4、5、6、8、10、16、32、64、128或1
‑
100 个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷,并且其中至少一个皂苷直接或经由至少一个接头共价结合至第一配体和/或第一效应子部分和/或第二配体和/或第二效应子部分,所述接头如例如基于 n
‑
ε
‑
马来酰亚胺基己酸酰肼和/或基于1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑ꢀ
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐)的双官能接头,或者当 w=1时的三官能接头l9和/或当j=1时的三官能接头l10,如方案ii 的三官能接头。
[0398]
一个实施方案是本发明的治疗性分子并且涵盖之前的实施方案,或涵盖之前实施方案的本发明的第二治疗性分子,其中当w=1时的三官能接头l9和/或当j=1时的三官能接头l10包含具有至少一个皂苷共价结合至其的第二化学基团、用于共价结合第一和/或第二配体的第三化学基团以及用于共价结合至少一个第一和/或第二效应子部分的第一化学基团,优选地,所述三官能接头是方案ii的三官能接头。
[0399]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中至少一个皂苷经由至少一个接头共价结合第一配体和/或第一效应子部分和/或第二配体和/或第二效应子部分,所述接头包括当j=1时的三官能接头l9和/或当w=1时的三官能接头l10,第一配体和至少一个第一效应子部分均结合所述三官能接头和/或第二配体和至少一个第二效应子部分均结合所述三官能接头,优选地,所述三官能接头是方案ii的三官能接头。
[0400]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中支架l3和/或l7的多聚结构或寡聚结构包括直链、支链和/或环状多聚物、寡聚物、树状物、树枝化基元、树枝化基元化多聚物、树枝化基元化寡聚物、dna、多肽、聚赖氨酸、聚乙二醇或者这些多聚结构或寡聚结构的组装体,所述组装体优选通过共价交联构建。
[0401]
一个实施方案是本发明的治疗性分子或本发明的第二治疗性分子,其中第一配体a1或b1和/或第一效应子部分b1或a1和/或第二配体 a2或b2和/或第二效应子部分b2或a2包含一种或多于一种共价连接的皂苷c,优选2、3、4、5、6、8、10、16、32、64、128或1
‑
100 个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷,并且其中第一配体a1或b1经由至少一个接头l1分别共价结合第一效应子部分b1 或a1,和/或其中第二配体a2或b2经由至少一个接头l5分别共价结合第二效应子部分b2或a2。
[0402]
一个实施方案是本发明的治疗性组合,其中第一配体a1是权利要求3
‑
7中任一项所述的单克隆抗体或者其至少一个结合片段或结合结构域,m=1,q=0,n=0,u=0,r=0,l3是根据本发明的支架并且s=1,或者l3不存在并且s=0,l4是根据本发明的接头或可裂解接头,v=1, p=2
‑
4并且如果s=1,则t=2
‑
4以及如果s=0,则t=0,以及皂苷c是根据本发明的皂苷,优选所述皂苷c是so1861和/或qs
‑
21,以及效应子部分a2是以下效应子部分,其选自寡核苷酸、核酸和异源核酸中的任何一种或多种,优选选自以下中的任何一种或多种:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸(rna)、反义寡核苷酸(aso,aon)、短干扰rna(sirna)、微小rna(mirna)、 dna适配体、rna适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)、桥接核酸(bna)、2
’‑
脱氧
ꢀ‑2’‑
氟阿拉伯糖核酸(fana)、2
’‑
o
‑
甲氧基乙基
‑
rna(moe)、2
’‑
o,4
’‑
氨基亚乙基桥接核酸、3
’‑
氟己糖醇核酸(fhna)、质粒、二醇核酸(gna) 和苏糖核酸(tna)、或它们的衍生物,更优选bna,例如用于沉默 hsp27蛋白表达的bna,a=1,d=0,b=0,h=0,e=0,f=0,i=0,c=0 并且g=0。
[0403]
一个实施方案是本发明的治疗性分子,其中r=0,s=0,v=0,s=0,v=0,p=0,t=0,配体a1是根据本发明中任一种的单克隆抗体或者其至少一个结合片段或结合结构域,m=1,效应子部分b1以下效应子部分,其选自寡核苷酸、核酸和异源核酸中的任何一种或多种,优选选自以下中的任何一种或多种:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸(rna)、反义寡核苷酸(aso,aon)、短干扰rna(sirna)、微小rna(mirna)、dna适配体、rna适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)、桥接核酸(bna)、2
’‑
脱氧
‑2’‑
氟阿拉伯糖核酸(fana)、 2
’‑
o
‑
甲氧基乙基
‑
rna(moe)、2
’‑
o,4
’‑
氨基亚乙基桥接核酸、3
’‑
氟己糖醇核酸(fhna)、质粒、二醇核酸(gna)和苏糖核酸(tna)、或它们的衍生物,更优选bna,例如用于沉默hsp27蛋白表达的bna,q=0, n=1并且u=2
‑
4,或者q=1,n=2
‑
4、u=2
‑
4并且接头l1是根据本发明的寡聚支架或多聚支架l3。
[0404]
一个实施方案是本发明的治疗性组合,其中治疗性分子是先前实施方案的治疗性分子,并且其中第二配体a2是根据本发明的单克隆抗体或者其至少一个结合片段或结合结构域,a=1,d=0,b=0,h=0, e=0,l7是根据本发明的支架并且f=1,或者l7不存在并且f=0,l8 是根据本发明的接头或可裂解接头,i=1,c=2
‑
4并且如果f=1,则g=2
‑ꢀ
4以及如果f=0,则g=0,以及皂苷c是根据本发明的皂苷,优选所述皂苷c是so1861和/或qs
‑
21,条件是配体a1和配体a2是相同的或不同的。
[0405]
本发明的一方面涉及治疗性组合,其中治疗性组合包含:(a)第一药物组合物,其包含根据本发明的具有化合物i的化学结构的治疗性分子,所述第一药物组合物任选地还包含药学上可接受的赋形剂;以及(b)第二药物组合物,其包含根据本发明的具有化合物ii的化学结构的第二治疗性分子,所述第二药物组合物任选地还包含药学上可接受的赋形剂。
[0406]
本发明的一方面涉及本发明的第一药物组合物,其用作药物。
[0407]
本发明的一方面涉及治疗性组合,其用于治疗或预防人类对象中的癌症,其中治疗性组合包含:(a)本发明的第一药物组合物;以及(b) 本发明的第二药物组合物,其中配体a1或b1和配体a2或b2能够结合肿瘤细胞表面分子,优选肿瘤细胞特异性表面分子上的肿瘤细胞表位,优选肿瘤细胞特异性表位,条件是配体a1或b1能够结合的肿瘤细胞表位或肿瘤细胞特异性表位与配体a2或b2能够结合的肿瘤细胞表位或肿瘤细胞特异性表位是相同的或不同的。
[0408]
本发明的一方面涉及本发明的第一药物组合物,其用于治疗或预防有需要的患者中的癌症,其中配体a1或b1能够结合肿瘤细胞表面分子,优选肿瘤细胞特异性表面分子上的肿瘤细胞表位,优选肿瘤细胞特异性表位。
[0409]
一个实施方案是根据本发明的用于所述用途的第一药物组合物或根据本发明的用于所述用途的治疗性组合,其中将本发明的第二药物组合物以及本发明的第一药物组合物施用至有需要的患者。
[0410]
本发明的一方面涉及本发明的第一药物组合物,其其还包含本发明的第二治疗性分子。
[0411]
本发明的一方面涉及用作药物用途的本发明的第一药物组合物,其还包含本发明的第二治疗性分子。
[0412]
本发明的一方面涉及用于治疗或预防人类对象中癌症的用途的本发明的第一药物组合物,其还包含本发明的第二治疗性分子。
[0413]
本发明的第一系列方面和实施方案中的本发明的一方面涉及适用于将至少一种生物活性分子共价结合至载体分子的支架,所述支架包含多聚结构或寡聚结构以及共价结合所述多聚结构或寡聚结构的所述生物活性分子中的至少一种,其中支架还包含用于将支架与载体分子共价偶联的第一化学基团。
[0414]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一种生物活性分子的分子质量是3.000道尔顿或更小,优选2.500道尔顿或更小,更优选2.300道尔顿或更小,最优选 2.000道尔顿或更小,如1.700道尔顿
‑
1.950道尔顿。
[0415]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一种生物活性分子是两亲性分子。
[0416]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中当多于一种生物活性分子共价结合支架包含的多聚结构或寡聚结构时,至少一种生物活性分子是单一特异性分子或是不同分子的混合物。
[0417]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一种生物活性分子是糖苷,优选双糖链三萜或三萜皂苷,更优选双糖链三萜皂苷,最优选属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的双糖链三萜皂苷。
[0418]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一种生物活性分子是可从石头花属物种和/或肥皂草属物种和/或麦仙翁属物种和/或皂皮树属物种,如皂皮树分离的皂苷或者是单一特异性皂苷或者是两个或更多个不同皂苷的混合物,如表 a1或方案i中的皂苷中的一种或多种、so1861、sa1657、ge1741、 sa1641、qs
‑
21、qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21a
‑
xyl、qs
‑
21b、qs
‑ꢀ
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、qs
‑
17
‑
xyl、 qs1861、qs1862、皂皮树皂苷、saponinum album、qs
‑
18、quil
‑
a、 gyp1、丝石竹皂苷a、ag1、ag2、so1542、so1584、so1658、so1674、 so1832,或者它们的立体异构体中的任一种和/或它们的任何组合,优选地,所述皂苷是so1861和/或ge1741和/或sa1641和/或qs
‑
21和 /或具有皂皮酸苷元核心、在c
‑
3β
‑
oh基团处具有gal
‑
(1
→
2)
‑
[xyl
‑ꢀ
(1
→
3)]
‑
glca碳水化合物取代基以及在c
‑
28
‑
oh基团处具有glc
‑ꢀ
(1
→
3)
‑
xyl
‑
(1
→
4)
‑
rha
‑
(1
→
2)
‑
[xyl
‑
(1
→
3)
‑4‑
oac
‑
qui
‑
(1
→
4)]
‑
fuc碳水化合物取代基的皂苷,和/或是3
‑
o
‑
β
‑
d
‑
半乳吡喃糖基
‑
(1
→
2)
‑
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)]
‑
β
‑
d
‑
葡糖醛酸吡喃糖基皂皮酸28
‑
o
‑
β
‑
d
‑
葡吡喃糖基
ꢀ‑
(1
→
3)
‑
β
‑
d
‑
木吡喃糖基
‑
(1
→
4)
‑
α
‑
l
‑
鼠李吡喃糖基
‑
(1
→
2)
‑
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)
‑4‑
oac
‑
β
‑
d
‑
奎诺吡喃糖基
‑
(1
→
4)]
‑
β
‑
d
‑
岩藻吡喃糖苷,更优选地,所述皂苷是so1861和/或qs
‑
21。
[0419]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一种生物活性分子是是具有至少1.500道尔顿的分子质量并且包含在c
‑
23位含有醛基团并且任选地在c
‑
16位含有羟基基团,以及在c
‑
3位具有第一支链碳水化合物侧链的齐墩果烷型三萜的双糖链皂苷,所述第一支链碳水化合物侧链任选地含有葡糖醛酸,其中所述皂苷含有在c
‑
28位具有第二支链碳水化合物侧链的酯基团,所述第二支链碳水化合物链优选包含至少四个碳水化合物单元,任选地含有至少一个乙酰基残基如两个乙酰基残基和/或任选地包含脱氧碳水化合物和/或任选地包含奎诺糖和/或任选地包含葡萄糖和/或任选地包含4
‑
甲氧基肉桂酸和/或任选地包含5
‑
o
‑
[5
‑
o
‑
ara/api
‑
3,5
‑
二羟基
ꢀ‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸和/或任选地包含经由酯键与碳水化合物结合的5
‑
o
‑
[5
‑
o
‑
rha
‑
(1
→
2)
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸,或者其中至少一个皂苷是qs
‑
21或者以下中的任何一种或多种:qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21a
‑
xyl、qs
‑ꢀ
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、qs
‑ꢀ
17
‑
xyl、qs
‑
18、qs1861、质子化的qs1861(qs1862)、quil
‑
a。
[0420]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一种生物活性分子经由不可裂解键或经由可裂解键共价结合多聚结构或寡聚结构,其中优选地,所述可裂解键在酸性条件、还原条件、酶促条件或光诱导条件下进行裂解,更优选可裂解键是在酸性条件下进行裂解的腙键或酰肼键和/或是易于蛋白质水解 (例如由组
织蛋白酶b进行蛋白质水解)的键和/或是易于在还原条件下裂解的键如二硫键。
[0421]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一种生物活性分子经由可裂解键共价结合多聚结构或寡聚结构,其中所述可裂解键在如存在于哺乳动物细胞,优选人类细胞的内体和/或溶酶体的酸性条件下,优选在ph 4.0
‑
6.5下并且更优选在ph≤5.5下在体内进行裂解。
[0422]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一种生物活性分子经由亚胺键、腙键、酰肼键、肟键、1,3
‑
二噁戊烷键、二硫键、硫醚键、酰胺键、肽键或酯键,优选经由至少一个接头共价结合支架的多聚结构或寡聚结构。
[0423]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一个皂苷的c
‑
23位的醛官能团参与共价键合至支架的多聚结构或寡聚结构,和/或如果存在,在至少一个皂苷的c
‑
3β
‑ꢀ
oh基团处的碳水化合物取代基中的葡糖醛酸官能团参与经由直接结合或经由至少一个接头共价键合至支架的多聚结构或寡聚结构。
[0424]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一个皂苷的c
‑
23位的醛官能团共价偶联至接头 n
‑
ε
‑
马来酰亚胺基己酸酰肼,所述接头经由硫醚键共价偶联至支架的多聚结构或寡聚结构中的巯基基团,如半胱氨酸的巯基基团。
[0425]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑ꢀ
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至支架的多聚结构或寡聚结构中的胺基团,如蛋白质分子的赖氨酸或n
‑
末端的胺基团。
[0426]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中用于将支架与载体分子的共价偶联的化学基团是点击化学基团。
[0427]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中点击化学基团是四嗪、叠氮化物、烯烃或炔烃或者这些基团中任一种的环状衍生物,优选叠氮化物。
[0428]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中支架是三官能接头,其包含具有至少一种生物活性分子共价结合至其的第二化学基团,包含用于共价结合分子的第三化学基团以及包含用于共价结合载体的第一化学基团,优选地,三官能接头是方案ii和结构b的三官能接头。
[0429]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中至少一种生物活性分子是限定数量的糖苷分子或限定范围的糖苷分子,优选1
‑
128或至少2、3、4、5、6、8、10、16、32、 64或128个糖苷分子,或其间的任何数量的糖苷分子,如7、9、12个糖苷分子。
[0430]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中多聚结构或寡聚结构包括直链、支链和/或环状多聚物、寡聚物、树状物、树枝化基元、树枝化基元化多聚物、树枝化基元化寡聚物、dna、多肽、聚赖氨酸、聚乙二醇或者这些多聚结构或寡聚结构的组装体,所述组装体优选通过共价交联构建。
[0431]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中
载体分子包括以下中的任一种或由以下中的任一种组成:蛋白质分子、蛋白质、肽、核酸、寡核苷酸、脂质、脂肪、脂肪酸、纳米颗粒、碳水化合物或任何共价结合的缀合物或其组合的共价结合的复合物。
[0432]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中载体分子包含以下或由以下组成:免疫球蛋白,免疫球蛋白的至少一个结合结构域和/或免疫球蛋白的至少一个结合片段,如抗体、igg、包含vhh结构域或vh结构域或者由其组成的分子、 fab、scfv、fv、dab、f(ab)2、fcab片段;或者包含以下或由以下组成:用于结合细胞表面分子的至少一种非蛋白质配体和/或至少一种蛋白质配体,如egf或细胞因子。
[0433]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中载体分子包含以下或由以下组成:用于结合细胞表面受体,如选自以下的肿瘤细胞特异性细胞表面受体的至少一个结合结构域和/或至少一个结合片段:cd71、ca125、epcam(17
‑
1a)、cd52、 cea、cd44v6、fap、egf
‑
ir、整合素、多配体蛋白聚糖
‑
1、血管整合素α
‑
vβ
‑
3、her2、egfr、cd20、cd22、叶酸受体1、cd146、cd56、 cd19、cd138、cd27l受体、psma、canag、整合素
‑
αv、ca6、cd33、间皮素、cripto、cd3、cd30、cd239、cd70、cd123、cd352、dll3、 cd25、肝配蛋白a4、muc1、trop2、ceacam5、ceacam6、her3、 cd74、ptk7、notch3、fgf2、c4.4a、flt3、cd38、fgfr3、cd7、 pd
‑
l1、ctla4、cd52、pdgfra、vegfr1、vegfr2,优选选自cd71、 egfr、her2。
[0434]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中载体分子包括以下中的任一种或由以下中的任一种组成:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗、莫西莫单抗、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型okt
‑
9抗cd71单克隆抗体、帕妥珠单抗、利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、okt
‑
10、抗cd38 单克隆抗体、表a2或表a3或表a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑
9,或者其至少一个肿瘤细胞受体结合片段和/或其至少一个肿瘤细胞受体结合结构域,如其至少一个肿瘤细胞特异性受体结合片段和/或其至少一个肿瘤细胞特异性受体结合结构域。
[0435]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中支架适用于与载体分子形成共价键,当载体分子至少包含半胱氨酸和/或赖氨酸时,所述共价键优选涉及载体分子的半胱氨酸侧链和/或载体分子的赖氨酸侧链。
[0436]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中载体分子包含至少一种效应分子或由其组成,或者其中载体还包含至少一种效应分子,其中效应分子是活性药物物质中的至少一种,如有效载荷、毒素、药物、多肽、寡核苷酸、核酸、异源核酸、酶如脲酶和cre重组酶、蛋白质毒素、核糖体失活蛋白中的任何一种或多种。
[0437]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中蛋白质毒素包含以下中的任何一种或多种或者由以下中的任何一种或多种组成:选自表a5的蛋白质毒素和/或病毒毒素,如凋亡蛋白;细菌毒素,如志贺毒素、志贺样毒素、铜绿假单胞菌外毒素(pe)或pe的外毒素a、全长或截短的白喉毒素(dt)、霍乱毒素;真菌毒素,如α
‑
帚曲菌素;植物毒素,包括核糖体失活蛋白和2型核糖体失活蛋白的a链,如石竹素例如石竹素
‑
30或石竹素
‑
32、皂草素例如皂草素
‑
s3或皂草素
‑
s6、三角梅核糖体失活蛋白或三角梅核糖体失活蛋白的去免疫化衍生物去三角梅核糖体失活蛋白、志贺样毒素a、商陆抗病毒蛋
白、蓖麻毒素、蓖麻毒素a链、蒴莲根毒素、蒴莲根毒素a链、相思子毒素、相思子毒素a链、蒴莲毒素、蒴莲毒素a链、槲寄生素、槲寄生素a链;或者动物或人类毒素,如蛙rna酶或者来自人类的颗粒酶b或血管生长素,或者它们的任何片段或衍生物;优选地,所述蛋白质毒素是石竹素和/或皂草素。
[0438]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中寡核苷酸、异源核酸或核酸包括以下中任何一种或多种或者由以下中任何一种或多种组成:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸(rna)、反义寡核苷酸(aso, aon)、短干扰rna(sirna)、微小rna(mirna)、dna适配体、rna 适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)、桥接核酸(bna)、2
’‑
脱氧
‑2’‑
氟阿拉伯糖核酸(fana)、2
’‑
o
‑
甲氧基乙基
‑
rna(moe)、2
’‑
o,4
’‑
氨基亚乙基桥接核酸、3
’‑
氟己糖醇核酸(fhna)、质粒、二醇核酸(gna)和苏糖核酸(tna)、或它们的衍生物,优选bna,例如用于沉默hsp27蛋白表达的bna。
[0439]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中效应分子包含至少一种有效载荷或由其组成,所述有效载荷优选选自以下中的任何一种或多种:靶向核糖体的毒素、靶向延长因子的毒素、靶向微管蛋白的毒素、靶向dna的毒素和靶向rna 的毒素,更优选选自以下中的任何一种或多种:美坦新、pasudotox、美登素衍生物dm1、美登素衍生物dm4、单甲基澳瑞他汀e(mmae,维多汀)、单甲基澳瑞他汀f(mmaf,莫福汀)、卡奇霉素、n
‑
乙酰基
‑ꢀ
γ
‑
卡奇霉素、吡咯并苯并二氮杂(pbd)二聚体、苯并二氮杂cc
‑ꢀ
1065类似物、倍癌霉素、多柔比星、紫杉醇、顺铂、环磷酰胺、依托泊苷、多西他赛、5
‑
氟尿嘧啶(5
‑
fu)、米托蒽醌、微管菌素、吲哚啉苯并二氮杂az13599185、念珠藻素、根瘤菌素、甲氨蝶呤、蒽环类药物、喜树碱类似物、sn
‑
38、dx
‑
8951f、甲磺酸依喜替康、截短形式的铜绿假单胞菌外毒素(pe38)、倍癌霉素衍生物、鹅膏蕈碱、α
‑
鹅膏蕈碱、斯考他汀、泰兰斯他汀、奥佐米星、特司林、amberstatin269和 soravtansine或它们的衍生物。
[0440]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的支架,其中载体分子包含以下或由以下组成:效应分子和单克隆抗体的共价连接的组合,优选选自吉妥珠单抗奥佐米星、本妥昔单抗维多汀、曲妥珠单抗美坦新、英妥珠单抗奥佐米星、moxetumomabpasudotox和泊洛妥珠单抗维多汀以及表a2和表a3的抗体
‑
药物缀合物。
[0441]
本发明的第一系列方面和实施方案中的本发明的一方面涉及用于产生适用于将至少一种生物活性分子共价结合至载体分子的支架的方法,所述方法包括:a)提供用于将多聚结构或寡聚结构与载体分子的共价偶联的第一化学基团,以及包含不同于第一化学基团的第二化学基团中的至少一个的多聚结构或寡聚结构,其中每个第二化学基团用于将至少一种生物活性分子中的一个共价偶联至寡聚结构或聚合结构;以及b)经由第二化学基团将至少一种生物活性分子共价偶联至所述多聚结构或寡聚结构,其中优选地,生物活性分子是本发明的生物活性分子中的任一种,更优选so1861和/或ge1741和/或sa1641和/或 qs
‑
21,随其一起提供了支架。
[0442]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的方法,包含至少一种共价结合的生物活性分子的支架,所述方法包括:a)提供包含共价结合所述支架中的多聚结构或寡聚结构的至少一种生物活性分子的支架,优选提供根据本发明的支架或由根据本发明的方法获得的支架;以及b)将a)的支架共价偶联至根据本发明的载体分子,
随其一起提供了共价结合载体分子的支架,所述支架包含至少一种共价结合的生物活性分子。
[0443]
本发明的第一系列方面和实施方案中的一个实施方案是根据本发明的方法,其中当任一所述效应分子共价结合支架并与哺乳动物细胞接触时或者当所述效应分子与哺乳动物细胞在支架存在下接触时,支架能够增加根据本发明的效应分子的内体逃逸和/或溶酶体逃逸。
[0444]
本发明的第二系列方面和实施方案中的本发明的一方面涉及包含用于结合第一细胞表面分子的第一表位的第一结合位点的第一蛋白质分子,所述第一蛋白质分子提供有经由至少一个接头和/或经由寡聚支架或多聚支架共价结合所述第一蛋白质分子的氨基酸残基或者直接共价结合所述第一蛋白质分子的氨基酸残基的至少一个皂苷。
[0445]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中第一结合位点包含以下或由以下组成:免疫球蛋白,或免疫球蛋白的至少一个结合结构域和/或免疫球蛋白的至少一个结合片段,如抗体、igg、包含vhh结构域或vh结构域或者由其组成的分子、fab、scfv、fv、dab、f(ab)2、fcab片段;和/或包含以下或由以下组成:用于结合细胞表面分子的至少一种配体,如egf 或细胞因子。
[0446]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中第一细胞表面分子的第一表位是第一肿瘤细胞表面分子的肿瘤细胞特异性第一表位,更优选特异性存在于肿瘤细胞上的第一肿瘤细胞表面受体的肿瘤细胞特异性第一表位。
[0447]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷,和/或是从石头花属物种和/或肥皂草属物种和/或麦仙翁属物种和/或皂皮树属物种如皂皮树分离的皂苷。
[0448]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中至少一个皂苷是单一特异性皂苷或者是两个或更多个不同皂苷的混合物,如表a1或方案i中的皂苷中的一种或多种、so1861、sa1657、ge1741、sa1641、qs
‑
21、qs
‑
21a、qs
‑ꢀ
21a
‑
api、qs
‑
21a
‑
xyl、qs
‑
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、qs
‑
17
‑
xyl、qs1861、qs1862、皂皮树皂苷、 saponinum album、qs
‑
18、quil
‑
a、gyp1、丝石竹皂苷a、ag1、ag2、 so1542、so1584、so1658、so1674、so1832,或者它们的立体异构体中的任一种和/或它们的任何组合,优选地,所述皂苷是so1861和 /或ge1741和/或sa1641和/或qs
‑
21和/或具有皂皮酸苷元核心、在 c
‑
3β
‑
oh基团处具有gal
‑
(1
→
2)
‑
[xyl
‑
(1
→
3)]
‑
glca碳水化合物取代基以及在c
‑
28
‑
oh基团处具有glc
‑
(1
→
3)
‑
xyl
‑
(1
→
4)
‑
rha
‑
(1
→
2)
‑
[xyl
‑ꢀ
(1
→
3)
‑4‑
oac
‑
qui
‑
(1
→
4)]
‑
fuc碳水化合物取代基的皂苷,和/或是3
‑
o
‑ꢀ
β
‑
d
‑
半乳吡喃糖基
‑
(1
→
2)
‑
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)]
‑
β
‑
d
‑
葡糖醛酸吡喃糖基皂皮酸28
‑
o
‑
β
‑
d
‑
葡吡喃糖基
‑
(1
→
3)
‑
β
‑
d
‑
木吡喃糖基
‑
(1
→
4)
‑
α
‑
l
‑ꢀ
鼠李吡喃糖基
‑
(1
→
2)
‑
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)
‑4‑
oac
‑
β
‑
d
‑
奎诺吡喃糖基
‑
(1
→
4)]
‑
β
‑
d
‑
岩藻吡喃糖苷,更优选地,所述皂苷是so1861和/或 qs
‑
21。
[0449]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白
质分子,其中至少一个皂苷是具有至少1.500道尔顿的分子质量并且包含在c
‑
23位含有醛基团并且任选地在c
‑
16位含有羟基基团,以及在c
‑
3位具有第一支链碳水化合物侧链的齐墩果烷型三萜的双糖链皂苷,所述第一支链碳水化合物侧链任选地含有葡糖醛酸,其中所述皂苷含有在c
‑
28位具有第二支链碳水化合物侧链的酯基团,所述第二支链碳水化合物链优选包含至少四个碳水化合物单元,任选地含有至少一个乙酰基残基如两个乙酰基残基和/或任选地包含脱氧碳水化合物和/或任选地包含奎诺糖和/或任选地包含葡萄糖和/或任选地包含4
‑
甲氧基肉桂酸和/或任选地包含5
‑
o
‑
[5
‑
o
‑
ara/api
‑
3,5
‑
二羟基
ꢀ‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸和/或任选地包含经由酯键与碳水化合物结合的5
‑
o
‑
[5
‑
o
‑
rha
‑
(1
→
2)
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸,或者其中至少一个皂苷是qs
‑
21或者以下中的任何一种或多种:qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21a
‑
xyl、qs
‑ꢀ
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、qs
‑ꢀ
17
‑
xyl、qs
‑
18、qs1861、质子化的qs1861(qs1862)、quil
‑
a。
[0450]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型的双糖链三萜皂苷,其中至少一个皂苷经由皂苷中的醛官能团,优选在c
‑
23位的所述醛官能团,优选经由至少一个接头,更优选经由至少一个可裂解接头共价偶联至第一蛋白质分子的氨基酸残基,其中氨基酸残基优选选自半胱氨酸和赖氨酸。
[0451]
一个实施方案是根据本发明的第一蛋白质分子,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中包含葡糖醛酸官能团的双糖链三萜皂苷,其中至少一个皂苷经由所述皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团,优选经由至少一个接头共价偶联至第一蛋白质分子的氨基酸残基,其中氨基酸残基优选选自半胱氨酸和赖氨酸。
[0452]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中至少一个皂苷的c
‑
23位的醛官能团共价偶联至接头n
‑
ε
‑
马来酰亚胺基己酸酰肼,所述接头经由硫醚键共价偶联至第一蛋白质分子中的巯基基团,如半胱氨酸的巯基基团。
[0453]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至第一蛋白质分子中的胺基团,如第一蛋白质分子的赖氨酸或n
‑
末端的胺基团。
[0454]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中第一蛋白质分子的第一结合位点所结合的第一细胞表面分子的第一表位是优选选自以下的肿瘤细胞特异性受体的肿瘤细胞特异性第一表位:cd71、ca125、epcam(17
‑
1a)、cd52、 cea、cd44v6、fap、egf
‑
ir、整合素、多配体蛋白聚糖
‑
1、血管整合素α
‑
vβ
‑
3、her2、egfr、cd20、cd22、叶酸受体1、cd146、cd56、 cd19、cd138、cd27l受体、psma、canag、整合素
‑
αv、ca6、cd33、间皮素、cripto、cd3、cd30、cd239、cd70、cd123、cd352、dll3、 cd25、肝配蛋白a4、muc1、trop2、ceacam5、ceacam6、her3、 cd74、ptk7、notch3、fgf2、c4.4a、flt3、cd38、fgfr3、cd7、 pd
‑
l1、ctla4、cd52、pdgfra、vegfr1、vegfr2,更优选选自 cd71、egfr、
her2。
[0455]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中肿瘤细胞特异性第一表位、第一肿瘤细胞表面分子或第一肿瘤细胞特异性受体是在本发明的第一蛋白质分子结合至第一表位或第一分子或第一受体之后由肿瘤细胞内化的第一表位或第一分子或第一受体,并且其中优选地,第一蛋白质分子在结合包括第一表位、肿瘤细胞表面分子或肿瘤细胞特异性受体的细胞表面分子时,进行如经由内吞作用进行的肿瘤细胞受体介导的内化,或如经由内吞作用进行的肿瘤细胞表面分子介导的内化。
[0456]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中第一蛋白质分子的第一结合位点包括以下中的任一种或由以下中的任一种组成:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗、莫西莫单抗、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型okt
‑
9抗cd71单克隆抗体、帕妥珠单抗、利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、okt
‑
10、抗cd38单克隆抗体、表a2或表a3或表a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑
9,或者其至少一个肿瘤细胞受体结合片段和/或其至少一个肿瘤细胞受体结合结构域,优选其至少一个肿瘤细胞特异性受体结合片段和/或其至少一个肿瘤细胞特异性受体结合结构域。
[0457]
本发明的第二系列方面和实施方案中的本发明的一方面涉及治疗性组合,其中治疗性组合包含:(a)第一药物组合物,其包含根据本发明的第一蛋白质分子和任选的药学上可接受的赋形剂;以及(b)第二药物组合物,其包含不同于第一蛋白质分子的第二蛋白质分子,所述第二蛋白质分子包含用于结合不同于第一细胞表面分子的第二细胞表面分子的第二表位的第二结合位点,以及包含效应子部分,所述第二药物组合物任选地还包含药学上可接受的赋形剂,其中第二表位不同于第一表位。
[0458]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合,其中治疗性组合包含:(a)根据本发明的第一药物组合物,其包含根据本发明的第一蛋白质分子,其中第一细胞表面分子上的第一表位是第一肿瘤细胞特异性表面分子上的肿瘤细胞特异性第一表位,优选特异性存在于肿瘤细胞的第一细胞表面受体上的肿瘤细胞特异性第一表位;以及(b)根据本发明的第二药物组合物,其中第二细胞表面分子是不同于第一肿瘤细胞特异性表面分子的第二肿瘤细胞特异性表面分子,优选不同于特异性存在于所述肿瘤细胞处的第一细胞表面受体的特异性存在于所述肿瘤细胞处的第二细胞表面受体,并且其中第二表位是肿瘤细胞特异性第二表位。
[0459]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合,其中治疗性组合包含:(a)根据本发明的第一药物组合物,其包含根据本发明的第一蛋白质分子以及包含用于结合第一细胞表面分子上的第一表位的第一结合位点,第一药物组合物任选地还包含药学上可接受的赋形剂;以及(b)第三药物组合物,其包含第三蛋白质分子,所述第三蛋白质分子包含用于结合(a)的细胞表面分子上的第一表位的第一结合位点和效应子部分,第三药物组合物任选地还包含药学上可接受的赋形剂,其中第一蛋白质分子的第一结合位点和第三蛋白质分子的第一结合位点是相同的,并且其中第一蛋白质分子能够结合的第一细胞表面分子和第一细胞表面分子上的第一表位与第三蛋白质分子能够结合的第一细胞表面分子和第一细胞表面分子上的第一表位相同。
[0460]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合,其中治疗性组合包含:(a)根据本发明的第一药物组合物;以及(b)根据本发明的第三药物组合物,其中第一细胞表面分子在肿瘤细胞表面上表达,并且优选地,第一细胞表面分子是肿瘤细胞特异性表面分子,并且其中优选地,第一表位是第一肿瘤细胞特异性表位。
[0461]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子或根据本发明的治疗性组合,其中用于结合第一细胞表面分子上的第一表位的第一结合位点是用于特异性存在于肿瘤细胞的第一细胞表面受体上的肿瘤细胞特异性第一表位的结合位点。
[0462]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合,其中第二蛋白质分子的第二结合位点和/或第三蛋白质分子的第一结合位点包含以下或由以下组成:免疫球蛋白,免疫球蛋白的至少一个结合结构域和/或免疫球蛋白的至少一个结合片段,如抗体、igg、包含vhh结构域或vh结构域或者由其组成的分子、fab、scfv、fv、dab、f(ab)2、fcab片段;和/或包含以下或由以下组成:用于结合细胞表面分子的至少一种配体,如egf或细胞因子。
[0463]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合,其中用于结合第二表位的第二蛋白质分子的第二结合位点是用于特异性存在于肿瘤细胞的第二细胞表面受体上的肿瘤细胞特异性第二表位的第二结合位点,其中第二结合位点不同于第一结合位点。
[0464]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子或根据本发明的治疗性组合,其中所述第一和第二蛋白质分子包含分别用于结合分别在第一和第二肿瘤细胞特异性受体上的第一和第二肿瘤细胞特异性表位的第一和第二结合位点,所述受体是不同的并且存在于相同的肿瘤细胞处,其中第一和第二结合位点是不同的并且第一和第二肿瘤细胞特异性表位是不同的。
[0465]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子或根据本发明的治疗性组合,其中所述第一和第三蛋白质分子包含用于结合第一肿瘤细胞特异性受体上的第一肿瘤细胞特异性表位的相同的第一结合位点。
[0466]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子或根据本发明的治疗性组合,其中第一受体和/或第二受体选自cd71、ca125、epcam(17
‑
1a)、cd52、cea、cd44v6、 fap、egf
‑
ir、整合素、多配体蛋白聚糖
‑
1、血管整合素α
‑
vβ
‑
3、her2、 egfr、cd20、cd22、叶酸受体1、cd146、cd56、cd19、cd138、 cd27l受体、psma、canag、整合素
‑
αv、ca6、cd33、间皮素、cripto、 cd3、cd30、cd239、cd70、cd123、cd352、dll3、cd25、肝配蛋白a4、muc1、trop2、ceacam5、ceacam6、her3、cd74、 ptk7、notch3、fgf2、c4.4a、flt3、cd38、fgfr3、cd7、pd
‑
l1、 ctla4、cd52、pdgfra、vegfr1、vegfr2,优选选自cd71、egfr 和her2。
[0467]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子和/或根据本发明的治疗性组合,其中第一和第二肿瘤细胞特异性受体在结合根据本发明的第一蛋白质分子和/或根据本发明的第二蛋白质分子之后由肿瘤细胞内化,并且其中优选地,第一蛋白质分子和/或第二蛋白质分子与第一和第二肿瘤细胞特异性受体的结合分别导致第一蛋白质分子和第一肿瘤细胞特异性受体的复合物以及第二蛋白质分子
和第二肿瘤细胞特异性受体的复合物如经由内吞作用进行的肿瘤细胞受体介导的内化。
[0468]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合或第一药物组合物,其中第一肿瘤细胞受体,优选第一肿瘤细胞特异性受体在结合根据本发明的第一蛋白质分子之后和/ 或在结合根据本发明的第三蛋白质分子之后由肿瘤细胞内化,并且其中优选地,第一蛋白质分子和/或第三蛋白质分子与第一肿瘤细胞受体 (如第一肿瘤细胞特异性受体)的结合之后是第一蛋白质分子和第一肿瘤细胞受体的复合物以及第三蛋白质分子和第一肿瘤细胞受体的复合物如经由内吞作用进行的肿瘤细胞受体介导的内化。
[0469]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子和/或根据本发明的治疗性组合,其中第一结合位点和/或第二结合位点是或包含单克隆抗体或其至少一个细胞表面分子结合片段和/或结合结构域,并且优选包含以下中的任一种或由以下中的任一种组成:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗、莫西莫单抗、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型okt
‑
9抗cd71单克隆抗体、帕妥珠单抗、利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、 okt
‑
10、抗cd38单克隆抗体和表a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑
9,或者其至少一个细胞表面分子结合片段或结合结构域,条件是第一蛋白质分子的第一结合位点不同于第二蛋白质分子的第二结合位点。
[0470]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合或根据本发明的第一药物组合物,其中第一蛋白质分子和第三蛋白质分子的第一结合位点包含单克隆抗体或者其细胞表面分子结合结构域和/或结合片段中的至少一种,并且优选包含以下中的任一种或由以下中的任一种组成:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗、莫西莫单抗、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型okt
‑
9抗cd71单克隆抗体、帕妥珠单抗、利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、okt
‑
10、抗cd38单克隆抗体、表a2或表a3或表 a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑
9,或者其至少一个细胞表面分子结合片段和/或结合结构域,条件是第一蛋白质分子的第一结合位点与第三蛋白质分子的第一结合位点相同。
[0471]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合,其中第二蛋白质分子的第二结合位点和/或第三蛋白质分子的第一结合位点是或包含单克隆抗体或者其至少一个细胞表面分子结合片段或结合结构域,并且优选包含以下中的任一种或由以下中的任一种组成:吉妥珠单抗奥佐米星、本妥昔单抗维多汀、曲妥珠单抗美坦新、英妥珠单抗奥佐米星、moxetumomab pasudotox和泊洛妥珠单抗维多汀以及表a2和表a3的抗体
‑
药物缀合物。
[0472]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合,其中第二蛋白质分子和/或第三蛋白质分子所包含的效应子部分包含寡核苷酸、核酸、外源核酸中的任何一种或多种或者由其组成,所述寡核苷酸、核酸、外源核酸优选选自以下中的任何一种或多种:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸(rna)、反义寡核苷酸(aso,aon)、短干扰rna(sirna)、微小rna(mirna)、dna适配体、rna适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)、桥接核酸(bna)、2
’‑
脱氧
‑2’‑
氟阿拉伯糖核酸(fana)、2
’‑
o
‑
甲氧基乙基
‑ꢀ
rna(moe)、2
’‑
o,4'
‑
氨基
亚乙基桥接核酸、3
’‑
氟己糖醇核酸(fhna)、质粒、二醇核酸(gna)和苏糖核酸(tna)、或它们的衍生物,更优选 bna,例如用于沉默hsp27蛋白表达的bna。
[0473]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合,其中第二蛋白质分子和/或第三蛋白质分子所包含的效应子部分包含至少一种蛋白质分子或由其组成,所述蛋白质分子优选选自以下中的任何一种或多种:肽、蛋白质、酶(如脲酶和cre重组酶)、核糖体失活蛋白、蛋白质毒素,更优选选自以下中的任何一种或多种:选自表a5的蛋白质毒素和/或病毒毒素,如凋亡蛋白;细菌毒素,如志贺毒素、志贺样毒素、铜绿假单胞菌外毒素(pe)或pe的外毒素a、全长或截短的白喉毒素(dt)、霍乱毒素;真菌毒素,如α
‑
帚曲菌素;植物毒素,包括核糖体失活蛋白和2型核糖体失活蛋白的a链,如石竹素例如石竹素
‑
30或石竹素
‑
32、皂草素例如皂草素
‑
s3或皂草素
‑
s6、三角梅核糖体失活蛋白或三角梅核糖体失活蛋白的去免疫化衍生物去三角梅核糖体失活蛋白、志贺样毒素a、商陆抗病毒蛋白、蓖麻毒素、蓖麻毒素a链、蒴莲根毒素、蒴莲根毒素a链、相思子毒素、相思子毒素a链、蒴莲毒素、蒴莲毒素a链、槲寄生素、槲寄生素a 链;或者动物或人类毒素,如蛙rna酶或者来自人类的颗粒酶b或血管生长素,或者它们的任何片段或衍生物;优选地,所述蛋白质毒素是石竹素和/或皂草素。
[0474]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合,其中第二蛋白质分子和/或第三蛋白质分子所包含的效应子部分包含至少一种有效载荷或由其组成,所述有效载荷优选选自以下中的任何一种或多种:靶向核糖体的毒素、靶向延长因子的毒素、靶向微管蛋白的毒素、靶向dna的毒素和靶向rna的毒素,更优选选自以下中的任何一种或多种:美坦新、pasudotox、美登素衍生物dm1、美登素衍生物dm4、单甲基澳瑞他汀e(mmae,维多汀)、单甲基澳瑞他汀f(mmaf,莫福汀)、卡奇霉素、n
‑
乙酰基
‑
γ
‑
卡奇霉素、吡咯并苯并二氮杂(pbd)二聚体、苯并二氮杂cc
‑
1065类似物、倍癌霉素、多柔比星、紫杉醇、多西他赛、顺铂、环磷酰胺、依托泊苷、多西他赛、5
‑
氟尿嘧啶(5
‑
fu)、米托蒽醌、微管菌素、吲哚啉苯并二氮杂az13599185、念珠藻素、根瘤菌素、甲氨蝶呤、蒽环类药物、喜树碱类似物、sn
‑
38、dx
‑
8951f、甲磺酸依喜替康、截短形式的铜绿假单胞菌外毒素(pe38)、倍癌霉素衍生物、鹅膏蕈碱、α
‑
鹅膏蕈碱、斯考他汀、泰兰斯他汀、奥佐米星、特司林、amberstatin269和 soravtansine或它们的衍生物。
[0475]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中第一蛋白质分子包含直接共价结合第一蛋白质分子的氨基酸残基,优选半胱氨酸和/或赖氨酸,和/或经由至少一个接头和/或经由至少一个可裂解接头和/或经由至少一个多聚支架或寡聚支架,优选1
‑
8个此类支架或2
‑
4个此类支架共价结合的多于一种的皂苷,优选2、3、4、5、6、8、10、16、32、64或1
‑
100个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷,其中至少一个支架任选地基于树枝化基元,其中1
‑
32个皂苷,如2、3、4、5、6、8、10、 16、32个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷共价结合至少一个支架。
[0476]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中至少一个接头是不可裂解接头或可裂解接头,其中可裂解接头例如在酸性条件、还原条件、酶促条件或光诱导条件下进行裂解,并且优选地,可裂解接头包含在酸性条件下进行裂解的腙键或酰肼键(当结合皂苷时)和/或包含易于蛋白质水解(例如由组织蛋白酶b进行蛋白质水解)的键和/或是易于在还原条件下裂解的键如二硫键(当结合皂苷
时)。
[0477]
本发明的第二系列的方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中当可裂解接头结合皂苷时,可裂解接头在如存在于哺乳动物细胞,优选人类细胞的内体和/或溶酶体的酸性条件下,优选在ph 4.0
‑
6.5下并且更优选在ph≤5.5下在体内进行裂解。
[0478]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中寡聚支架或多聚支架包含多聚结构或寡聚结构并且包含化学基团,所述化学基团用于支架与所述第一蛋白质分子的氨基酸残基的共价偶联。
[0479]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中至少一个皂苷经由根据本发明的至少一个可裂解接头共价结合寡聚支架或多聚支架的多聚结构或寡聚结构。
[0480]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中寡聚支架或多聚支架的用于所述寡聚支架或多聚支架与所述第一蛋白质分子的氨基酸残基的共价偶联的化学基团是点击化学基团,其优选选自四嗪、叠氮化物、烯烃或炔烃或者这些基团的环状衍生物,更优选地,所述化学基团是叠氮化物。
[0481]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的第一蛋白质分子,其中寡聚支架或多聚支架的多聚结构或寡聚结构包括直链、支链和/或环状多聚物、寡聚物、树状物、树枝化基元、树枝化基元化多聚物、树枝化基元化寡聚物、dna、多肽、聚赖氨酸、聚乙二醇或者这些多聚结构或寡聚结构的组装体,所述组装体优选通过共价交联构建。
[0482]
本发明的第二系列方面和实施方案中的本发明的一方面涉及组合物,其包含根据本发明的第一蛋白质分子以及根据本发明的第二蛋白质分子。
[0483]
本发明的第二系列方面和实施方案中的本发明的一方面涉及组合物,其包含根据本发明的第一蛋白质分子以及根据本发明的第三蛋白质分子。
[0484]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的组合物,其中第二蛋白质分子或第三蛋白质分子所包含的效应子部分是根据本发明的效应子部分中的任一种,优选bna。
[0485]
本发明的第二系列方面和实施方案中的本发明的一方面涉及组合物,其包含根据本发明的第一蛋白质分子以及寡核苷酸、核酸和异源核酸中的任何一种或多种,所述寡核苷酸、核酸和异源核酸优选选自以下中的至少一种:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸(rna)、反义寡核苷酸(aso,aon)、短干扰 rna(sirna)、微小rna(mirna)、dna适配体、rna适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸 (lna)、桥接核酸(bna)、2
’‑
脱氧
‑2’‑
氟阿拉伯糖核酸(fana)、2
’‑
o
‑
甲氧基乙基
‑
rna(moe)、2
’‑
o,4'
‑
氨基亚乙基桥接核酸、3
’‑
氟己糖醇核酸 (fhna)、质粒、二醇核酸(gna)和苏糖核酸(tna)、或它们的衍生物,更优选bna,例如用于沉默hsp27蛋白表达的bna。
[0486]
本发明的第二系列方面和实施方案中的本发明的一方面涉及抗体
‑
药物缀合物或配体
‑
药物缀合物,其包含根据本发明的第一蛋白质分子和效应子部分。
[0487]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的抗体
‑
药物缀合物或配体
‑
药物缀合物,其中抗体能够结合以下中的任一种:cd71、ca125、epcam(17
‑
1a)、cd52、cea、cd44v6、 fap、egf
‑
ir、整合素、多配体蛋白聚糖
‑
1、血管整合素α
‑
vβ
‑
3、her2、 egfr、cd20、cd22、叶酸受体1、cd146、cd56、cd19、cd138、 cd27l受体、psma、canag、整合素
‑
αv、ca6、cd33、间皮素、cripto、 cd3、cd30、cd239、cd70、cd123、cd352、dll3、cd25、肝配蛋白a4、muc1、trop2、ceacam5、ceacam6、her3、cd74、 ptk7、notch3、fgf2、c4.4a、flt3、cd38、fgfr3、cd7、pd
‑
l1、 ctla4、cd52、pdgfra、vegfr1、vegfr2,优选cd71、her2、 egfr,和/或是或包含以下中的任一种:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗、莫西莫单抗、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型okt
‑
9抗cd71单克隆抗体、帕妥珠单抗、利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、okt
‑
10、抗cd38单克隆抗体、表a2或表a3或表a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑
9或者其至少一个肿瘤细胞受体结合片段和/或其至少一个肿瘤细胞受体结合结构域,和/或其中抗体
‑
药物缀合物包含以下中的任一种:吉妥珠单抗奥佐米星、本妥昔单抗维多汀、曲妥珠单抗美坦新、英妥珠单抗奥佐米星、 moxetumomab pasudotox和泊洛妥珠单抗维多汀以及表a2和表a3的抗体
‑
药物缀合物,或者其中配体
‑
药物缀合物包含至少一个用于结合细胞表面分子的配体,如egf或细胞因子。
[0488]
本发明的第二系列方面和实施方案中的一个实施方案是根据本发明的抗体
‑
药物缀合物或配体
‑
药物缀合物,其中效应子部分是根据本发明的效应子部分中的任何一种或多种。
[0489]
本发明的第二系列方面和实施方案中的本发明的一方面涉及药物组合物,其包含根据本发明的组合物或根据本发明的抗体
‑
药物缀合物或根据本发明的配体
‑
药物缀合物以及任选地还包含药学上可接受的赋形剂。
[0490]
本发明的第二系列方面和实施方案中的本发明的一方面涉及根据本发明的治疗性组合或组合物或抗体
‑
药物缀合物或配体
‑
药物缀合物或药物组合物,其用作药物。
[0491]
本发明的第二系列方面和实施方案中的本发明的一方面涉及根据本发明的治疗性组合或组合物或抗体
‑
药物缀合物或配体
‑
药物缀合物或药物组合物,其用于治疗或预防癌症或自身免疫疾病。
[0492]
本发明的第三系列方面和实施方案中的本发明的一方面涉及当存在于哺乳动物细胞内部时能够诱导细胞内效应的效应子部分,所述效应子部分与至少一个皂苷缀合,其中至少一个皂苷经由至少一个接头共价结合效应子部分,或者直接共价结合所述效应子部分。
[0493]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其包括寡核苷酸、核酸、外源核酸中的至少一种或者由其组成,所述寡核苷酸、核酸、外源核酸优选选自以下中的任何一种或多种:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸(rna)、反义寡核苷酸(aso,aon)、短干扰rna(sirna)、微小rna(mirna)、dna适配体、rna适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)、桥接核酸(bna)、2
’‑
脱氧
‑2’‑
氟阿拉伯糖核酸(fana)、2
’‑
o
‑
甲氧基乙基
‑ꢀ
rna(moe)、2
’‑
o,4'
‑
氨基亚乙基桥接核酸、3
’‑
氟己糖醇核酸(fhna)、质粒、二醇核酸(gna)和苏糖核酸(tna)、或它们的衍生物,更优选 bna,例如用于沉默hsp27蛋白表达的bna。
[0494]
本发明的第三系列方面和实施方案中的一个实施方案是包含至少一个蛋白质分
子的根据本发明的效应子部分,所述蛋白质分子优选选自以下中的任何一种或多种:肽、蛋白质、酶(如脲酶和cre重组酶)、核糖体失活蛋白、蛋白质毒素如选自表a5的蛋白质毒素中的任何一种或多种和/或细菌毒素或植物毒素,更优选选自以下中的任何一种或多种:病毒毒素,如凋亡蛋白;细菌毒素,如志贺毒素、志贺样毒素、铜绿假单胞菌外毒素(pe)或pe的外毒素a、全长或截短的白喉毒素 (dt)、霍乱毒素;真菌毒素,如α
‑
帚曲菌素;植物毒素,包括核糖体失活蛋白和2型核糖体失活蛋白的a链,如石竹素例如石竹素
‑
30或石竹素
‑
32、皂草素例如皂草素
‑
s3或皂草素
‑
s6、三角梅核糖体失活蛋白或三角梅核糖体失活蛋白的去免疫化衍生物去三角梅核糖体失活蛋白、志贺样毒素a、商陆抗病毒蛋白、蓖麻毒素、蓖麻毒素a链、蒴莲根毒素、蒴莲根毒素a链、相思子毒素、相思子毒素a链、蒴莲毒素、蒴莲毒素a链、槲寄生素、槲寄生素a链;或者动物或人类毒素,如蛙rna酶或者来自人类的颗粒酶b或血管生长素,或者它们的任何片段或衍生物;优选地,所述蛋白质毒素是石竹素和/或皂草素。
[0495]
本发明的第三系列方面和实施方案中的一个实施方案是包含至少一种有效载荷的根据本发明的效应子部分,所述有效载荷优选选自以下中的任何一种或多种:靶向核糖体的毒素、靶向延长因子的毒素、靶向微管蛋白的毒素、靶向dna的毒素和靶向rna的毒素,更优选选自以下中的任何一种或多种:美坦新、pasudotox、美登素衍生物dm1、美登素衍生物dm4、单甲基澳瑞他汀e(mmae,维多汀)、单甲基澳瑞他汀f(mmaf,莫福汀)、卡奇霉素、n
‑
乙酰基
‑
γ
‑
卡奇霉素、吡咯并苯并二氮杂(pbd)二聚体、苯并二氮杂cc
‑
1065类似物、倍癌霉素、多柔比星、紫杉醇、多西他赛、顺铂、环磷酰胺、依托泊苷、多西他赛、5
‑
氟尿嘧啶(5
‑
fu)、米托蒽醌、微管菌素、吲哚啉苯并二氮杂az13599185、念珠藻素、根瘤菌素、甲氨蝶呤、蒽环类药物、喜树碱类似物、sn
‑
38、dx
‑
8951f、甲磺酸依喜替康、截短形式的铜绿假单胞菌外毒素(pe38)、倍癌霉素衍生物、鹅膏蕈碱、α
‑
鹅膏蕈碱、斯考他汀、泰兰斯他汀、奥佐米星、特司林、amberstatin269和soravtansine 或它们的衍生物。
[0496]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的 12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷,和/或是从石头花属物种和/或肥皂草属物种和/或麦仙翁属物种和/ 或皂皮树属物种如皂皮树分离的皂苷。
[0497]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷是单一特异性皂苷或者两个或更多个不同皂苷的混合物。
[0498]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中皂苷是表a1或方案i中的皂苷中的一种或多种、so1861、sa1657、ge1741、sa1641、qs
‑
21、qs
‑
21a、qs
‑
21a
‑ꢀ
api、qs
‑
21a
‑
xyl、qs
‑
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑ꢀ7‑
api、qs
‑
17
‑
api、qs
‑
17
‑
xyl、qs1861、qs1862、皂皮树皂苷、saponinumalbum、qs
‑
18、quil
‑
a、gyp1、丝石竹皂苷a、ag1、ag2、so1542、 so1584、so1658、so1674、so1832,或者它们的立体异构体中的任一种和/或它们的任何组合,优选地,所述皂苷是so1861和/或ge1741 和/或sa1641和/或qs
‑
21和/或具有皂皮酸苷元核心、在c
‑
3β
‑
oh基团处具有gal
‑
(1
→
2)
‑
[xyl
‑
(1
→
3)]
‑
glca碳水化合物取代基以及在c
‑ꢀ
28
‑
oh基团处具有glc
‑
(1
→
3)
‑
xyl
‑
(1
→
4)
‑
rha
‑
(1
→
2)
‑
[xyl
‑
(1
→
3)
‑4‑ꢀ
oac
‑
qui
‑
(1
→
4)]
‑
fuc碳水化合物取代基的皂苷,和/或是3
‑
o
‑
β
‑
d
‑
半乳吡喃糖基
‑
(1
→
2)
‑
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)]
‑
β
‑
d
‑
葡糖醛酸吡喃糖基皂
皮酸28
‑
o
‑
β
‑
d
‑
葡吡喃糖基
‑
(1
→
3)
‑
β
‑
d
‑
木吡喃糖基
‑
(1
→
4)
‑
α
‑
l
‑
鼠李吡喃糖基
‑
(1
→
2)
‑
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)
‑4‑
oac
‑
β
‑
d
‑
奎诺吡喃糖基
‑ꢀ
(1
→
4)]
‑
β
‑
d
‑
岩藻吡喃糖苷,更优选地,所述皂苷是so1861和/或qs
‑ꢀ
21。
[0499]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中皂苷是具有至少1.500道尔顿的分子质量并且包含在c
‑
23位含有醛基团并且任选地在c
‑
16位含有羟基基团,以及在c
‑
3位具有第一支链碳水化合物侧链的齐墩果烷型三萜的双糖链皂苷,所述第一支链碳水化合物侧链任选地含有葡糖醛酸,其中所述皂苷含有在c
‑
28位具有第二支链碳水化合物侧链的酯基团,所述第二支链碳水化合物链优选包含至少四个碳水化合物单元,并且任选地含有至少一个乙酰基残基如两个乙酰基残基和/或任选地包含一个或多个脱氧碳水化合物和/或奎诺糖和/或葡萄糖和/或4
‑
甲氧基肉桂酸和/或任选地包含5
‑
o
‑
[5
‑
o
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑ꢀ6‑
甲基
‑
辛酸和/或任选地包含经由酯键结合碳水化合物的5
‑
o
‑
[5
‑
o
‑ꢀ
rha
‑
(1
→
2)
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸,或者其中至少一个皂苷是qs
‑
21或者以下中的任何一种或多种: qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21a
‑
xyl、qs
‑
21b、qs
‑
21b
‑
api、qs
‑
21b
‑ꢀ
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、qs
‑
17
‑
xyl、qs
‑
18、qs1861、质子化的qs1861(qs1862)、quil
‑
a。
[0500]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的 12,13
‑
脱氢齐墩果烷类型的双糖链三萜皂苷,其中至少一个皂苷经由皂苷中的醛官能团,优选在c
‑
23位的所述醛官能团,优选经由至少一个接头,更优选经由至少一个可裂解接头共价偶联至效应子部分的氨基酸残基(当存在时),其中氨基酸残基优选选自半胱氨酸和赖氨酸。
[0501]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的 12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中包含葡糖醛酸官能团的双糖链三萜皂苷,其中至少一个皂苷经由皂苷的c
‑
3β
‑
oh基团处碳水化合物取代基中的葡糖醛酸官能团,优选经由至少一个接头共价偶联至效应子部分的氨基酸残基(当存在时),其中氨基酸残基优选选自半胱氨酸和赖氨酸。
[0502]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个接头包含至少一个不可裂解接头和/或至少一个可裂解接头,其中任选地所述可裂解接头在酸性条件、还原条件、酶促条件或光诱导条件下进行裂解,并且优选地,可裂解接头包含选自以下的可裂解键:在酸性条件下进行裂解的腙键和酰肼键,和/或易于蛋白质水解(例如由组织蛋白酶b进行蛋白质水解)的键,和 /或易于在还原条件下裂解的键,如二硫键。
[0503]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个接头包括至少一个可裂解接头,所述可裂解接头在如存在于哺乳动物细胞,优选人类细胞的内体和/或溶酶体的酸性条件下在体内,优选在ph 4.0
‑
6.5下并且更优选在ph≤5.5 下进行裂解。
[0504]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷共价结合赖氨酸侧链,形成酰胺键,和/或共价结合半胱氨酸侧链,形
成硫醚连键或二硫键,其中效应子部分包含赖氨酸和/或半胱氨酸,并且其中皂苷直接结合效应子部分,或者经由至少一个接头结合效应子部分。
[0505]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷经由至少一个接头共价结合效应子部分,其中接头是或包含支架,所述支架包含多聚结构或寡聚结构以及用于将支架与效应子部分的共价偶联的化学基团。
[0506]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷经由可裂解键共价结合支架的多聚结构或寡聚结构。
[0507]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中可裂解键在酸性条件、还原条件、酶促条件和光诱导条件中的任一种下进行裂解,更优选地,可裂解键是在酸性条件下进行裂解的腙键或酰肼键和/或是易于蛋白质水解(例如由组织蛋白酶b进行蛋白质水解)的键和/或是易于在还原条件下裂解的键,如二硫键。
[0508]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中可裂解键在如存在于哺乳动物细胞,优选人类细胞的内体和/或溶酶体的酸性条件下,优选在ph 4.0
‑
6.5下并且更优选在ph≤5.5下在体内进行裂解。
[0509]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷经由亚胺键、腙键、酰肼键、肟键、1,3
‑
二噁戊烷键、二硫键、硫醚键、酰胺键、肽键和/或酯键,优选经由至少一个接头,优选酰胺键、酰肼键、硫醚键和/或腙键共价结合支架的多聚结构或寡聚结构。
[0510]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷共价结合支架的多聚结构或寡聚结构,在共价键中涉及至少一个皂苷的c
‑
23位的醛官能团(当存在时),所述共价键优选为亚胺键或腙键或酰胺键或硫醚键或二硫键,和/或在共价键中涉及至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团(当存在时),其中优选地,共价键是酰胺键或二硫键或硫醚键。
[0511]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷的c
‑
23位的醛官能团共价偶联至接头n
‑
ε
‑
马来酰亚胺基己酸酰肼,所述接头经由硫醚键共价偶联至支架的多聚结构或寡聚结构中的巯基基团,如半胱氨酸的巯基基团。
[0512]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至支架的多聚结构或寡聚结构中的胺基团,如支架的多聚结构或寡聚结构的赖氨酸或n
‑
末端的胺基团。
[0513]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中支架的用于将支架与效应子部分的共价偶联的化学基团是点击化学基团,其优选选自四嗪、叠氮化物、烯烃或炔烃或者这些基团的环状衍生物,更优选地,点击化学基团是叠氮化物。
[0514]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中支架的多聚结构或寡聚结构包括直链、支链和/ 或环状多聚物、寡聚物、树状物、树枝化基元、树枝化基元化多聚物、树枝化基元化寡聚物、dna、多肽、聚赖氨酸、聚乙二醇或者
这些多聚结构或寡聚结构的组装体,所述组装体优选通过共价交联构建。
[0515]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中至少一个皂苷是限定数量的皂苷或限定范围的皂苷,优选1
‑
128个皂苷或者至少2、3、4、5、6、8、10、16、32、 64或128个皂苷,或者其间的任何数量的皂苷,如7、9、12个皂苷。
[0516]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的效应子部分,其中效应子部分包含直接共价结合效应子部分的氨基酸残基,优选半胱氨酸和/或赖氨酸,和/或权利要求14
‑
23中任一项所述的经由至少一个接头和/或经由至少一个可裂解接头和/或经由至少一个多聚支架或寡聚支架,优选1
‑
8个此类支架或2
‑
4个此类支架共价结合的多于一个的皂苷,优选2、3、4、5、6、8、10、16、32、 64或1
‑
100个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷,其中1
‑
32个皂苷,优选2、3、4、5、6、8、10、16或32个皂苷共价结合至少一个支架。
[0517]
本发明的第三系列方面和实施方案中的本发明的一方面涉及包含根据本发明的效应子部分的抗体
‑
药物缀合物,或者包含根据本发明的效应子部分的配体
‑
药物缀合物。
[0518]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的抗体
‑
药物缀合物或配体
‑
药物缀合物,其中抗体能够结合以下中的任一种:cd71、ca125、epcam(17
‑
1a)、cd52、cea、cd44v6、 fap、egf
‑
ir、整合素、多配体蛋白聚糖
‑
1、血管整合素α
‑
vβ
‑
3、her2、 egfr、cd20、cd22、叶酸受体1、cd146、cd56、cd19、cd138、 cd27l受体、psma、canag、整合素
‑
αv、ca6、cd33、间皮素、cripto、 cd3、cd30、cd239、cd70、cd123、cd352、dll3、cd25、肝配蛋白a4、muc1、trop2、ceacam5、ceacam6、her3、cd74、 ptk7、notch3、fgf2、c4.4a、flt3、cd38、fgfr3、cd7、pd
‑
l1、 ctla4、cd52、pdgfra、vegfr1、vegfr2,优选cd71、her2、 egfr,和/或其中抗体
‑
药物缀合物的抗体是或包含以下中的任一种:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗、莫西莫单抗、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型okt
‑
9抗cd71单克隆抗体、帕妥珠单抗、利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、okt
‑
10、抗cd38 单克隆抗体、表a2或表a3或表a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑
9或者其至少一个肿瘤细胞特异性受体结合片段和/或其至少一个肿瘤细胞特异性受体结合结构域,和/或其中抗体
‑
药物缀合物包含以下中的任一种:吉妥珠单抗奥佐米星、本妥昔单抗维多汀、曲妥珠单抗美坦新、英妥珠单抗奥佐米星、moxetumomab pasudotox和泊洛妥珠单抗维多汀以及表a2和表a3的抗体
‑
药物缀合物,和/或其中配体
‑
药物缀合物包含以下或由以下组成:用于结合细胞表面分子的至少一个非蛋白质配体和/或至少一个蛋白质配体,如egf或细胞因子。
[0519]
本发明的第三系列方面和实施方案中的本发明的一方面涉及治疗性组合,其包含:(a)根据本发明的效应子部分和任选的药学上可接受的赋形剂;以及(b)抗体
‑
药物缀合物或配体
‑
药物缀合物和任选的药学上可接受的赋形剂。
[0520]
本发明的第三系列方面和实施方案中的一个实施方案是根据本发明的治疗性组合,其中抗体
‑
药物缀合物能够结合以下中的任一种:肿瘤细胞受体cd71、ca125、epcam(17
‑
1a)、cd52、cea、cd44v6、 fap、egf
‑
ir、整合素、多配体蛋白聚糖
‑
1、血管整合素α
‑
vβ
‑
3、her2、 egfr、cd20、cd22、叶酸受体1、cd146、cd56、cd19、cd138、 cd27l受体、psma、canag、整合素
‑
αv、ca6、cd33、间皮素、cripto、 cd3、cd30、cd239、cd70、cd123、cd352、dll3、cd25、肝配蛋白a4、muc1、trop2、ceacam5、ceacam6、her3、cd74、 ptk7、notch3、fgf2、c4.4a、flt3、
cd38、fgfr3、cd7、pd
‑
l1、 ctla4、cd52、pdgfra、vegfr1、vegfr2,优选cd71、her2、 egfr,和/或其中抗体
‑
药物缀合物的抗体是或包含以下中的任一种:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗、莫西莫单抗、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型okt
‑
9抗cd71单克隆抗体、帕妥珠单抗,利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、okt
‑
10、抗cd38单克隆抗体、表a2或表a3或表a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑
9或者其至少一个肿瘤细胞特异性受体结合片段和/或其至少一个肿瘤细胞特异性受体结合结构域,和/或其中抗体
‑
药物缀合物包含以下中的任一种:吉妥珠单抗奥佐米星、本妥昔单抗维多汀、曲妥珠单抗美坦新、英妥珠单抗奥佐米星、moxetumomab pasudotox和泊洛妥珠单抗维多汀以及表a2和表a3的抗体
‑
药物缀合物,和/或其中配体
‑
药物缀合物包含以下或由以下组成:用于结合细胞表面分子的至少一个非蛋白质配体和/或至少一个蛋白质配体,如egf或细胞因子。
[0521]
本发明的第三系列方面和实施方案中的本发明的一方面涉及药物组合物,其包含根据本发明的效应子部分或根据本发明的抗体
‑
药物缀合物或根据本发明的配体
‑
药物缀合物及任选的药学上可接受的赋形剂。
[0522]
本发明的第三系列方面和实施方案中的本发明的一方面涉及根据本发明的效应子部分或根据本发明的抗体
‑
药物缀合物或根据本发明的治疗性组合或根据本发明的配体
‑
药物缀合物或根据本发明的药物组合物,其用作药物。
[0523]
本发明的第三系列方面和实施方案中的本发明的一方面涉及根据本发明的效应子部分或根据本发明的抗体
‑
药物缀合物或根据本发明的配体
‑
药物缀合物或根据本发明的治疗性组合或根据本发明的药物组合物,其用于治疗或预防癌症或自身免疫疾病。
[0524]
本发明的第四系列方面和实施方案中的本发明的一方面涉及缀合物,其包含细胞表面分子靶向分子和至少一个效应子部分并且还包含至少一个共价结合的皂苷。
[0525]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷,和/或是从石头花属物种和/或肥皂草属物种和/或麦仙翁属物种和/或皂皮树属物种如皂皮树中的任何一种或多种分离的皂苷。
[0526]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷是单一特异性皂苷或者两个或更多个不同皂苷的混合物。
[0527]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷的分子质量是3.000道尔顿或更小,优选 2.500道尔顿或更小,更优选2.300道尔顿或更小,最优选2.000道尔顿或更小,如1.500道尔顿
‑
1.900道尔顿。
[0528]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷是表a1或方案i中的皂苷中的一种或多种、so1861、sa1657、ge1741、sa1641、qs
‑
21、qs
‑
21a、qs
‑
21a
‑ꢀ
api、qs
‑
21a
‑
xyl、qs
‑
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑ꢀ7‑
api、qs
‑
17
‑
api、qs
‑
17
‑
xyl、qs1861、qs1862、皂皮树皂苷、saponinumalbum、qs
‑
18、quil
‑
a、gyp1、丝石竹皂苷a、ag1、ag2、so1542、 so1584、so1658、so1674、so1832,或者它们的立体异构体中的任一种和/或它们的任何组合,优选地,所述皂苷是so1861和/或ge1741 和/或sa1641和/或qs
‑
21和/或具有皂皮酸苷元核心、在c
‑
3β
‑
oh基团处具有gal
‑
(1
→
2)
‑
[xyl
‑
(1
→
3)]
‑
glca碳水化合物取代基以及在c
‑ꢀ
28
‑
oh基团处具有glc
‑
(1
→
3)
‑
xyl
‑
(1
→
4)
‑
rha
‑
(1
→
2)
‑
[xyl
‑
(1
→
3)
‑4‑ꢀ
oac
‑
qui
‑
(1
→
4)]
‑
fuc碳水化合物取代基的皂苷,和/或是3
‑
o
‑
β
‑
d
‑
半乳吡喃糖基
‑
(1
→
2)
‑
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)]
‑
β
‑
d
‑
葡糖醛酸吡喃糖基皂皮酸28
‑
o
‑
β
‑
d
‑
葡吡喃糖基
‑
(1
→
3)
‑
β
‑
d
‑
木吡喃糖基
‑
(1
→
4)
‑
α
‑
l
‑
鼠李吡喃糖基
‑
(1
→
2)
‑
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)
‑4‑
oac
‑
β
‑
d
‑
奎诺吡喃糖基
‑ꢀ
(1
→
4)]
‑
β
‑
d
‑
岩藻吡喃糖苷,更优选地,至少一个皂苷是so1861和/或 qs
‑
21。
[0529]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷是具有至少1.500道尔顿的分子质量并且包含在c
‑
23位含有醛基团并且任选地在c
‑
16位含有羟基基团,以及在c
‑
3位具有第一支链碳水化合物侧链的齐墩果烷型三萜的双糖链皂苷,所述第一支链碳水化合物侧链任选地含有葡糖醛酸,其中皂苷含有在c
‑
28位具有第二支链碳水化合物侧链的酯基团,所述第二支链碳水化合物链优选包含至少四个碳水化合物单元,任选地含有至少一个乙酰基残基如两个乙酰基残基和/或任选地包含脱氧碳水化合物和/或任选地包含奎诺糖和/或任选地包含葡萄糖和/或任选地包含4
‑
甲氧基肉桂酸和/或任选地包含5
‑
o
‑
[5
‑
o
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑ꢀ
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸和/或任选地包含经由酯键与碳水化合物结合的5
‑
o
‑
[5
‑
o
‑
rha
‑
(1
→
2)
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸,或者其中至少一个皂苷是qs
‑
21或者以下中的任何一种或多种:qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21a
‑
xyl、qs
‑
21b、qs
‑
21b
‑ꢀ
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、qs
‑
17
‑
xyl、qs
‑
18、 qs1861、质子化的qs1861(qs1862)、quil
‑
a。
[0530]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中细胞表面分子靶向分子包含以下或由以下组成:用于结合细胞表面分子的配体或蛋白质配体或蛋白质结合分子。
[0531]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中细胞表面分子靶向分子包含以下或由以下组成:用于结合细胞表面分子的非蛋白质配体和/或蛋白质配体,如egf或细胞因子,和/或包含以下或由以下组成:免疫球蛋白,免疫球蛋白的至少一个结合结构域和/或免疫球蛋白的至少一个结合片段,如抗体、igg、包含vhh结构域或vh结构域或者由其组成的分子、fab、scfv、fv、 dab、f(ab)2、fcab片段,其能够结合细胞表面分子。
[0532]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中细胞表面分子靶向分子能够结合肿瘤细胞表面分子,优选肿瘤细胞受体,如肿瘤细胞特异性受体,更优选选自以下的受体: cd71、ca125、epcam(17
‑
1a)、cd52、cea、cd44v6、fap、egf
‑ꢀ
ir、整合素、多配体蛋白聚糖
‑
1、血管整合素α
‑
vβ
‑
3、her2、egfr、 cd20、cd22、叶酸受体1、cd146、cd56、cd19、cd138、cd27l 受体、psma、canag、整合素
‑
αv、ca6、cd33、间皮素、cripto、cd3、 cd30、cd239、cd70、cd123、cd352、dll3、cd25、肝配蛋白a4、 muc1、trop2、ceacam5、ceacam6、her3、cd74、ptk7、notch3、 fgf2、c4.4a、flt3、cd38、fgfr3、cd7、pd
‑
l1、ctla4、cd52、 pdgfra、vegfr1、vegfr2,优选选自cd71、her2和egfr。
[0533]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中肿瘤细胞受体在结合本发明的细胞表面分子靶向分子和本发明的缀合物之后被肿瘤细胞内化,以及其中优选地缀合物与肿瘤细胞受体的结合之后是如经由缀合物和肿瘤细胞受体的
复合物的内吞作用进行的肿瘤细胞受体介导的内化。
[0534]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中细胞表面分子靶向分子是或包含单克隆抗体或者其至少一个细胞表面分子结合片段或结合结构域,并且优选包含以下中的任一种或由以下中的任一种组成:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗、莫西莫单抗、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型okt
‑
9抗cd71单克隆抗体、帕妥珠单抗、利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、okt
‑
10、抗cd38单克隆抗体、表a2或表a3或表a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑
9,或者其至少一个细胞表面分子结合片段和/或结合结构域。
[0535]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个效应子部分包含寡核苷酸、核酸和外源核酸中的任何一种或多种或者由其组成,所述寡核苷酸、核酸和外源核酸优选选自以下中的任何一种或多种:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸(rna)、反义寡核苷酸(aso,aon)、短干扰rna(sirna)、微小rna(mirna)、dna适配体、rna适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)、桥接核酸(bna)、2
’‑
脱氧
‑2’‑
氟阿拉伯糖核酸(fana)、 2
’‑
o
‑
甲氧基乙基
‑
rna(moe)、2
’‑
o,4
’‑
氨基亚乙基桥接核酸、3
’‑
氟己糖醇核酸(fhna)、质粒、二醇核酸(gna)和苏糖核酸(tna)、或它们的衍生物,更优选bna,例如用于沉默hsp27蛋白表达的bna。
[0536]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个效应子部分包含至少一种蛋白质分子或由其组成,所述蛋白质分子优选选自以下中的任何一种或多种:肽、蛋白质、酶(如脲酶和cre重组酶)、核糖体失活蛋白、选自表a5的蛋白质毒素以及更优选选自以下中的任何一种或多种:病毒毒素,如凋亡蛋白;细菌毒素,如志贺毒素、志贺样毒素、铜绿假单胞菌外毒素(pe)或pe 的外毒素a、全长或截短的白喉毒素(dt)、霍乱毒素;真菌毒素,如α
‑
帚曲菌素;植物毒素,包括核糖体失活蛋白和2型核糖体失活蛋白的a链,如石竹素例如石竹素
‑
30或石竹素
‑
32、皂草素例如皂草素
‑
s3 或皂草素
‑
s6、三角梅核糖体失活蛋白或三角梅核糖体失活蛋白的去免疫化衍生物去三角梅核糖体失活蛋白、志贺样毒素a、商陆抗病毒蛋白、蓖麻毒素、蓖麻毒素a链、蒴莲根毒素、蒴莲根毒素a链、相思子毒素、相思子毒素a链、蒴莲毒素、蒴莲毒素a链、槲寄生素、槲寄生素a链;或者动物或人类毒素,如蛙rna酶或者来自人类的颗粒酶b或血管生长素,或者它们的任何片段或衍生物;优选地,所述蛋白质毒素是石竹素和/或皂草素。
[0537]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个效应子部分包含至少一种有效载荷或由其组成,所述有效载荷优选选自以下中的任何一种或多种:靶向核糖体的毒素、靶向延长因子的毒素、靶向微管蛋白的毒素、靶向dna的毒素和靶向 rna的毒素,更优选选自以下中的任何一种或多种:美坦新、pasudotox、美登素衍生物dm1、美登素衍生物dm4、单甲基澳瑞他汀e(mmae,维多汀)、单甲基澳瑞他汀f(mmaf,莫福汀)、卡奇霉素、n
‑
乙酰基
‑ꢀ
γ
‑
卡奇霉素、吡咯并苯并二氮杂(pbd)二聚体、苯并二氮杂cc
‑ꢀ
1065类似物、倍癌霉素、多柔比星、紫杉醇、多西他赛、顺铂、环磷酰胺、依托泊苷、多西他赛、5
‑
氟尿嘧啶(5
‑
fu)、米托蒽醌、微管菌素、吲哚啉苯并二氮杂az13599185、念
珠藻素、根瘤菌素、甲氨蝶呤、蒽环类药物、喜树碱类似物、sn
‑
38、dx
‑
8951f、甲磺酸依喜替康、截短形式的铜绿假单胞菌外毒素(pe38)、倍癌霉素衍生物、鹅膏蕈碱、α
‑
鹅膏蕈碱、斯考他汀、泰兰斯他汀、奥佐米星、特司林、amberstatin269 和soravtansine或它们的衍生物。
[0538]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个效应子部分经由至少一个接头共价结合细胞表面分子靶向分子,或者直接结合细胞表面分子靶向分子。
[0539]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个效应子部分共价结合细胞表面分子靶向分子,从而形成抗体
‑
药物缀合物吉妥珠单抗奥佐米星、本妥昔单抗维多汀、曲妥珠单抗美坦新、英妥珠单抗奥佐米星、moxetumomab pasudotox和泊洛妥珠单抗维多汀以及表a2和表a3的抗体
‑
药物缀合物中的任一种。
[0540]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷经由皂苷中的醛官能团共价结合细胞表面分子靶向分子,优选细胞表面分子靶向分子的氨基酸残基,和/或优选经由至少一个效应子部分中的氨基酸残基,经由皂苷中的醛官能团,优选属于12,13
‑
脱氢齐墩果烷类型的双糖链三萜皂苷中c
‑
23位的醛官能团共价结合至少一个效应子部分。
[0541]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷中的醛官能团,优选至少一个皂苷的c
‑
23 位的醛官能团共价偶联至接头n
‑
ε
‑
马来酰亚胺基己酸酰肼,所述接头经由硫醚键共价偶联至细胞表面分子靶向分子和/或至少一个效应子部分中的巯基基团,如半胱氨酸的巯基基团。
[0542]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中包含葡糖醛酸官能团的双糖链三萜皂苷,其中皂苷经由所述葡糖醛酸官能团和优选经由至少一个效应子部分中的氨基酸残基共价结合细胞表面分子靶向分子的氨基酸残基和/或共价结合至少一个效应子部分。
[0543]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑ꢀ
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至细胞表面分子靶向分子和/或至少一个效应子部分中的胺基团,如细胞表面分子靶向分子和/或至少一个效应子部分的赖氨酸或 n
‑
末端的胺基团。
[0544]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷直接或经由至少一个接头共价结合细胞表面分子靶向分子和/或至少一个效应子部分,所述接头如例如基于n
‑
ε
‑ꢀ
马来酰亚胺基己酸酰肼和/或基于1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑ꢀ
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐的双官能接头或者三官能机头,如方案ii和结构b的三官能接头。
[0545]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中三官能接头包含具有至少一个皂苷共价结合至其的第二化学基团、用于共价结合细胞表面分子靶向分子的第三化学基团以及用于共价结合至少一个效应子部分的第一化学基团,优选三官能接头是方案ii和结构b的三官能接头。
[0546]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至
少一个皂苷经由包括三官能接头(细胞表面分子靶向分子和至少一个效应子部分两者结合的三官能接头)的至少一个接头共价结合细胞表面分子靶向分子和至少一个效应子部分,优选地,三官能接头是方案ii和结构b的三官能接头。
[0547]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个接头包含至少一个可裂解接头,其中任选地所述可裂解接头在酸性条件、还原条件、酶促条件或光诱导条件下进行裂解,并且优选地,可裂解接头包含选自以下的可裂解键:在酸性条件下进行裂解的腙键或酰肼键,和/或易于蛋白质水解(例如由组织蛋白酶b进行蛋白质水解)的键,和/或易于在还原条件下裂解的键,如二硫键。
[0548]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个接头包含至少一个可裂解接头,其中所述可裂解接头在如存在于哺乳动物细胞,优选人类细胞的内体和/或溶酶体的酸性条件下,优选在ph 4.0
‑
6.5下并且更优选在ph≤5.5下在体内进行裂解。
[0549]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷共价结合赖氨酸侧链,形成酰胺键,和/或共价结合半胱氨酸侧链,形成硫醚连键或二硫键,其中细胞表面分子靶向分子包含和/或至少一个效应子部分包含赖氨酸和/或半胱氨酸,并且其中至少一个皂苷直接结合赖氨酸和/或半胱氨酸或者经由任选包含可裂解接头和/或三官能接头(如方案ii和结构b的三官能接头)的至少一个接头结合。
[0550]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中接头基于n
‑
ε
‑
马来酰亚胺基己酸酰肼和/或基于1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,三官能接头如方案ii和结构b的三官能接头,可裂解接头,和/或涉及二硫键、硫醚键、酰胺键、酰肼键中的任何一种或多种。
[0551]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷经由至少一个接头共价结合细胞表面分子靶向分子和/或至少一个效应子部分,其中接头是或包含支架,所述支架包含多聚结构或寡聚结构并且还包含用于共价偶联支架到细胞表面分子靶向分子和/或到至少一个效应子部分的至少一个第四化学基团。
[0552]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷经由可裂解键和/或经由不可裂解键共价结合支架的多聚结构或寡聚结构。
[0553]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中可裂解键在酸性条件、还原条件、酶促条件和光诱导条件中的任一种下进行裂解,并且优选地,可裂解键包括在酸性条件下进行裂解的腙键或酰肼键和/或易于蛋白质水解(例如由组织蛋白酶b 进行蛋白质水解)的键和/或易于在还原条件下裂解的键,如二硫键。
[0554]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中可裂解键在如存在于哺乳动物细胞,优选人类细胞的内体和/或溶酶体的酸性条件下,优选在ph 4.0
‑
6.5下并且更优选在ph≤ 5.5下在体内进行裂解。
[0555]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷经由亚胺键、腙键、酰肼键、肟键、1,3
‑
二噁戊烷键、二硫键、硫醚键、酰胺键、肽键或酯键中的任何一种或多种,优选经由至少一个接头共价结合支架的多聚结构或寡聚结构。
[0556]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至
少一个皂苷经由亚胺键、腙键和酰肼键中的任何一种或多种共价结合支架的多聚结构或寡聚结构,根据本发明,所述键优选是可裂解的,其中优选地,至少一个皂苷经由至少一个皂苷的c
‑
23 位的醛官能团共价结合支架的多聚结构或寡聚结构。
[0557]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷的c
‑
23位的醛官能团共价偶联至接头n
‑ꢀ
ε
‑
马来酰亚胺基己酸酰肼,所述接头经由硫醚键共价偶联至支架的多聚结构或寡聚结构中的巯基基团,如半胱氨酸的巯基基团。
[0558]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷经由酰胺键共价结合支架的多聚结构或寡聚结构,其中优选地,至少一个皂苷经由至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团(当存在时)共价结合支架的多聚结构或寡聚结构。
[0559]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑ꢀ
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至支架的多聚结构或寡聚结构中的胺基团,如支架的多聚结构或寡聚结构的赖氨酸或n
‑
末端的胺基团。
[0560]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷共价结合支架的多聚结构或寡聚结构,在共价键中涉及至少一个皂苷的c
‑
23位的醛官能团(当存在时),和/或在共价键中涉及至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团(当存在时)。
[0561]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中支架的用于支架与细胞表面分子靶向分子和/或至少一个效应子部分的共价偶联的至少一个第四化学基团是点击化学基团,优选选自以下中的任何一种或多种:四嗪、叠氮化物、烯烃或炔烃或者这些基团的环状衍生物,优选叠氮化物基团。
[0562]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中支架的多聚结构或寡聚结构包括直链、支链和/或环状多聚物、寡聚物、树状物、树枝化基元、树枝化基元化多聚物、树枝化基元化寡聚物、dna、多肽、聚赖氨酸、聚乙二醇或者这些多聚结构或寡聚结构的组装体,所述组装体优选通过共价交联构建。
[0563]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷是限定数量的皂苷或限定范围的皂苷,优选1
‑
128个皂苷或者至少2、3、4、5、6、8、10、16、32、64或128 个皂苷,或者其间的任何数量的皂苷,如7、9、12个皂苷。
[0564]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中缀合物包含直接共价结合细胞表面分子靶向分子的氨基酸残基和/或至少一个效应子部分,优选经由至少一个效应子部分中的氨基酸残基,优选共价结合半胱氨酸和/或赖氨酸,和/或经由至少一个接头和/或经由至少一个可裂解接头和/或经由本发明的至少一个多聚支架或寡聚支架,优选1
‑
8个此类支架或2
‑
4个此类支架共价结合的多于一种的皂苷,优选2、3、4、5、6、8、10、16、32、64或1
‑
100 个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷,其中1
‑
32个皂苷,优选2、3、4、5、6、8、10、16或32个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷共价结合至少一个支架。
[0565]
本发明的第四系列方面和实施方案中的一个实施方案是本发明的缀合物,其中至少一个皂苷经由三官能接头共价结合细胞表面分子靶向分子以及共价结合至少一个效应子部分,所述三官能接头包含含有至少一个皂苷直接或经由接头(如可裂解接头)和/或经由包含多聚结构或寡聚结构和根据本发明的第四化学基团(用于支架与三官能接头的共价偶联)的支架共价结合至其的第二化学基团,所述三官能接头还包含用于共价结合细胞表面分子靶向分子的第三化学基团以及包含用于共价结合至少一个效应子部分的第一化学基团,其中细胞表面分子靶向分子结合第三化学基团和/或至少一个效应子部分结合第一化学基团,优选地,三官能接头是方案ii和结构b的三官能接头。
[0566]
本发明的第四系列方面和实施方案中的本发明的一方面涉及药物组合物,其包含本发明的缀合物和任选的药学上可接受的赋形剂和/或药学上可接受的稀释剂。
[0567]
本发明的第四系列方面和实施方案中的本发明的一方面涉及本发明的缀合物或本发明的药物组合物,其用作药物。
[0568]
本发明的第四系列方面和实施方案中的本发明的一方面涉及本发明的缀合物或本发明的药物组合物,其用于治疗或预防癌症或自身免疫疾病。
[0569]
本发明的第五系列方面和实施方案中的本发明的一方面涉及用作药物的治疗性组合,其中治疗性组合包含:(a)第四药物组合物,其包含含有用于结合细胞表面分子上的表位的结合位点的第四蛋白质分子和共价结合所述第四蛋白质分子,优选所述第四蛋白质分子的氨基酸残基的至少一个皂苷,所述第四药物组合物任选地还包含药学上可接受的赋形剂;和(b)第五药物组合物,其包含第五蛋白质分子,所述第五蛋白质分子包含用于结合(a)的细胞表面分子上的表位的结合位点和效应子部分,所述第五药物组合物任选地还包含药学上可接受的赋形剂,其中第四蛋白质分子的结合位点和第五蛋白质分子的结合位点是相同的,并且其中第四蛋白质分子能够结合的细胞表面分子和细胞表面分子上的表位以及第五蛋白质分子能够结合的细胞表面分子和细胞表面分子上的表位是相同的。
[0570]
本发明的第五系列方面和实施方案中的本发明的一方面涉及本发明的第四药物组合物,其用作药物。
[0571]
本发明的第五系列方面和实施方案中的本发明的一方面涉及治疗性组合,其用于治疗或预防人类对象中的癌症,其中治疗性组合包含: (a)本发明的第四药物组合物;和(b)本发明的第五药物组合物,其中细胞表面分子在肿瘤细胞表面上表达,并且优选地,细胞表面分子是肿瘤细胞特异性表面分子,并且其中优选地,表位是肿瘤细胞特异性表位。
[0572]
本发明的第五系列方面和实施方案中的本发明的一方面涉及本发明的第四药物组合物,其用于治疗或预防有需要的患者中的癌症,其中细胞表面分子在肿瘤细胞表面上表达,并且优选地,细胞表面分子是肿瘤细胞特异性表面分子,并且其中优选地,表位是肿瘤细胞特异性表位。
[0573]
本发明的第五系列方面和实施方案中的一个实施方案是用于根据本发明的用途的第四药物组合物或用于本发明的用途的治疗性组合,其中本发明的第五药物组合物和第四药物组合物施用至有需要的患者。
[0574]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中第四蛋白质分子和第五蛋白质分子的结合位点包含以下或由以下组成:免疫球蛋白或者所述免疫球蛋白的结合片段或结
合结构域,如以下中任何一种或多种:抗体、igg、包含vhh结构域或vh结构域或者由其组成的分子、fab、scfv、fv、dab、f(ab)2、fcab片段;和/或包含至少一种配体或由其组成,所述配体如egf或细胞因子用于结合细胞表面分子。
[0575]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中肿瘤细胞表面分子是特异性存在于肿瘤细胞上的细胞表面受体,并且其中优选地,表位是肿瘤细胞特异性表位。
[0576]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中任选地包含葡糖醛酸官能团的三萜皂苷和/或双糖链三萜皂苷,和/或是从石头花属物种和/或肥皂草属物种和/或麦仙翁属物种和/或皂皮树属物种如皂皮树分离的皂苷。
[0577]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中至少一个皂苷是单一特异性皂苷或者是两个或更多个不同皂苷的混合物,如表a1或方案i中的皂苷中的一种或多种、so1861、sa1657、 ge1741、sa1641、qs
‑
21、qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21a
‑
xyl、qs
‑ꢀ
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、qs
‑ꢀ
17
‑
xyl、qs1861、qs1862、皂皮树皂苷、saponinum album、qs
‑
18、 quil
‑
a、gyp1、丝石竹皂苷a、ag1、ag2、so1542、so1584、so1658、 so1674、so1832,或者它们的立体异构体中的任一种和/或它们的任何组合,优选地,所述皂苷是so1861和/或ge1741和/或sa1641和/ 或qs
‑
21和/或具有皂皮酸苷元核心、在c
‑
3β
‑
oh基团处具有gal
‑ꢀ
(1
→
2)
‑
[xyl
‑
(1
→
3)]
‑
glca碳水化合物取代基以及在c
‑
28
‑
oh基团处具有glc
‑
(1
→
3)
‑
xyl
‑
(1
→
4)
‑
rha
‑
(1
→
2)
‑
[xyl
‑
(1
→
3)
‑4‑
oac
‑
qui
‑
(1
→
4)]
‑ꢀ
fuc碳水化合物取代基的皂苷,和/或是3
‑
o
‑
β
‑
d
‑
半乳吡喃糖基
‑
(1
→
2)
‑ꢀ
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)]
‑
β
‑
d
‑
葡糖醛酸吡喃糖基皂皮酸28
‑
o
‑
β
‑
d
‑
葡吡喃糖基
‑
(1
→
3)
‑
β
‑
d
‑
木吡喃糖基
‑
(1
→
4)
‑
α
‑
l
‑
鼠李吡喃糖基
‑
(1
→
2)
‑
[β
‑ꢀ
d
‑
木吡喃糖基
‑
(1
→
3)
‑4‑
oac
‑
β
‑
d
‑
奎诺吡喃糖基
‑
(1
→
4)]
‑
β
‑
d
‑
岩藻吡喃糖苷,更优选地,所述皂苷是so1861和/或qs
‑
21。
[0578]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中至少一个皂苷是具有至少1.500道尔顿的分子质量并且包含在c
‑ꢀ
23位含有醛基团以及在c
‑
16位任选地含有羟基基团以及在c
‑
3位具有第一支链碳水化合物侧链的齐墩果烷型三萜的双糖链皂苷,所述第一支链碳水化合物侧链任选地含有葡糖醛酸,其中所述皂苷含有在c
‑ꢀ
28位具有第二支链碳水化合物侧链的酯基团,所述第二支链碳水化合物链优选包含至少四个碳水化合物单元,任选地含有至少一个乙酰基残基如两个乙酰基残基和/或任选至少一个脱氧碳水化合物和/或奎诺糖和/或葡萄糖和/或4
‑
甲氧基肉桂酸和/或任选地包含5
‑
o
‑
[5
‑
o
‑ꢀ
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸和/或任选地包含经由酯键与碳水化合物结合的5
‑
o
‑
[5
‑
o
‑
rha
‑
(1
→
2)
‑
ara/api
‑ꢀ
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸,或者其中至少一个皂苷是qs
‑
21或者以下中的任何一种或多种:qs
‑
21a、qs
‑
21a
‑
api、 qs
‑
21a
‑
xyl、qs
‑
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑ꢀ
api、qs
‑
17
‑
api、qs
‑
17
‑
xyl、qs
‑
18、qs1861、质子化的qs
‑
1861(qs1862)、 quil
‑
a。
[0579]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治
疗性组合或用于根据本发明的用途的第四药物组合物,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型,其中皂苷共价偶联至第四蛋白质分子,优选经由皂苷中的醛官能团,优选在c
‑
23位的所述醛官能团,优选经由至少一个接头和 /或经由至少一个可裂解接头共价偶联至第四蛋白质分子的氨基酸残基,其中氨基酸残基优选选自半胱氨酸和赖氨酸。
[0580]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中至少一个皂苷的c
‑
23位的醛官能团共价偶联至接头n
‑
ε
‑
马来酰亚胺基己酸酰肼,所述接头经由硫醚键共价偶联至第四蛋白质分子中的巯基基团,如半胱氨酸的巯基基团。
[0581]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中包含葡糖醛酸官能团的双糖链三萜皂苷,其中皂苷经由皂苷的c
‑
3β
‑
oh基团处碳水化合物取代基中的葡糖醛酸官能团,优选经由至少一个接头共价偶联至第四蛋白质分子的氨基酸残基,其中氨基酸残基优选选自半胱氨酸和赖氨酸。
[0582]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并 [4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至第四蛋白质分子中的胺基团,如第四蛋白质分子的赖氨酸或n
‑
末端的胺基团。
[0583]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中表位是肿瘤细胞受体上的表位,优选肿瘤细胞特异性表位,并且其中受体优选是肿瘤细胞特异性受体,更优选选自以下的受体:cd71、 ca125、epcam(17
‑
1a)、cd52、cea、cd44v6、fap、egf
‑
ir、整合素、多配体蛋白聚糖
‑
1、血管整合素α
‑
vβ
‑
3、her2、egfr、cd20、 cd22、叶酸受体1、cd146、cd56、cd19、cd138、cd27l受体、 psma、canag、整合素
‑
αv、ca6、cd33、间皮素、cripto、cd3、cd30、 cd239、cd70、cd123、cd352、dll3、cd25、肝配蛋白a4、muc1、 trop2、ceacam5、ceacam6、her3、cd74、ptk7、notch3、fgf2、c4.4a、flt3、cd38、fgfr3、cd7、pd
‑
l1、ctla4、cd52、pdgfra、 vegfr1、vegfr2,最优选选自cd71、her2和egfr。
[0584]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中肿瘤细胞受体,优选肿瘤细胞特异性受体在结合本发明的第四蛋白质分子之后和/或在结合本发明的第五蛋白质分子之后由肿瘤细胞内化,并且其中优选地,第四蛋白质分子和/或第五蛋白质分子与肿瘤细胞受体(如肿瘤细胞特异性受体)的结合之后是如经由第四蛋白质分子和肿瘤细胞受体的复合物以及第五蛋白质分子和肿瘤细胞受体的复合物的内吞作用进行的肿瘤细胞受体介导的内化。
[0585]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第四药物组合物,其中第四蛋白质分子和第五蛋白质分子的结合位点包括单克隆抗体或者其细胞表面分子结合结构域和/或结合片段中的至少
一种,并且优选包含以下中的任一种或由以下中的任一种组成:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗、莫西莫单抗、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型okt
‑
9抗 cd71单克隆抗体、帕妥珠单抗、利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、okt
‑
10、抗cd38单克隆抗体、表a2或表a3或表a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑
9,或者其至少一个细胞表面分子结合片段和/或结合结构域,条件是第四结合位点与第五结合位点相同。
[0586]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中第五蛋白质分子所包含的效应子部分包含寡核苷酸、核酸和异源核酸中的任何一种或多种或者由其组成,所述寡核苷酸、核酸和异源核酸优选选自以下中的任何一种或多种:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸 (rna)、反义寡核苷酸(aso,aon)、短干扰rna(sirna)、微小 rna(mirna)、dna适配体、rna适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)、桥接核酸(bna)、2
’‑
脱氧
‑2’‑
氟阿拉伯糖核酸(fana)、2
’‑
o
‑
甲氧基乙基
‑ꢀ
rna(moe)、2
’‑
o,4
’‑
氨基亚乙基桥接核酸、3
’‑
氟己糖醇核酸(fhna)、质粒、二醇核酸(gna)和苏糖核酸(tna)、或它们的衍生物,更优选 bna,例如用于沉默hsp27蛋白表达的bna。
[0587]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中第五蛋白质分子所包含的效应子部分包含至少一种蛋白质分子或由其组成,所述蛋白质分子优选选自以下中的任何一种或多种:肽、蛋白、酶(如脲酶和cre重组酶)、蛋白质毒素、核糖体失活蛋白、选自表a5的蛋白质毒素和/或细菌毒素、植物毒素,更优选选自以下中的任何一种或多种:病毒毒素,如凋亡蛋白;细菌毒素,如志贺毒素、志贺样毒素、铜绿假单胞菌外毒素(pe)或pe的外毒素a、全长或截短的白喉毒素(dt)、霍乱毒素;真菌毒素,如α
‑
帚曲菌素;植物毒素,包括核糖体失活蛋白和2型核糖体失活蛋白的a 链,如石竹素例如石竹素
‑
30或石竹素
‑
32、皂草素例如皂草素
‑
s3或皂草素
‑
s6、三角梅核糖体失活蛋白或三角梅核糖体失活蛋白的去免疫化衍生物去三角梅核糖体失活蛋白、志贺样毒素a、商陆抗病毒蛋白、蓖麻毒素、蓖麻毒素a链、蒴莲根毒素、蒴莲根毒素a链、相思子毒素、相思子毒素a链、蒴莲毒素、蒴莲毒素a链、槲寄生素、槲寄生素a链;或者动物或人类毒素,如蛙rna酶或者来自人类的颗粒酶 b或血管生长素,或者它们的任何片段或衍生物;优选地,所述蛋白质毒素是石竹素和/或皂草素。
[0588]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中第五蛋白质分子所包含的效应子部分包含至少一种有效载荷或由其组成,所述有效载荷优选选自以下中的任何一种或多种:靶向核糖体的毒素、靶向延长因子的毒素、靶向微管蛋白的毒素、靶向dna的毒素和靶向rna的毒素,更优选选自以下中的任何一种或多种:美坦新、pasudotox、美登素衍生物dm1、美登素衍生物dm4、单甲基澳瑞他汀e(mmae,维多汀)、单甲基澳瑞他汀f(mmaf,莫福汀)、卡奇霉素、n
‑
乙酰基
‑
γ
‑
卡奇霉素、吡咯并苯并二氮杂(pbd)二聚体、苯并二氮杂cc
‑
1065类似物、倍癌霉素、多柔比星、紫杉醇、多西他赛、顺铂、环磷酰胺、依托泊苷、多西他赛、5
‑
氟尿嘧啶(5
‑
fu)、米托蒽醌、微管菌素、吲哚啉苯并二氮杂 az13599185、念珠藻素、根瘤菌素、甲氨蝶呤、蒽环类药物、喜树碱类似物、sn
‑
38、dx
‑
8951f、甲磺酸依喜替康、截短形式的铜绿假单胞菌外毒素(pe38)、倍癌霉素衍生物、鹅膏蕈碱、α
‑
鹅膏蕈碱、斯考他汀、泰兰斯他汀、奥佐米星、特司林、amberstatin269
和soravtansine或它们的衍生物。
[0589]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中第五蛋白质分子包含以下中的任一种或由以下中的任一种组成:吉妥珠单抗奥佐米星、本妥昔单抗维多汀、曲妥珠单抗美坦新、英妥珠单抗奥佐米星、moxetumomab pasudotox和泊洛妥珠单抗维多汀以及表a2和表a3的抗体
‑
药物缀合物,或者其至少一个肿瘤细胞受体结合结构域和/或至少一个肿瘤细胞受体结合片段,其中所述结构域或片段包括效应子部分并且优选是肿瘤细胞特异性受体结合结构域和/或肿瘤细胞特异性受体结合片段。
[0590]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中第四蛋白质分子包含多于一种共价结合的皂苷,优选2、3、4、5、6、8、10、16、32、64、128或1
‑
100个皂苷,或者其间的任何数量的皂苷,如7、9、12个皂苷。
[0591]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中至少一个皂苷直接共价结合第四蛋白质分子的氨基酸残基,优选半胱氨酸和/或赖氨酸,和/或经由至少一个接头和/或经由至少一个可裂解接头和/或经由至少一个寡聚支架或多聚支架,优选1
‑
8个此类支架或2
‑
4个此类支架共价结合,其中至少一个支架任选地基于树枝化基元,其中1
‑
32个皂苷,如2、3、4、5、6、 8、10、16、32个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷共价结合至少一个支架。
[0592]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中可裂解接头在酸性条件、还原条件、酶促条件或光诱导条件下进行裂解,并且优选可裂解接头包含选自以下的可裂解键:在酸性条件下进行裂解的腙键和酰肼键和/或易于蛋白质水解(例如由组织蛋白酶b进行蛋白质水解)的键和/或易于在还原条件下裂解的键,如二硫键。
[0593]
本发明的第五系列的方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中可裂解接头在如存在于哺乳动物细胞,优选人类细胞的内体和/或溶酶体的酸性条件下,优选在ph 4.0
‑
6.5下并且更优选在ph≤5.5下在体内进行裂解。
[0594]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中寡聚支架或多聚支架包含多聚结构或寡聚结构并包含至少一个化学基团,所述至少一个化学基团用于支架与所述第四蛋白质分子的氨基酸残基的共价偶联。
[0595]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中至少一个皂苷经由根据本发明的可裂解接头共价结合支架的多聚结构或寡聚结构。
[0596]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中至少一个皂苷经由亚胺键、腙键、酰肼键、肟键、1,3
‑
二噁戊烷键、二硫键、硫醚键、酰胺键、肽键或酯键中的任何一种或多种,优选经由至少一个接头共价结合支架的多聚结构或寡聚结构。
[0597]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中至少一个皂苷包含在c
‑
23位的醛官能团和任选地在至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团,所述醛官能团参与共价键合至支架的多聚结构或寡聚结构,和/或,如果存在,葡糖醛酸官能团参与共价键合至支架的多聚结构
或寡聚结构(经由直接结合或经由至少一个接头)。
[0598]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中至少一个皂苷的c
‑
23位的醛官能团共价偶联至接头n
‑
ε
‑
马来酰亚胺基己酸酰肼,所述接头经由硫醚键共价偶联至支架的多聚结构或寡聚结构中的巯基基团,如半胱氨酸的巯基基团。
[0599]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头1
‑
[双(二甲基氨基) 亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至支架的多聚结构或寡聚结构中的胺基团,如蛋白质分子的赖氨酸或n
‑
末端的胺基团。
[0600]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中支架的用于寡聚支架或多聚支架与所述第四蛋白质分子的氨基酸残基的共价偶联的至少一个化学基团是点击化学基团,其优选选自四嗪、叠氮化物、烯烃或炔烃或者这些基团的环状衍生物,更优选地,点击化学基团是叠氮化物。
[0601]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中支架的多聚结构或寡聚结构包括直链、支链和/或环状多聚物、寡聚物、树状物、树枝化基元、树枝化基元化多聚物、树枝化基元化寡聚物、dna、多肽、聚赖氨酸、聚乙二醇或者这些多聚结构或寡聚结构的组装体,所述组装体优选通过共价交联构建。
[0602]
本发明的第五系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中第四药物组合物和第五药物组合物施用至有需要的患者。
[0603]
本发明的第五系列方面和实施方案中的本发明的一方面涉及本发明的第四药物组合物,其还包含本发明的第五蛋白质分子。
[0604]
本发明的第五系列方面和实施方案中的本发明的一方面涉及用作药物用途的本发明的第四药物组合物,其还包含本发明的第五蛋白质分子。
[0605]
本发明的第五系列方面和实施方案中的本发明的一方面涉及用于治疗或预防有需要的患者中的癌症的用途的本发明的第四药物组合物,其还包含本发明的第五蛋白质分子。
[0606]
本发明的第六系列方面和实施方案中的本发明的一方面涉及治疗性组合,其用作药物,其中治疗性组合包含:(a)第六药物组合物,其包含含有用于结合第六细胞表面分子的第六结合位点的第六蛋白质分子和共价结合所述第六蛋白质分子,优选共价结合所述第六蛋白质分子的氨基酸残基的至少一个皂苷,所述第六药物组合物任选地还包含药学上可接受的赋形剂;以及(b)第七药物组合物,其包含优选不同于第六蛋白质分子的第七蛋白质分子,所述第七蛋白质分子包含用于结合不同于第六细胞表面分子的第七细胞表面分子的第七结合位点和效应子部分,所述第七药物组合物任选地还包含药学上可接受的赋形剂。
[0607]
本发明的第六系列方面和实施方案中的本发明的一方面涉及本发明的第六药物组合物,其用作药物。
[0608]
本发明的第六系列方面和实施方案中的本发明的一方面涉及治疗性组合,其用于治疗或预防人类对象中的癌症,其中治疗性组合包含: (a)本发明的第六药物组合物,其中第六细胞表面分子是第六肿瘤细胞表面分子,优选第六肿瘤细胞特异性表面分子;以及(b)
本发明的第七药物组合物,其中第七细胞表面分子是不同于第六肿瘤细胞表面分子的第七肿瘤细胞表面分子,优选第七细胞表面分子是不同于第六肿瘤细胞特异性表面分子的第七肿瘤细胞特异性表面分子。
[0609]
本发明的第六系列方面和实施方案中的本发明的一方面涉及本发明的第六药物组合物,其用于治疗或预防有需要的患者中的癌症,其中第六细胞表面分子是第六肿瘤细胞表面分子,优选第六肿瘤细胞特异性表面分子。
[0610]
本发明的第六系列方面和实施方案中的一个实施方案是用于根据本发明的用途的第六药物组合物或本发明的治疗性组合,其中本发明的第七药物组合物和第六药物组合物施用至有需要的患者,并且其中第七肿瘤细胞表面分子不同于第六肿瘤细胞表面分子,优选地,第七肿瘤细胞特异性表面分子不同于第六肿瘤细胞特异性表面分子。
[0611]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中第六蛋白质分子的第六结合位点包含以下或由以下组成:用于结合第六细胞表面分子的免疫球蛋白或者所述免疫球蛋白的至少一个结合片段或结合结构域,如抗体、igg、包含vhh结构域或vh结构域或者由其组成的分子、fab、scfv、fv、dab、f(ab)2、fcab片段中的任何一种或多种,和/或包含以下或由以下组成:至少一种配体,优选用于结合第六细胞表面分子的至少一种配体,如egf或细胞因子。
[0612]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中用于结合第六肿瘤细胞表面分子,优选肿瘤细胞特异性表面分子的第六结合位点是针对存在于肿瘤细胞,优选特异性存在于肿瘤细胞上的第六细胞表面受体的第六结合位点。
[0613]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且任选地在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中包含葡糖醛酸官能团的三萜皂苷或双糖链三萜皂苷,和/或是从石头花属物种和/或肥皂草属物种和/或麦仙翁属物种和/或皂皮树属物种如皂皮树分离的皂苷。
[0614]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中至少一个皂苷是单一特异性皂苷或者是两个或更多个不同皂苷的混合物,如表a1或方案i中的皂苷中的一种或多种、so1861、sa1657、 ge1741、sa1641、qs
‑
21、qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21a
‑
xyl、qs
‑ꢀ
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、qs
‑ꢀ
17
‑
xyl、qs1861、qs1862、皂皮树皂苷、saponinum album、qs
‑
18、 quil
‑
a、gyp1、丝石竹皂苷a、ag1、ag2、so1542、so1584、so1658、 so1674、so1832,或者它们的立体异构体中的任一种和/或它们的任何组合,优选地,所述皂苷是so1861和/或ge1741和/或sa1641和/ 或qs
‑
21和/或具有皂皮酸苷元核心、在c
‑
3β
‑
oh基团处具有gal
‑ꢀ
(1
→
2)
‑
[xyl
‑
(1
→
3)]
‑
glca碳水化合物取代基以及在c
‑
28
‑
oh基团处具有glc
‑
(1
→
3)
‑
xyl
‑
(1
→
4)
‑
rha
‑
(1
→
2)
‑
[xyl
‑
(1
→
3)
‑4‑
oac
‑
qui
‑
(1
→
4)]
‑ꢀ
fuc碳水化合物取代基的皂苷,和/或是3
‑
o
‑
β
‑
d
‑
半乳吡喃糖基
‑1→
2)
‑ꢀ
[β
‑
d
‑
木吡喃糖基
‑
(1
→
3)]
‑
β
‑
d
‑
葡糖醛酸吡喃糖基皂皮酸28
‑
o
‑
β
‑
d
‑
葡吡喃糖基
‑
(1
→
3)
‑
β
‑
d
‑
木吡喃糖基
‑
(1
→
4)
‑
α
‑
l
‑
鼠李吡喃糖基
‑
(1
→
2)
‑
[β
‑ꢀ
d
‑
木吡喃糖基
‑
(1
→
3)
‑4‑
oac
‑
β
‑
d
‑
奎诺吡喃糖基
‑
(1
→
4)]
‑
β
‑
d
‑
岩藻吡喃糖苷,更优选地,所述皂苷是so1861和/或qs
‑
21。
[0615]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中至少一个皂苷是具有至少1.500道尔顿的分子质量并且包含在c
‑ꢀ
23位含有醛基团并且在任选地c
‑
16位含有羟基基团,以及在c
‑
3位具有第一支链碳水化合物侧链的齐墩果烷型三萜的双糖链皂苷,所述第一支链碳水化合物侧链任选地含有葡糖醛酸,其中所述皂苷含有在 c
‑
28位具有第二支链碳水化合物侧链的酯基团,所述第二支链碳水化合物链优选包含至少四个碳水化合物单元,任选地含有至少一个乙酰基残基如两个乙酰基残基和/或至少一个脱氧碳水化合物和/或奎诺糖和/或葡萄糖和/或4
‑
甲氧基肉桂酸和/或任选地包含5
‑
o
‑
[5
‑
o
‑
ara/api
‑ꢀ
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸和/或任选地包含经由酯键与碳水化合物结合的5
‑
o
‑
[5
‑
o
‑
rha
‑
(1
→
2)
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸,或者其中至少一个皂苷是 qs
‑
21或者以下中的任何一种或多种:qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21 a
‑
xyl、qs
‑
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑ꢀ
17
‑
api、qs
‑
17
‑
xyl、qs
‑
18、qs1861、质子化的qs
‑
1861(qs1862)、quil
‑ꢀ
a。
[0616]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型的双糖链三萜皂苷,其中皂苷优选经由皂苷中的醛官能团,优选在c
‑
23位的所述醛官能团,优选经由至少一个接头和/或经由至少一个可裂解接头共价偶联至第六蛋白质分子,优选共价偶联至第六蛋白质分子的氨基酸残基,其中氨基酸残基优选选自半胱氨酸和赖氨酸。
[0617]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中至少一个皂苷的c
‑
23位的醛官能团共价偶联至接头n
‑
ε
‑
马来酰亚胺基己酸酰肼,所述接头经由硫醚键共价偶联至第六蛋白质分子中的巯基基团,如半胱氨酸的巯基基团。
[0618]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中至少一个皂苷是属于在c
‑
23位具有醛官能团的12,13
‑
脱氢齐墩果烷类型并且在皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中包含葡糖醛酸官能团的双糖链三萜皂苷,其中皂苷经由皂苷的c
‑
3β
‑
oh基团处碳水化合物取代基中的葡糖醛酸官能团,优选经由至少一个接头共价偶联至第六蛋白质分子的氨基酸残基,其中氨基酸残基优选选自半胱氨酸和赖氨酸。
[0619]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并 [4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至第六蛋白质分子中的胺基团,如第六蛋白质分子的赖氨酸或n
‑
末端的胺基团。
[0620]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中第七蛋白质分子的第七结合位点包含以下或由以下组成:用于结合第七细胞表面分子的免疫球蛋白、所述免疫球蛋白的至少一个结合结构域和/或所述免疫球蛋白的至少一个结合片段,如抗体、igg、包含 vhh结
构域或vh结构域或者由其组成的分子、fab、scfv、fv、dab、 f(ab)2、fcab片段;和/或包含以下或由以下组成:至少一种配体,优选用于结合第七细胞表面分子的配体,如egf或细胞因子。
[0621]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中用于结合第七肿瘤细胞表面分子,优选第七肿瘤细胞特异性表面分子的第七结合位点是针对存在于肿瘤细胞,优选特异性存在于肿瘤细胞上的第七细胞表面受体的第七结合位点。
[0622]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中第六结合位点和第七结合位点是分别用于结合至第六和第七肿瘤细胞受体,优选分别用于结合第六和第七肿瘤细胞特异性受体,优选存在于相同肿瘤细胞上的结合位点,并且其中第六和第七肿瘤细胞受体优选是肿瘤细胞特异性受体,和/或选自cd71、ca125、epcam(17
‑ꢀ
1a)、cd52、cea、cd44v6、fap、egf
‑
ir、整合素、多配体蛋白聚糖
‑
1、血管整合素α
‑
vβ
‑
3、her2、egfr、cd20、cd22、叶酸受体1、 cd146、cd56、cd19、cd138、cd27l受体、psma、canag、整合素
‑
αv、ca6、cd33、间皮素、cripto、cd3、cd30、cd239、cd70、 cd123、cd352、dll3、cd25、肝配蛋白a4、muc1、trop2、ceacam5、 ceacam6、her3、cd74、ptk7、notch3、fgf2、c4.4a、flt3、 cd38、fgfr3、cd7、pd
‑
l1、ctla4、cd52、pdgfra、vegfr1、 vegfr2,更优选选自her2、cd71和egfr,条件是第六结合位点不同于第七结合位点并且条件是第六和第七肿瘤细胞特异性受体,优选第六和第七肿瘤细胞特异性受体是不同的受体。
[0623]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中第六结合位点和第七结合位点包含以下或由以下组成:西妥昔单抗、达雷木单抗、吉妥珠单抗、曲妥珠单抗、帕尼单抗、本妥昔单抗、英妥珠单抗、莫西莫单抗、泊洛妥珠单抗、奥滨尤妥珠单抗、igg型 okt
‑
9抗cd71单克隆抗体、帕妥珠单抗、利妥昔单抗、奥法木单抗、赫塞汀、阿仑单抗、匹那妥珠单抗、okt
‑
10、抗cd38单克隆抗体、表a2或表a3或表a4的抗体,优选西妥昔单抗或曲妥珠单抗或okt
‑ꢀ
9,或其至少一个肿瘤细胞受体结合结构域和/或至少一个肿瘤细胞受体结合片段,条件是第六结合位点不同于第七结合位点。
[0624]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中第六肿瘤细胞受体在结合本发明的第六蛋白质分子之后由肿瘤细胞内化,并且其中优选地,第六蛋白质分子与第六肿瘤细胞受体结合之后是如经由第六蛋白质分子和第六肿瘤细胞受体的复合物的内吞作用进行的肿瘤细胞受体介导的内化,其中第六肿瘤细胞受体优选是第六肿瘤细胞特异性受体。
[0625]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中第七肿瘤细胞受体在结合本发明的第七蛋白质分子之后由肿瘤细胞内化,并且其中优选地,第七蛋白质分子与第七肿瘤细胞受体的结合之后是如经由第七蛋白质分子和第七肿瘤细胞受体的复合物的内吞作用进行的肿瘤细胞受体介导的内化,其中第七肿瘤细胞受体优选是第七肿瘤细胞特异性受体。
[0626]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中第七蛋白质分子所包含的效应
子部分包含寡核苷酸、核酸和异源核酸中的至少一种或由其组成,所述寡核苷酸、核酸和异源核酸优选选自以下中的任何一种或多种:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸(rna)、反义寡核苷酸(aso,aon)、短干扰rna(sirna)、微小rna(mirna)、dna适配体、rna适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)、桥接核酸(bna)、2
’‑
脱氧
‑2’‑
氟阿拉伯糖核酸(fana)、 2
’‑
o
‑
甲氧基乙基
‑
rna(moe)、2
’‑
o,4
’‑
氨基亚乙基桥接核酸、3
’‑
氟己糖醇核酸(fhna)、质粒、二醇核酸(gna)和苏糖核酸(tna)、或它们的衍生物,更优选bna,例如用于沉默hsp27蛋白表达的bna。
[0627]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中第七蛋白质分子所包含的效应子部分包含至少一种蛋白质分子或由其组成,所述蛋白质分子优选选自以下中的任何一种或多种:肽、蛋白、酶(如脲酶和cre重组酶)、蛋白质毒素、核糖体失活蛋白、选自表a5的蛋白质毒素和/或细菌毒素、植物毒素,更优选选自以下中的任何一种或多种:病毒毒素,如凋亡蛋白;细菌毒素,如志贺毒素、志贺样毒素、铜绿假单胞菌外毒素(pe)或pe的外毒素a、全长或截短的白喉毒素(dt)、霍乱毒素;真菌毒素,如α
‑
帚曲菌素;植物毒素,包括核糖体失活蛋白和2型核糖体失活蛋白的a链,如石竹素例如石竹素
‑
30或石竹素
‑
32、皂草素例如皂草素
‑
s3或皂草素
‑
s6、三角梅核糖体失活蛋白或三角梅核糖体失活蛋白的去免疫化衍生物去三角梅核糖体失活蛋白、志贺样毒素a、商陆抗病毒蛋白、蓖麻毒素、蓖麻毒素a链、蒴莲根毒素、蒴莲根毒素a链、相思子毒素、相思子毒素a 链、蒴莲毒素、蒴莲毒素a链、槲寄生素、槲寄生素a链;或者动物或人类毒素,如蛙rna酶或者来自人类的颗粒酶b或血管生长素,或者它们的任何片段或衍生物;优选地,所述蛋白质毒素是石竹素和/ 或皂草素。
[0628]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六药物组合物,其中第七蛋白质分子所包含的效应子部分包含至少一种有效载荷或由其组成,所述有效载荷优选选自以下中的任何一种或多种:靶向核糖体的毒素、靶向延长因子的毒素、靶向微管蛋白的毒素、靶向dna的毒素和靶向rna的毒素,更优选选自以下中的任何一种或多种:美坦新、pasudotox、美登素衍生物dm1、美登素衍生物dm4、单甲基澳瑞他汀e(mmae,维多汀)、单甲基澳瑞他汀f(mmaf,莫福汀)、卡奇霉素、n
‑
乙酰基
‑
γ
‑
卡奇霉素、吡咯并苯并二氮杂(pbd)二聚体、苯并二氮杂cc
‑
1065类似物、倍癌霉素、多柔比星、紫杉醇、多西他赛、顺铂、环磷酰胺、依托泊苷、多西他赛、5
‑
氟尿嘧啶(5
‑
fu)、米托蒽醌、微管菌素、吲哚啉苯并二氮杂az13599185、念珠藻素、根瘤菌素、甲氨蝶呤、蒽环类药物、喜树碱类似物、sn
‑
38、dx
‑
8951f、甲磺酸依喜替康、截短形式的铜绿假单胞菌外毒素(pe38)、倍癌霉素衍生物、鹅膏蕈碱、α
‑
鹅膏蕈碱、斯考他汀、泰兰斯他汀、奥佐米星、特司林、amberstatin269和soravtansine或它们的衍生物。
[0629]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中第七蛋白质分子包含以下中的任一种或由以下中的任一种组成:吉妥珠单抗奥佐米星、本妥昔单抗维多汀、曲妥珠单抗美坦新、英妥珠单抗奥佐米星、moxetumomab pasudotox和泊洛妥珠单抗维多汀以及表a2和表a3的抗体
‑
药物缀合物,或其至少一个肿瘤细胞受体结合结构域和/或至少一个肿瘤细胞受体结合片段,其中所述结构域或片段包含效应子部分并且优选是肿瘤细胞特异性受体结合结构域和/或肿瘤细胞特异性受体结合片段。
[0630]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六蛋白质分子,其中第六蛋白质分子包含多于一种共价结合的皂苷,优选2、3、4、5、 6、8、10、16、32、64、128或1
‑
100个皂苷,或者其间的任何数量的皂苷,如7、9、12个皂苷。
[0631]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合或用于根据本发明的用途的第六蛋白质分子,其中多于一种共价结合的皂苷直接共价结合第六蛋白质分子的氨基酸残基,优选半胱氨酸和/或赖氨酸,和/或经由至少一个接头和/或经由至少一个可裂解接头和/或经由至少一个寡聚支架或多聚支架,优选1
‑ꢀ
8个此类支架或2
‑
4个此类支架共价结合,其中至少一个支架任选地基于树枝化基元,其中1
‑
32个皂苷,优选2、3、4、5、6、8、10、16、 32个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷共价结合至少一个支架。
[0632]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中可裂解接头在酸性条件、还原条件、酶促条件或光诱导条件下进行裂解,并且优选地,可裂解接头包含选自以下的可裂解键:在酸性条件下进行裂解的腙键和酰肼键和/或易于蛋白质水解(例如由组织蛋白酶b进行蛋白质水解)的键和/或易于在还原条件下裂解的键,如二硫键。
[0633]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的治疗性组合,其中可裂解接头在如存在于哺乳动物细胞,优选人类细胞的内体和/或溶酶体的酸性条件下,优选在ph 4.0
‑
6.5下并且更优选在ph≤5.5下在体内进行裂解。
[0634]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中寡聚支架或多聚支架包含多聚结构或寡聚结构并且包含至少一个化学基团,所述至少一个化学基团用于支架与所述第六蛋白质分子的氨基酸残基的共价偶联。
[0635]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中至少一个皂苷经由根据本发明的可裂解接头共价结合支架的多聚结构或寡聚结构。
[0636]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中至少一个皂苷经由亚胺键、腙键、酰肼键、肟键、1,3
‑
二噁戊烷键、二硫键、硫醚键、酰胺键、肽键或酯键中的任何一种或多种,优选经由至少一个接头共价结合支架的多聚结构或寡聚结构。
[0637]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中至少一个皂苷包含在c
‑
23位的醛官能团和任选地在至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团,所述醛官能团参与共价键合至支架的多聚结构或寡聚结构,和/或,如果存在,葡糖醛酸官能团参与共价键合至支架的多聚结构或寡聚结构(经由直接结合或经由至少一个接头)。
[0638]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中至少一个皂苷的c
‑
23位的醛官能团共价偶联至接头n
‑
ε
‑
马来酰亚胺基己酸酰肼,所述接头经由硫醚键共价偶联至支架的多聚结构或寡聚结构中的巯基基团,如半胱氨酸的巯基基团。
[0639]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治
疗性组合,其中至少一个皂苷的c
‑
3β
‑
oh基团处的碳水化合物取代基中的葡糖醛酸官能团共价偶联至接头1
‑
[双(二甲基氨基) 亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐,所述接头经由酰胺键共价偶联至支架的多聚结构或寡聚结构中的胺基团,如蛋白质分子的赖氨酸或n
‑
末端的胺基团。
[0640]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中多聚支架或寡聚支架的用于支架与第六蛋白质分子的氨基酸残基的共价偶联的化学基团是点击化学基团,其优选选自四嗪、叠氮化物、烯烃或炔烃或者这些基团的环状衍生物,更优选地,点击化学基团是叠氮化物。
[0641]
本发明的第六系列方面和实施方案中的一个实施方案是用于本发明的用途的治疗性组合,其中支架的多聚结构或寡聚结构包括直链、支链和/或环状多聚物、寡聚物、树状物、树枝化基元、树枝化基元化多聚物、树枝化基元化寡聚物、dna、多肽、聚赖氨酸、聚乙二醇或者这些多聚结构或寡聚结构的组装体,所述组装体优选通过共价交联构建。
[0642]
本发明的第六系列方面和实施方案中的本发明的一方面涉及用于本发明的用途的治疗性组合,其中第六药物组合物和第七药物组合物施用至有需要的患者。
[0643]
本发明的第六系列方面和实施方案中的本发明的一方面涉及本发明的第六药物组合物,其还包含本发明的第七蛋白质分子。
[0644]
本发明的第六系列方面和实施方案中的本发明的一方面涉及用作药物的本发明的第六药物组合物,其还包含本发明的第七蛋白质分子。
[0645]
本发明的第六系列方面和实施方案中的本发明的一方面涉及用于治疗或预防有需要的患者中癌症的本发明的第六药物组合物,其还包含本发明的第七蛋白质分子。
[0646]
当然,a、b、c、d、e、f、g、h、i、j、k、m、n、p、q、r、s、t、 u、v、w和/或x中的任一个和全部对于根据本发明的任何和全部上述方面和实施方案,具有根据本发明的每个单独实施方案和方面的值。此外,(三官能)接头l1、l2、l4、l5、l6、l8、l9和/或l10,如果存在于本发明的分子或缀合物或部分中,则为本发明的上述方面和实施方案中的每一个和任一个所示的(三官能)接头,如技术人员容易理解的。寡聚支架或多聚支架l3和/或l7,如果存在于本发明的分子或缀合物或部分中,则为本发明的上述方面和实施方案中的每一个和任一个所示的寡聚支架或多聚支架,也如技术人员容易理解的。此外,第一配体a1和第一效应子部分b1(如果存在),以及第二配体a2和第二效应子部分b2(如果存在),以及第一效应子部分a1和第一配体 b1(如果存在),以及第二效应子部分a2和第二配体b2(如果存在)是选定和指定的配体和效应子部分,如针对本发明的第一、第二、第三、第四、第五和第六系列的实施方案和方面以及上文列出的本发明的所有另外的实施方案和方面所公开的。皂苷c是本发明的上述方面和实施方案的任一个中提及和列出的皂苷中的任何一种或多种,特别是选自方案i和/或表a1的一种或多种皂苷。
[0647]
一个实施方案是本发明的第一、第四或第六蛋白质分子,其包含含有方案i中的结构a的皂苷的一个或几个或所有指定结构特征的皂苷,结构a的皂苷被称为当内体逃逸增强活性针对存在于与第一蛋白质分子接触的细胞的内体中的效应子部分时具有
‘
理想’结构的皂苷,和/或选自方案i的另外皂苷中的任何一种或多种的皂苷:
[0648]
方案i
[0649][0650]
方案i(续)
[0651][0652]
方案i(续)
[0653]
[0654][0655]
方案i(续)
[0656][0657]
根据本发明,具有用于增强结合至本发明的第二或第三或第五或第七蛋白质分子的效应分子的内体逃逸的目的的
‘
理想’结构的结合至本发明的第一、第四和/或第六蛋白质分子的糖苷(如根据本发明的皂苷c)是具有至少1.500道尔顿的分子质量并且包含在c
‑
23位含有醛基团并且在c
‑
16位任选地含有羟基基团以及在c
‑
3位具有第一支链碳水化合物侧链的齐墩果烷型三萜的根据方案i的结构a的双糖链皂苷,所述第一支链碳水化合物侧链任选地含有葡糖醛酸,其中皂苷含有在c
‑
28位具有第二支链碳水化合物侧链的酯基团,所述第二支链碳水化合物链优选包含至少四个碳水化合物单元,任选地含有至少一个乙酰基残基如两个乙酰基残基和/或任选地包含脱氧碳水化合物和/或任选地包含奎诺糖和/或任
选地包含葡萄糖和/或任选地包含4
‑
甲氧基肉桂酸和/或任选地包含5
‑
o
‑
[5
‑
o
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑ꢀ
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸和/或任选地包含经由酯键与碳水化合物结合的5
‑
o
‑
[5
‑
o
‑
rha
‑
(1
→
2)
‑
ara/api
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酰基]
‑
3,5
‑
二羟基
‑6‑
甲基
‑
辛酸。
[0658]
so1861不同于方案i,结构a中显示的“理想结构”,仅在奎诺糖处具有仅一个乙酰基残基并且具有另外的木糖。用于增强效应分子或效应子部分或有效载荷的内体逃逸的皂苷的“理想结构”是优选具有方案i中的结构a的皂苷,以及表现出内体逃逸增强活性的皂苷具有方案i的结构a中展现的一个或多个结构特征。不希望受任何理论的束缚,发明人相信方案i的结构a表示用于内体逃逸增强活性的“理想皂苷”(并且不是最低要求皂苷),这意味着并非所有的结构(化学基团)可以或必须存在于具有至少足够的内体逃逸增强活性以促进效应子部分在胞质中累积的每个皂苷中,并且意味着一些皂苷可具有其它结构元件如乙酰基链,和/或对于表现出内体逃逸增强活性的其它皂苷,糖可以不同于方案i中表现出的糖。例如,当考虑方案i中的结构a的理想结构:例如在qs
‑
21中存在乙酰基链时,qs
‑
21皂苷和皂皮树水溶性级分中的一些皂苷(皂皮树皂苷;quil
‑
a)在c
‑
28处的碳水化合物修饰方面不同。在皂皮树的水溶性级分中,皂苷,如qs
‑
7、qs1862类似于理想结构a,并且类似于so1861。
[0659]
一个实施方案是本发明的第一、第四或第六蛋白质分子,其包含根据方案ii的寡聚三官能接头作为支架核心结构:
[0660][0661]
其中皂苷经由不稳定的、可裂解的腙接头(酸敏感)和/或经由包含马来酰亚胺的键共价结合三官能接头支架l9和/或l10,然而支架与结合位点如抗体的结合是经由不稳定的、可裂解的腙接头(酸敏感)和/ 或经由包含马来酰亚胺的键与结合位点中的半胱氨酸
(如1、2、3或4 个半胱氨酸)建立的,从而形成结构b:
[0662][0663]
使得1
‑
4个支架共价结合单个例如抗体,如单克隆抗体。根据本发明,结构b中两种显示的皂苷中的一个也可以被共价偶联的效应子部分、效应分子、有效载荷替代或者两个皂苷都不存在偶联至三官能接头的两个共价偶联的效应子部分、效应分子、有效载荷。结合位点例如是抗体或其结合片段或结合结构域。
[0664]
发明人确定抗体药物缀合物的治疗窗,如本发明的第二或第三或第五或第七药物组合物中的第二和第三和第五和第七蛋白质分子,当施用至还施用了第一、第四或第六药物组合物的荷瘤哺乳动物(小鼠) 时,分别增加。第一、第四或第六蛋白质蛋白具有至少一种糖苷,如优选共价,更优选经由可裂解接头与其结合的皂苷c。皂苷c可能通过增强效应子部分内体逃逸进入胞质(在那期望效应子部分的活性),增强了结合至第二和第三和第五和第七蛋白质分子的效应子部分a1、 b1、a2或b2的治疗功效。这样,已经在比adc的常规剂量更低的剂量,即第二或第三或第五或第七蛋白质分子,在靶向细胞附近、处和/或内部包含皂苷c的第一、第四或第六蛋白质分子的存在的影响下确立治疗效果。靶向细胞是例如患病细胞,如肿瘤细胞或自身免疫细胞或b细胞疾病相关的b细胞等。效应子部分a1、b1、a2或b2是例如作为adc的一部分的毒素,或作为根据本发明的aoc的一部分的寡核苷酸如bna。
[0665]
通过用第一和第二、或第四和第五、或第六和第七蛋白质分子靶向(两个)不同的细胞表面分子,皂苷c和效应分子a1、b1、a2或b2 在细胞表面上暴露(两者不同)细胞表面分子的非常相同的靶向细胞的胞质处和内部的递送,与将此类细胞仅暴露于第二、第三、第五或第七蛋白质分子如不存在细胞靶向的皂苷c的adc或aoc(第一、第四或第六蛋白质分子)相比时,得到改善并且更有特异性。当考虑患者中的(相邻)健康细胞时,经选择由第一、第四或第六蛋白质分子的结合位点和由第二、第三、第五或第七蛋白质分子的结合位点单独靶向的异常细胞(其中结合位点是不同的并且其中第一和第二蛋白质分子结合的表位是不同的并且位于不同种类和类型的细胞表面分子如两个不同的受体中/上)以较高程度(即与
非靶向细胞,如例如健康细胞上的表达相比,靶向细胞(如例如肿瘤细胞或自身免疫细胞)上两种独特且不同的细胞表面分子的相对更高表达)理想地携带分别在第一细胞表面分子和第二细胞表面分子上的第一表位和第二表位,和/或特异性地暴露第一和第二细胞表面分子。优选地,与不旨在用本发明的分子和缀合物靶向的健康细胞相比,第一和第二结合位点所靶向的两个细胞表面分子在靶向(患病,肿瘤)细胞上相对高度和/或特异性表达。一个实施方案是药物组合,其中当与健康(邻近)细胞表面上的第一(第四、第六) 细胞表面分子和/或第二(第五、第七)细胞表面分子的表达比较时,第一(第四、第六)和第二(第五、第七)结合位点中的至少一个并且因此第一(第四、第六)和第二(第五、第七)细胞表面分子如第一(第四、第六) 和第二(第五、第七)肿瘤细胞受体中的至少一个特异性表达或以相对更高程度表达。因此,在靶向细胞表面分子上的第一(第四、第六)表位或第二(第五、第七)表位,优选第一(第四、第六)表位和第二(第五、第七)表位对于靶向的患病细胞是理想独特的,并且在靶向细胞的表面至少特异性地存在和暴露。第一(第四、第六)和第二(第五、第七)蛋白质分子与靶向细胞上的其相应的第一(第四、第六)和第二(第五、第七)表位的结合之后是第一(第四、第六)蛋白质分子和第一(第四、第六)靶细胞表面分子以及第二(第五、第七)蛋白质分子和第二(第五、第七)靶细胞表面分子的复合物的内吞作用。由于与不应被靶向的健康细胞相比,第一和第二蛋白质分子必须通过与均以足够的程度表达或在靶向细胞上独特表达的两种不同的细胞表面分子的结合相互作用进入相同的靶细胞,因此治疗活性量的第一和第二蛋白质分子在靶细胞内的累积只有两种不同靶向细胞表面分子的表达水平均高于某个最小表达阈值时,才是可能的并且发生。同时,当第一和第二蛋白质分子均能够通过结合足够暴露和表达的第一和第二细胞表面分子以足够的量而进入靶细胞时,与第二(第五、第七)蛋白质分子结合的效应子部分仅能够在携带共价结合的皂苷的第一(第四、第六)蛋白质分子的存在下发挥其细胞内(如细胞毒性或基因沉默)活性的事实,还提供了当第一和第二细胞表面分子中的至少一个的表达在健康细胞中足够低时并且优选地当第一和第二靶向细胞表面分子两者的表达在健康细胞中足够低时,针对例如不意在由效应子部分靶向和影响的健康细胞和健康组织的负面和不期望的副作用的保护措施。也就是说,对于第一和第二细胞表面分子以及至少第一细胞表面分子或第二细胞表面分子(分别由第一和第二蛋白质分子的第一和第二结合位点结合的)来说,足够低表达的或甚至不存在暴露的第一和第二细胞表面分子,在理想情况下不允许第一和第二蛋白质分子两者以将在结合第一蛋白质分子的皂苷的影响下共同导致效应子部分的内体逃逸的量进入(非靶向的)健康细胞。由于与未将第一蛋白质分子添加到治疗方案时相比adc或aoc可以以更低的剂量使用,因此例如当考虑靶向和杀伤靶标患病细胞如肿瘤细胞和自身免疫细胞时,adc或aoc以较低程度进入健康细胞已经具有较低的发生不需要的副作用的风险。
[0666]
同步是成功的小鼠递送策略与其在人类中的应用之间缺失的环节。事实上,发明人在一系列体内小鼠肿瘤模型中确定:与未用adc和游离皂苷处理的对照动物相比,分别施用至小鼠一定剂量的游离皂苷和一定剂量的adc(根据本发明的第二或第三或第五或第七蛋白质分子) 没有导致任何期望的抗肿瘤活性,如延迟肿瘤生长、肿瘤消退、减小和减慢肿瘤生长。与施用adc的时刻(在施用adc之前、期间和之后施用游离皂苷)相比,使用各种施用途径和使用各个施用游离皂苷的时间点来施用游离皂苷。体内肿瘤模型中测试的adc为西妥昔单抗
‑
石竹素(含游离so1861)或曲妥珠单抗
‑
皂草素(含游离so1861)。改变游离皂苷的
剂量并不提供有效的抗肿瘤活性。所提及的adc以本身不会对荷瘤动物产生任何有益的抗肿瘤作用的剂量施用。令人惊讶的是,发明人现在确定,在各种体外基于哺乳动物细胞的生物测定和/或各种体内动物肿瘤模型中的有益抗肿瘤活性可以通过用任选包含根据本发明的支架的根据本发明的缀合物(即本发明的第一和第二或第一和第三或第四和第五或第六和第七蛋白质分子的组合)处理动物来实现。例如,支架是具有经由可裂解或不可裂解的连键共价结合的皂苷(如 so1861、qs
‑
21)和/或具有经由可裂解或不可裂解的键共价结合的效应子部分(如石竹素,沉默bna(hsp27)和/或具有共价结合的单克隆抗体 (如西妥昔单抗、曲妥珠单抗、okt
‑
9)的三官能接头,或者支架是树枝化基元,如例如四个部分能够结合的树枝化基元(如四个皂苷分子),或用于例如结合两个皂苷和两个效应分子的树枝化基元,所述树枝化基元包含用于(共价)偶联至配体或者抗体或者其片段或结构域的化学基团。参考实施例部分,例示了根据本发明的各种这些支架,显示了当考虑由如蛋白质毒素施加的细胞毒性时或当考虑肿瘤细胞中基因沉默时的体内和/或体外抗肿瘤细胞活性。
[0667]
不希望受任何理论的束缚,鉴于在考虑用adc连同游离皂苷一起处理荷瘤动物时观察到的失败,优选使两者(即至少一个皂苷和效应子部分)在靶细胞(如肿瘤细胞或自身免疫细胞)的内吞途径的区室或囊泡中的存在同步。根据发明人的尝试,在具有adc(和/或aoc)和游离皂苷的情况下,同步晚期内体中分子的存在以在体内获得协同效应并不是有益可获得的。在一方面,本发明优选地解决了至少就组合第二、第三、第五或第七蛋白质分子所包含的效应子部分与第一、第四或第六蛋白质分子所包含的皂苷而言的以下问题:不希望受任何理论的束缚,例如,可用于(共价),特别是单一和可裂解的可保留偶联的皂苷内的唯一合理的化学基团是内体逃逸活性所需的。已知的限制最可能是皂苷在除了皂苷在疫苗接种方案中的应用之外的临床研究中未曾与药学活性物质组合使用的原因,其中暗示了使用免疫增强佐剂物质,尽管例如列于表a1和方案i中的皂苷的显著的内体逃逸增强剂效应已知10多年了。例如,提供具有共价缀合的支架的本发明的第一、第四或第六蛋白质分子至少部分地解决了这些困难。令人惊讶的是,先前在涉及皂苷作为佐剂组分的疫苗接种环境中应用其免疫增强活性的皂苷,现在也适用于(共价)偶联至本发明的第一、第四或第六蛋白质分子,用于体外和体内抗肿瘤活性。
[0668]
可用于本发明中的效应子部分优选依赖于晚期内体逃逸以发挥其作用。一些效应子,诸如例如假单胞菌外毒素,在“晚期内体阶段”之前被改道到(rerouted)其它细胞器,因此通常不会受益于与根据本发明的第二蛋白质分子的偶联。然而,此类毒素可以适合与本发明一起使用,如通过缺失负责改道的信号肽。特别地,具有高度毒性并且仅需要一个分子即可逃逸出内体以杀伤细胞的毒素可能会被修饰为效力较低。如果至少2个、更优选至少5个、更优选至少10个、更优选至少 20个、更优选至少50个、最优选至少100个毒素分子逃逸出内体,则优选使用杀伤细胞的毒素。进一步优选的是,本发明的第二蛋白质分子包含共价缀合的官能化支架,即包含用于在靶细胞(如肿瘤细胞或自身免疫细胞)处靶向包含结合的效应子部分的支架的共价结合的效应子部分的支架。此外,为了降低脱靶毒性,细胞膜不可渗透性小分子毒素是优于细胞膜可渗透性毒素的优选效应分子。
[0669]
如在本发明中使用的术语“配体”具有其普通含义,并且优选地意指能够结合靶细胞细胞表面上的另一分子或结构的分子或结构,其中细胞表面上的所述分子或结构可被内吞并且优选在脱靶细胞上不存在或不那么突出。优选地,细胞表面上的所述分子或结构被
组成性内吞。更优选地,本发明中的配体在与所述分子或结构结合后诱导靶细胞的细胞表面上所述分子或结构的内吞作用。这是例如存在于多种癌细胞表面上的表皮生长因子受体(egfr)的情况。组成型内吞的靶细胞的细胞表面上的分子或结构的实例是例如密封蛋白
‑
1或主要组织相容性复合体ii类糖蛋白。配体可以是如抗体、生长因子或细胞因子。将毒素与配体组合到载体分子中是一种产生靶向毒素的可能性。只在靶细胞中有毒的毒素(因为它只干扰靶细胞中发生的过程),也可以被看作是靶向毒素(因为在脱靶细胞中它不能发挥其毒性作用,如凋亡蛋白)。优选地,靶向毒素是与配体或如单克隆抗体组合的毒素,以便在靶细胞中有活性而在脱靶细胞没有活性(因为它只与靶细胞结合并被靶细胞内吞)。在包含含有配体和效应子部分的载体分子的官能化支架(即第二或第三蛋白质分子)中,配体或单克隆抗体将效应子部分和支架引导至靶细胞。内化后,至少一种糖苷,优选第一蛋白质分子和皂苷的缀合物所包含的皂苷,介导效应子部分的内体逃逸。皂苷通常是表a1和方案i中列出的皂苷,并且优选地,所述皂苷是so1861和/或qs
‑
21,和/或sa1641和/或ge1741。
[0670]
发明人确定了免疫球蛋白、其结构域、配体等特别适合应用为包含(第一)结合位点的第一、第四或第六蛋白质分子的(第一)结合位点 (和第三蛋白质分子的相同结合位点)。类似地,发明人确定了免疫球蛋白、其结构域、配体等特别适合应用为包含第二、第五或第七结合位点的第二、第五或第七蛋白质分子的第二、第五或第七结合位点。例如,抗体和抗体的结合结构域适用于靶向选定细胞表面分子的暴露表面上的表位,导致第一和第三和第四和第六(以及分别是第二、第五和第七)蛋白质分子靶向表达由第一和第三和第四和第六蛋白质分子靶向的细胞表面分子的靶细胞,和/或还有表达由第二或第五或第七蛋白质分子靶向的第二或第五或第七细胞表面分子的靶细胞,这些细胞还表达第一和第三和第四和第六细胞表面分子(其是相同的细胞表面分子)并在其细胞表面上具有所述细胞表面分子。类似地,靶向靶细胞上的egfr的配体如egf适合应用为第一和第三和第四和第六蛋白质分子中的结合位点,或应用为第二或第五或第七蛋白质分子中的第二或第五或第七结合位点,条件是第二、第五和第七结合位点不同于第一和第三和第四和第六结合位点,所述第一和第三结合位点是相同的。优选的是针对第一和第三、第四、第六表位或针对第二、第五、第七表位的结合位点,其对于第一和第三、第四、第六蛋白质分子与第一、第四、第六细胞表面分子的结合和/或对于第二、第五、第七蛋白质分子与第二、第五、第七细胞表面分子的结合是具有特异性的,所述第一(第三、第四、第六)和第二(第五、第七)细胞表面分子暴露于非常相同的靶细胞上。基于抗体或其结构域或结合片段的结合位点例如提供了针对在用于靶向如患病细胞、肿瘤细胞、自身免疫细胞等的的选定细胞的选定的第一或第二或第三(与第一相同)、第四、第五、第六、第七细胞表面分子上的选定的第一、第二、第三、第四、第五、第六、第七表位的此类期望的特异性。因此,对于第一和第二和第三和第四和第五和第六和第七蛋白质分子,基于抗体或结合分子(片段、结构域) 的第一、第二和第三和第四、第五、第六和第七结合位点是优选的。
[0671]
通过用第一和第三和第四和第五蛋白质分子靶向相同的细胞表面分子,皂苷c和效应子部分a1、b1、a2或b2在非常相同的靶向细胞的胞质处和内部的递送得到了改善并且更有特异性。当考虑患者中的(相邻)健康细胞时,选定由第一和第三或第四和第五蛋白质分子的结合位点靶向的异常细胞高度理想地携带细胞表面分子和/或特异性地携带细胞表面分子。因此,靶向细胞表面分子上的表位对于靶向患病细胞是理想地独特的,并且是至少
特异性地存在并暴露于靶向细胞的表面处。第一和第三或第四和第五蛋白质分子的结合之后是第一蛋白质分子和靶细胞表面分子及第三蛋白质分子和靶细胞表面分子的复合物或者第四蛋白质分子和靶细胞表面分子及第五蛋白质分子和靶细胞表面分子的复合物的内吞作用。由于第一和第三或第四和第五蛋白质分子必须通过与非常相同的细胞表面分子的结合相互作用进入相同的靶细胞,因此治疗有效量的第一和第三、或第四和第五蛋白质分子在靶分子内的累积只有在靶向细胞表面分子的表达水平高于某个最小表达阈值时才有可能并且发生。同时,当第一和第三蛋白质分子两者、或第四和第五蛋白质分子两者能够通过结合足够暴露和表达的细胞表面分子以足够的量进入靶细胞时,与第三和第五蛋白质分子结合的效应子部分仅能够在携带共价结合的皂苷的第一或第四蛋白质分子的存在下发挥其细胞内(如细胞毒性或基因沉默)活性的事实,还提供了当靶向细胞表面分子的表达在健康细胞中足够低时,针对例如不意在由效应子部分靶向和影响的健康细胞和健康组织的负面和不期望的副作用的保护措施。也就是说,由第一和第三、或第四和第五蛋白质分子的结合位点结合的细胞表面分子的低表达不允许第一和第三蛋白质分子两者、或第四和第五蛋白质分子两者以将在结合第一和第四蛋白质分子的皂苷的影响下共同导致效应子部分的内体逃逸的量进入。由于与未将第一或第四蛋白质分子添加到治疗方案时相比adc或aoc可以以更低的剂量使用,因此例如当考虑靶向和杀伤靶标患病细胞如肿瘤细胞和自身免疫细胞时,adc或aoc以较低程度进入健康细胞已经具有较低的发生不需要的副作用的风险。
[0672]
在整个说明书和权利要求书(整个申请)中,术语
‘
第一’和
‘
第三’在考虑第一和第三表位、第一和第三结合位点、第一和第三细胞表面分子时具有相同的含义。也就是说,对于第一和第三蛋白质分子,靶向表位是相同的,结合位点是相同的,靶向的细胞表面分子如肿瘤细胞 (特异性)受体是相同的。对于术语
‘
第四’和
‘
第五’是相同的。
[0673]
表a2、a3和a4列出了包含针对第一(第三、第四和第六)蛋白质分子的第一(第三、第四和第六)结合位点的第一(第三、第四和第六)表位的第一、第三、第四和第六细胞表面分子的优选实例。此外,表a2、 a3和a4还列出了包含针对第二、第五、第七蛋白质分子的第二、第五和第七结合位点的第二、第五和第七表位的第二、第五和第七细胞表面分子的优选实例。当第一、第三、第四、第六和/或第二、第五、第七细胞表面分子,优选第一、第三、第四和第六和第二、第五、第七细胞表面分子,在靶细胞上特异性表达时,以及当第一结合位点和/ 或第二结合位点能够分别结合的分别的第一和第二细胞表面分子上的第一和第二表位特异性存在于第一和/或第二细胞表面分子中时,促进了第一、第三和/或第二蛋白质分子特异性靶向相同的期望靶细胞(如暴露第一和第二肿瘤细胞表面分子的肿瘤细胞),然而与靶向(异常)细胞上的细胞表面分子的表达相比,不表达第一和/或第二细胞表面分子或者的确以较低程度表达第一和/或第二细胞表面分子,优选不表达第一和第二细胞表面分子或者的确以较低程度表达第一和第二细胞表面分子的其它细胞(如健康细胞)不由第一、第三和第二蛋白质分子靶向,或以较低程度被靶向。
[0674]
本发明中的药学活性物质是用于在有机体内,优选脊椎动物,更优选人类如癌症患者或自身免疫患者中实现有益结果的效应子部分。益处包括疾病和/或症状的诊断、预后、治疗、治愈和/或预防。药学活性物质还可导致不期望的有害副作用。在这种情况下,必须权衡利弊,以决定药学活性物质是否适用于特定情况。如果细胞内的药学活性物质的效应主要是对整个有机体有益,则该细胞被称为靶细胞。如果细胞内的效应主要对整个有机
体有害,则该细胞被称为脱靶细胞。在人工系统如细胞培养和生物反应器中,靶细胞和脱靶细胞取决于目的并且由用户定义。
[0675]
作为多肽的效应子部分可以是如恢复失去的功能,诸如例如酶替代、基因调控功能的多肽,或毒素。
[0676]
一个实施方案是本发明的第一、第四或第六蛋白质分子,其中第一、第四或第六蛋白质分子包含直接共价结合第一、第四或第六蛋白质分子的氨基酸残基,优选半胱氨酸和/或赖氨酸,和/或经由至少一个接头和/或经由至少一个可裂解接头和/或经由至少一个多聚支架或寡聚支架l3、l7,优选1
‑
8个此类支架或2
‑
4个此类支架共价结合的多于一种的皂苷c,优选2、3、4、5、6、8、10、16、32、64或1
‑
100 个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷,其中至少一个支架任选地基于树枝化基元,其中1
‑
32个皂苷,如2、3、4、5、6、 8、10、16、32个皂苷或者其间任何数量的皂苷,如7、9、12个皂苷共价结合至少一个支架。
[0677]
表a1和方案i以及上述实施方案总结了一系列皂苷c,其在以游离形式与第二分子(如,效应子部分或效应分子,有效载荷,如毒素,寡核苷酸)一起与哺乳动物细胞,特别是人类肿瘤细胞接触时,鉴定了其内体逃逸增强活性。事实上,在使用人类肿瘤细胞的基于细胞的生物测定中,对于表a1中制表的皂苷及本文所述的本发明的方案i和各个实施方案中的那些皂苷,确定了在这些皂苷的影响下,当结合第一、第四或第六蛋白质分子时,结合第二或第三或第五或第七蛋白质分子的第二分子(效应子部分),如核酸和/或毒素,如蛋白质毒素(如表 a5中列出的一种或多种蛋白质毒素)被以增加的效率和/或功效递送至胞质中,推测是通过从(晚期)内体和溶酶体的胞内释放进行的。也就是说,此类第二分子(与本发明的第二或第三或第五或第七蛋白质分子结合的效应子部分)如核酸和/或毒素的内体和/或溶酶体逃逸在没有皂苷的情况下效率更低。
[0678]
令人惊讶的是,发明人现在证明了来自皂皮树的水溶性皂苷级分 (包括qs
‑
21及其家族成员qs
‑
21a、qs
‑
21a
‑
api、qs
‑
21a
‑
xyl、qs
‑ꢀ
21b、qs
‑
21b
‑
api、qs
‑
21b
‑
xyl、qs
‑7‑
xyl、qs
‑7‑
api、qs
‑
17
‑
api、qs
‑ꢀ
17
‑
xyl、qs1861、qs1862、qs
‑
18和quil
‑
a)当以包含单克隆抗体(本发明的第一、第四、第六蛋白质分子)连同包含效应子部分的第二和/或第三和/或第五和/或第七蛋白质分子(上述第二和/或第三和/或第五和/或第七蛋白质分子)和由作为共价缀合物的第一、第四或第六蛋白质分子所包含的至少一种糖苷(如由此类qs
‑
21制剂(如,皂皮树的水溶性级分)涵盖的qs
‑
21及其家族成员皂苷)的共价缀合物的形式施用至哺乳动物物种(人类)的肿瘤细胞时,也表现出增强如结合至单克隆抗体的核酸或结合至单克隆抗体的蛋白质毒素(包含共价结合的寡核苷酸或有效载荷如(蛋白质)毒素的本发明的第二和/或第三和/或第五和/或第七蛋白质分子的实例)的体外生物效应的能力,其中效应分子和糖苷 (如皂皮树的皂苷级分、qs
‑
21、so1861、sa1641、ge1741)直接或经由接头或经由多聚支架或寡聚支架、直接或经由至少一个接头共价结合例如蛋白质分子。不希望受任何理论的束缚,在体外源自皂皮树的皂苷存在下,观察到的例如反义bna介导的肿瘤细胞hsp27表达减少(hsp27基因沉默)的刺激或增强可能(也)与皂苷激活肿瘤细胞中的炎性体有关,例如导致肿瘤细胞焦亡。发明人确定缀合至例如反义 bna或石竹素或皂草素的第二和第三和第五和第七蛋白质分子当在生物基细胞测定中与细胞接触时,当在包含皂苷并且靶向与由第二和 /或第三和/或第五和/或第七蛋白质分子靶向的细胞表面分子相同的 (肿瘤)细胞的本发明的第一或第四或第六蛋白质分子的存在下时,在体外完全发挥任何抗
肿瘤细胞活性或改善抗肿瘤细胞活性,然而在第一、第四、第六蛋白质分子的不存在下并且因此在皂苷的不存在下,未观察到对肿瘤细胞的此类活性。
[0679]
qs
‑
21,以及还有来自皂皮树的包含qs
‑
21的水溶性皂苷级分早已为人所知,并且先前因其增强免疫的能力而被广泛应用,如作为如亚单位疫苗中的佐剂。例如,qs
‑
21应用于人类患者的两种iii期临床试验中,所述患者疫苗接种了与包含qs
‑
21的佐剂混合的亚单位疫苗 (glaxo
‑
smith
‑
kline,magrit试验,derma研究),其中亚单位是由肿瘤细胞特异性表达并呈递的mage
‑
a3蛋白。qs
‑
21增强的抗肿瘤疫苗接种旨在延长癌症患者(黑色素瘤;非小细胞肺癌)的无病存活期。此外,qs
‑
21已在临床试验中作为佐剂进行测试,用于使用glaxo
‑ꢀ
smith
‑
kline的包含qs
‑
21的佐剂as01和as02,开发抗癌疫苗治疗、用于hiv
‑
1感染的疫苗、开发针对乙型肝炎的疫苗以及用于抗疟疾疫苗开发。先前的研究揭示了,在包含qs
‑
21皂苷的佐剂(as15;gsk)的影响下,针对在癌细胞表面呈递的mage
‑
a3肽引发的免疫应答。令发明人惊讶的是,皂皮树的皂苷级分以及因此可能是qs
‑
21(作为皂皮树的水溶性皂苷级分的一部分)增强了如结合至第二、第五、第七蛋白质分子(如配体egf)的有效载荷(如蛋白质毒素(石竹素))的抗肿瘤细胞活性。
[0680]
发明人显示,与对照相比以及与仅aoc(第三和第五蛋白质分子)(不存在具有偶联皂苷的第一或第四蛋白质分子)相比,提供有共价偶联的反义bna(如bna(hsp27))并且连同具有共价偶联的皂苷(如 so1861、qs
‑
21)的本发明的第一或第四蛋白质分子一起与肿瘤细胞接触的肿瘤细胞靶向单克隆抗体(bna和皂苷两者经由可裂解键偶联至第一和第三或第四和第五蛋白质分子的相应抗体(如西妥昔单抗)能够在肿瘤中在体内沉默hsp27。adc或抗体
‑
寡核苷酸缀合物(aoc)(如抗体
‑
bna缀合物)与具有皂苷的第一或第四蛋白质分子的共施用因此赋予adc或aoc抗肿瘤细胞活性,这在相同剂量下的仅adc或仅 aoc未观察到。值得注意的是,与对照组(仅施用媒介物)相比,aoc(第二或第三或第五蛋白质分子)和具有共价偶联的皂苷的单克隆抗体(第一或第四蛋白质分子)在施用至分别在不同小鼠组中的荷瘤小鼠时增加了肿瘤细胞中的hsp27表达。当与对照相比时,仅包含本发明的效应子部分的aoc(第二或第三或第五蛋白质分子)和具有共价偶联的皂苷的第一或第四蛋白的共施用表现出降低的hsp27表达。反义bna(hsp27)是根据zhang等人(2011)的具有寡聚核酸序列5
’‑ꢀ
ggcacagccagtggcg
‑3’
的bna[y zhang,z qu,s kim,v shi,b liao1,p kraft,r bandaru,y wu,lm greenberger and id horak,down
‑
modulationof cancer targets using locked nucleic acid(lna)
‑
based antisenseoligonucleotides without transfection,gene therapy(2011)18,326
‑
333]。值得注意的是,据发明人所知,bna被设计作为游离核酸应用。发明人现在是第一个证明反义bna可以通过(不
‑
)可裂解接头与配体或抗体以一定方式共价偶联,使得在体外保留了基因沉默活性并且更重要的是在体内保留在荷瘤动物的肿瘤细胞中。这种提供基于bna的aoc 的方法开辟了向有需要的人类(癌症)患者施用靶向bna的新方法。
[0681]
发明人在本文公开了,诸如经由三官能接头(如方案ii和结构b的三官能接头)或经由包含共价结合的皂苷的支架的寡聚结构或多聚结构将皂苷(如皂皮树的水溶性级分中的皂苷、qs
‑
21、sa1641、so1861、表a1、方案i)共价偶联至第一或第四或第六蛋白质分子,导致在第一、第四或第六蛋白质分子中共价偶联的皂苷的影响下由第二和/或第三和/或第五和/或第七蛋白质分子所包含的效应子部分如毒素所发挥的细胞毒性得到改善。
[0682]
根据本发明,通常皂苷是表a1,方案i中列出的皂苷。已经证明皂苷的活性是有益的,如考虑当共价偶联至第二或第三或第五或第七蛋白质分子的效应子部分进入细胞中并累积在胞质内时,当皂苷共价偶联至涉及腙键和/或酰肼键和/或二硫键的第一或第四或第六蛋白质分子时,细胞内的内体逃逸增强活性。此类键类型在哺乳动物细胞(如人类细胞)的(晚期)内体和溶酶体内的酸性条件下和/或在还原条件下很容易裂解。可替代地,发明人还证明皂苷经由在细胞(如(晚期)内体、溶酶体、胞质)内的生理条件下不容易裂解的键共价偶联至第一、第四或第六蛋白质分子还有益于皂苷对例如效应子部分如核酸(如bna沉默hsp27)和蛋白质毒素如皂草素的生物效应的增强活性。在包括权利要求书在内的整个申请中,术语
‘
可裂解接头’、
‘
可裂解键’等,也被称为
‘
不稳定接头’(
‘
l’)和
‘
不稳定键’,例如在(晚期)内体和/或溶酶体中此类键或接头的裂解的上下文中,当提及本发明的缀合物,如任选包含含有通过接头和/或经由支架经由腙键或二硫键偶联至第一、第四或第六蛋白质分子的皂苷的支架的第一、第四或第六蛋白质分子时。例如,图1
‑
1、图6
‑
2、图2
‑
4和图1
‑
5显示了在小鼠中的人类肿瘤内的体内 hsp27基因沉默。例如用由单克隆抗体组成的第一蛋白质分子与经由根据本发明的不稳定接头(腙键)结合至其的皂苷处理荷瘤小鼠,然而包含用于沉默肿瘤细胞中的hsp27基因的结合的反义bna的第三蛋白质分子经由二硫键共价偶联至单克隆抗体(与第一单克隆抗体相同类型)。也就是说,不希望受任何理论的束缚,一旦本发明的治疗性组合通过例如内吞作用被内化,腙键和二硫键在靶向肿瘤细胞的(晚期) 内体和/或溶酶体中被裂解,所述靶向肿瘤细胞在细胞表面的靶向细胞表面分子(在本文是egfr)上表达表位。当考虑到bna从内体和/或溶酶体进入胞质时,键的裂解可能有助于皂苷的内体逃逸增强活性,尽管此类裂解对于观察本发明的西妥昔单抗
‑
so1861缀合物和西妥昔单抗
‑
bna缀合物的组合的基因沉默效果不是必需的。
[0683]
技术人员将理解三官能接头是适合于共价偶联一个、两个或三个皂苷部分的本发明的支架。对于三官能接头,一个或两个皂苷部分的共价偶联是优选的。第二和/或第三结合位点例如适合于共价偶联蛋白质配体如第一、第四或第六蛋白质分子。典型的蛋白质配体是用于靶向在细胞表面处表达egfr的(肿瘤)细胞的egf,以及用于靶向肿瘤细胞或自身免疫细胞的细胞因子。此外,三官能接头的第二或第三结合位点适合于免疫球蛋白如单克隆抗体,即用于结合细胞表面分子,如肿瘤细胞表面分子,优选肿瘤细胞特异性分子,更优选在肿瘤细胞表面特异性(过
‑
)表达的肿瘤细胞受体的第一、第四或第六蛋白质分子的共价偶联。类似地,免疫球蛋白或涵盖免疫球蛋白结合特异性的其任何片段和/或结构域,适合于结合在自身免疫细胞的表面表达的细胞表面分子如受体。因此,在一个实施方案中,第一、第四或第六蛋白质分子包含三官能接头,所述接头包含以下或由以下组成:共价结合的皂苷,如qs
‑
21、so1861,以及共价结合的结合位点,如细胞靶向部分,如用于(特异性)结合肿瘤细胞、自身免疫细胞、患病细胞、异常细胞、非健康细胞、b细胞疾病的配体或抗体。
[0684]
因此,根据本发明的第一、第四或第六蛋白质分子包含至少一个皂苷。在该上下文中的“至少一个”意指第一、第四或第六蛋白质分子包含一个皂苷分子,但也可以包含几个(如两个、三个或四个)皂苷或多种(如10、20或100种)皂苷。根据应用,第一、第四或第六蛋白质分子可包含具有共价结合的皂苷的共价结合的支架,其中可将支架设计为包含限定数量的皂苷。优选地,根据本发明的第一、第四或第六蛋白质分子包含限定数量或范围的皂苷,而不是随机数量。这对于与上市许可相关的药物开发尤其有利。在这方面限定的数量意
味着第一、第四或第六蛋白质分子优选地包含先前限定数量的皂苷。这是例如通过设计包含聚合结构的支架来实现的,所述聚合结构具有一定数量的用于皂苷连接的可能部分。在理想情况下,所有这些部分都偶联至皂苷,并且支架则包含先前限定数量的皂苷。设想提供一组标准的支架,其包含如两个、四个、八个、十六个、三十二个、六十四个等皂苷,以便用户根据他的需要可以容易地测试最佳数量。一个实施方案是包含本发明的支架的本发明的第一、第四或第六蛋白质分子,其中皂苷存在于限定的范围内,如在非理想情况下,并非聚合结构中存在的所有部分都结合皂苷。例如,此类范围可以是每个支架2
‑
4个皂苷分子、每个支架3
‑
6个皂苷分子、每个支架4
‑
8个皂苷分子、每个支架6
‑
8个皂苷分子、每个支架6
‑
12个皂苷分子等等。在此类情况下,如果范围被限定为2
‑
4,则包含根据本发明的支架的第一蛋白质分子因此包含2、 3或4个皂苷。
[0685]
支架基本上与共价结合支架的皂苷的类型无关,所述支架随后(以依序顺序)共价偶联至第一、第四或第六蛋白质分子。因此,包含支架的第一、第四或第六蛋白质分子是新平台技术的基础产物。由于至少一种共价结合的皂苷介导结合第二、第三、第五或第七蛋白质分子的效应子部分的细胞内递送,根据本发明的支架技术是已知介导由皂苷进行的受控细胞内效应子部分递送的第一系统。支架提供了优化的和有功能活性的单元,其可以在单个和限定位置处连接至皂苷以及由第一、第四或第六蛋白质分子所包含的结合位点,如配体、抗体等。
[0686]
一个实施方案是包含根据本发明的支架的第一、第四或第六蛋白质分子,其中多聚结构或寡聚结构的单体的数量是精确限定的数量或范围。优选地,多聚结构或寡聚结构包括诸如聚(胺),如聚乙烯亚胺和聚(酰氨基胺)的结构,或者诸如聚乙二醇、聚(酯)、如聚(丙交酯)、聚 (内酰胺)、聚丙交酯
‑
共
‑
乙交酯共聚物、聚(糊精)或肽或蛋白的结构,或者诸如天然和/或人工聚氨基酸如聚赖氨酸、dna聚合物、稳定化 rna聚合物或pna(肽核酸)聚合物的结构,其作为直链、支链或环状多聚物、寡聚物、树状物、树枝化基元、树枝化基元化多聚物、树枝化基元化寡聚物或这些结构的组装体(无论是纯的还是混合的)出现。优选地,多聚结构或寡聚结构是生物相容的,其中生物相容意指多聚结构或寡聚结构在有机体内不显示出显著的急性或慢性毒性并且可以原样排泄或者通过身体的代谢完全降解为可排泄和/或生理化合物。组装体可以通过共价交联或非共价键和/或吸引来构建。因此它们也可以形成纳米凝胶、微凝胶或水凝胶,或者它们可以连接至载体,如无机纳米颗粒、胶体、脂质体、胶束或颗粒状结构(包含胆固醇和/或磷脂)。所述多聚结构或寡聚结构优选携带精确限定数量或范围的偶联部分用于糖苷分子(和/或效应分子和/或载体分子如配体、单克隆抗体或其片段)的偶联。多聚结构或寡聚结构中优选至少50%、更优选至少75%、更优选至少85%、更优选至少90%、更优选至少95%、更优选至少98%、更优选至少99%,最优选100%的精确限定数量或范围的偶联部分被根据本发明的支架中的糖苷分子占据。
[0687]
优选地,树枝化基元是在树的起点(称为焦点)处具有单个化学可寻址基团的分支的、明确限定的树状聚合物。树状物是两个或更多个树枝化基元在其焦点处的连接。树枝化基元化多聚物是一个或多个树枝化基元的焦点与聚合物的连接。在优选的实施方案中,提供了根据本发明的支架,其中多聚结构或寡聚结构包括直链、支链或环状多聚物、寡聚物、树状物、树枝化基元、树枝化基元化多聚物、树枝化基元化寡聚物或这些结构的组装体(无论是纯的还是混合的),其中组装体可通过共价交联或非共价吸引构建并且可形成纳米凝
胶、微凝胶或水凝胶,并且其中,优选地,聚合物是聚(胺)的衍生物,如聚乙烯亚胺和聚(酰氨基胺),以及诸如聚乙二醇、聚(酯),如聚(丙交酯)、聚(内酰胺)、聚丙交酯
‑
共
‑
乙交酯共聚物和聚(糊精)的结构,以及诸如天然和/ 或人工聚氨基酸如聚赖氨酸,或肽或蛋白质或dna聚合物,稳定的 rna聚合物或pna(肽核酸)聚合物的结构。优选地,多聚结构或寡聚结构是生物相容的。
[0688]
一个实施方案是本发明的治疗性组合或用于根据本发明的用途的治疗性组合,其中第一、第四或第六蛋白质分子包含多于一个共价结合的皂苷,优选2、3、4、5、6、8、10、16、32、64、128或1
‑
100个皂苷,或者其间的任何数量的皂苷,如7、9、12个皂苷。
[0689]
一个实施方案是本发明的第一、第四或第六蛋白质分子,其中至少一个皂苷经由根据本发明的至少一个可裂解接头共价结合寡聚支架或多聚支架的多聚结构或寡聚结构。
[0690]
一个实施方案是本发明的第一、第四或第六蛋白质分子,其中寡聚支架或多聚支架的用于寡聚支架或多聚支架与所述第一、第四或第六蛋白质分子的氨基酸残基的共价偶联的化学基团是点击化学基团,其优选选自四嗪、叠氮化物、烯烃或炔烃或者这些基团的环状衍生物,更优选地,所述化学基团是叠氮化物。
[0691]
一个实施方案是本发明的第一、第四或第六蛋白质分子,其中寡聚支架或多聚支架的多聚结构或寡聚结构包括直链、支链和/或环状多聚物、寡聚物、树状物、树枝化基元、树枝化基元化多聚物、树枝化基元化寡聚物、dna、多肽、聚赖氨酸、聚乙二醇或者这些多聚结构或寡聚结构的组装体,所述组装体优选通过共价交联构建。
[0692]
发明人确定,根据上文实施方案中的任一个,皂苷与第一、第四或第六蛋白质分子优选经由可裂解键或接头的共价偶联提供了与第二和第三和第五和第七蛋白质分子结合的效应子部分的活性的有效且细胞靶向的增强,其中第一和第三和第四和第六蛋白质分子包含相同的第一、第四或第六结合位点并且其中第一(第四、第六)和第二(第五、第七)蛋白质分子包含不同的第一(第三、第四、第六)和第二(第五、第七)结合位点。当效应子部分也通过使用包含与第一蛋白质分子相同的第一结合位点(当考虑第三或第五蛋白质分子时)以及分别包含不同的第一和第二、第七结合位点(当考虑第一和第二、第七蛋白质分子时)的第二和/或第三和/或第五和/或第七蛋白质分子在相同靶细胞中递送时,直接或经由接头将皂苷偶联至第一、第四或第六蛋白质分子如单克隆抗体的半胱氨酸侧链或赖氨酸侧链被证明是在靶细胞内特异性和有效递送效应子部分增强活性的有益方式。
[0693]
为了更详细地解释本发明,描述了物质的细胞摄取过程(尽管发明人不希望受任何理论的束缚)和本发明中使用的术语。通过囊泡出芽将细胞外物质摄入到细胞中被称为内吞作用。所述囊泡出芽的特征可以在于:(1)由胞质蛋白网格蛋白介导的受体依赖性配体摄取,(2)胆固醇结合蛋白小窝蛋白介导的脂筏摄取,(3)非特异性液体摄取(胞饮作用),或(4)非特异性颗粒摄取(吞噬作用)。所有类型的内吞作用都会进入以下称为内吞途径的囊泡运输和物质分选的细胞过程。内吞途径很复杂,并且尚未完全了解。不希望受任何理论的束缚,细胞器可以从头形成并沿着内吞途径成熟为下一个细胞器。然而,现在假设内吞途径涉及通过囊泡运输连接的稳定区室。区室是一种复杂的多功能膜细胞器,其专门用于细胞的一组特定基本功能。囊泡被认为是短暂的细胞器,组成更简单,并被定义为膜封闭的容器,其通过从预先存在的区室出芽而从头形成。与区室相比,囊泡可以经历成熟,这是一系列生理上不可逆的生化变化。早期内体和晚期内体代表内吞途径中的稳定区室,而初级内
吞囊泡、吞噬体、多泡体(也称为内体载体囊泡)、分泌颗粒和甚至溶酶体代表囊泡。内吞囊泡出现在质膜处,最突出的是来自网格蛋白包被的小窝,首先与早期内体融合,早期内体是大约ph 6.5的主要分选区室。大部分内化的货物和膜通过再循环囊泡(再循环途径)循环回到质膜。应降解的组分经由多泡体运输到酸性晚期内体(ph低于 6)。溶酶体是可以储存成熟的溶酶体酶并在需要时将它们递送至晚期内体区室的囊泡。所得细胞器称为杂合细胞器(hybrid organelle)或内溶酶体。溶酶体在称为溶酶体再生(lysosome reformation)的过程中从杂合细胞器中出芽。晚期内体、溶酶体和杂合细胞器是极其动态的细胞器,并且通常很难区分它们。内吞分子的降解发生在内溶酶体或溶酶体内。内体逃逸是物质从内吞途径,优选从网格蛋白介导的内吞作用,或再循环途径从任何种类的区室或囊泡的内腔主动或被动释放到溶质中。因此,内体逃逸包括但不限于从内体、内溶酶体或溶酶体(包括它们的中间和杂合细胞器)中释放。
[0694]
除非另有特别说明并且特别是在涉及糖苷分子(如本发明的皂苷) 的内体逃逸机制时,每当本文中使用措辞“内体”或“内体逃逸”时,其还包括内溶酶体和溶酶体,以及分别从内溶酶体和溶酶体中逃逸。进入胞质后,所述物质可能会移动到其它细胞单元如细胞核。
[0695]
在正式用语中,糖苷是其中经由糖苷键将糖基团通过其异头碳结合至另一个基团的任何分子。在本发明的上下文中,糖苷分子如皂苷是这样的分子,其能够进一步增强效应子部分的效果,不希望受任何理论的束缚,特别是通过促进效应子部分的内体逃逸。不希望受任何理论的束缚,糖苷分子(皂苷,如表a1中列出的那些)与内吞和再循环途径的区室和囊泡的膜相互作用并使它们对于所述效应子部分渗漏,导致增加的内体逃逸。术语“支架能够增加效应子部分的内体逃逸”意指当两个分子在内体(如晚期内体)中时,任选地和优选地在至少一种糖苷如皂苷从第一、第四、第六蛋白质分子(如从由所述第一、第四、第六蛋白质分子所包含的接头或多聚结构或寡聚结构)中释放,如通过裂解至少一种糖苷(皂苷)与第一、第四、第六蛋白质分子(例如经由支架的多聚结构或寡聚结构和/或经由接头)之间的可裂解键之后,与支架的多聚结构或寡聚结构偶联的至少一个皂苷(糖苷分子)能够增强效应子部分的内体逃逸。即使在至少一种糖苷如根据本发明的皂苷与第一、第四、第六蛋白质分子之间的键(任选地经由接头或支架)可以是“稳定键”,这并不意味着此类键不能在内体中被如酶裂解。例如,可以从剩余的接头片段或寡聚结构或多聚结构中裂解下来糖苷或皂苷,任选地连同接头或者支架的一部分寡聚结构或多聚结构。例如,可以是蛋白酶切割(蛋白质)接头或蛋白质聚合结构,如白蛋白,从而释放至少一种糖苷、皂苷。然而,优选的是糖苷分子(优选皂苷)以活性形式释放,优选以其在(被制备为)任选地经由接头和/或寡聚支架或多聚支架偶联至第一、第四、第六蛋白质分子之前的原始形式释放;因此,糖苷(皂苷)在此类裂解后具有其天然结构或糖苷(皂苷)在此类裂解后具有结合至其的(部分)化学基团或接头,而糖苷生物活性(皂苷生物活性),如针对存在于相同内体或溶酶体中的效应子部分的内体/溶酶体逃逸增强活性在糖苷(皂苷)与载体分子(即任选包含本发明的接头和/或支架的第一、第四、第六蛋白质分子)之间的键的所述裂解后得以维持或恢复。关于本发明,就如皂苷与第一、第四、第六蛋白质分子的氨基酸残基、接头、(支架的)多聚结构或寡聚结构、配体、(单克隆)免疫球蛋白或其结合结构域或结合片段和/或效应子(效应子部分、效应分子) 之间的键而言的术语“稳定的”意指该键不容易断裂或者至少并未被设计成容易被如ph差、盐浓度或紫外线、还原条件断裂。关于本发明,就如皂苷与第一、第四、第六蛋白
质分子、接头、氨基酸残基、支架的多聚结构或寡聚结构、配体、抗体和/或效应子之间的键而言的术语“可裂解的”意指该键被设计成容易被如ph差、盐浓度、在还原条件下等断裂。技术人员非常了解此类可裂解的键以及如何制备它们。
[0696]
在本发明之前,在市场上引入adc和aoc的主要障碍之一是小的治疗窗:治疗有效剂量的adc或aoc伴随着(不可接受的)副作用,阻碍了用adc治疗患者的发展和暗示。通过应用本发明的第一、第四或第六蛋白质分子,现在可以将一个或多个糖苷分子(皂苷)连同携带有效载荷的adc或连同与根据本发明的寡核苷酸如bna缀合的(单克隆)抗体(即,本发明的特定的第二或第三或第五或第七蛋白质分子) 一起引导至(靶)细胞。特别地,先前不可能同时将第二或第三或第五或第七蛋白质分子的效应子部分和每效应子部分(预定的、可控的)特定数量或范围的糖苷分子(皂苷)如经由细胞的内吞途径特异性引导至细胞胞质。
[0697]
本发明提供的解决方案包括至少一个皂苷共价结合第一或第四或第六蛋白质分子。本发明提供的另一种解决方案包括使用寡聚支架或多聚支架(首先)使糖苷分子(皂苷)聚合,以及为第一或第四或第六蛋白质分子提供一簇共价结合的皂苷,使得例如在内吞作用之后一个或多个皂苷能够在细胞内位点(在那需要皂苷的作用模式)处重新单体化。“聚合”在本上下文中意指皂苷分子与第一、第四、第六蛋白质分子经由接头或者直接或者经由多聚结构或寡聚结构以形成支架的可逆和/ 或不可逆的多重缀合,或者(修饰的)皂苷的可逆和/或不可逆的多重缀合从而形成多聚结构或寡聚结构以形成支架。“再单体化”在本上下文中意指例如在内吞作用之后皂苷从第一、第四、第六蛋白质分子、从将皂苷连接至第一、第四、第六蛋白质分子的接头或者从支架裂解,并恢复未结合的皂苷的(天然)化学状态,所述未结合的皂苷可以包含或可以不包含另外的化学基团,如用于将皂苷连接至接头、第一蛋白质分子的氨基酸残基或支架的化学基团,和/或结合皂苷的化学基团 (如醛基团或羧酸基团)的(化学)接头。由于皂苷的复杂化学性质,例如皂苷在支架或其它连接接头处的
‘
聚合’以及它们在期望位置处(如胞内,在内吞作用后)的
‘
再单体化’是一项具有挑战性的任务。特别地,用于提供接头和支架(包含用于共价结合第一、第四、第六蛋白质分子的共价连接的糖苷,如三萜皂苷(糖苷的聚合))的化学反应通常发生在无水有机溶剂中,但皂苷和例如应用为用于携带结合的皂苷的支架的生物相容性聚合物是水溶性分子。未修饰的皂苷的化学性质进一步禁止其自身聚合,并且一种其它可能的解决方案是将多个皂苷(直接)结合至效应分子,据估计不是非常有前途,因为效应分子(药物、毒素、多肽或多核苷酸)通常不提供足够的结合位点并且因为偶联产物将变得非常异质的和/或偶联生物活性分子如皂苷和如肽、毒素、核酸在一起承担影响和阻碍此类含皂苷的缀合物中一个或甚至结合在一起的两个分子的活性的风险。此外,以下有相当大的风险:由第二或第三或第五或第七蛋白质分子所包含的效应子部分在皂苷偶联至如adc或抗体
‑
寡核苷酸缀合物(aoc)后丧失其功能。本发明的实施方案解决了这些缺点中的至少一个。
[0698]
本发明的一方面涉及包含本发明的第一或第四或第六蛋白质分子和本发明的第二、第三、第五或第七蛋白质分子的组合物。
[0699]
本发明的一方面涉及包含本发明的第一、第四或第六蛋白质分子和本发明的第三或第五蛋白质分子的组合物。
[0700]
一个实施方案是包含本发明的第一、第四或第六蛋白质分子和本发明的第二或第七蛋白质分子的组合物,或者是包含本发明的第一、第四或第六蛋白质分子和本发明的第
三或第五蛋白质分子的组合物,其中第二、第五或第七蛋白质分子或者第三蛋白质分子所包含的效应子部分是根据本发明的效应子部分中任一种,优选bna。
[0701]
本发明的一方面涉及包含本发明的第一、第四或第六蛋白质分子和寡核苷酸、核酸和异源核酸中的任何一种或多种的组合物,所述寡核苷酸、核酸和异源核酸优选选自以下中的任一种:载体、基因、诱导细胞自杀的转基因、脱氧核糖核酸(dna)、核糖核酸(rna)、反义寡核苷酸(aso,aon)、短干扰rna(sirna)、微小rna(mirna)、dna 适配体、rna适配体、mrna、微环dna、肽核酸(pna)、氨基磷酸酯吗啉代寡聚物(pmo)、锁核酸(lna)、桥接核酸(bna)、2
’‑
脱氧
‑2’‑ꢀ
氟阿拉伯糖核酸(fana)、2
’‑
o
‑
甲氧基乙基
‑
rna(moe)、2
’‑
o,4
’‑
氨基亚乙基桥接核酸、3
’‑
氟己糖醇核酸(fhna)、质粒、二醇核酸(gna)和苏糖核酸(tna)、或它们的衍生物,更优选bna,例如用于沉默hsp27 蛋白表达的bna(反义bna(hsp27))。
[0702]
效应分子或效应子部分在本发明的上下文中是通过与细胞内效应分子靶标相互作用影响细胞代谢的任何物质,其中该效应分子靶标是除了内吞和循环途径的区室和囊泡的腔之外但包括这些区室和囊泡的膜的细胞内的任何分子或结构。因此,细胞内的所述结构包括细胞核、线粒体、叶绿体、内质网、高尔基体、其它转运囊泡、质膜的内部和胞质。在本发明的上下文中,效应子部分的胞质递送优选意指效应子部分能够逃逸出内体(和/或溶酶体),其如先前所定义的还包括逃逸出内体和溶酶体,并且优选地能够到达如本文所述的效应子部分靶标。本发明还涵盖了一种新型分子(被称为支架),当效应子部分由本发明的第二或第三或第五或第七蛋白质分子所包含并且皂苷由第一、第四或第六蛋白质分子所包含时,其用于以预定的比率同时将效应子部分和至少一种糖苷分子(如本发明的皂苷)两者带入内体。在本发明的上下文中,支架的多聚结构或寡聚结构是结构有序的形成,如多聚物、寡聚物、树状物、树枝化基元化多聚物或树枝化基元化寡聚物,或者其是组装的聚合结构,如水凝胶、微凝胶、纳米凝胶、稳定的聚合物胶束或脂质体,但不包括由单体的非共价组装体(如胆固醇/磷脂混合物)组成的结构。术语“多聚物、寡聚物、树状物、树枝化基元化多聚物或树枝化基元化寡聚物”具有其普通含义。特别地,多聚物是分子结构主要或完全由大量相同或相似的键合在一起的单元构成的物质,寡聚物是分子由相对较少的重复单元组成的聚合物。对于多聚物和低聚物的上述定义中分别使用的“许多”和“几个”的一个具体截止值没有达成共识。然而,由于支架可包含多聚结构或寡聚结构或两者,因此键合在一起的相似单元的完整数量范围适合于此类结构,即2个单体单元至100个单体单元、1000个单体单元和更多。例如,包含5个或更少个的结构可以被称为寡聚结构,然而包含50个单体单元的结构可以被称为多聚结构。10个单体单元的结构可以被称为寡聚物或多聚物。如本文所定义的支架还包含至少一种糖苷分子,如本发明的皂苷。支架优选包含多聚结构或寡聚结构,如聚(胺)或寡聚(胺),如聚乙烯亚胺和聚(酰氨基胺),以及生物相容结构,如聚乙二醇,聚(酯)或寡聚(酯),如聚(丙交酯)、聚(内酰胺)、聚丙交酯
‑
共
‑
乙交酯共聚物和聚(糊精),聚糖或寡糖,如环糊精或聚葡萄糖,和聚氨基酸或寡氨基酸,如聚赖氨酸或肽或蛋白质,或者dna寡聚物或多聚物。如本文所定义的组装聚合结构包括至少一个支架和任选的其它单独的多聚结构或寡聚结构。所述组装体的其它单独的多聚结构或寡聚结构可以是(a)支架(因此包含至少一种糖苷分子,如本发明的皂苷),(b)官能化支架(因此包含至少一种糖苷分子,如皂苷以及配体、抗体等作为第一蛋白质分子,(c) 不含糖苷分子(如本发明的皂苷(参见例如表a1)),不含配体、抗体等作为第一蛋白质分子的多聚结构或寡聚结构。官能化组装
的聚合结构是含有(a)至少一个官能化支架或(b)至少一个支架和至少一个聚合结构的组装聚合结构,其包含至少一个配体、抗体等作为第一蛋白质分子。在组装的聚合结构中的不包含以上提及的分子(即,无糖苷如皂苷,无第一蛋白质分子如配体、抗体)中的任一种的多聚结构或寡聚结构特别作为组装结构的结构组分添加,所述组装结构有助于构建或稳定组装结构(“胶状”)。不希望受任何理论的束缚,酸性环境似乎是糖苷(皂苷)与效应子部分之间的协同作用的先决条件。
[0703]
进一步包含或者不进一步包含一个或多个(可裂解)接头和/或任选的支架的包含皂苷的第一、第四或第六蛋白质分子是否能够扰乱酸性环境并抑制至少一种糖苷(皂苷)的内体逃逸功能可以用本领域已知的测定容易地确定。抑制被描述为“诱导50%细胞杀伤所必需的糖苷倍增量”。优选的是,支架不会导致增加,其至少为当使用氯喹作为阳性对照时观察到获得50%细胞杀伤所必需的糖苷分子(皂苷)增加。可替代地并且优选地,进一步包含或不进一步包含一个或多个(可裂解)接头和/或任选的支架的包含皂苷的第一、第四、第六蛋白质分子不导致糖苷分子至少4倍增加以诱导50%细胞杀伤,更优选不导致至少2倍增加。倍数增加待在测定中测量,其中氯喹作为阳性对照诱导糖苷量,优选皂苷量的2倍增加,其中皂苷是本发明的皂苷中的任何一种或多种(参见表a1,方案i,先前的实施方案)以观察50%的细胞杀伤。
[0704]
术语“改善或增强效应子部分的效果”意指糖苷分子,优选本发明的皂苷,优选通过启用或改善其靶标接合(target engagement)来增加此效应子部分的功能功效(如,毒素或药物或寡核苷酸如bna的治疗指数;生物技术过程中修饰剂的代谢功效;细胞培养研究实验中基因的转染功效)。优选不包括抗原特异性免疫应答的加速、延长或增强。治疗功效包括但不限于更强的治疗效果,优选具有更低的剂量和/或更少的副作用。“改善效应子部分的效果”也可意指因为缺乏效果而不能使用(和如不被称为是效应子部分)的效应子部分当与本发明组合使用时变得有效。如由本发明提供的有益的或期望的并且可以归因于效应子部分和第二或第三蛋白质分子的组合的任何其它效果都被认为是“改善的效果”。在实施方案中,包含结合的皂苷并且第一、第四或第六蛋白质分子所包含的支架增强了第二、第三、第五或第七蛋白质分子所包含的效应子部分的效果,所述效果是预期的和/或期望的。在包含结合至蛋白质支架的皂苷的第一、第四或第六蛋白质分子的情况下,支架的蛋白质聚合结构本身可对血流中的胶体渗透压具有例如效果。如果此类效果不是第一、第四或第六蛋白质分子所包含的此类官能化支架的预期的或期望的效果,则支架的蛋白质结构不是如本发明中定义的效应子部分。或者,例如在携带结合的皂苷并且第一蛋白质分子所包含的基于dna或rna的支架情况下,此dna或rna的部分可能具有(非预期的)功能,如通过干扰表达。如果此类干扰不是最终官能化支架的预期或期望的效果,则支架的dna聚合结构或rna聚合结构不是如本发明中定义的效应子部分。
[0705]
许多优选的特征可以被配制用于第一、第四或第六蛋白质分子所包含的内体逃逸增强剂,即糖苷或皂苷,优选根据本发明的皂苷:(1) 它们优选无毒且不引起免疫应答,(2)它们优选不介导效应子部分胞质摄取至脱靶细胞中,(3)它们在作用位点的存在优选与效应子部分的存在同步,(4)它们优选是可生物降解的或可排泄的,以及(5)它们优选基本上不干扰与内体逃逸增强剂所组合的效应分子的生物活性无关的有机体生物过程,如与激素相互作用。糖苷分子的实例如至少在一定程度上满足上述标准的本发明的皂苷是双糖链三
萜,优选双糖链三萜皂苷,如so1861、sa1641、qs
‑
21、ge1741以及表a1,方案i中的皂苷。
[0706]
本发明的一方面涉及包含本发明的第一、第四或第六蛋白质分子和效应子部分的抗体
‑
药物缀合物或抗体
‑
寡核苷酸缀合物或配体
‑
药物缀合物。
[0707]
如前所述,根据本发明的第一、第四或第六蛋白质分子所包含的至少一个皂苷增加了如在本发明中定义的至少当前和新的效应子部分的功效。由于降低了第二或第三或第五或第七蛋白质分子所包含的效应子部分的剂量,将减少潜在的副作用,而不会降低功效。因此,本发明提供了根据本发明的第一、第四或第六蛋白质分子,其用于医药或用作药物。因此,本发明的一方面涉及用作药物的根据本发明的第一、第四或第六蛋白质分子,所述第一、第四或第六蛋白质分子包含至少一个皂苷。还提供了根据本发明的第一、第四或第六蛋白质分子用于生产药物的用途。尤其是癌症药物并且特别是经典的化疗药物,因其副作用而著名。由于第二或第三或第五或第七蛋白质分子所包含的药学活性物质和第一、第四或第六蛋白质分子所包含的皂苷两者的靶向以及时间和位置和同步,因为第一和第三和/或第四和第五蛋白质分子携带针对相同细胞表面分子上的相同表位的相同结合位点或者因为第一和第二或第六和第七蛋白质分子携带分别针对第一和第二或第六和第七细胞表面分子上的不同的第一和第二或第六和第七表位的不同结合位点,根据本发明的治疗性组合对于用作药物,特别是用于治疗癌症的方法中尤其有价值。因此,本发明提供了根据本发明的治疗性组合或者本发明的第一、第四或第六蛋白质分子,其用于治疗癌症的方法中。本发明还提供了根据本发明的治疗性组合或者本发明的第一、第四或第六蛋白质分子,其用于治疗获得性或遗传性病症,特别是单基因缺陷病症的方法中。因此,治疗性组合包含第一和第二蛋白质分子和/或包含第一和第三蛋白质分子,和/或第四和第五蛋白质分子,和/或第六和第七蛋白质分子。因此,本发明的一方面涉及用于治疗癌症或自身免疫疾病的方法中的根据本发明的治疗性组合,其中第二或第三或第五或第七蛋白质分子包含共价结合的效应子部分。
[0708]
本发明的第一、第二和第三、第四、第五、第六、第七蛋白质分子在医学中的进一步应用是替代以不足的量或不足的功能产生这些酶的靶细胞中的细胞内酶。所得疾病可能是遗传性的或获得性的。在大多数情况下,仅对症治疗是可能的,并且对于一些罕见疾病,治疗选项不足导致相关患者的寿命缩短。此类疾病的一个实例是苯丙酮尿症,这是一种先天性代谢错误,导致氨基酸苯丙氨酸的代谢降低。该疾病的特征在于肝酶苯丙氨酸羟化酶的基因突变。苯丙酮尿症目前无法治愈。发病率约为1:10,000,在土耳其已知的最高发病率为1:2,600。具有结合的苯丙氨酸羟化酶或具有结合的编码苯丙氨酸羟化酶的多核苷酸的第二或第三或第五或第七蛋白质分子,优选抗体,可用于通过使用适合的特异性抗体靶向肝细胞,并替代肝细胞中的缺陷酶。这是使用本发明的治疗性组合用于替代疗法或基因疗法的一个实例,本发明的治疗性组合包含第一、第四或第六蛋白质分子(其具有结合至其的皂苷),以及第二或第三或第五或第七蛋白质分子(其具有根据本发明的结合至其的酶或寡核苷酸)。在优选的实施方案中,提供了用于基因疗法或替代疗法的方法中的根据本发明的治疗性组合。
[0709]
本发明还提供了治疗癌症的方法,所述方法包括向有需要的患者施用包含根据本发明的治疗性组合的药物,优选向有需要的患者,优选人类癌症患者施用有效剂量的所述药物。
[0710]
关于适合施用的形式的考虑是本领域已知的,并且包括毒性效应、溶解性、施用途径和维持活性。例如,注射到血流中的药理学组合物应该是可溶的。
[0711]
适合的剂型部分取决于用途或进入途径,例如透皮或通过注射。无论靶细胞是否存在于多细胞宿主中,此类剂型都应允许化合物到达靶细胞。其它因素在本领域中是已知的,并且包括对阻止化合物或组合物发挥其作用的诸如毒性和剂型的考虑。
[0712]
一个实施方案是根据本发明的内体逃逸增强缀合物和结合部分的组合,所述内体逃逸增强缀合物包含含有至少一个共价结合的皂苷的第一、第四或第六蛋白质分子,其中结合部分包含至少一个效应子部分,所述结合部分是包含结合的效应子部分的第二或第三或第五或第七蛋白质分子,其中内体逃逸增强缀合物和结合部分是彼此独立地能够特异性结合靶细胞特异性表面分子或结构,从而诱导内体逃逸增强缀合物和靶细胞特异性表面分子的复合物以及结合部分和靶细胞特异性表面分子的复合物的受体介导的内吞作用,其中内体逃逸增强缀合物和结合部分能够经由其相同结合位点结合相同的靶细胞特异性表面分子,或者其中内体逃逸增强缀合物和结合部分能够经由其不同的结合位点结合不同的靶细胞特异性表面分子。一个实施方案是根据本发明的组合,其中内体逃逸增强缀合物能够与结合部分竞争结合靶细胞特异性表面分子或结构。一个实施方案是根据本发明的组合,其中内体逃逸增强缀合物和结合部分彼此独立地能够特异性结合相同表位或不同表位。一个实施方案是用于治疗异常(如根据本发明的癌症)的方法中的组合,其中所述内体逃逸增强缀合物和所述结合部分同时或依序、优选同时施用。
[0713]
本发明的一方面涉及试剂盒,其包含含有根据本发明的内体逃逸增强缀合物(即,第一、第四或第六蛋白质分子)的第一容器和含有根据本发明的结合部分(即第二和/或第三和/或第五和/或第七蛋白质分子) 的第二容器,所述试剂盒还包含使用结合分子(即,包含第一和第二或第一和第三或第四和第五或第六和第七药物组合物的治疗性组合)的说明书。
[0714]
本发明的一部分是本发明的治疗性组合、第一药物组合物、第一蛋白质分子、第二或第三药物组合物或者第二或第三蛋白质分子还与结合分子或结合部分和皂苷的共价缀合物(复合物)组合,或者还与药物化合物、抗体等组合,随其提供了包含三种或更多种增强剂、药物活性成分等的组合物,如本发明的缀合物(如第一蛋白质分子和/或第二或第三蛋白质分子)组合与效应分子复合的结合部分,还组合连接或不连接至皂苷并且偶联或不偶联至配体(如靶向的免疫球蛋白、其结构域或片段)的药物。此外,一个实施方案是本发明的治疗性组合、第一药物组合物、第一蛋白质分子、第二或第三药物组合物或者第二或第三蛋白质分子,其中第二或第三蛋白质分子提供有两个或更多个效应子部分,如毒素或免疫毒素,其中两个或更多个效应子部分是相同的或不同的。
[0715]
[0716]
[0717]
[0718]
[0719]
[0720][0721]
表a2
‑
先前在人类临床环境中研究,随后从进一步临床研究中撤回的 adc
[0722]
[0723]
[0724]
[0725]
[0726]
[0727]
[0728]
[0729]
[0730]
[0731]
[0732]
[0733]
[0734]
[0735]
[0736][0737]
表a3
‑
达到iii期临床开发的adc
[0738]
[0739][0740]
[0741][0742]
表a5:来自植物的rip*
[0743]
[0744]
[0745]
[0746]
[0747]
[0748]
[0749]
[0750]
[0751]
[0752]
[0753]
[0754][0755]
本发明的一方面涉及缀合物,其包含共价连接在一起的抗体和反义寡核苷酸,如反义bna,或者由其组成。在图1
‑
5中,描绘了此类缀合物的基因沉默活性(在动物肿瘤模型中的体内测试)。还参考实施例部分。
[0756]
本发明的一方面涉及第一组合物和第二组合物的组合,所述第一组合物包含缀合物,所述缀合物包含共价连接在一起的抗体和反义寡核苷酸,如反义bna,或者由其组成,所述第二组合物包含本发明的游离皂苷(参见表a1,方案i)。在图1
‑
7a和图1
‑
7c中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0757]
本发明的一方面涉及药物组合,其包含第一组合物和第二组合物,或者由其组成,所述第一组合物包含第一缀合物,所述第一缀合物包含抗体和反义寡核苷酸,如反义bna,或者由其组成,所述第二组合物包含第一缀合物,所述第一缀合物包含相同抗体和本发明的至少一个皂苷,或者由其组成。在图1
‑
5中,描绘了此类缀合物的基因沉默活性(在动物肿瘤模型中的体内测试)。在图8
‑
5中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0758]
本发明的一方面涉及药物组合,其包含第四组合物和第五组合物,或者由其组成,所述第四组合物包含第四缀合物,所述第四缀合物包抗体和反义寡核苷酸,如反义bna,或者由其组成,所述第五组合物包含第一缀合物,所述第一缀合物包含不同抗体和本发明的至少一个皂苷,或者由其组成。在图10
‑
6a和图10
‑
6c中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0759]
本发明的一方面涉及缀合物,其包含共价连接至本发明的至少一个皂苷的反义寡核苷酸如反义bna,或者由其组成。在图1
‑
3中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0760]
本发明的一方面涉及缀合物,其包含共价偶联至聚合支架如树枝化基元(如g4
‑
树枝化基元)的反义寡核苷酸(如反义bna),或者由其组成,其中聚合支架与本发明的一个或多个皂苷分子(如四个皂苷分子) 共价缀合。在图1
‑
3中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0761]
本发明的一方面涉及缀合物,其包含以下或由以下组成:共价连接至至少一个反义寡核苷酸分子(如反义bna)以及共价连接至本发明的至少一个皂苷分子的对肿瘤标志物或肿瘤细胞受体具有特异性的抗体(如单克隆抗体)。在图2
‑
4中,描绘了此类缀合物的基因
沉默活性(在动物肿瘤模型中的体内测试)。还参考实施例部分。
[0762]
本发明的一方面涉及缀合物,其包含以下或由以下组成:经由三官能接头(如方案ii的接头)共价连接至至少一个反义寡核苷酸分子(如反义bna)以及经由相同的三官能接头共价连接至本发明的至少一个皂苷分子的对肿瘤标志物或肿瘤细胞受体具有特异性的抗体(如单克隆抗体)。在图1
‑
1中,描绘了此类缀合物的基因沉默活性(在动物肿瘤模型中的体内测试)。还参考实施例部分。
[0763]
本发明的一方面涉及治疗性组合,其包含以下或由以下组成:第八组合物,所述第八组合物包含缀合物,所述缀合物包含以下或由以下组成:优选经由至少一个接头,优选在生理酸性条件下可裂解的至少一个可裂解接头共价连接至本发明的至少一个皂苷分子的对肿瘤标志物或肿瘤细胞受体具有特异性的抗体,优选单克隆抗体;以及还包含第九组合物,所述第九组合物包含反义寡核苷酸(如反义bna分子)。在图6
‑
2中,描绘了此类缀合物的基因沉默活性(在动物肿瘤模型中的体内测试)。在图5
‑
2a和图5
‑
2c中,描绘了此类缀合物的基因沉默活性(用人类肿瘤细胞进行的体外基于细胞的生物测定)。还参考实施例部分。
[0764]
本发明的一方面涉及上述缀合物或组合物或治疗性组合中的任一种,其用作药物。
[0765]
本发明的一方面涉及上述缀合物或组合物或治疗性组合中的任一种,其用于治疗或预防癌症。
[0766]
当然,如前所述,a、b、c、d、e、f、g、h、i、j、k、m、n、p、 q、r、s、t、u、v、w和/或x中的任一个和全部对于根据本发明的任何和全部上述方面和实施方案,具有根据本发明的每个单独实施方案和方面的值。此外,(三官能)接头l1、l2、l4、l5、l6、l8、l9和/或 l10,如果存在于本发明的分子或缀合物或部分中,则为本发明的上述方面和实施方案中的每一个和任一个所示的(三官能)接头,如技术人员容易理解的。寡聚支架或多聚支架l3和/或l7,如果存在于本发明的分子或缀合物或部分中,则为本发明的上述方面和实施方案中的每一个和任一个所示的寡聚支架或多聚支架,也如技术人员容易理解的。此外,第一配体a1和第一效应子部分b1(如果存在),以及第二配体 a2和第二效应子部分b2(如果存在),以及第一效应子部分a1和第一配体b1(如果存在),以及第二效应子部分a2和第二配体b2(如果存在)是选定和指定的配体和效应子部分,如针对本发明的第一、第二、第三、第四、第五和第六系列的实施方案和方面以及上文列出的本发明的所有另外的实施方案和方面所公开的。皂苷c是本发明的上述方面和实施方案的任一个中提及和列出的皂苷中的任何一种或多种,特别是选自方案i和/或表a1的一种或多种皂苷。
[0767]
通过以下实施例来进一步说明本发明,这些实施例不应被理解为以任何方式限制本发明。
实施例
[0768]
实施例a
‑
用本发明的缀合物组合adc处理哺乳动物荷瘤动物导致存活和肿瘤消退
[0769]
用人类a431肿瘤细胞悬液皮下注射雌性balb/c裸小鼠。在小鼠的皮肤下,在异种移植动物肿瘤模型中发展了人类表皮癌。注射肿瘤细胞后,使异种移植的肿瘤发展到大约170
‑
180mm3的大小。a431肿瘤细胞具有以下特征:高egfr表达子、中cd71表达子、低her2 表达子。
[0770]
在表a中,呈现了对照小鼠和荷瘤小鼠的处理结果。用针对人类 her2/neu、人类egfr或人类cd71(作为异种移植肿瘤上的细胞表面受体)的指定抗体处理荷瘤小鼠。西妥昔单抗与皂苷so1861共价缀合。 so1861首先提供有接头emch(n
‑
ε
‑
马来酰亚胺基己酸酰肼),所述 emch是用于将巯基(抗体的还原的半胱氨酸))共价缀合至羰基(醛或酮;在本文中是皂苷的c
‑
23位的醛的羰基)的马来酰亚胺
‑
和
‑
酰肼交联剂。皂苷
‑
emch共价偶联至西妥昔单抗的还原的半胱氨酸,在emch 与半胱氨酸侧链之间形成共价硫醚键。在小鼠中测试了adc曲妥珠单抗
‑
皂草素(共价缀合物)和抗cd71 mab(okt
‑
9,igg)
‑
皂草素(共价缀合物)的肿瘤攻击功效,测量为在用adc开始处理之后时间内的肿瘤体积。adc的剂量在肿瘤模型中是次优的。也就是说,根据先前的实验,确定了在次优剂量的adc下不会观察到肿瘤消退或肿瘤生长停滞。
[0771][0772][0773]
这些结果证明,当考虑用单独的adc处理荷瘤小鼠时无效剂量的adc(肿瘤生长,未防止小鼠的死亡(安乐死))与由共价结合皂苷(即 so1861)的肿瘤细胞特异性受体靶向抗
体组成的本发明的缀合物(所述共价缀合物以单独施用时无效的剂量(肿瘤生长,未防止小鼠死亡(安乐死))施用至患有癌症的小鼠)的组合疗法提供了高效且有效的治疗方案,表示为处理的动物中消退的肿瘤和延长的存活(超过实验的持续时间)。次优剂量的adc组合本发明的包含共价结合的皂苷的缀合物(当单独施用时,其不具有抗肿瘤活性),从而为癌症患者提供了有效的治疗选项,其中相对低剂量的adc是有效的。较低剂量的adc有望降低不良事件的风险,或甚至根本没有副作用。此外,考虑到adc的功效时,本发明的携带皂苷的缀合物的刺激效应表明,当关注肿瘤患者治疗时先前已被证明缺乏功效的adc可获得加倍重视和价值,因为 adc功效在组合疗法环境中得到提高,如当前实施例所示。参考表a2 和表a3,总结了先前在人类临床环境中研究过的adc,但针对从进一步临床研究中撤回的一些adc。尤其是由于观察到缺乏功效和/或由于发生不可接受的不良事件而终止临床开发的adc是当与本发明的包含共价结合的皂苷的缀合物(如测试的西妥昔单抗
‑
皂苷)组合时可使癌症患者获得加倍价值的adc。
[0774]
实施例b
‑
具有内体/溶酶体逃逸增强活性的包含qs
‑
21的皂皮树的皂苷混合物
[0775]
方案i展现了一系列qs
‑
21皂苷的常见分子结构(部分改编自: conrado pedebos,la
é
rcio pol
‑
fachin,ramon pons,cilaine v.teixeirahugo verli,atomic model and micelle dynamics of qs
‑
21 saponin, molecules 2014,19,3744
‑
3760)。基于混合物中存在的至少一种单独皂苷(如qs
‑
21)的内体/溶酶体逃逸增强特性,或者基于混合物所包含的两种或更多种皂苷的组合(如qs
‑
21和qs
‑
7),从皂皮树获得的水溶性皂苷的混合物(sigma
‑
aldrich,产品号:s4521;roth,项号(item no.): 6857;invivogen,产品
‘
quil
‑
a’)可应用于本发明的内体/溶酶体逃逸增强缀合物、组合物、组合。
[0776]
发明人证明,如在基于细胞的生物测定中用哺乳动物肿瘤细胞测试的,2.5微克/ml剂量的来自皂皮树的皂苷的混合物能够增强石竹素的内体逃逸。暴露至细胞的效应子部分是共价偶联至配体egf的石竹素:egf
‑
石竹素。测试的细胞为用于游离皂苷的肿瘤细胞系hela,以及用于测试当共价偶联至西妥昔单抗时皂苷的a431、mda
‑
mb
‑
468、 caski和a2058。
[0777]
实施例1
[0778]
设计并产生了三官能接头支架,其具有特异性化学末端基团 (dbco,tco),用于在一个臂上与so1861分子缀合(不稳定,(l)缀合)并且在另一个臂上与反义hsp27bna寡核苷酸(在癌细胞中靶向并诱导肿瘤靶标(onco
‑
target)hsp27 mrna降解)缀合,以产生so1861
‑
l
‑ꢀ
三官能接头
‑
l
‑
hsp27bna(图16
‑
1)。so1861
‑
l
‑
三官能接头
‑
l
‑ꢀ
hsp27bna用其第三臂(马来酰亚胺)缀合至抗egfr抗体西妥昔单抗(西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna)4)的半胱氨酸残基(cys)。
[0779]
在a431异种移植
‘
裸’鼠肿瘤模型中测试了这种包含支架的缀合物的egfr介导的肿瘤靶向基因沉默活性。在第12天开始给药,此时肿瘤大小达到~170mm3,在第一次给药后72h收集肿瘤样品,并分析与细胞对照mrna表达(参考基因)相比的hsp27基因表达。这揭示了与单次给药西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
或西妥昔单抗
‑
(lys
‑
l
‑ꢀ
hsp27bna)4单一疗法相比,1次给药25mg/kg西妥昔单抗
‑
cys
‑ꢀ
(so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna)
3,7
导致肿瘤中hsp27基因表达降低40%(图1
‑
1)。与媒介物对照肿瘤相比,观察到25%的基因沉默降低。这表明并使得缀合的so1861可以在体内有效地诱导治疗性寡聚物核苷酸在肿瘤中的靶向递送。
[0780]
为了进一步加强这一点,在体外测试了西妥昔单抗
‑
cys
‑
(so1861
‑ꢀ
l
‑
三官能接头
‑
l
‑
hsp27bna dar4)4在表达egfr的(a431)中增强的 hsp27基因沉默,如图2
‑
1所示。与单独的西妥昔单抗
‑
(lys
‑
l
‑ꢀ
hsp27bna)4或西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
相比,西妥昔单抗
‑
cys
‑ꢀ
(so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna)
3,7
在a431细胞中有效地诱导了 hsp27基因沉默(ic50=
…
)(图2
‑
1)。
[0781]
实施例2
[0782]
1靶标2
‑
组分系统是如图11
‑
1中所示的mab1
‑
(树枝化基元 (so1861)
n
)
n
和mab1
‑
效应子的组合治疗,然而2靶标2
‑
组分系统是如图12
‑
1中所示的mab1
‑
(树枝化基元(so1861)
n
)
n
mab2
‑
效应子的组合。
[0783]
树枝化基元(
‑
l
‑
so1861)4经由与半胱氨酸残基(cys)缀合而以 dar3,9缀合至抗egfr抗体西妥昔单抗,即西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
,并在表达egfr的细胞(mda
‑
mb
‑
468)中测试其与抗egfr抗体
‑
蛋白质毒素缀合物(西妥昔单抗
‑
皂草素)的组合的增强的细胞杀伤活性。西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
10 pm西妥昔单抗
‑
皂草素在高表达egfr的细胞中有效地诱导毒素介导的细胞杀伤(ic50=
…
),然而这并未由西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑ꢀ
so1861)4)
3,9
或西妥昔单抗(等效物) 10pm西妥昔单抗
‑
皂草素或西妥昔单抗诱导(图3
‑
1a)。在表达低水平egfr的细胞(hela)中的类似实验揭示了西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
没有活性(图3
‑ꢀ
1c),表明在缺乏足够的egfr受体表达的情况下下,没有达到有效的细胞内so1861浓度(阈值)来诱导内体蛋白质毒素逃逸和毒素介导的细胞杀伤。
[0784]
接下来,树枝化基元(
‑
l
‑
so1861)4经由半胱氨酸(cys)缀合而以 dar4缀合至抗her2抗体曲妥珠单抗,即曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4,并在表达her2的细胞(sk
‑
br
‑
3)中测试其与抗 her2抗体
‑
蛋白质毒素缀合物(曲妥珠单抗
‑
皂草素)组合的增强的细胞杀伤活性。曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4 50pm曲妥珠单抗
‑
皂草素有效地诱导毒素介导的细胞杀伤(ic50=
…
),然而这并未由曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4或曲妥珠单抗(等效物) 50nm曲妥珠单抗
‑
皂草素或曲妥珠单抗诱导(图3
‑
1b)。在表达低水平her2的细胞(jimt
‑
1)中的类似实验揭示了曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4没有活性(图3
‑
1d),表明在足够her2受体表达的不存在下,没有达到有效的细胞内so1861浓度(阈值)来诱导内体蛋白质毒素逃逸和毒素介导的细胞杀伤。
[0785]
接下来,在egfr
/cd71
细胞(mda
‑
mb
‑
468)中,在2靶标2组分系统中,测试西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
或西妥昔单抗
‑
lys
‑
(树枝化基元(
‑
l
‑
so1861)4)
4,4
(lys=缀合至抗体的赖氨酸的树枝化基元(
‑
l
‑
so1861)4)与10pm cd71mab
‑
皂草素的组合。这显示了两种缀合物对细胞杀伤活性的强烈增强(分别是ic50=..ic50=..),然而这并未由西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
或西妥昔单抗
‑ꢀ
lys
‑
(树枝化基元(
‑
l
‑
so1861)4)
4,4
或西妥昔单抗(等效物) 10pmcd71mab
‑
皂草素或西妥昔单抗诱导(图4
‑
1a)。在表达较低水平的 egfr的细胞(caski,egfr
/cd71
)中的类似实验揭示了西妥昔单抗
ꢀ‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
或西妥昔单抗
‑
lys
‑
(树枝化基元(
‑
l
‑ꢀ
so1861)4)
4,4
两者(图4
‑
1c)与在高表达子中的活性(图4
‑
1a)相比活性降低,表明在具有较低egfr受体表达水平的细胞中,有效的细胞内 so1861浓度更低,从而导致毒素介导的细胞杀伤活性降低。
[0786]
用曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4或曲妥珠单抗
‑
lys
‑ꢀ
(树枝化基
元(
‑
l
‑
so1861)4)
4,7
与cd71mab
‑
皂草素的组合在 her2
/cd71
(sk
‑
br
‑
3)细胞系上进行相同的实验,揭示了与对照相比强烈的细胞杀伤活性(图4
‑
1b)。当在her2
/
‑
/cd71
(jimt
‑
1)上测试曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4或曲妥珠单抗
‑
lys
‑
(树枝化基元(
‑
l
‑
so1861)4)
4,7
与10pm cd71mab
‑
皂草素的组合时,不可观察到细胞杀伤活性,表明在足够her2受体表达的不存在下,没有达到有效的细胞内so1861浓度(阈值)以诱导内体蛋白质毒素逃逸和毒素介导的细胞杀伤。
[0787]
接下来,在表达her2的细胞(sk
‑
br
‑
3)中测试曲妥珠单抗
‑
cys
‑ꢀ
(树枝化基元(
‑
l
‑
so1861)4)4 曲妥珠单抗
‑
美坦新(t
‑
dm1,抗体
‑
小分子毒素缀合物)的增强的细胞杀伤活性。与单独的t
‑
dm1或t
‑
dm1 等效的曲妥珠单抗相比,用这种组合没有观察到增强的细胞杀伤,因为内体膜没有形成小分子到达细胞质的屏障(图5
‑
1)。
[0788]
实施例3
[0789]
材料和方法
[0790]
树枝化基元(so1861)4‑
bna寡核苷酸合成(图17
‑
1)
[0791]
将hsp27bna寡核苷酸二硫化物(1.1mg,0.187μmol)溶解在具有 1.0mm tcep的20mm nh4hco3(500μl)中,将混合物摇动1min并在室温下静置。1小时后,通过使用截留分子量为3000da的离心过滤器(14000
×
g,持续30min)过滤反应混合物。将残余物溶液用具有1.0 mm tcep的20mm nh4hco3(500μl)稀释,并在上述相同条件下再次过滤所得混合物。将残余物溶液用20mm nh4hco3/乙腈(3:1,v/v,1.0 ml)稀释,并将所得混合物加入到树枝化基元(so1861)4
‑
马来酰亚胺 1(3.54mg,0.375μmol)中(图17
‑
1)。将反应混合物摇动1分钟并在室温下静置。10min后,将反应混合物进行制备型lc
‑
ms
4a
。将对应于产物的级分立即汇集在一起,冷冻并冻干过夜,得到呈白色蓬松固体的标题化合物(1.25mg,85%)。基于lc
‑
ms的纯度:94%
[0792]
lrms(m/z):1896[m
‑
8]8‑
,2167[m
‑
7]7‑
[0793]
lc
‑
ms r.t.(min):3.77
6b
[0794]
结果
[0795]
将hsp27bna寡核苷酸(靶向癌症靶标的mrna转录物的反义 bna寡核苷酸,即热休克蛋白27(hsp27bna))缀合至树枝化基元(
‑
l
‑ꢀ
so1861)4(hsp27bna
‑
树枝化基元(
‑
l
‑
so1861)4,图17
‑
1)并共施用至 a431癌细胞。作为读出,测定a431细胞中hsp27 mrna的基因沉默。这揭示了hsp27bna
‑
树枝化基元(
‑
l
‑
so1861)4处理导致与单独的 hsp27bna相比hsp27基因沉默活性的改善(图6
‑
1)。
[0796]
实施例4
[0797]
方法
[0798]
so1861释放测定
[0799]
向树枝化基元(so1861)4‑
cbz(0.05mg)(图7
‑
1)添加含水/乙腈 /tfa(1.00ml/1.00ml/4滴)的50μl溶液。将反应混合物摇动1分钟并在室温下静置。通过使用uplc
‑
ms4,经时间推移追踪so1861释放。
[0800]
结果
[0801]
已经测定了在酸性条件下so1861分子从树枝化基元(
‑
l
‑
so1861)4中的释放效率(图7
‑
1)。
[0802]
接下来,测试了树枝化基元(
‑
l
‑
so1861)4的对表达egfr的细胞 (a431和hela)上的靶向毒素egf石竹素的增强递送。这显示了树枝化基元(l
‑
so1861)4 10pm egf石竹素可以诱导增强的毒素介导的细胞杀伤(ic50
…
nm),然而
‘
裸’的树枝化基元(树枝化基元(nem)4)或树枝化基元(
‑
l
‑
so1861)4或树枝化基元(nem)4 10pm egf石竹素在这些浓度下未显示出增强的细胞杀伤(图8
‑
1a,8
‑
1b)。
[0803]
实施例5
[0804]
材料和方法
[0805]
在我们目前的工作中,我们研究了模型支架,其由用于经由希夫碱(亚胺)结合皂苷的四个分子臂和用于点击化学的一个臂组成。聚合结构(图19
‑
1)是购自iris biotech gmbh(marktredwitz,germany)的第一代(即重复支化循环数)五价的基于聚乙二醇的树状物。皂苷(在本实施例中为sa1641)是从获自merck(darmstadt,germany)的来自称为 saponinum album的石头花属物种的皂苷复合粗提取物纯化的。将粉末状粗提取物(2.5g)在水(100ml)中用氢氧化钠(0.2g)水解。将溶液在40℃下搅拌20h,然后补充冰醋酸直至达到ph 5.0。为了去除丹宁酸,将溶液在分液漏斗中与30ml丁醇一起摇动。回收水相并重复丁醇萃取两次。用无水硫酸钠补充丁醇相,过滤并汇集。蒸发丁醇,并将剩余的皂苷粉末溶解在20%甲醇中,至最终浓度为30mg/ml。短时间超声处理后,通过高效液相色谱(hplc)分离不同的皂苷。将管(不包括柱) 用温水(40℃)以1.5ml/min的流速冲洗,然后包括用异丙醇(100%)冲洗eurospher rp
‑
c18
‑
柱(5μm,250
×
8mm)。将皂苷施加至柱并用甲醇梯度洗脱(30min内在补充有0.01%三氟乙酸的水中以1.5ml/min从 20%甲醇到70%甲醇,然后用70%甲醇再洗脱60min)(sama等人,2018)。通过电喷雾电离质谱(esi
‑
ms)分析级分的等分试样的sa1641含量。汇集含有纯sa1641的级分并蒸发甲醇。通过使用干冰将水溶液在旋转圆底烧瓶中冷冻成薄膜。在
‑
80℃下储存16h后,将样品冻干。为了产生如本发明中定义的支架,将聚合结构(0.2mm)和sa1641(3.2mm) 溶解在水(约ph 8)中,将等体积混合并在26℃下摇动24h。然后添加相对于sa1641的4倍摩尔过量的氰基硼氢化钠(nacnbh3;0,1m),并将样品再孵育24h。然后通过超高效液相色谱(uplc)/esi
‑
ms验证结构。将样品施加至rp
‑
c4柱并用甲醇梯度洗脱(15min内在补充有 0.01%三氟乙酸的水中从25%甲醇到80%甲醇,然后用80%甲醇再洗脱10min)。通过使用lockspray
tm
分析级分,lockspray
tm
是一种被专门设计为用于使用来自waters corporation的lc
‑
飞行时间(lc
‑
tof) 质谱仪通过电喷雾电离进行精确质量测量的离子源。
[0806]
实施例6
[0807]
材料和方法
[0808]
作为药物活性物质的实例,我们使用了靶向的毒素石竹素
‑
表皮生长因子(石竹素
‑
egf)。将质粒his
‑
石竹素
‑
egf
‑
pet11d(weng等人, 2009)(100ng)添加到20μl大肠杆菌rosetta
tm
2(de3)plyss感受态细胞(novagen,san diego,ca,usa)中。通过热休克(在冰上30分钟,在 42℃下90s和在冰上1min)转化细胞。此后,添加300μl溶原性肉汤(lb),并将悬浮液在以200rpm摇动同时在37℃下孵育1h。用100μl 细菌悬浮液接种含有50μg/ml氨苄青霉素的预热溶原性肉汤琼脂板,并将板在37℃下孵育过夜。用来自板的菌落接种含有50μg/ml氨苄青霉素的溶原性肉汤(3ml),并将细菌在37℃和200rpm下孵育8h。将悬浮液(50μl)添加到500ml含50μg/ml氨苄青霉素的溶原性肉汤中,并在37℃和200rpm下孵育过夜。随后,体积
扩大到2.0l,并且细菌在相同条件下生长,直到在600nm波长处的光密度达到0.9。此后,通过添加终浓度为1mm的异丙基β
‑
d
‑1‑
硫代吡喃半乳糖苷(iptg) 诱导蛋白质表达。蛋白质表达在37℃和200rpm下持续3h。最后,将细菌悬浮液在5,000
×
g和4℃下离心5min,重悬在20ml pbs(137mmnacl,2.7mm kcl,8.1mm na2hpo4,1.47mm kh2po4)中,并在
‑
20℃下储存直至使用。为了纯化,将细菌悬浮液解冻并通过超声处理裂解。将裂解物离心(15,800
×
g,4℃,30min)并添加咪唑至终浓度为20mm。在20mm咪唑存在下,在4℃下,在连续摇动下,将上清液与2mlni
‑
次氮基三乙酸琼脂糖孵育30min。随后,将材料倒入20ml柱中,用10ml洗涤缓冲液(50mm nah2po4,300mm nacl,20mm咪唑)洗涤3次,并将石竹素
‑
egf用在洗涤缓冲液中10ml
‑
部分的渐增浓度的咪唑(31、65、125和250mm)洗脱。将洗脱液级分(2ml)在4℃下针对 2.0l pbs透析过夜。脱盐的石竹素
‑
egf通过ultra
‑
15(10kda) 浓缩,并定量蛋白浓度。
[0809]
为了将适合的点击化学基团引入石竹素
‑
egf中,将相对于石竹素
ꢀ‑
egf 8倍摩尔过量的炔烃
‑
peg5‑
n
‑
羟基琥珀酰亚胺酯溶解在二甲基亚砜中,并添加到9体积的石竹素
‑
egf(1mg于0.2m nah2po4/na2hpo4中,ph 8)中。在室温下孵育4h后,通过使用pd10柱(ge
‑
healthcare, freiburg,germany)分离未结合的炔烃。具有聚合结构的点击化学是通过铜(i)
‑
催化的炔烃
‑
叠氮化物环加成进行的。将炔烃
‑
石竹素
‑ꢀ
egf(0.02mm)、树状物(0.05mm)、cuso4(0.1mm)、三(3
‑
羟丙基三唑基甲基)胺(0.5mm)和抗坏血酸钠(5mm)在室温下在温和搅动下在 0.1m nah2po4/na2hpo4,ph 8中孵育1h。然后使用pd10柱分离低分子量物质。
[0810]
为了测试本发明的功效,我们用her14细胞进行了活力测定。这些细胞是用人表皮生长因子受体稳定转染的成纤维细胞,因此是靶向毒素石竹素
‑
egf的靶细胞。将her14细胞(2,000个细胞/100μl/孔) 接种到96孔细胞培养板的孔中,并在补充有10%胎牛血清和1%青霉素/链霉素的dmem培养基中于37℃、5%co2和98%湿度下孵育 24h。然后将不同的测试物质(参见结果和图21
‑
1)以25μl的体积一式三份添加,并补充另外25μl培养基。孵育72h后,每孔加入30μl 的3
‑
(4,5
‑
二甲基噻唑
‑2‑
基)
‑
2,5
‑
二苯基溴化四唑(0.5mg/ml于水中)并孵育2h。此后,小心移除培养基,用含有10%(v/v)异丙醇、5%(w/v) 十二烷基硫酸钠和400mm hcl的水溶液代替,并孵育5min。在酶标仪(spectra max 340pc,molecular devices,sunnyvale,ca,usa)中在 570nm下光度学定量溶解的甲臜。将未处理的细胞归一化至1,并且所有样品均参照未处理的对照。通过未配对的两样品t检验确定显著性。
[0811]
结果
[0812]
聚合结构,在该实施例中是五聚物的树状物(pentrimer),在不存在或存在sa1641的情况下对靶细胞都没有任何细胞毒性效应(图21
‑
1,第2列和第3列)。在不存在支架的情况下,靶向毒素(石竹素
‑
egf)在 0.1nm浓度下显示出半数最大毒性(第4列)。在sa1641的存在下,相同的浓度导致所有细胞死亡,表明sa1641用作内体逃逸增强剂的一般能力(第5列)。聚合结构的存在在存在或不存在sa1641的情况下均不影响石竹素
‑
egf的毒性(第6列和第7列),表明支架不影响石竹素
ꢀ‑
egf的毒性。为了经由点击化学将模型聚合结构偶联至石竹素
‑
egf 的示例性药物活性物质,该物质必须先与炔烃基团偶联。药物活性物质的制造商可以在合成期间在他选择的物质活性保持不受影响的位置处将点击位置直接引入到该物质中。当点击炔烃修饰的药物活性物质至聚合结构时没有另外的活性损失,表明聚合结
构本身是无毒的。
[0813]
实施例7
[0814]
考虑到可用于与so1861分子发生缀合反应的化学基团,已经鉴定了四种化学基团。糖残基的醇和二醇、三萜主链上的醛基团、糖残基之一上的羧酸(葡糖醛酸)和三萜主链上的烯烃基团,如图19
‑
1中突出显示。
[0815]
鉴于每种鉴定的化学基团的优点和缺点(表1),醛基团和醇基团最适合于可逆缀合反应,而烯烃和羧酸(葡糖醛酸)是最适合于不可逆/稳定缀合反应的基团。然而,与醇相比,so1861分子结构内的醛基团最适合于可逆缀合反应。一方面,因为结构中仅存在一种醛,其允许进行化学选择性反应。另一方面,因为醛可以与多种化学基团,如胺、酰肼和羟胺进行可逆缀合反应,形成酸可裂解的部分,像亚胺、腙和肟。该因素使得可以自由选择化学基团以进行期望的可逆缀合反应。相反,醇也是经由形成缩醛和缩酮进行可逆缀合反应的良好候选物,但缺乏化学选择性,因为它们大量存在于糖苷结构上。
[0816]
对于不可逆和稳定键的形成,羧酸是最适合的,因为它可以用肽化学中使用的常用工具形成酰胺和酯(如,经由碳二亚胺介导的酰胺形成与胺反应)。
[0817]
表1.可用于皂苷缀合反应的官能团
[0818][0819][0820]
关于用于与聚合结构缀合的理想emch间隔子长度,计算机模拟 (perkinelmer,chembio3d,ver.13.0.0.3015)显示so1861
‑
emch上的马来酰亚胺基团位于分子的外围,因
此应可以接近带有硫醇的聚合结构 (图27
‑
1)。
[0821]
作为聚合结构,具有16个官能性氨基末端基团和一个叠氮基基团(在焦点处)的g4
‑
树枝化基元(pfd
‑
g4
‑
叠氮化物
‑
nh
‑
boc,polymerfactory)用于与so1861缀合(图24
‑
1)。与树状物相比,使用树枝化基元的优势是树枝化基元结构表现出的焦点。
[0822]
在所讨论的聚合物中开发so1861支架的另一种方法和蛋白质方法是聚(so1861)方法。该方法的理念是生成仅由so1861分子组成的聚合物,其具有释放so1861的ph敏感性可裂解键。此外,聚(so1861) 应该能够与毒素和生物聚合物进行缀合反应。这种方法的主要目标是使其尽可能简单和具有成本效益。由于已经开发了用于生成酸可裂解的so1861的方案(so1861
‑
emch方法),因此有趣的是看看是否有可能通过简单添加聚合引发剂来聚合so1861
‑
emch而无需进一步修饰 so1861或鉴定so1861分子上的其它缀合位点。过去,有几篇论文讨论了通过使用自由基引发剂来聚合马来酰亚胺基团,所述自由基引发剂攻击马来酰亚胺基团的双键,从而引发沿着马来酰亚胺双键的自由基聚合。由于so1861
‑
emch在其结构中揭示了马来酰亚胺基团,该基团可潜在地被探索用于自由基聚合反应,以产生具有酸可裂解官能团的聚(so1861)。如果聚合反应具有合理的反应时间,则生成的 so1861聚合物可以用自由基淬灭剂淬灭,所述自由基淬灭剂不仅淬灭反应,而且还生成用于毒素或生物聚合物缀合的官能团。在这里,过硫酸铵(aps)和四甲基乙二胺(tmeda)的系统以示例性方式表示为自由基生成器,并且氨基丙硫醇用作模型自由基淬灭剂。使用氨基丙硫醇作为示例性淬灭剂,生成的胺基团可被进一步特异性地修饰为可点击基团,或者用于将聚(so1861)直接缀合至毒素。
[0823]
另一种用于开发so1861支架的方法是dna方法。这种方法的理念是利用所谓的dna
‑
折纸(dna
‑
origami)的概念(kolb等人,2004; bird等人,1988)。dna
‑
折纸作为用于缀合皂苷至其上的聚合物或组装的聚合结构,可以提供几个固有的优点,包括稳定性、可扩展性以及对所得dna
‑
皂苷支架的最终尺寸和形状的精确控制。由于这些 dna纳米载体由天然dna组成,因此它们是生物相容的并且对活细胞不显示毒性,并且可以减轻货物从内部细胞区室释放。此类结构的多价性可以进一步允许各种有效载荷(如荧光团和毒素)的微调靶向能力和高容量。因此,在该方法中,鉴定出的dna链分别在3’和5’末端上提供化学官能团,并且能够仅在序列的某些所需区域中进行杂交,从而允许控制构建体的最终形状。应该利用化学基团来偶联皂苷,例如通过已经开发的so1861
‑
emch与3’和5’dna链之一上的硫醇基团之间的硫醇
‑
烯反应。互补dna链可以提供可用于与靶向毒素偶联的点击官能团。该概念在图23
‑
1中示出。
[0824]
通过使用能够结合并释放皂苷以及可以聚合,从而形成大的聚(肽) 样结构的特定肽序列而不是dna链,可以想象到类似的方法。在该方法中,已经鉴定并购买了长度与so1861
‑
emch分子的计算大小相匹配的肽序列,其在序列中间提供了半胱氨酸残基并在n末端和c末端处获得了胺基团。半胱氨酸残基可用于经由so1861
‑
emch的马来酰亚胺基团和半胱氨酸残基的硫醇基团的硫醇
‑
烯反应缀合so1861
‑ꢀ
emch。两个胺基团可用于聚合肽
‑
so1861缀合物与适合的交联剂。
[0825]
实施例8
[0826]
so1861
‑
bna寡核苷酸缀合
[0827]
将hsp27 bna寡核苷酸二硫化物(1.10mg,0.187μmol)溶解在具有1.0mm tcep的
20mm nh4hco3(500μl)中,将混合物摇动1min 并在室温下静置。1小时后,通过使用截留分子量为3000da的离心过滤器(14000
×
g,持续30min)过滤反应混合物。将残余物溶液用具有1.0mm tcep的20mm nh4hco3(500μl)稀释,并在上述相同条件下再次过滤所得混合物。将残余物溶液用20mm nh4hco3/乙腈(3:1, v/v,1.00ml)稀释,并将所得混合物加入到so1861
‑
emch(3.54mg, 0.375μmol)中。将反应混合物摇动1分钟并在室温下静置。10min后,将反应混合物进行制备型lc
‑
ms
4a
。将对应于产物的级分立即汇集在一起,冷冻并冻干过夜,得到呈白色蓬松固体的标题化合物 (1.25mg,85%)。基于lc
‑
ms的纯度:100%。
[0828]
lrms(m/z):1561[m
‑
5]5‑
,1951[m
‑
4]4‑
[0829]
lc
‑
ms r.t.(min):2.46
6b
[0830]
树枝化基元(so1861)4‑
bna寡核苷酸缀合
[0831]
将hsp27 bna寡核苷酸二硫化物(1.1mg,0.187μmol)溶解在具有 1.0mm tcep的20mm nh4hco3(500μl)中,将混合物摇动1min并在室温下静置。1小时后,通过使用截留分子量为3000da的离心过滤器(14000
×
g,持续30min)过滤反应混合物。将残余物溶液用具有 1.0mm tcep的20mm nh4hco3(500μl)稀释,并在上述相同条件下再次过滤所得混合物。将残余物溶液用20mm nh4hco3/乙腈(3:1, v/v,1.0ml)稀释,并将所得混合物加入到树枝化基元(so1861)4
‑
马来酰亚胺1(3.54mg,0.375μmol)中。将反应混合物摇动1分钟并在室温下静置。10min后,将反应混合物进行制备型lc
‑
ms
4a
。将对应于产物的级分立即汇集在一起,冷冻并冻干过夜,得到呈白色蓬松固体的标题化合物(1.25mg,85%)。基于lc
‑
ms的纯度:94%。
[0832]
lrms(m/z):1896[m
‑
8]8‑
,2167[m
‑
7]7‑
[0833]
lc
‑
ms r.t.(min):3.77
6b
[0834]
细胞培养
[0835]
将细胞以5,000c/w接种在96孔板中的补充有10%胎牛血清(pan
‑ꢀ
biotech gmbh)和1%青霉素/链霉素(pan
‑
biotech gmbh)的 dmem(pan
‑
biotech gmbh)(100μl/孔)中,并在37℃和5%co2下孵育过夜。第二天,在dmem中制备样品并处理细胞。
[0836]
基因沉默
[0837]
根据标准程序和方案进行rna分离和qpcr分析。
[0838]
hsp27引物:f:r:
[0839]
hsp27bna寡核苷酸
[0840]
hsp27bna(
‑
硫醇)寡核苷酸(序列5
’‑
ggcacagccagtggcg
‑ꢀ3’
)(zhang等人,2011)在bio
‑
synthesis inc.(lewisville,texas)订购。
[0841]
结果
[0842]
将bna寡核苷酸,即靶向癌症靶标(在癌细胞中上调)的mrna转录物的反义bna寡核苷酸热休克蛋白27(hsp27bna)缀合至so1861
‑ꢀ
emch(hsp27bna
‑
l
‑
so1861)或树枝化基元(
‑
l
‑ꢀ
so1861)4(hsp27bna
‑
树枝化基元(
‑
l
‑
so1861)4),并共施用至根据本发明的a431癌细胞系。作为读出,测定a431细胞中hsp27 mrna的基因沉默。这揭示了与单独的hsp27bna相比,hsp27bna
‑
l
‑
so1861 处理导致hsp27基因沉默活性的改善,然而与单独的hsp27bna的基因沉默活性相比,hsp27bna
‑
树枝化基元(
‑
l
‑
so1861)4(4个so1861 分子/bna)的活性甚至更强(3倍)(图1
‑
3)。这显示了由于so1861介导的内体逃逸和反义bna的细胞质递送的增强,一个或多个so1861分子的缀合改善了治疗性bna寡核苷酸的基因沉默活性。
[0843]
实施例9
[0844]
so1861经由半胱氨酸残基(cys)缀合(不稳定的)并且石竹素(蛋白质毒素)经由赖氨酸残基(lys)缀合(稳定的)至西妥昔单抗(识别和结合人egfr的单克隆抗体),导致西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
s
‑
石竹素)2的产生。在a431(egfr
)异种移植小鼠肿瘤模型中测试缀合物的egfr肿瘤靶向的细胞杀伤,如图9
‑
4所示。在第12天开始给药,此时肿瘤大小达到~150mm3,并且在每次给药后测定肿瘤体积。在第 12天用0.5mg/kg,第15天用1mg/kg和第24天用1.5mg/kg的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
s
‑
石竹素)2或西妥昔单抗
‑
(lys
‑
s
‑
石竹素)
1,6
处理小鼠(n=3)(腹膜内;i.p.;剂量递增)。在第26天,与对照组相比,在用西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
s
‑
石竹素)2处理的荷瘤小鼠中可以观察到肿瘤体积减小(图1
‑
4a)。这显示了so1861与抗体
‑ꢀ
蛋白质毒素(稳定的)缀合物的不稳定缀合可以增强肿瘤靶向抗体
‑
蛋白质毒素的靶向治疗功效,从而诱导更有效的肿瘤靶向疗法。
[0845]
接下来,so1861经由半胱氨酸残基(cys)缀合(不稳定的)并且石竹素(蛋白质毒素)经由赖氨酸残基(lys)缀合(不稳定的)至西妥昔单抗(识别和结合人egfr的单克隆抗体),导致西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,9
(lys
‑
l
‑
石竹素)2的产生。在a431(egfr
)异种移植小鼠肿瘤模型中测试缀合物的egfr肿瘤靶向的细胞杀伤,如图9
‑
4所示。在第12天开始给药,此时肿瘤大小达到~150mm3,并且在每次给药后测定肿瘤体积。在第12天用0.5mg/kg,第15天用1mg/kg,第24天用1.5mg/kg的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
l
‑
石竹素)2或西妥昔单抗
‑
(lys
‑
l
‑
石竹素)
1,6
处理小鼠(n=3)(腹膜内;i.p.;剂量递增)。这揭示了,35天后,与对照相比,用西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑ꢀ
l
‑
石竹素)2处理的荷瘤小鼠显示出肿瘤生长抑制(图1
‑
4b)。当在第12 天用0.5mg/kg,第15天用1mg/kg,第18天用2mg/kg,第24天用 2.5mg/kg的根据本发明的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
l
‑
石竹素)2处理小鼠(n=3;(静脉内,i.v.;剂量递增)时,还可观察到与对照相比肿瘤生长抑制(数据表示1只小鼠,因为2只小鼠在处理期间死亡)。这显示了so1861与抗体
‑
蛋白质毒素(不稳定的)缀合物的不稳定缀合可以增强肿瘤靶向抗体
‑
蛋白质毒素的靶向治疗功效,从而诱导更有效的肿瘤靶向疗法。
[0846]
接下来,so1861
‑
emch经由半胱氨酸残基(cys)以dar 3,9缀合至西妥昔单抗(识别并结合人类egfr的单克隆抗体),并且反义 hsp27bna寡核苷酸(靶向并诱导癌细胞中肿瘤靶标hsp27 mrna降解(基因沉默))经由不稳定(l)接头以dar 1,8缀合至抗体的赖氨酸残基(lys),导致西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
l
‑
hsp27bna)
1,8
的产生。根据本发明,在a431异种移植
‘
裸’鼠肿瘤模型中测试西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
l
‑
hsp27bna)
1,8
的egfr介导的肿瘤靶向的hsp27基因沉默,如图10
‑
4所示。在第12天开始给药,此时肿瘤大小达到~150mm3,并且测定hsp27 mrna表达。为此,在第一次给药后72小时收集肿瘤样品并分析与细胞对照mrna表达水平(参考基因) 相比的hsp27基因表达水平。与单次给药西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
或西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)
1,5
相比,用30mg/kg西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
l
‑
hsp27bna)
1,8
处理(腹膜内;i.p.)的荷瘤小鼠(n=3)显示了在一次给药后肿瘤细胞中hsp27 mrna表达降低40%(图2
‑
4)。与媒介物对照肿瘤相比,观察到25%的hsp27基因表达降低。这显示并使得根据本发明的so1861和hsp27bna与相同靶向抗体的缀合有效地诱导了so1861介导的治疗性反义寡核苷酸在荷瘤小鼠的实体瘤中的细胞质递送增强,从而诱导肿瘤靶向基因沉默。
[0847]
在另一个实例中,设计并产生了三官能接头支架,其具有3个特异性化学末端基团,用于在一个臂上与so1861缀合并且在另一个臂上与hsp27bna缀合以产生so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna。接下来,so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna用其第三臂缀合至抗egfr 抗体西妥昔单抗(西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑ꢀ
hsp27bna)
3,7
)的半胱氨酸残基(cys),并且根据本发明,在a431异种移植
‘
裸’鼠肿瘤模型中测试egfr介导的肿瘤靶向基因沉默活性,如图11
‑
4中所示。在第12天开始给药,此时肿瘤大小达到~150mm3,并且测定hsp27 mrna表达。为此,在第一次给药后72小时收集肿瘤样品并分析与细胞对照mrna表达水平(参考基因)相比的hsp27基因表达水平。这揭示了与单次给药25mg/kg西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
或25mg/kg西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4单一疗法相比, 1次给药30mg/kg西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑ꢀ
hsp27bna)
3,7
导致肿瘤中hsp27基因表达降低40%(图3
‑
4)。与媒介物对照肿瘤相比,在用1剂量的西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna)
3,7
处理的荷瘤小鼠中观察到hsp27基因表达降低 25%。这显示并使得西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑ꢀ
hsp27bna)
3,7
有效地诱导了so1861介导的治疗性反义核苷酸在荷瘤小鼠的实体瘤中的细胞质递送增强,从而在体内诱导靶向基因沉默。
[0848]
实施例10
[0849]
在根据本发明的另一个实例中,so1861(不稳定的)和蛋白质毒素石竹素(不稳定或稳定的)缀合至her2靶向抗体曲妥珠单抗。产生曲妥珠单抗
‑
(cys
‑
l
‑
so1861)
3,8
(lys
‑
l
‑
石竹素)
1,7
或曲妥珠单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
(lys
‑
s
‑
石竹素)
1,7
,并在sk
‑
br
‑
3(her2
)和mda
‑
mb
‑ꢀ
468(her2
‑
)细胞中测试其增强的细胞杀伤,如图9
‑
4所示。曲妥珠单抗
‑
(cys
‑
l
‑
so1861)
3,8
(lys
‑
l
‑
石竹素)
1,7
(ic50=0,8nm)和曲妥珠单抗
‑ꢀ
(cys
‑
l
‑
so1861)
3,8
(lys
‑
s
‑
石竹素)
1,7(
ic50=0,8nm)两者有效地诱导了 sk
‑
br
‑
3细胞(her2
)的细胞杀伤(图4
‑
4a)。这在用单独的曲妥珠单抗、曲妥珠单抗
‑
(lys
‑
l
‑
石竹素)
1,7
、曲妥珠单抗
‑
(lys
‑
s
‑
石竹素)
1,7
或曲妥珠单抗
‑
(l
‑
so1861)
3,8
处理的sk
‑
br
‑
3细胞中未观察到(图4
‑
4a)。在 mda
‑
mb
‑
468细胞(her2
‑
)中,未能观察到根据本发明的任何缀合物的细胞杀伤活性(图4
‑
4b)。这显示了so1861与her靶向抗体
‑
蛋白质毒素缀合物的缀合有效地诱导了so1861介导的蛋白质毒素在靶细胞中的细胞质递送增强,从而导致靶细胞死亡。
[0850]
在根据本发明的另一个实例中,so1861(不稳定的)和蛋白质毒素石竹素(不稳定或稳定的)缀合至egfr靶向抗体西妥昔单抗。在a431 细胞(egfr
)和a2058细胞(egfr
‑
)中测试西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,9
(lys
‑
l
‑
石竹素)2或西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
s
‑
石竹素)2的细胞杀伤增强,如图9
‑
4所示。与单独的西妥昔单抗
‑
(lys
‑
l
‑ꢀ
石竹素)
1,6(
ic50=2pm)、西妥昔单抗
‑
(lys
‑
s
‑
石竹素)
1,6
(ic5=2pm)相比,西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
(lys
‑
l
‑
石竹素)2(ic50=0,3nm)和西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
(lys
‑
s
‑
石竹素)
1,7(
ic50=0,3nm)两者显示出在 a431细胞(egfr
)中增强的细胞杀伤(图4
‑
4c)。在a2058细胞(egfr
‑
) 中,根据本发明的组合未显示出任何细胞杀伤活性(ic50>200nm;图 4
‑
4d)。这显示了so1861与egfr靶向抗体
‑
蛋白质毒素缀合物的缀合有效地增强了so1861介导的蛋白质毒素在靶细胞中的细胞质递送,从而导致增强的靶细胞死亡。
[0851]
实施例11
[0852]
在根据本发明的另一个实例中,so1861(不稳定的)和hsp27bna 寡核苷酸(不稳定
的)缀合至egfr靶向抗体西妥昔单抗。根据本发明,在a431细胞(egfr
)和a2058(egfr
‑
)细胞中测试西妥昔单抗
‑
(cys
‑ꢀ
l
‑
so1861)
3,8
(lys
‑
l
‑
hsp27bna)
3,8
的增强的hsp27基因沉默,如图10
‑ꢀ
4所示。与单独的西妥昔单抗、西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)
3,9
或西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
相比,西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
(lys
‑ꢀ
l
‑
hsp27bna)
3,8
在a431细胞中有效地诱导了hsp27基因沉默 (ic50=3nm)(图5
‑
4a)。在a2058细胞(egfr
‑
)中,用西妥昔单抗
‑
(cys
‑ꢀ
l
‑
so1861)
3,8
(lys
‑
l
‑
hsp27bna)
3,8
未观察到基因沉默活性(ic50> 100nm;图5
‑
4b)。这显示并使得根据本发明的so1861和hsp27bna 与相同靶向抗体的缀合有效地诱导了so1861介导的治疗性反义寡核苷酸在靶细胞中的细胞质递送增强,从而诱导靶向基因沉默。
[0853]
在根据本发明的另一个实例中,so1861(不稳定的)和hsp27bna 寡核苷酸(不稳定的)缀合至her2靶向抗体曲妥珠单抗。根据本发明,在sk
‑
br
‑
3细胞(her2
)细胞中测试曲妥珠单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
(lys
‑
l
‑
hsp27bna)
3,5
的增强的hsp27基因沉默,如图10
‑
4 所示。与单独的曲妥珠单抗
‑
(lys
‑
l
‑
hsp27bna)
4,4
相比,曲妥珠单抗
‑ꢀ
(cys
‑
l
‑
so1861)
3,8
(lys
‑
l
‑
hsp27bna)
3,5
在sk
‑
br
‑
3细胞中有效地诱导了hsp27基因沉默(ic50=9nm)(图6
‑
4)。这显示并使得根据本发明的 so1861和hsp27bna与her2靶向抗体的缀合有效地诱导了so1861
‑ꢀ
介导的治疗性反义寡核苷酸在靶细胞中的细胞质递送增强,从而诱导靶向基因沉默。
[0854]
在另一个实例中,根据本发明,在a431(egfr
)和a2058(egfr
‑
) 细胞中测试西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna)
3,7
的增强的hsp27基因沉默,如图11
‑
4所示。与单独的西妥昔单抗
‑
(lys
‑ꢀ
l
‑
hsp27bna)4或西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
相比,西妥昔单抗
‑ꢀ
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna)
3,7
在a431细胞中有效地诱导了hsp27基因沉默(ic50=2nm)(图7
‑
4a)。在a2058细胞(egfr
‑
)中,基因沉默活性仅在高(>80nm)浓度的西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna)
3,7
下观察到(ic50=100nm;图7
‑
4b)。这显示并使得在高表达egfr的细胞中,西妥昔单抗
‑
cys
‑
(so1861
‑
l
‑
三官能接头
‑
l
‑
hsp27bna)
3,7
有效地诱导了so1861介导的治疗性反义寡核苷酸在靶细胞中的细胞质递送增强,从而诱导靶向基因沉默。
[0855]
实施例12
[0856]
图8
‑
4a
‑
d显示了当将曲妥珠单抗(图8
‑
4a)、西妥昔单抗(图8
‑
4b) 或t
‑
dm1(图8
‑
4c)、未缀合的蛋白质毒素皂草素、石竹素和缀合至(非细胞结合)igg抗体的皂草素(图8
‑
4d)施用至各种癌细胞系sk
‑
br
‑
3、 jimt
‑
1、mda
‑
mb
‑
468、a431、caski、hela、a2058时的相对细胞活力。
[0857]
曲妥珠单抗和西妥昔单抗在暴露于大多数细胞系时不影响或几乎不影响细胞活力,而当曲妥珠单抗以相对高剂量暴露于sk
‑
br
‑
3细胞时,经由阻断her2生长因子受体的功能而对细胞生长抑制具有一些作用,以及当西妥昔单抗以相对高剂量暴露于mda
‑
mb
‑
468细胞时经由阻断egfr生长因子受体的功能而对细胞生长抑制具有一些作用。
[0858]
tdm
‑
1或ado
‑
曲妥珠单抗美坦新是美国食品药品管理局(u.s. food and drug administration)批准用于治疗以下疾病的靶向治疗:先前已经用赫塞汀(化学名称:曲妥珠单抗)和紫杉烷化疗进行治疗的 her2阳性转移性乳腺癌;如果用赫塞汀和紫杉烷化疗进行新辅助(手术前)治疗后发现残留疾病,手术后早期her2阳性乳腺癌。tdm
‑
1是赫塞汀(曲
妥珠单抗)和化疗药物美坦新的组合。图8
‑
4c显示了tdm
‑ꢀ
1导致在>1000pm浓度下测试的所有细胞系的细胞活力降低。
[0859]
游离毒素皂草素和石竹素以及偶联至对照igg的毒素皂草素(对测试的细胞系上的任何细胞表面分子没有亲和力)在测试的宽范围的毒素浓度(高达100.000pm)下对细胞活力没有或几乎没有任何影响(图 8
‑
4d)。
[0860]
实施例13(实施例1发明5)
[0861]
1靶标2
‑
组分系统(1t2c)是mab1
‑
蛋白质毒素和mab1
‑
so1861的组合治疗,如图13
‑
5所示。so1861
‑
emch经由半胱氨酸残基(cys)缀合缀合至西妥昔单抗(识别并结合人类egfr的单克隆抗体)并且 hsp27bna寡核苷酸经由赖氨酸残基缀合至西妥昔单抗(识别并结合人类egfr的单克隆抗体),两者的dar均为4,导致产生2种缀合物:西妥昔单抗
‑
(cys
‑
l
‑
so1861)4和西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4。在a431异种移植小鼠肿瘤模型中测试西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)4(腹膜内施用,(i.p.))和西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4(静脉内施用,(i.v.))的组合的egfr肿瘤靶向基因沉默活性。在第12天开始给药,此时肿瘤大小达到~150mm3,并在第一次给药后72h收集肿瘤样品,并分析与对照基因mrna表达(参考基因)相比的hsp27基因表达。这揭示了,与单次给药西妥昔单抗
‑
(cys
‑
l
‑
so1861)4或西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4单一疗法相比,1次给药50mg/kg西妥昔单抗
ꢀ‑
(cys
‑
l
‑
so1861)
4
25mg/kg西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4导致 a431肿瘤中hsp27基因表达降低50%(图1
‑
5)。与媒介物对照肿瘤相比,观察到40%的hsp27基因沉默降低。这显示并且使得根据1t2c 发明的西妥昔单抗
‑
缀合的so1861 西妥昔单抗
‑
缀合的hsp27bna寡核苷酸的组合诱导治疗性反义寡核苷酸在实体瘤细胞的细胞质中的有效靶向递送,从而在体内诱导肿瘤靶向基因沉默。
[0862]
接下来,so1861
‑
emch经由半胱氨酸残基(cys)以dar 4缀合至曲妥珠单抗(识别并结合人类her2的单克隆抗体),导致产生曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4。在具有高her2表达水平并对曲妥珠单抗单一疗法具有抗性的小鼠肿瘤模型(患者来源的异种移植肿瘤模型,pdx) 中,测试了曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4和曲妥珠单抗
‑
皂草素(曲妥珠单抗蛋白质毒素缀合物)的组合。根据1t2c发明的40mg/kg曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4(腹膜内施用,(i.p.)) 0.03(第1天、第8天)/0.02(第 15天、第22天、第30天、第36天、第43天)mg/kg曲妥珠单抗
‑
皂草素(静脉内施用,(i.v.))的组合揭示了与媒介物对照和40mg/kg曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4或0.03/0.02mg/kg曲妥珠单抗
‑
皂草素单一疗法相比,强烈的肿瘤生长抑制(图2
‑
5)。此外,在用更低给药组合(40 mg/kg曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4 0.01mg/kg曲妥珠单抗
‑
皂草素)处理的荷瘤小鼠中,未观察到肿瘤生长抑制活性(图2
‑
5)。这显示并使得曲妥珠单抗缀合的so1861 曲妥珠单抗缀合的蛋白质毒素的1t2c组合诱导治疗性蛋白质毒素在实体瘤细胞的细胞质中的有效靶向递送,从而在体内诱导肿瘤细胞死亡和肿瘤生长抑制。
[0863]
实施例14
[0864]
1靶标2
‑
组分系统(1t2c)是mab1
‑
so1861和mab1
‑
蛋白质毒素的组合治疗(图13
‑
5)
[0865]
so1861
‑
emch经由半胱氨酸残基(cys)以dar 3,7缀合至西妥昔单抗(识别并结合人类egfr的单克隆抗体)(西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,7
)。在固定浓度的10pm西妥昔单抗
‑
皂草素(缀合至蛋白质毒素皂草素的西妥昔单抗)上滴定西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
,
并且测定靶向蛋白质毒素介导的对表达egfr的细胞(a431,egfr
; caski,egfr
)的细胞杀伤。这揭示了在低浓度的西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,7
下强烈的细胞杀伤(a431:ic50=0,6nm和caski ic50=1nm;图5a,3
‑
5b),而西妥昔单抗、西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
或西妥昔单抗 10pm西妥昔单抗
‑
皂草素不能在表达egfr的细胞中诱导任何细胞杀伤活性。这显示了西妥昔单抗缀合的so1861有效地增强了西妥昔单抗缀合的蛋白质毒素(在非有效浓度下)的内体逃逸,从而诱导表达egfr的细胞的细胞杀伤。与在这些细胞系中同egfr表达水平相关的caski相比,在a431中的细胞杀伤活性更有效。当西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
浓度增加,细胞杀伤活性由于超过西妥昔单抗
ꢀ‑
皂草素的受体结合和内化而下降时,也观察到1t2c内的两种缀合物之间的egfr受体结合竞争(图3
‑
5a,3
‑
5b)。
[0866]
接下来,在固定浓度的75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
上滴定西妥昔单抗
‑
皂草素,并且测定靶向蛋白质毒素介导的对表达egfr 的细胞的细胞杀伤。这揭示了75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
与低浓度西妥昔单抗
‑
皂草素的组合在表达egfr的细胞中诱导已经有效的细胞杀伤(a431:ic50=0.4pm;和caski:(ic50=2pm;图3
‑
5c和 3
‑
5d),然而单独的西妥昔单抗
‑
皂草素或西妥昔单抗
‑
皂草素 75nm西妥昔单抗仅在高浓度的西妥昔单抗
‑
皂草素下在两种细胞系中显示出细胞杀伤(分别为ic50=40pm,ic50=1000pm)(图3
‑
5c,3
‑
5d)。所有这些都显示,相对低浓度的西妥昔单抗
‑
皂草素只有与低浓度的西妥昔单抗
‑
so1861组合才可以在高表达egfr的细胞中有效并诱导细胞杀伤。当比较具有和没有75nm西妥昔单抗的情况下西妥昔单抗
‑
皂草素的细胞杀伤活性时,也在西妥昔单抗
‑
毒素滴定处理中观察到在1t2c 系统内两种缀合物之间的受体竞争(图3
‑
5c,3
‑
5d)。
[0867]
接下来,在固定浓度的10pm西妥昔单抗
‑
皂草素上滴定西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
,并且测定靶向蛋白质毒素介导的对低表达 egfr的细胞或无egfr表达的细胞(hela,egfr
/
‑
;a2058,egfr
‑
)的细胞杀伤。具有低(hela)或无(a2058)egfr表达的细胞对西妥昔单抗
ꢀ‑
(cys
‑
l
‑
so1861)
3,7
10pm西妥昔单抗
‑
皂草素的任何组合根本不敏感 (hela:ic50>1000nm;a2058:ic50>1000nm;图4
‑
5a,4
‑
5b)。这显示了在足够egfr受体表达的不存在下,有效细胞内递送的so1861 浓度不是诱导内体蛋白质毒素逃逸和毒素介导的细胞杀伤的最佳浓度 (阈值)。接下来,在固定浓度的75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
上滴定西妥昔单抗
‑
皂草素,并且测定靶向蛋白质毒素介导的对低 (hela)或无(a2058)表达egfr的细胞的细胞杀伤。低表达egfr的细胞(hela)仅在高西妥昔单抗
‑
皂草素浓度与75nm西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,7
的组合下显示出细胞杀伤(hela:ic50=60pm),图4
‑
5c),而a2058细胞(egfr
‑
)在任何测试的浓度下均不敏感(a2058:ic50> 10.000pm;图4
‑
5d)。所有这些都显示,具有低或无egfr受体表达的细胞对西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
西妥昔单抗
‑
皂草素的组合不敏感,这是由于缺乏足够的egfr受体,其促进抗体介导的足够的 so1861在内溶酶体区室中的递送,以促进蛋白质毒素的逃逸。
[0868]
接下来,so1861
‑
emch经由半胱氨酸残基(cys)以dar 4缀合至曲妥珠单抗(识别并结合人类her2的单克隆抗体,(曲妥珠单抗
‑
(cys
‑ꢀ
l
‑
so1861)4)。在固定浓度的50pm曲妥珠单抗
‑
皂草素(缀合至蛋白质毒素皂草素的曲妥珠单抗)上滴定曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4,并且测定靶向蛋白质毒素介导的对表达her2的细胞(sk
‑
br
‑
3,her2
) 的细胞杀伤。这揭示了在低浓度的曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4下强烈的细胞杀伤(sk
‑
br
‑
3:ic50=0,8nm;图5
‑
5a),然而等浓度的曲妥珠单抗、曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4或曲妥珠单抗
50pm曲妥珠单抗
‑
皂草素不能在表达her2的细胞中诱导任何细胞杀伤活性。这显示了曲妥珠单抗缀合的so1861有效地增强了曲妥珠单抗缀合的蛋白质毒素(在非有效浓度下)的内体逃逸,从而诱导表达her2的细胞的细胞杀伤。当曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4浓度增加,细胞杀伤活性由于超过曲妥珠单抗
‑
皂草素的受体结合和内化而下降时,也观察到1t2c内的两种缀合物之间的受体竞争(图5
‑
5a)。
[0869]
接下来,根据本发明,在固定浓度的2,5nm曲妥珠单抗
‑
(cys
‑
l
‑ꢀ
so1861)4上滴定曲妥珠单抗
‑
皂草素,并且测定靶向蛋白质毒素介导的对表达her2的细胞的细胞杀伤。这揭示了2,5nm曲妥珠单抗
‑
(cys
‑ꢀ
l
‑
so1861)4与低浓度曲妥珠单抗
‑
皂草素的组合在表达her2的细胞中诱导已经有效的细胞杀伤(sk
‑
br
‑
3:ic50=2pm;图5
‑
5b),然而单独的曲妥珠单抗
‑
皂草素或曲妥珠单抗
‑
皂草素 2,5nm曲妥珠单抗仅在高浓度的曲妥珠单抗
‑
皂草素下显示出细胞杀伤(图5
‑
5b)。所有这些都显示,相对低浓度的曲妥珠单抗
‑
皂草素只有与低浓度的曲妥珠单抗
ꢀ‑
(cys
‑
l
‑
so1861)4组合才可以在高表达her2的细胞中有效并诱导细胞杀伤。
[0870]
接下来,根据本发明,在固定浓度的50pm曲妥珠单抗
‑
皂草素上滴定曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4,并且测定靶向蛋白质毒素介导的对低表达her2的细胞(a431 her2
/
‑
)或无her2表达的细胞(jimt
‑
1: her2
;mda
‑
mb
‑
468:her2
‑
)的细胞杀伤。具有低或无her2表达的细胞对曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4 50pm曲妥珠单抗
‑
皂草素的任何组合根本不敏感(jimt
‑
1:ic50>1000nm;mda
‑
mb
‑
468:ic50>1000 nm;图6
‑
5a,6
‑
5b)。这显示了在足够her2受体表达的不存在下,有效细胞内递送的so1861浓度不是诱导内体蛋白质毒素逃逸和毒素介导的细胞杀伤的最佳浓度(阈值)。接下来,在固定浓度的2,5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4上滴定曲妥珠单抗
‑
皂草素,并且测定靶向蛋白质毒素介导的对低或无表达her2的细胞的细胞杀伤。低表达 her2的细胞(jimt
‑
1)仅在高的曲妥珠单抗
‑
皂草素浓度与2,5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4的组合下显示出细胞杀伤(jimt
‑
1:ic50> 10.000pm;图6
‑
5c),然而mda
‑
mb
‑
468细胞(her2
‑
)在任何测试浓度下都不敏感(mda
‑
mb
‑
468:ic50>10.000pm;图6
‑
5d)。
[0871]
所有这些都显示,具有低或无her2受体表达的细胞对曲妥珠单抗
‑
(cys
‑
l
‑
so1861)
3,7
曲妥珠单抗
‑
皂草素的组合不敏感,这是由于缺乏足够的egfr受体,其促进抗体介导的足够的so1861在内溶酶体区室中的递送,以促进蛋白质毒素的逃逸。
[0872]
实施例15
[0873]
为了显示1t2c系统的活性是由内溶酶体区室的酸化驱动的,根据本发明的1t2c系统与内体酸化抑制剂氯喹组合进行了测试。将曲妥珠单抗
‑
皂草素与5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4(与氯喹组合或没有氯喹)的组合滴定。曲妥珠单抗
‑
皂草素 5nm曲妥珠单抗
‑
(cys
‑
l
‑ꢀ
so1861)4在高表达her2的细胞中显示了强烈的细胞杀伤活性(sk
‑ꢀ
br
‑
3,her2
;ic50=0.2pm;),然而曲妥珠单抗
‑
皂草素 5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)
4
0.5μm氯喹导致在sk
‑
br
‑
3(her2
)细胞中对1t2c细胞杀伤活性的强烈抑制(ic50=40pm)。这显示了当抑制内溶酶体的酸化时,降低/阻断抗体缀合的so1861的活性(图7
‑
5a)。与西妥昔单抗
‑
皂草素 5nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
0.5μm氯喹 (ic50=200pm)相比,在表达egfr的细胞(a431,egfr
;图7
‑
5b) 中用根据本发明的西妥昔单抗
‑
皂草素 5nm西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,8
(ic50=1pm)的1t2c组合得出相同结果。
[0874]
实施例16
[0875]
1靶标2
‑
组分系统(1t2c)还可以是mab1
‑
so1861和mab1
‑
反义 bna寡核苷酸的组合治疗,如图14
‑
5所示。为此,我们使用了针对癌症特异性靶基因(在癌细胞中上调)热休克蛋白27(hsp27)的mrna的反义bna寡核苷酸。释放到细胞质中后,反义bna识别并结合编码 hsp27的mrna,靶向mrna以进行破坏,从而耗损癌细胞内的hsp27 mrna表达。hsp27bna以dar4缀合至西妥昔单抗(西妥昔单抗
‑ꢀ
(lys
‑
l
‑
hsp27bna)4)并根据本发明在表达egfr的细胞(a431,egfr
) 和不表达egfr的细胞(a2058,egfr)中测试了其与西妥昔单抗
‑
(cys
‑ꢀ
l
‑
so1861)
3,8
的组合的增强的hsp27基因沉默活性(图14
‑
5)。西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
100nm西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4在表达egfr的细胞中显示了强烈的hsp27基因沉默(a431:ic50=nm,图8
‑
5a),然而单独的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
未显示任何基因沉默活性。在a2058细胞(egfr
‑
)中,未在1t2c组合中观察到基因沉默活性(图8
‑
5b)。接下来,西妥昔单抗
‑
(lys
‑
l
‑
hsp27bna)4 76.9nm 西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
在表达egfr的细胞中显示了强烈的 hsp27基因沉默活性(a431:ic50=4nm,图8
‑
5c),然而西妥昔单抗
‑ꢀ
(lys
‑
l
‑
hsp27bna)4或西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,8
或西妥昔单抗
‑ꢀ
(lys
‑
l
‑
hsp27bna)
4
77nm西妥昔单抗的组合并未揭示任何显著的基因沉默活性(ic50>100nm)。当在不表达egfr的细胞(a2058)中进行实验时,在1t2c组合中未观察到基因沉默活性(ic50>100nm;图8
‑ꢀ
5d)。这一切显示,1t2c系统将反义bna寡核苷酸有效地递送至高表达egfr的细胞的细胞质,从而诱导bna靶标mrna的mrna降解,导致靶基因沉默。
[0876]
实施例17
[0877]
1靶标2
‑
组分系统(1t2c)还可以是mab1
‑
(支架(
‑
so1861)
n
)
n
和 mab1
‑
蛋白质毒素的组合治疗,如图15
‑
5所示。树枝化基元(
‑
l
‑ꢀ
so1861)4经由半胱氨酸残基(cys)缀合以dar3,9缀合至西妥昔单抗,并在表达egfr的细胞(mda
‑
mb
‑
468)中测试西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
与抗egfr抗体
‑
蛋白质毒素缀合物(西妥昔单抗
‑ꢀ
皂草素)的组合的增强的细胞杀伤活性。西妥昔单抗
‑
cys
‑
(树枝化基元 (
‑
l
‑
so1861)4)
3,9
10pm西妥昔单抗
‑
皂草素在高表达egfr的细胞中有效地诱导了毒素介导的细胞杀伤(ic50=0.4nm;图9
‑
5a),然而这未由西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
或西妥昔单抗 10pm 西妥昔单抗
‑
皂草素或西妥昔单抗诱导(图9
‑
5a)。这显示了根据1t2c 发明,西妥昔单抗缀合的树枝化基元(
‑
l
‑
so1861)4有效地增强了西妥昔单抗缀合的蛋白质毒素(在非有效浓度下)的内体逃逸,从而诱导高表达her2的细胞的细胞杀伤。在表达低水平的egfr的细胞(hela, egfr
/
‑
)中进行类似的1t2c实验,并且这揭示了当使用根据本发明的 1t2c组合时无细胞杀伤活性(ic50>100pm;图9
‑
5b),表明在足够 egfr受体表达的不存在下,有效的细胞内so1861浓度不是诱导细胞质递送导致毒素介导的细胞杀伤的蛋白质毒素的最佳浓度(阈值)。
[0878]
接下来,树枝化基元(
‑
l
‑
so1861)4经由半胱氨酸缀合以dar4缀合至抗her2抗体曲妥珠单抗,即曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑ꢀ
so1861)4)4,并在表达her2的细胞(sk
‑
br
‑
3,her2
)中测试其与抗 her2抗体
‑
蛋白质毒素缀合物(曲妥珠单抗
‑
皂草素)的组合的增强的细胞杀伤活性。曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4 50pm曲妥珠单抗
‑
皂草素有效地诱导了毒素介导的细胞杀伤(ic50=2nm,图9
‑ꢀ
5c),然而这未由曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4或曲妥珠单抗 50nm曲妥珠单抗
‑
皂草素或曲妥珠单抗诱导(图
9
‑
5c)。这显示了曲妥珠单抗缀合的树枝化基元(
‑
l
‑
so1861)4有效地增强了曲妥珠单抗缀合的蛋白质毒素(在非有效浓度下)的内体逃逸,从而诱导高表达 her2的细胞的细胞杀伤。在表达低水平her2的细胞(jimt
‑
1,her2
/
‑
) 中的类似实验揭示了曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4 50 pm曲妥珠单抗
‑
皂草素(ic50>200nm;图9
‑
5d)无活性,表明在足够 her2受体表达的不存在下,有效的细胞内so1861浓度不是诱导内体蛋白质毒素逃逸和毒素介导的细胞杀伤的最佳浓度(阈值)。
[0879]
实施例18
[0880]
临床批准的adc,曲妥珠单抗
‑
美坦新(t
‑
dm1)是抗her2抗体曲妥珠单抗和小分子毒素美坦新的缀合物(dar3.5)。t
‑
dm1与曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4组合滴定,并与根据本发明的抗体蛋白质毒素缀合物,即曲妥珠单抗
‑
皂草素 曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4比较。然而,与曲妥珠单抗
‑
皂草素 2.5nm曲妥珠单抗或单独的曲妥珠单抗
‑
皂草素比较时,曲妥珠单抗
‑
皂草素 2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4显示了增强的活性(ic50=2pm,图10
‑
5),t
‑
dm1 25.6nm曲妥珠单抗
‑ꢀ
(cys
‑
l
‑
so1861)4显示了没有增强的细胞杀伤活性(ic50>100pm;图10
‑
5)。这显示了根据本发明的1t2c系统不能增强抗体小分子缀合物的递送,因为小分子可能已经被动地穿过(内溶酶体)膜。
[0881]
实施例19
[0882]
图11
‑
5a
‑
d显示了当将曲妥珠单抗(图11
‑
5a)、西妥昔单抗(图11
‑ꢀ
5b)或t
‑
dm1(图11
‑
5c)、未缀合的蛋白质毒素皂草素、石竹素和缀合至(非细胞结合)igg抗体的皂草素(图11
‑
5d)施用至各种癌细胞系sk
‑ꢀ
br
‑
3、jimt
‑
1、mda
‑
mb
‑
468、a431、caski、hela、a2058时的相对细胞活力。
[0883]
曲妥珠单抗和西妥昔单抗在暴露于大多数细胞系时不影响或几乎不影响细胞活力,而当曲妥珠单抗以相对高剂量暴露于sk
‑
br
‑
3细胞时,经由阻断her2生长因子受体的功能而对细胞生长抑制具有一些作用,以及当西妥昔单抗以相对高剂量暴露于mda
‑
mb
‑
468细胞时经由阻断egfr生长因子受体的功能而对细胞生长抑制具有一些作用。
[0884]
tdm
‑
1或ado
‑
曲妥珠单抗美坦新是美国食品药品管理局批准用于治疗以下疾病的靶向治疗:先前已经用赫塞汀(化学名称:曲妥珠单抗) 和紫杉烷化疗进行治疗的her2阳性转移性乳腺癌;如果用赫塞汀和紫杉烷化疗进行新辅助(手术前)治疗后发现残留疾病,手术后早期 her2阳性乳腺癌。tdm
‑
1是赫塞汀(曲妥珠单抗)和化疗药物美坦新的组合。图11
‑
5c显示了tdm
‑
1导致在>1000pm浓度下测试的所有细胞系的细胞活力降低。
[0885]
游离毒素皂草素和石竹素以及偶联至对照igg的毒素皂草素(对测试的细胞系上的任何细胞表面分子没有亲和力)在测试的宽范围的毒素浓度(高达100.000pm)下对细胞活力没有或几乎没有任何影响(图 11
‑
5d)。
[0886]
实施例20
[0887]
1靶标2
‑
组分系统(1t2c)还可以是mab1
‑
qsmix(来自皂皮树的皂苷的混合物)和mab1
‑
蛋白质毒素的组合治疗。
[0888]
qsmix
‑
emch经由半胱氨酸残基(cys)以dar 4.1缀合至西妥昔单抗(识别并结合人类egfr的单克隆抗体)(西妥昔单抗
‑
(cys
‑
l
‑ꢀ
qsmix)
4,1
)。在固定浓度的10pm西妥昔单抗
‑
皂草素或10pm西妥昔单抗
‑
石竹素上滴定西妥昔单抗
‑
(cys
‑
l
‑
qsmix)
4,1
,并且测定靶向蛋白质毒素介导的对a431(egfr
)、caski(egfr
)和a2058(egfr
‑
)细胞的细胞杀伤。这揭示了
在a431(egfr
)和caski(egfr
)细胞中在低浓度的西妥昔单抗
‑
(cys
‑
l
‑
qsmix)
4.1
10pm西妥昔单抗
‑
皂草素或 10pm西妥昔单抗
‑
石竹素下的强烈细胞杀伤(a431:ic50=3nm,图12
‑ꢀ
5a;caski:ic50=1nm,图12
‑
5b),然而所有对照处理不能在表达egfr 的细胞中诱导任何细胞杀伤。在不表达egfr的细胞(a2058;egfr
‑
) 中,用根据本发明的组合未观察到hsp27基因沉默(ic50>1000nm;图12
‑
5c)。这显示了西妥昔单抗缀合的qs21mix有效地增强了西妥昔单抗缀合的蛋白质毒素(在非有效浓度下)的内体逃逸,从而诱导仅在表达egfr的细胞中的细胞杀伤。
[0889]
实施例21:2靶标2
‑
组分系统(体内)
[0890]
2靶标2
‑
组分系统(2t2c)是mab1
‑
so1861和mab2
‑
蛋白质毒素的组合治疗(图15
‑
6;16
‑
6;17
‑
6)。so1861
‑
emch经由半胱氨酸残基 (cys)以dar 4缀合至西妥昔单抗(识别并结合人类egfr的单克隆抗体),导致产生西妥昔单抗
‑
(cys
‑
l
‑
so1861)4。在a431(egfr
/her2
/
‑ /cd71
)异种移植
‘
裸’鼠肿瘤模型中测试西妥昔单抗
‑
(cys
‑
l
‑
so1861)4和曲妥珠单抗
‑
皂草素或cd71mab
‑
皂草素的组合的egfr肿瘤靶向细胞杀伤,如图1
‑
6和图2
‑
6所示。进行剂量递增以测定治疗功效(第9 天:0.3mg/kg曲妥珠单抗
‑
皂草素或0.1mg/kg cd71mab
‑
皂草素 5 mg/kg西妥昔单抗
‑
(cys
‑
l
‑
so1861)4;第14天、第18天:0.1mg/kg曲妥珠单抗
‑
皂草素或0.05mg/kg cd71mab
‑
皂草素 5mg/kg西妥昔单抗
ꢀ‑
(cys
‑
l
‑
so1861)4;第21天:0.05mg/kg曲妥珠单抗
‑
皂草素或0.05 mg/kg cd71mab
‑
皂草素 15mg/kg西妥昔单抗
‑
(cys
‑
l
‑
so1861)4;第28 天:0.02mg/kg曲妥珠单抗
‑
皂草素或0.02mg/kg cd71mab
‑
皂草素 15 mg/kg西妥昔单抗
‑
(cys
‑
l
‑
so1861)4曲妥珠单抗
‑
皂草素/西妥昔单抗
‑ꢀ
so1861。对照分别根据相同的给药方案,每个处理日仅给予25mg/kg 西妥昔单抗(i.v.)。在第32天(虚线)、第35天和第39天,我们开始根据2t2c发明的25mg/kg西妥昔单抗
‑
(cys
‑
l
‑
so1861)4(腹膜内注射 (i.p.) 0.02mg/kg曲妥珠单抗
‑
皂草素或0.02cd71mab
‑
皂草素(静脉内施用,(i.v.))的组合,并且这揭示了与媒介物对照、25mg/kg西妥昔单抗
‑
(cys
‑
l
‑
so1861)4或0.02mg/kg曲妥珠单抗
‑
皂草素/cd71mab
‑
皂草素单一疗法相比,两个2t2c组合组的强烈肿瘤消退(图1
‑
6、2
‑
6)。 2t2c系统甚至超过西妥昔单抗,即临床上使用的针对egfr的单克隆抗体。接下来,我们进行了相同的实验,但我们用25mg/kg西妥昔单抗
‑
(cys
‑
l
‑
so1861)4(腹膜内注射(i.p.) 0.03mg/kg曲妥珠单抗
‑
皂草素或0.03cd71mab
‑
皂草素(静脉内施用,(i.v.))开始治疗,在第9天和第14天给药并且之后每周给药1次。根据本发明的2t2c系统显示了在两个2t2c组中的所有小鼠并且甚至1只小鼠的肿瘤消退,完全肿瘤根除(肿瘤体积=0mm3)(图2
‑
6)。在此对照还显示了肿瘤体积强烈增加,然而该a431小鼠模型的阳性对照西妥昔单抗仅显示了肿瘤生长抑制,但无消退(图2
‑
6)。这显示并且使得西妥昔单抗缀合的so1861 曲妥珠单抗缀合的蛋白质毒素或cd71mab缀合的蛋白质毒素的根据本发明的2t2c系统方法在体内诱导治疗性蛋白毒素在荷瘤小鼠的实体肿瘤的细胞质中的高效靶向递送,从而在一些小鼠中诱导甚至完全的肿瘤根除以及在其它小鼠中诱导甚至大尺寸肿瘤(2000mm3)的强烈的肿瘤消退。
[0891]
实施例22:2靶标2
‑
组分系统(体外)
[0892]
结果
[0893]
2靶标2
‑
组分系统(2t2c)是mab1
‑
so1861和mab2
‑
蛋白质毒素的组合治疗(还参见图1
‑
6、2
‑
6、15
‑
6、16
‑
6、17
‑
6)。so1861
‑
emch经由半胱氨酸残基(cys)以dar 3,7缀合至西妥
昔单抗(识别并结合人类 egfr的单克隆抗体)(西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
)。在固定浓度的 50pm曲妥珠单抗
‑
皂草素(缀合至蛋白质毒素皂草素的曲妥珠单抗)上滴定西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
,并且测定靶向蛋白质毒素介导的对表达egfr/her2的细胞(a431,egfr
/her2
/
‑
;caski, egfr
/her2
/
‑
)的细胞杀伤,如图3
‑
6所示。这揭示了在低浓度的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
下强烈的细胞杀伤(a431:ic50=3nm和 caski ic50=10nm;图3
‑
6a,3
‑
6b),然而等浓度的西妥昔单抗、西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
或西妥昔单抗 50pm曲妥珠单抗
‑
皂草素不能在表达egfr/her2的细胞中诱导任何细胞杀伤活性。这显示了相对低浓度的西妥昔单抗
‑
so1861缀合物有效地增强了曲妥珠单抗缀合的蛋白质毒素(在非有效浓度下)的内体逃逸,从而诱导高表达egfr/ 低表达her2的细胞的有效细胞杀伤。
[0894]
接下来,在固定浓度的75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
上滴定曲妥珠单抗
‑
皂草素,并且测定靶向蛋白质毒素介导的对表达 egfr/her2的细胞的细胞杀伤。这揭示了75nm西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,7
与低浓度曲妥珠单抗
‑
皂草素的组合在表达egfr/her2的细胞中诱导已经有效的细胞杀伤(a431:ic50=5pm;和caski:ic50=1 pm;图3
‑
6c和3
‑
6d),然而单独的曲妥珠单抗
‑
皂草素或曲妥珠单抗
‑ꢀ
皂草素 75nm西妥昔单抗在两种细胞系中未显示显著的细胞杀伤活性(ic50>10.000pm)(图3
‑
6c,3
‑
6d)。所有这些都显示,相对低浓度的曲妥珠单抗
‑
皂草素与低浓度的西妥昔单抗
‑
so1861缀合物的组合在高表达egfr/低表达her2的细胞中可以有效并诱导细胞杀伤。
[0895]
接下来,在固定浓度的50pm曲妥珠单抗
‑
皂草素上滴定西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
,并且测定靶向蛋白质毒素介导的对 hela(egfr
/
‑
/her2
/
‑
)或a2058(egfr
‑
/her2
/
‑
)的细胞杀伤,如图4
‑ꢀ
6、15
‑6‑
17
‑
6所示。hela(egfr
/
‑
/her2
/
‑
)和a2058(egfr
‑
/her2
/
‑
)细胞两者在低浓度西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
50pm曲妥珠单抗
‑
皂草素下不显示出细胞杀伤(hela:ic50=400nm;a2058:ic50>400nm;图4
‑
6a,4
‑
6b)。这显示了在足够受体表达的不存在下,没有达到有效细胞内递送的so1861浓度(阈值)以诱导蛋白质毒素的内体逃逸和细胞质递送。接下来,在固定浓度的75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
上滴定曲妥珠单抗
‑
皂草素,并且测定靶向蛋白质毒素介导的对 hela(egfr
/
‑
/her2
/
‑
)或a2058(egfr
‑
/her2
/
‑
)的细胞杀伤。 hela(egfr
/
‑
/her2
/
‑
)和a2058(egfr
‑
/her2
/
‑
)细胞两者未显示出细胞杀伤活性(hela:ic50>10.000pm;a2058:ic50>10.000pm;图4
‑ꢀ
6c,4
‑
6d)。所有这些都显示,具有低或无egfr受体表达的细胞对西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
曲妥珠单抗
‑
皂草素的组合不敏感,这是由于缺乏足够的egfr受体,其促进抗体介导的足够so1861(阈值)的递送,从而确保在细胞的细胞质中的毒素的内体逃逸。
[0896]
接下来,so1861
‑
emch经由半胱氨酸残基(cys)以dar 4缀合至曲妥珠单抗(识别并结合人类her2的单克隆抗体)(曲妥珠单抗
‑
(cys
‑ꢀ
l
‑
so1861)4)。在固定浓度的1.5pm egf石竹素(egfr靶向的配体毒素融合蛋白)上滴定曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4,并且测定靶向蛋白质毒素介导的对表达her2/egfr的细胞(sk
‑
br
‑
3:her2
/egfr
/
‑
) 的细胞杀伤。这揭示了在低浓度的曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4 1.5 pm egf石竹素下强烈的细胞杀伤(sk
‑
br
‑
3:ic50=1nm;图5
‑
6a),然而等浓度的曲妥珠单抗、曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4或曲妥珠单抗 1.5pm egf石竹素不能在表达her2
/egfr
/
‑
的细胞中诱导任何细胞杀伤活性。这显示了曲妥珠单抗缀合的so1861有效地增强了egf 融合蛋白质毒素(在非有效浓度下)的内体逃逸,从而诱导高表达her2/ 低表达egfr的细胞的细胞杀伤。
[0897]
接下来,在固定浓度的2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4上滴定egf石竹素,并且测定靶向蛋白质毒素介导的对表达sk
‑
br
‑ꢀ
3(her2
/egfr
/
‑
)的细胞的细胞杀伤。这揭示了2.5nm曲妥珠单抗
‑ꢀ
(cys
‑
l
‑
so1861)4与低浓度egf石竹素的组合在表达her2/egfr的细胞中诱导已经有效的细胞杀伤(sk
‑
br
‑
3:ic50=1pm)(图5
‑
6b),然而单独的egf石竹素或egf石竹素 2.5nm曲妥珠单抗未显示出细胞杀伤活性(ic50>10.000pm)(图5
‑
6b)。所有这些都显示,相对低浓度的egf石竹素只有与低浓度的曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4组合才可以在高表达her2/低表达egfr的细胞中有效并诱导细胞杀伤。
[0898]
接下来,在固定浓度的1.5pm egf石竹素上滴定曲妥珠单抗
‑ꢀ
(cys
‑
l
‑
so1861)4,并且测定靶向蛋白质毒素介导的对jimt
‑
1(her2
/
‑ /egfr
/
‑
)或mda
‑
mb
‑
468(her2
‑
/egfr
)的细胞杀伤。两种细胞系对曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4 1.5pm egf石竹素的任何组合都不敏感 (jimt
‑
1:ic50>1000nm;mda
‑
mb
‑
468:ic50>1000nm;图6
‑
6a,6
‑
6b)。这显示了在足够her2受体表达的不存在下,没有达到有效细胞内递送的so1861浓度(阈值)以诱导蛋白质毒素的内体逃逸和细胞质递送。
[0899]
接下来,在固定浓度的2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4上滴定egf石竹素,并且测定靶向蛋白质毒素介导的对jimt
‑
1(her2
/
‑ /egfr
/
‑
)或mda
‑
mb
‑
468(her2
‑
/egfr
)的细胞杀伤。在具有或没有 2,5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4的情况下,两种细胞系在高egf 石竹素浓度下显示出细胞杀伤(jimt
‑
1:ic50=10.000pm;mda
‑
mb
‑ꢀ
468:ic50=200pm,图6
‑
6c,6
‑
6d)。
[0900]
所有这些都显示,具有低或无her2受体表达的细胞对曲妥珠单抗
‑
(cys
‑
l
‑
so1861)
3,7
1.5pm egf石竹素的组合不敏感,这是由于缺乏足够的her2受体,其促进抗体介导的足够so1861(阈值)的递送,从而确保在细胞的细胞质中的毒素的内体逃逸。
[0901]
接下来,so1861
‑
emch经由半胱氨酸残基(cys)以dar 4缀合至曲妥珠单抗(识别并结合人类her2的单克隆抗体)(曲妥珠单抗
‑
(cys
‑ꢀ
l
‑
so1861)4)。在固定浓度的5pm西妥昔单抗
‑
皂草素(egfr靶向抗体
ꢀ‑
蛋白质毒素缀合物)上滴定曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4,并且测定靶向蛋白质毒素介导的对表达her2/egfr的细胞(sk
‑
br
‑
3: her2
/egfr
/
‑
)的细胞杀伤,如图15
‑6‑
17
‑
6所示。这揭示了在低浓度的曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4 5pm西妥昔单抗
‑
皂草素下强烈的细胞杀伤(sk
‑
br
‑
3:ic50=1nm;图7
‑
6a),然而等浓度的曲妥珠单抗、曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4或曲妥珠单抗 5pm西妥昔单抗
‑
皂草素不能在表达her2
/egfr
/
‑
的细胞中诱导任何细胞杀伤活性。这显示了曲妥珠单抗缀合的so1861有效地增强了西妥昔单抗缀合的蛋白质毒素(在非有效浓度下)的内体逃逸,从而诱导表达her2
/egfr
/
‑
的细胞的细胞杀伤。
[0902]
接下来,在固定浓度的2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4和75 nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4上滴定西妥昔单抗
‑
皂草素,并且测定靶向蛋白质毒素介导的对表达her2/egfr的细胞(sk
‑
br
‑
3: her2
/egfr
/
‑
)的细胞杀伤。这揭示了2.5nm曲妥珠单抗
‑
(cys
‑
l
‑ꢀ
so1861)4与低浓度的西妥昔单抗
‑
皂草素的组合在sk
‑
br
‑
3细胞中诱导已经有效的细胞杀伤(sk
‑
br
‑
3:ic50=1pm;图7
‑
6b),然而单独的西妥昔单抗
‑
皂草素或西妥昔单抗
‑
皂草素 2.5nm曲妥珠单抗仅在高浓度的曲妥珠单抗
‑
皂草素下显示出细胞杀伤(sk
‑
br
‑
3:ic50>4000 pm;图7
‑
6b)。所有这些都显示,相对低浓度的西妥昔单抗
‑
皂草素只有与低浓度的曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4组合才可以在表达 her2
/egfr
/
‑
的细胞中有效并诱导细
胞杀伤。
[0903]
接下来,在固定浓度的5pm西妥昔单抗
‑
皂草素上滴定曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4,并且测定靶向蛋白质毒素介导的对jimt
‑ꢀ
1(her2
/
‑
/egfr
/
‑
)和mda
‑
mb
‑
468(her2
‑
/egfr
)细胞的细胞杀伤。两种细胞系对曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4 5pm西妥昔单抗
‑
皂草素的组合都不敏感(jimt
‑
1:ic50>1000nm;mda
‑
mb
‑
468:ic50>1000 nm;图8
‑
6a,8
‑
6b)。这显示了在足够her2受体表达的不存在下,没有达到有效细胞内递送的so1861浓度(阈值)以诱导蛋白质毒素的内体逃逸和细胞质递送。
[0904]
接下来,在固定浓度的2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4上滴定西妥昔单抗
‑
皂草素,并且测定靶向蛋白质毒素介导的对jimt
‑ꢀ
1(her2
/
‑
/egfr
/
‑
)和mda
‑
mb
‑
468(her2
‑
/egfr
)细胞的细胞杀伤。在具有或没有2.5nm曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4的情况下,两种细胞系在类似的西妥昔单抗
‑
皂草素浓度下显示出细胞杀伤(jimt
‑
1: ic50=80pm;mda
‑
mb
‑
468:ic50=100pm;图8
‑
6c,8
‑
6d)。
[0905]
所有这些都显示,具有低或无her2受体表达的细胞对曲妥珠单抗
‑
(cys
‑
l
‑
so1861)4 西妥昔单抗
‑
皂草素的组合不敏感,这是由于缺乏足够的her2受体,其促进抗体介导的足够so1861(阈值)的递送,从而确保在细胞的细胞质中毒素的内体逃逸。
[0906]
实施例23
[0907]
为了显示缀合的so1861的活性是由内体区室的酸化驱动的,根据本发明的2t2组分系统与内体酸化抑制剂氯喹组合进行了测试。曲妥珠单抗
‑
皂草素 77nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
或曲妥珠单抗
‑
石竹素 77nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
在 a431(egfr
/her2
/
‑
)细胞中显示了强烈的细胞杀伤活性,然而当将 800nm氯喹共施用至两个组合时抑制根据本发明的该2t2c活性(图 9
‑
6a)。当在a431(egfr
/cd71
)和mda
‑
mb
‑
468(egfr
/cd71
)细胞中测试cd71mab
‑
皂草素 10.5nm西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3,9
500nm氯喹时(图9
‑
6b、9c)或当在sk
‑
br
‑ꢀ
3(her2
/cd71
)细胞中测试cd71mab
‑
皂草素 5nm曲妥珠单抗
‑ꢀ
(cys
‑
l
‑
so1861)4 500nm氯喹时(图9
‑
6d),观察到相同结果。这显示了当阻断内体酸化时,可以抑制缀合的so1861在2t2c系统中的细胞内活性。
[0908]
实施例24
[0909]
2靶标2
‑
组分系统(2t2c)也是mab1
‑
so1861和mab2
‑
反义bna 寡核苷酸的组合治疗(图16
‑
6)。因此,还与反义bna寡核苷酸组合针对癌症特异性靶基因热休克蛋白27(hsp27)的mrna测试了2t2c系统。释放到细胞质中后,反义bna识别并结合编码hsp27的mrna,靶向mrna以进行破坏,从而耗损癌细胞内的hsp27表达。hsp27bna 以dar4.4缀合至曲妥珠单抗(曲妥珠单抗
‑
(lys
‑
l
‑
hsp27bna)
4,4
),并与西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
组合在a431(egfr
/her2
/
‑
)细胞和 a2058(egfr
‑
/her2
/
‑
)细胞中测试增强的hsp27基因沉默活性,如图16
‑
6所示。在固定浓度的100nm曲妥珠单抗
‑
(lys
‑
l
‑
hsp27bna)
4,4
上滴定西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
,并且测定靶向的hsp27bna介导的基因沉默活性。与单独的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
相比,西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
100nm曲妥珠单抗
‑
(lys
‑
l
‑
hsp27bna)
4,4
在a431细胞(egfr
/her2
/
‑
)中显示强烈的基因沉默活性(a431: ic50=1nm;图10
‑
6a)。在a2058细胞(egfr
‑
/her2
/
‑
)中,根据本发明的组合未显示hsp27基因沉默(a2058:ic50>100nm;图10
‑
6b)。这显示了西妥昔单抗缀合的so1861有效地增强了曲妥珠单抗缀合的 bna寡核苷酸(在非有效浓度下)的内体逃逸,从而在表达 egfr
/her2
/
‑
的细胞
中诱导靶基因沉默。
[0910]
接下来,在固定浓度的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
上滴定曲妥珠单抗
‑
(lys
‑
l
‑
hsp27bna)
4,4
,并且在a431(egfr
/her2
/
‑
)细胞和 a2058(egfr
‑
/her2
/
‑
)细胞中测定靶向的hsp27bna介导的基因沉默活性,如图16
‑
6所示。曲妥珠单抗
‑
(lys
‑
l
‑
hsp27bna)
4,4
77nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
在a431细胞(egfr
/her2
/
‑
)中显示强烈的基因沉默活性(a431:ic50=1nm;图10
‑
6c),然而单独的曲妥珠单抗
ꢀ‑
(lys
‑
l
‑
hsp27bna)
4,4
或单独的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
或曲妥珠单抗
‑
(lys
‑
l
‑
hsp27bna)
4,4
77nm西妥昔单抗并未揭示任何显著的基因沉默活性(ic50>100nm)。a2058(egfr
‑
/her2
/
‑
)细胞在根据本发明的组合中未显示任何基因沉默活性(a2058:ic50>100nm;图10
‑
6d)。所有这些都显示,相对低浓度的曲妥珠单抗
‑
hsp27bna只有与低浓度的西妥昔单抗
‑
(
‑
l
‑
so1861)组合才可以在表达her2
/egfr
/
‑
的细胞中有效并诱导细胞杀伤。
[0911]
实施例25
[0912]
2靶标2
‑
组分系统(2t2c)还可以是mab1
‑
(树枝化基元(
‑ꢀ
so1861)
n
)
n
和mab2
‑
蛋白质毒素的组合治疗。树枝化基元(
‑
l
‑
so1861)4经由半胱氨酸残基(cys)以dar3,9缀合至抗egfr抗体西妥昔单抗, (西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
),并在表达mda
‑
mb
‑ꢀ
468(egfr
/cd71
)的细胞中测试其与抗cd71抗体蛋白质毒素缀合物(cd71mab
‑
皂草素)的组合增强的细胞杀伤活性,如图17
‑
6所示。西妥昔单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
10pm cd71mab
‑
皂草素在表达mda
‑
mb
‑
468(egfr
/cd71
)的细胞中有效地诱导了毒素介导的细胞杀伤(ic50=0.4nm,图11
‑
6a),然而这不能由西妥昔单抗
‑
cys
‑ꢀ
(树枝化基元(
‑
l
‑
so1861)4)
3,9
)或西妥昔单抗 10pm cd71mab
‑
皂草素或西妥昔单抗诱导(图11
‑
6a)。这显示了西妥昔单抗缀合的树枝化基元 (
‑
l
‑
so1861)4有效地增强了cd71mab
‑
蛋白质毒素(在非有效浓度下)的内体逃逸,从而诱导表达egfr
/cd71
的细胞的细胞杀伤。在hela 细胞(her2
/
‑
/cd71
)中进行类似的实验,并且这揭示了西妥昔单抗
‑ꢀ
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)
3,9
) 10pm cd71mab
‑
皂草素无活性(ic50>100nm,图11
‑
6b),表明在足够egfr受体表达的不存在下,未达到有效细胞内so1861浓度以诱导蛋白质毒素的内体逃逸和细胞质递送。
[0913]
接下来,树枝化基元(
‑
l
‑
so1861)4经由半胱氨酸缀合(cys)以dar4 缀合至抗her2抗体曲妥珠单抗,即曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑ꢀ
so1861)4)4,并且在sk
‑
br
‑
3细胞(her2
/cd71
)表达细胞中测试其与抗cd71抗体蛋白质毒素缀合物(cd71mab
‑
皂草素)的组合的增强的细胞杀伤活性。曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4 10pmcd71mab
‑
皂草素在sk
‑
br3细胞中有效地诱导了毒素介导的细胞杀伤(ic50=3nm,图11
‑
6c),然而这未由曲妥珠单抗
‑
cys
‑
(树枝化基元(
‑ꢀ
l
‑
so1861)4)4或曲妥珠单抗(等效) 10pm cd71mab
‑
皂草素或曲妥珠单抗诱导(图11
‑
6c)。这显示了根据本发明的曲妥珠单抗缀合的树枝化基元(
‑
l
‑
so1861)4有效地增强了cd71mab
‑
蛋白质毒素(在非有效浓度下) 的内体逃逸,从而诱导表达her2
/cd71
的细胞的细胞杀伤。在jimt
‑ꢀ
1细胞(her2
/
‑
/cd71
)中进行类似实验,并且这揭示了曲妥珠单抗
‑ꢀ
cys
‑
(树枝化基元(
‑
l
‑
so1861)4)4 10pm cd71mab
‑
皂草素无活性(ic50> 100nm,图11
‑
6c),表明在足够her2受体表达的不存在下,未达到有效细胞内so1861浓度以诱导蛋白质毒素的内体逃逸和细胞质递送。
[0914]
实施例26
[0915]
临床批准的adc,曲妥珠单抗
‑
美坦新(t
‑
dm1)是抗her2抗体、曲妥珠单抗和小分子毒素美坦新的缀合物(dar3
‑
4)。t
‑
dm1在根据本发明的2t2c系统内与西妥昔单抗
‑
(cys
‑
l
‑
so1861)4组合测试。与单独的t
‑
dm1或t
‑
dm1 77nm西妥昔单抗相比,t
‑
dm1 77nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
显示了没有增强的细胞杀伤活性(ic50=80.000 pm,图12
‑
6),然而与曲妥珠单抗
‑
皂草素 75nm西妥昔单抗或单独的曲妥珠单抗
‑
皂草素相比,根据本发明的曲妥珠单抗
‑
皂草素 75nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,7
显示出增强的细胞杀伤活性(ic50=3pm,图12
‑
6)。所有这些都显示,2t2c系统不增强已经能够被动穿过细胞 (内体)膜的抗体缀合的小分子的递送。
[0916]
实施例1
‑2[0917]
在bt474(her2 )异种移植小鼠模型中测试各种浓度的曲妥珠单抗
‑
皂草素(her2靶向的蛋白质
‑
毒素缀合物;静脉内)与1.5mg/kgso1861(抗体
‑
毒素注射之前1小时;皮下)的组合的增强的功效。在第 13天开始给药,此时肿瘤大小达到~150mm3,并且在每次处理后测定肿瘤体积。尽管在用1mg/kg和0.3mg/kg曲妥珠单抗
‑
皂草素处理的小鼠中观察到肿瘤生长抑制,但在用曲妥珠单抗
‑
皂草素 so1861的组合处理的小鼠中观察到没有增强的肿瘤生长抑制。这显示了未缀合的 so1861不能增强当前设置和小鼠模型中的抗体
‑
蛋白质毒素。
[0918]
实施例2
‑2[0919]
材料:
[0920]
qsmix(1):s4521(sigma aldrich);qsmix(2):6857.1(carl roth) qsmix(3):adjuvant:vac
‑
quil(invivogen/brenntag)。
[0921]
先前,各种皂苷(so1861,so1642)的功效作为
‘
游离’未缀合的分子与配体毒素融合物(如egf石竹素)或抗体
‑
蛋白质毒素缀合物组合共施用至细胞,从而导致增强的对靶标表达细胞的细胞杀伤活性。在此,在存在和不存在非有效固定浓度的1.5pm egf石竹素下在 hela(egfr
)细胞上滴定从肥皂草(saponaria officinalis)的根提取物中分离的三种不同的皂苷分子(so1861、so1862(so1861的异构体)、 so1832和so1904)。这揭示了与没有egf石竹素的处理相比,所有测试的皂苷变体的细胞杀伤活性强烈增强(ic50=300nm;图2
‑
2a)。接下来,用固定浓度的皂苷(~1000nm)滴定egf石竹素,这揭示了对所有使用的皂苷so1861、so1862(so1861的异构体)、so1832和so1904 观察到的在低pm浓度的egf石竹素下强烈的靶向细胞杀伤增强 (ic50=0.4pm;图2
‑
2b)。单独的egf
‑
石竹素仅可在非常高的浓度下诱导细胞杀伤(ic50=10.000pm)。这显示了这些特定类型的皂苷在仅具有可用的非常低量的靶向毒素的情况下都具有有效诱导内体逃逸的固有能力。
[0922]
为了延伸该测试,分析了来自其它来源的皂苷。在存在和不存在 1.5pm egf石竹素下在hela细胞上滴定从圆锥丝石竹的根提取物纯化的皂苷(ge1741),并与纯化的so1861进行比较。ge1741还增强了 egf石竹素诱导的hela细胞杀伤,但与so1861相比显示了略微更小的功效(ge1741 ic50=800nm;图2
‑
2c)并且还显示了更高的一般毒性(在egf石竹素的不存在下ic50=5.000nm;图2
‑
2c)。其中对hela 细胞共施用不同部分纯化的皂皮树皂苷混合物(qsmix 1
‑
3)与1.5pmegf石竹素的类似测试,并且这揭示了3种中有2种(qsmix 1和qsmix 3)与so1861具有类似活性(ic50 qsmix/qsmix3=300nm;图2
‑
2d)。 qsmix(2)在增强1.5pm egf石竹素诱导的细胞杀伤中效率较低 (ic50=2000nm;图2
‑
2d),然而,未观察到一
般毒性。这显示了在qs 提取物中也可获得特定类型的皂苷,其有效地诱导靶向配体毒素egf 石竹素的内体逃逸。
[0923]
实施例3
‑2[0924]
为了将so1861分子缀合至抗体,根据本发明,将不稳定的/酸敏感的接头(
‑
emch或
‑
n3)经由醛基团缀合至so1861,产生so1861
‑ꢀ
emch或so1861
‑
n3。为了验证so1861
‑
emch的活性,在存在和不存在固定非有效(1.5pm)egf石竹素浓度下在表达egfr(a431、hela) 和不表达egfr(a2058)的细胞上滴定分子。在所有三种细胞系中,单独的so1861显示出强的细胞活力降低,然而作为单一化合物的 so1861
‑
emch在高达25.000nm下显示无毒性(图3
‑
2a
‑
c)。当 so1861
‑
emch与1.5pm egf石竹素组合时,在egfr
a431和hela 细胞中观察到强烈的靶标特异性细胞活力降低(ic50=3.000nm;图3
‑ꢀ
2a,b),而egfr
‑
a2058细胞根本不受影响(图3
‑
2c)。对so1861
‑
n3 获得类似结果。与1.5pm egf石竹素共施用的so1861
‑
n3也显示了对a431和hela细胞的有效细胞杀伤(ic50=3.000nm),但在没有egf 石竹素的情况下,在高于10.000nm下观察到一般毒性(图3
‑
2d,3
‑ꢀ
2e)。
[0925]
对于so1861与抗体的稳定缀合,根据本发明,稳定接头(hatu) 经由so1861的羧酸基团缀合至so1861,产生so1861
‑
(s)。为了测定活性,在表达egfr的hela细胞中共施用不同浓度的so1861
‑
(s)与 1.5pm egf石竹素并且测试其细胞杀伤活性。so1861
‑
(s)显示出与 so1861类似的活性,表明缀合至羧酸不影响分子的内体逃逸增强效力,如用so1861
‑
emch所观察到的(图4
‑
2)。
[0926]
实施例4
‑2[0927]
不稳定的so1861经由半胱氨酸残基(cys)以dar3.9缀合至抗 egfr抗体西妥昔单抗(识别并结合人类egfr的单克隆抗体),即(西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3.9
),并且测试其增强的反义bna寡核苷酸递送,导致增强的靶基因沉默。在本研究中,我们使用了针对癌症特异性靶基因热休克蛋白27(hsp27)的mrna的反义bna寡核苷酸。在细胞的细胞质内,hsp27bna结合编码hsp27的mrna,靶向mrna 以进行破坏,从而降低癌细胞内的hsp27表达。在egfr
(a431)和 egfr
‑
(a2058)细胞上的固定浓度的100nm hsp27bna上滴定西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3.9
。根据本发明的组合显示了对a431的有效的 hsp27沉默(ic50=2nm;图5
‑
2a),而对单独的西妥昔单抗
‑
(cys
‑
l
‑ꢀ
so1861)
3.9
未观察到沉默。西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3.9
100nmhsp27bna在egfr
‑
细胞(a2058)中未显示基因沉默活性(图5
‑
2b)。这显示了低浓度的抗体缀合的so1861可以有效地增强反义bna寡核苷酸的细胞质递送和内溶酶体逃逸,从而在靶标表达细胞中诱导有效的基因沉默。
[0928]
接下来,在egfr
(a431)和egfr
‑
(a2058)细胞上滴定 hsp27bna与固定浓度的西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3.9
的组合。这显示了hsp27bna与28.6nm西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3.9
或77nm 西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3.9
的组合在a431细胞中非常有效地增强了hsp27基因沉默(ic50=10nm;图5
‑
2c)。单独的或与固定当量的77 nm西妥昔单抗组合的hsp27bna效率较低(ic50=1.000nm;图5
‑
2c)。还在egfr
‑
细胞(a2058)上测试了hsp27bna 77nm西妥昔单抗
‑ꢀ
(cys
‑
l
‑
so1861)
3.9
的组合治疗,并且这揭示了无hsp27基因沉默增强(ic50=1.000nm;图5
‑
2d)。这显示了具有低或无egfr受体表达的细胞对西妥昔单抗
‑
(cys
‑
l
‑
so1861)
3,9
hsp27bna的组合不敏感,而西妥昔单抗靶向的so1861可在高egfr细胞中在低浓度的非靶向的 hs27bna下有效增强hsp27基因沉默。
hsp27基因沉默。这些缀合物显示了在4000nmso1861
‑
emch的存在下在a431(egfr
)细胞中非常有效的hsp27基因沉默需要更少的hsp27bna寡核苷酸(ic50=0.04nm;图1
‑
7c),而靶向的hsp27bna样品的沉默与在so1861
‑
emch的不存在下非靶向的hsp27bna相当。总之,a2058(egfr
‑
)细胞中的沉默与非靶向的hsp27bna相比未得以改善。甚至看起来在so1861的存在下需要约10倍以上的hsp27bna寡核苷酸,而在没有so1861的情况下观察到没有显著沉默(图1
‑
7d)。这显示了使用靶向的hsp27bna与so1861的组合可以实现具有高egfr受体表达的细胞的非常有效的靶向hsp27基因沉默。
[0936]
参考文献
[0937]
weng,a.;thakur,m.;beceren
‑
braun,f.;bachran,d.;bachran,c.;riese,s.b.;jenett
‑
siems,k.;gilabert
‑
oriol,r.;melzig,m.f.;fuchs,h.thetoxincomponentoftargetedanti
‑
tumortoxinsdeterminestheirefficacyincreasebysaponins.molecularoncology2012,6,323
‑
332.
[0938]
saponinumalbuminasynergisticway.journalofimmunotherapy2009,32,713
‑
725.
[0939]
sama,s.;jerz,g.;schmieder,p.;joseph,j.f.;melzig,m.f.;weng,a.plantderived
[0940]
triterpenesfromgypsophilaelegansm.bieb.enablenon
‑
toxicdeliveryofgene
[0941]
loadednanoplexes.journalofbiotechnology2018,284,131
‑
139
[0942]
kolb,h.c.;finn,m.g.;sharpless,k.b.clickchemistry:diversechemicalfunctionfromafewgoodreactions.angewandtechemie2001,40,2004
‑
2021.
[0943]
bird,r.e.;hardmann,k.d.;jacobson,j.w.;johnson,s.;kaufman,b.m.;lee,s.m.;lee,t.;pope,s.h.;riordan,g.s.;whitlow,m.single
‑
chainantigen
‑
bindingproteins.science1988,242,423
‑
426.
[0944]
yzhang,zqu,skim,vshi,bliao1,pkraft,rbandaru,ywu,lmgreenbergerandidhorak,down
‑
modulationofcancertargetsusinglockednucleicacid(lna)
‑
basedantisenseoligonucleotideswithouttransfection,genetherapy(2011)18,326
‑
333。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。