1.本发明涉及蜂蜜检测技术领域,具体地说,涉及一种判断枇杷蜂蜜是否成熟的方法与应用。
背景技术:
2.蜂蜜是蜜蜂采集植物的花蜜、分泌物或蜜露,与自身分泌物结合后,在巢脾内转化、脱水、贮存至成熟的天然甜物质(章彬佳, 程春生, 胡福良. 蜂蜜中几种常见酶的研究进展. 蜜蜂杂志,2007(06):11
‑
13)。枇杷蜜是蜜蜂采集枇杷花蜜酿造而成的蜂蜜,色白浓稠,口感滋润,具有浓郁的杏仁味,具有润肺祛痰、止咳定喘的功效,是冬蜜中的佳品(欧阳军. 不同蜜源蜂蜜的医疗保健功效. 中国蜂业,2018,69(12):50
‑
52)。
3.目前,养蜂业存在以生产加工蜂蜜作为主流的情况,即将未成熟蜂蜜进行加工以满足标准,进而进入流通领域销售给消费者的现象。在蜂蜜加工过程中容易引起美拉德反应产生有害产物,同时也会造成蜂蜜中的营养成分被破坏,从而降低了蜂蜜的质量。加工蜂蜜的生产销售不仅扰乱了蜂产品市场的健康发展,也降低了蜂蜜的在国际市场上的品牌形象。2019年和2020年,国际蜂联连续两年发布公告并将“收获未成熟蜂蜜,利用技术设备(包括但不限于真空干燥机)对蜂蜜进行进一步主动脱水”的行为定义为蜂蜜造假行为(龙学军. 国际养蜂联合会关于蜂蜜欺诈的声明.蜜蜂杂志,2019(8):3
‑
6)。为规范蜂蜜行业健康发展,提升蜂蜜品质,应鼓励生产天然成熟蜂蜜。在蜂巢内充分酿造的成熟蜂蜜,口感醇厚,成分丰富,具有较高的营养价值。
4.蜂蜜的酿造过程是复杂的生物转化过程,蜜蜂对花蜜的吞吐酿造时蜜蜂分泌的酶对物质进行分解转化,随着蜜蜂的振翅扇风活动脱除水分,用蜂蜡封盖蜂巢贮存蜂蜜防止外部水分向蜂蜜中迁移(eyer m, neumann p, dietemann v. a look into the cell: honey storage in honey bees, apis mellifera. plos one, 2016,11(8):1
‑
20)。目前已有对蜂蜜成熟过程中水分、单糖、蔗糖及酶值等物质变化规律的研究,表明随酿造时间延长,蜂蜜中水分和蔗糖含量降低,单糖及酶值等含量增加(于泽浩. 蜂蜜成熟过程中成分变化的研究.福建农林大学,2017),但目前缺少判别蜂蜜是否成熟的特征成分指标,难以鉴定蜂蜜是否成熟。
5.具体地,现有的关于鉴别蜂蜜是否成熟的相关成分指标不完善。
6.1、水分含量小于18%判定蜂蜜是否成熟具有局限性:短时酿造的未成熟蜂蜜通过加工手段脱水处理,可以降低水分含量。
7.2、蔗糖含量小于5%判定蜂蜜是否成熟具有局限性:蜂蜜通过加工处理可以降低其中的蔗糖含量。
8.3、果糖和葡萄糖总含量判定蜂蜜是否成熟具有局限性:通过添加糖浆可以提高果糖和葡萄糖含量。
9.4、以封盖率达到80%判定蜂蜜是否成熟具有局限性:封盖行为受蜜源植物、产地和蜂种及环境影响很大,导致其封盖行为存在差异,如南方部分地区蜂蜜水分偏高且巢脾难
以实现封盖率达80%。此外,无法从流通领域的商品蜜中观察到蜂蜜的封盖率是否达到80%。
10.5、以脯氨酸含量高于180mg/kg判定蜂蜜是否成熟具有局限性:不同蜜源植物、产地和蜂种来源的蜂蜜中脯氨酸含量差异较大。已有研究测定部分蜂蜜(包括未成熟蜂蜜中)中脯氨酸含量高于180 mg/kg。
11.因此,仍需进一步找寻更有效的蜂蜜成熟判断方法。
技术实现要素:
12.针对现有技术的问题,本发明的目的在于提供一种可有效判定蜂蜜是否成熟的方法。
13.为了实现该目的,本发明的技术方案如下:一种判断枇杷蜂蜜是否成熟的方法,其根据待测枇杷蜂蜜中α,β
‑
海藻糖、黑曲霉二糖、松二糖和蔗糖的含量来判断枇杷蜂蜜是否成熟。
14.蜂蜜中主要成分是糖类物质,低聚糖是蜂蜜中重要的营养功效成分。蜂蜜的酿造过程是复杂的生物转化过程,在蜂蜜成过程中,在淀粉酶、蔗糖转换酶、β
‑
葡萄糖苷酶的作用下可使糖类物质发生分解和转化,其中蔗糖可发生转糖基化产生新的低聚糖(因此相比于花蜜中,在蜂蜜中发现了更多种类和更高含量的低聚糖),致使其碳水化合物组成及含量在成熟过程中存在变化。本发明研究发现特定低聚糖的含量可作为枇杷蜂蜜成熟度评价的依据。
15.具体地本发明方法中,当所述待测枇杷蜂蜜中的t值≥5%时,判定所述待测枇杷蜂蜜为成熟蜂蜜;;;其中,w
(干重)
公式中的低聚糖分别指代α,β
‑
海藻糖、黑曲霉二糖、松二糖或蔗糖,由此计算获得w
(干重)黑曲霉二糖
、w
(干重)松二糖
、w
(干重)α,β
‑
海藻糖
和w
(干重)蔗糖
。
16.即,w
(干重)黑曲霉二糖
=蜂蜜湿重中黑曲霉二糖含量测定值/(100%
‑
蜂蜜水分含量(%));w
(干重)松二糖
=蜂蜜湿重中松二糖含量测定值/(100%
‑
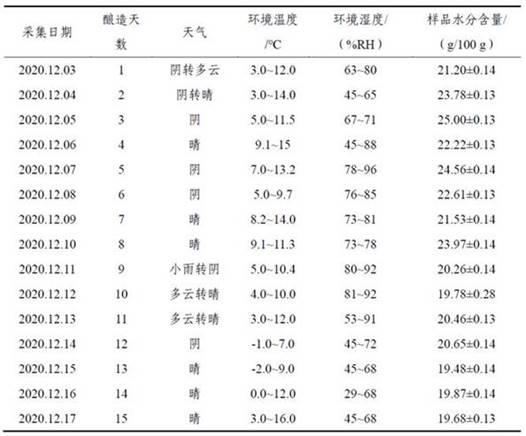
蜂蜜水分含量(%)),依次类推。
17.本发明经研究发现在枇杷蜂蜜中的众多低聚糖中,将α,β
‑
海藻糖、黑曲霉二糖、松二糖和蔗糖的含量经特定运算后获得的t值,在酿造成熟的枇杷蜂蜜中与酿造不成熟的枇杷蜂蜜中具有显著性差异,可用来高效、稳定、重复性好地准确鉴别枇杷蜂蜜成熟与否。
18.本发明方法中,在进行蜂蜜湿重中α,β
‑
海藻糖、黑曲霉二糖、松二糖和蔗糖的含量测定时,包括:(1)蜂蜜样品前处理:将所述α,β
‑
海藻糖、黑曲霉二糖、松二糖和蔗糖从蜂蜜样品中提取出来;(2)将步骤(1)获得的提取物与内标物质混合后,进行肟化和硅烷化;(3)通过气相色谱
‑
质谱联用法进行定性测定和内标法定量测定。
19.本发明方法中,蜂蜜样品前处理时,以80%的乙醇水溶液作为提取液,所述蜂蜜样
品与所述提取液的质量体积比为1:(40
‑
200)g/ml,将所述蜂蜜样品与所述提取液混合后,涡旋震荡提取3~5 min,于8000 r/min,4 ℃离心8~15 min。
20.本发明方法中,优选,蜂蜜样品前处理时,称取蜂蜜样品0.20~0.50 g,与20~50 ml 80%的乙醇水溶液混合,涡旋震荡提取3~5 min,于8000 r/min,4 ℃离心8~15 min,吸取上清液并用80%乙醇水溶液进行稀释,在进行α,β
‑
海藻糖、黑曲霉二糖、松二糖测定时,稀释倍数为5~15倍,在进行蔗糖测定时稀释倍数为20~50倍,最后将稀释液以氮吹至干;更优选,称取混合均匀的蜂蜜样品0.25
±
0.005 g,与25 ml 80%的乙醇水溶液混合,涡旋震荡提取5 min,于8000 r/min,4 ℃离心10 min,吸取上清液1 ml,用80%乙醇水溶液进行稀释,在进行α,β
‑
海藻糖、黑曲霉二糖、松二糖测定时,稀释倍数为10倍,在进行蔗糖测定时稀释倍数为50倍,最后吸取稀释液100 μl于1.5 ml离心管,于40 ℃氮吹至干。
21.本发明的前处理方式可更加充分地将目标物质提取出来,以利于后续对蜂蜜成熟度进行准确判断。
22.本发明方法中,所述内标物质为苯基
‑
β
‑
d
‑
葡萄糖苷,肟化时采用盐酸羟胺吡啶,硅烷化时采用六甲基二硅胺烷和三氟乙酸酐。
23.本发明研究发现由于枇杷蜂蜜中不含有苯基
‑
β
‑
d
‑
葡萄糖苷,且其与低聚糖的化学性质相似,分子量近似,能够通过色谱实现苯基
‑
β
‑
d
‑
葡萄糖苷与低聚糖的分离,因此,选用其作为内标物质可校正前处理过程中以及测定分析中的质量损失,实现准确定量。
24.本发明方法中,气相色谱
‑
质谱联用条件包括:升温程序:初始温度180 ℃,保持5 min,以10 ℃/min升温至255 ℃,保持1 min,然后以15 ℃/min升温至265 ℃,保持1 min,然后以1 ℃/min升温至270 ℃,然后以10 ℃/min升温至300 ℃,然后以20 ℃/min升温至320 ℃,保持10~15min;进样量1 μl。
25.气相色谱
‑
质谱联用条件进一步包括:色谱柱:db
‑
5;载气为氦气,流量0.95~1 ml/min;进样口温度300 ℃,不分流进样;电子电离源;离子源温度230 ℃;电离能量70 ev;溶剂延迟10 min;扫描类型:选择离子监测模式。
26.优选,气相色谱
‑
质谱联用条件为:色谱柱:db
‑
5;载气为氦气,流量0.99 ml/min;进样口温度300 ℃,不分流进样;升温程序:初始温度180 ℃,保持5 min,以10 ℃/min升温至255 ℃,保持1 min,然后以15 ℃/min升温至265 ℃,保持1 min,然后以1 ℃/min升温至270 ℃,然后以10 ℃/min升温至300 ℃,然后以20 ℃/min升温至320 ℃,保持10 min,进样量1 μl;电子电离源;离子源温度230 ℃;电离能量70 ev;溶剂延迟10 min;扫描类型:选择离子监测模式。
27.采用本发明的特定气相色谱
‑
质谱联用条件进行检测,各物质分离度高,检测灵敏度高,利于后续对枇杷蜂蜜成熟与否的准确判断。
28.优选,本发明的方法中所述待测枇杷蜂蜜为江苏中蜂枇杷蜂蜜。
29.本发明采用80%乙醇水溶液提取蜂蜜中的低聚糖,经盐酸羟胺肟化
‑
六甲基二硅胺烷和三氟乙酸酐硅烷化生成特定易挥发的低聚糖衍生产物,利用气相色谱
‑
质谱联用法对蜂蜜中α,β
‑
海藻糖、黑曲霉二糖、松二糖和蔗糖衍生物进行准确的定性和定量分析。之后,由α,β
‑
海藻糖、黑曲霉二糖、松二糖和蔗糖含量经特定运算计算蜂蜜的t值。最后,根据t值鉴别枇杷蜂蜜是否成熟。
30.作为一个优选方式,具体包括如下步骤:标准贮备液的配制:准确称取100 mg苯基
‑
β
‑
d
‑
葡萄糖苷,用吡啶溶解并定容至5 ml配制成20 mg/ml的标准储备液;准确称取各100 mg蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖标准品,用超纯水溶解并定容至5 ml配制成20 mg/ml的标准储备液。标准储备液均于4 ℃下避光储存,可稳定保存3个月。
31.低聚糖标准工作溶液配制及预处理:吸取适量蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖标准储备液,混匀,配制蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖均为200 μg/ml的混合标准储备液。用80%乙醇稀释混合标准储备液为0.1、0.5、1、5、20 μg/ml系列混合标准工作溶液。吸取各浓度混合标准工作溶液100 μl于1.5 ml离心管,于40 ℃氮吹至干,待衍生。
32.样品前处理方法:称取混合均匀的蜂蜜样品0.25
±
0.005 g,置于50 ml离心管中,加入25 ml 80%的乙醇水溶液,涡旋震荡提取5 min,于8000 r/min,4 ℃离心10 min,吸取上清液1ml,用80%乙醇水溶液稀释10倍(在测定蔗糖时吸取上清液1ml用80%乙醇水溶液稀释50倍),吸取稀释液100 μl于1.5 ml离心管,于40 ℃氮吹至干。
33.样品提取物及标准品衍生化处理:在上述低聚糖混合标准工作溶液和样品提取物的1.5 ml离心管中分别加入浓度为0.025 mg/ml苯基
‑
β
‑
d
‑
葡萄糖苷溶液20 μl,涡旋混匀,加入浓度为25 mg/ml的盐酸羟胺吡啶溶液350 μl,涡旋混匀,于75 ℃下进行肟化反应30 min,之后加入350 μl六甲基二硅胺烷和35 μl三氟乙酸酐,涡旋混匀,在45 ℃下进行硅烷化反应30 min,12000 r/min,4 ℃离心10 min,取上清液至进样瓶内,用于gc
‑
ms分析。
34.气相色谱
‑
质谱联用条件:日本岛津公司qp 2010 气相色谱
‑
质谱联用仪,配备db
‑
5(30 m
×
0.25 mm,0.25 μm);载气为氦气,流量为0.99 ml/min;进样口温度300 ℃,不分流进样;气相色谱升温条件为:初始温度180 ℃(保持5 min),以10 ℃/min升温速率升温至255 ℃(保持1 min),然后以15 ℃/min升温至265 ℃(保持1 min),随后以1 ℃/min升温至270 ℃,之后以10 ℃/min升温至300 ℃,最后以20 ℃/min升温至320 ℃(保持10 min),进样体积为1 μl。电子电离源;离子源温度230 ℃;电离能量70 ev;溶剂延迟时间为10 min;扫描类型:选择离子监测(sim)模式。
35.混合标准工作溶液总离子色谱图见图1,各低聚糖的离子质谱图见图2和图3。
36.测定方法:定性测定:通过色谱保留时间与质谱特征定量和定性离子实现共同定性。
37.定量测定:以内标法定量,配制含同浓度内标的系列溶剂标准工作液,对标准溶液进行衍生化处理后上机测定。以低聚糖与内标物苯基
‑
β
‑
d
‑
葡萄糖苷的峰面积比为纵坐标,以低聚糖浓度为横坐标绘制标准曲线。
38.鉴别方法:由以下公式计算蜂蜜的t值:;其中低聚糖在蜂蜜干重质量中的含量w
(干重)
以下式计算:
;规定当t值≥5%,判定为成熟蜂蜜,否则为不成熟蜂蜜。
39.本发明的方法中:1) 衍生化反应需在无水条件下进行,因此蜂蜜提取物在衍生化前应氮吹至干。
40.2) 衍生化试剂用量应过量,以确保糖类物质被完全衍生。
41.3) 衍生化完成后需在16小时内完成上机测定,以确保低聚糖衍生物在分析过程中保持稳定状态。
42.本发明还提供一种上述方法在进行枇杷蜂蜜品质检测中的应用。
43.本发明的有益效果至少在于:本发明研究发现成熟枇杷蜂蜜和未成熟枇杷蜂蜜在t值上存在显著性差异,依据t值可有效鉴别枇杷成熟蜂蜜和未成熟蜂蜜。本发明首次提供了一种通过gc
‑
ms法测定蔗糖、α,β
‑
海藻糖、黑曲霉二糖和松二糖含量来区分枇杷成熟蜂蜜和未成熟蜂蜜的方法。该方法高效、稳定、重复性好,可准确鉴别成熟枇杷蜂蜜,对鼓励生产天然成熟蜜、完善蜂蜜品质检测、规范蜂蜜市场的管理具有重要的意义。
附图说明
44.图1为混合标准工作溶液总离子色谱图,即苯基
‑
β
‑
d
‑
葡萄糖苷、蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖标准品的总离子流色谱图。其中,1号峰代表苯基
‑
β
‑
d
‑
葡萄糖苷衍生物;2号峰代表蔗糖衍生物;3号峰代表α,β
‑
海藻糖衍生物;4号峰代表黑曲霉二糖衍生物1;5号峰代表松二糖衍生物1;6号峰代表麦芽糖衍生物1;7号峰代表松二糖衍生物2;8号峰代表麦芽糖衍生物2;9号峰代表黑曲霉二糖衍生物2;10号峰代表异麦芽酮糖衍生物1;11号峰代表异麦芽酮糖衍生物2;12号峰代表异麦芽糖衍生物1;13号峰代表异麦芽糖衍生物2。横坐标为保留时间,单位为min。
45.图2为苯基
‑
β
‑
d
‑
葡萄糖苷(图中的a)、蔗糖(图中的b)、α,β
‑
海藻糖(图中的c)、黑曲霉二糖(图中的d)、松二糖(图中的e)标准品的离子质谱图。
46.图3为麦芽糖(f)、异麦芽酮糖(g)、异麦芽糖(h)标准品的离子质谱图。
47.图4为实施例3中酿造14天的成熟蜂蜜(图中的a)和实施例3酿造1天的未成熟蜂蜜(图中的b)的总离子流色谱图。其中,1号峰代表苯基
‑
β
‑
d
‑
葡萄糖苷衍生物;2号峰代表蔗糖衍生物;3号峰代表α,β
‑
海藻糖衍生物;4号峰代表黑曲霉二糖衍生物1;5号峰代表松二糖衍生物1;6号峰代表松二糖衍生物2;7号峰代表黑曲霉二糖衍生物2;8号峰代表麦芽糖衍生物2;9号峰代表黑曲霉二糖衍生物2;10号峰代表异麦芽酮糖衍生物1;11号峰代表异麦芽酮糖衍生物2;12号峰代表异麦芽糖衍生物1;13号峰代表异麦芽糖衍生物2。横坐标为保留时间,单位为min。
具体实施方式
48.下面将结合实施例对本发明的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技
术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。
49.下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
50.本发明具体实施方式部分t值和w
(干重)
的计算公式如下:的计算公式如下:。
51.实施例1中枇杷蜂蜜样品区别于实施例2和实施例3中蜂蜜样品,相同酿造天数的蜂蜜样品采自于不同蜂箱。实施例1中蜂蜜样品分别采集于a组15个蜂箱;实施例2中蜂蜜样品分别采集于b组15个蜂箱;实施例3中蜂蜜样品分别采集于c组15个蜂箱。
52.实施例1本实验例提供一种本发明的蜂蜜成熟与否鉴别方法。具体如下:以采集于江苏省苏州市的酿造1~15天的枇杷蜂蜜作为样品进行蜂蜜成熟与否的鉴定。蜂蜜样品于
‑
20℃贮存直至分析。依据《进出口蜂蜜检验规程》(sn/t0852
‑
2012),采用阿贝折光仪测定蜂蜜中的水分含量(重复测定3次)。样品采集信息及水分含量见表1。
53.表1a组蜂箱枇杷蜂蜜样品信息表低聚糖标准贮备液的配制:准确称取苯基
‑
β
‑
d
‑
葡萄糖苷100mg,用吡啶溶解并定容至5ml配制成20mg/ml的标准储备液;准确称取蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、
麦芽糖、异麦芽酮糖、异麦芽糖标准品各100 mg,用超纯水溶解并定容至5 ml配制成20 mg/ml的标准储备液,标准储备液均于4 ℃下避光储存,可稳定保存3个月。
54.低聚糖标准工作溶液配制及预处理:吸取适量蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖标准储备液,混匀,配制蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖均为200 μg/ml的混合标准储备液。用80%乙醇稀释混合标准储备液为0.1、0.5、1、5、20 μg/ml系列混合标准工作溶液。吸取各浓度混合标准工作溶液100 μl于1.5 ml离心管,于40 ℃氮吹至干,待衍生。
55.样品前处理方法:称取混合均匀的蜂蜜样品0.25
±
0.005 g,置于50 ml离心管中,加入25 ml 80%的乙醇水溶液,涡旋震荡提取5 min,于8000 r/min,4 ℃离心10 min,吸取上清液1ml,用80%乙醇水溶液稀释10倍(测定蔗糖含量时稀释50倍),吸取稀释液100 μl于1.5 ml离心管,于40 ℃氮吹至干,待衍生。
56.样品提取物及标准品衍生化处理:在上述低聚糖混合标准工作溶液和样品提取物的1.5 ml离心管中分别加入20 μl苯基
‑
β
‑
d
‑
葡萄糖苷溶液(0.025 mg/ml),涡旋混匀,加入350 μl盐酸羟胺吡啶溶液(25 mg/ml),涡旋混匀,75 ℃反应30 min,随后加入350 μl六甲基二硅胺烷和35 μl三氟乙酸酐,涡旋混匀,45 ℃反应30 min,12000 r/min,4 ℃离心10 min,取上清液至进样瓶内,用于gc
‑
ms分析。
57.气相色谱
‑
质谱联用条件:采用日本岛津公司qp 2010 气相色谱
‑
质谱联用仪,色谱柱:db
‑
5(30 m
×
0.25 mm,0.25 μm);载气为氦气,流量0.99 ml/min;进样口温度300 ℃,不分流进样;升温程序:初始温度180 ℃,保持5 min,以10 ℃/min升温至255 ℃,保持1 min,然后以15 ℃/min升温至265 ℃,保持1 min,然后以1 ℃/min升温至270 ℃,然后以10 ℃/min升温至300 ℃,然后以20 ℃/min升温至320 ℃,保持10 min,进样量1 μl。电子电离源;离子源温度230 ℃;电离能量70 ev;溶剂延迟10 min;扫描类型:选择离子监测(sim)模式。
58.混合标准工作溶液总离子色谱图见图1,各低聚糖的离子质谱图见图2和图3:苯基
‑
β
‑
d
‑
葡萄糖苷衍生物的色谱保留时间为12.763 min,特征定量离子为217,特征定性离子为361、204、319;蔗糖衍生物的色谱保留时间为16.297 min,特征定量离子为217,特征定性离子为361、204、319;α,β
‑
海藻糖衍生物的色谱保留时间为18.223 min,特征定量离子为217,特征定性离子为361,204,319;黑曲霉二糖衍生物1的色谱保留时间为19.343 min,特征定量离子为204,特征定性离子为361,217,319;黑曲霉二糖衍生物2的色谱保留时间为19.976 min,特征定量离子为204,特征定性离子为361,217,319;松二糖衍生物1的色谱保留时间为19.400 min,特征定量离子为217,特征定性离子为361,204,319;松二糖衍生物2的色谱保留时间为19.551 min,特征定量离子为217,特征定性离子为361,204,319。麦芽糖衍生物1的色谱保留时间为19.471 min,特征定量离子为217,特征定性离子为361,204,319;麦芽糖衍生物2的色谱保留时间为19.788 min,特征定量离子为217,特征定性离子为361,204,319。异麦芽酮糖衍生物1的色谱保留时间为20.550 min,特征定量离子为204,特征定性离子为217,361,319;异麦芽酮糖衍生物2的色谱保留时间为20.848 min,特征定量离子为204,特征定性离子为217,361,319。异麦芽糖衍生物1的色谱保留时间为21.479 min,特征定量离子为204,特征定性离子为217,361,319;异麦芽糖衍生物2的色谱保留时间为22.043 min,特征定量离子为204,特征定性离子为217,361,319。
59.其中,黑曲霉二糖衍生物1和黑曲霉二糖衍生物2为同分异构体。松二糖衍生物1和松二糖衍生物2为同分异构体。麦芽糖衍生物1和麦芽糖衍生物2为同分异构体。异麦芽酮糖衍生物1和异麦芽酮糖衍生物2为同分异构体。异麦芽糖衍生物1和异麦芽糖衍生物2为同分异构体。
60.测定方法:1、定性测定:通过色谱保留时间与质谱特征定量和定性离子共同定性。
61.2、低聚糖(蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖和异麦芽糖)湿重含量测定:以内标法定量,配制系列溶剂标准工作液,各标准溶液衍生化处理前均加入20μl的内标物苯基
‑
β
‑
d
‑
葡萄糖苷溶液(0.025 mg/ml),之后进行两步衍生化处理,最后经gc
‑
ms测定(具体步骤参见上文所述)。以低聚糖与内标物苯基
‑
β
‑
d
‑
葡萄糖苷的峰面积比为纵坐标,以低聚糖浓度为横坐标绘制标准曲线。结果在0.07~2.65 μg/ml的测定浓度范围内,蔗糖标准曲线方程为y = 0.4905x 0.018,r2=0.9995,检出限为0.01 g/100 g,定量限为0.05 g/100 g;α,β
‑
海藻糖标准曲线方程为y = 1.1474x 0.0134,r2=0.9999,检出限为0.005 g/100 g,定量限为0.01 g/100 g;黑曲霉二糖标准曲线方程为y = 1.9778x 0.0441,r2=0.9999 检出限为0.005 g/100 g,定量限为0.01 g/100 g;松二糖标准曲线方程为y = 1.4057x 0.0137,r2=0.9990,检出限为0.01 g/100 g,定量限为0.05g/100 g。麦芽糖标准曲线方程为 y = 3.1138x 0.078,r2=0.9999,检出限为0.010 g/100 g,定量限为0.02 g/100 g。异麦芽酮糖标准曲线方程为y = 2.1534x 0.023,r2=0.9998,检出限为0.010 g/100 g,定量限为0.05 g/100 g。异麦芽糖标准曲线方程为y = 2.2377x 0.0104,r2=0.9998,检出限为0.010 g/100 g,定量限为0.05 g/100 g。根据各标准曲线获得待测样品蜂蜜湿重中各低聚糖的含量。
62.3、低聚糖(蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖和异麦芽糖)干重含量计算:据蜂蜜湿重中各低聚糖的含量计算获得各低聚糖在蜂蜜干重中含量。
63.本实施例采用上述检测方法,测定并计算酿造1~15天中江苏中蜂枇杷蜂蜜中α,β
‑
海藻糖、黑曲霉二糖、松二糖和蔗糖在蜂蜜干重中的含量(重复测定3次,结果以平均值
±
标准偏差表示),由不同各低聚糖在蜂蜜干重中平均含量计算t值(参见表2)。由t值可知,酿造1~13天蜂蜜t值为2.40%~3.95%,均不超过5%,判定为未成熟蜂蜜;酿造14天和酿造15天蜂蜜t值分别为6.97%和5.39%,均≥5%,判定为成熟蜂蜜。
64.表2 蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖在蜂蜜干重中含量及t值
蜂蜜成熟过程中糖类物质在酶作用下发生转化现象,糖类物质含量在酿造前期处于动态变化中,当蜂蜜充分成熟时,蜂蜜中物质呈稳定状态,糖类物质含量保持稳定。本实施例通过测定不同成熟天数的蜂蜜中糖类物质含量的变化情况,以验证本发明方法的判断准确性。
65.本实施例进一步对枇杷蜂蜜干重中的蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖含量,进行单因素方差分析,多重比较结果见表3。
66.表3 蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖在蜂蜜干重中含量(平均值
±
标准偏差)注:同列含相同字母表示差异不显著(p<0.05)。
67.由表3可看出,随酿造时间延长,蔗糖含量呈降低趋势,α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖呈增加趋势。同时,据方差分析结果显示,酿造14天
和酿造15天的蜂蜜中蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、异麦芽酮糖、异麦芽糖含量差异不显著。麦芽糖含量波动上升,在酿造15天蜂蜜中含量上升至最高。由以上结果表明酿造前期蜂蜜中低聚糖含量呈波动性变化,随酿造时间延长大多数低聚糖含量保持稳定,同时多数低聚糖含量在酿造14天后低聚糖含量保持稳定,说明其已达到成熟,与本发明的上述方法判断结果一致。
68.实施例2本实验例提供一种本发明的蜂蜜成熟与否鉴别方法。具体如下:以采集于江苏省苏州市的酿造1~15天的枇杷蜂蜜作为样品进行蜂蜜成熟与否的鉴定。蜂蜜样品于
‑
20℃贮存直至分析。依据《进出口蜂蜜检验规程》(sn/t0852
‑
2012),采用阿贝折光仪测定蜂蜜中的水分含量(重复测定3次)。样品采集信息及水分含量见表4。
69.表4b组蜂箱枇杷蜂蜜样品信息表低聚糖标准贮备液的配制:准确称取苯基
‑
β
‑
d
‑
葡萄糖苷100mg,用吡啶溶解并定容至5ml配制成20mg/ml的标准储备液;准确称取蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖标准品各100mg,用超纯水溶解并定容至5ml配制成20mg/ml的标准储备液,标准储备液均于4℃下避光储存,可稳定保存3个月。
70.低聚糖标准工作溶液配制及预处理:吸取适量蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖标准储备液,混匀,配制蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖均为200μg/ml的混合标准储备液。用80%乙醇稀释混合标准储备液为0.1、0.5、1、5、20μg/ml系列混合标准工作溶液。吸取各浓度混合标准工作溶液100μl于1.5ml离心管,于40℃氮吹至干,待衍生。
71.样品前处理方法:称取混合均匀的蜂蜜样品0.40
±
0.005 g,置于50 ml离心管中,加入50 ml 80%的乙醇水溶液,涡旋震荡提取3 min,于8000 r/min,4 ℃离心10 min,吸取上清液2 ml,用80%乙醇水溶液稀释10倍(测定蔗糖含量时稀释50倍),吸取稀释液100 μl于1.5 ml离心管,于40 ℃氮吹至干,待衍生。
72.衍生化处理:在上述低聚糖混合标准工作溶液和样品提取物的1.5 ml离心管中分别加入20 μl苯基
‑
β
‑
d
‑
葡萄糖苷溶液(0.025 mg/ml),涡旋混匀,加入350 μl盐酸羟胺吡啶溶液(25 mg/ml),涡旋混匀,75 ℃反应40 min,随后加入350 μl六甲基二硅胺烷和35 μl三氟乙酸酐,涡旋混匀,45 ℃反应40 min,12000 r/min,4 ℃离心10 min,取上清液至进样瓶内,用于gc
‑
ms分析。
73.气相色谱
‑
质谱联用条件:采用日本岛津公司qp 2010 气相色谱
‑
质谱联用仪,色谱柱:db
‑
5(30 m
×
0.25 mm,0.25 μm);载气为氦气,流量0.99 ml/min;进样口温度300 ℃,不分流进样;升温程序:初始温度180 ℃,保持5 min,以10 ℃/min升温至255 ℃,保持1 min,然后以15 ℃/min升温至265 ℃,保持1 min,然后以1 ℃/min升温至270 ℃,然后以10 ℃/min升温至300 ℃,然后以20 ℃/min升温至320 ℃,保持10 min,进样量1 μl。电子电离源;离子源温度230 ℃;电离能量70 ev;溶剂延迟10 min;扫描类型:选择离子监测(sim)模式。
74.苯基
‑
β
‑
d
‑
葡萄糖苷衍生物的色谱保留时间为12.805min,特征定量离子为217,特征定性离子为361、204、319;蔗糖衍生物的色谱保留时间为16.292 min,特征定量离子为217,特征定性离子为361、204、319;α,β
‑
海藻糖衍生物的色谱保留时间为18.291 min,特征定量离子为217,特征定性离子为361,204,319;黑曲霉二糖衍生物1的色谱保留时间为19.404 min,特征定量离子为204,特征定性离子为361,217,319;黑曲霉二糖衍生物2的色谱保留时间为20.050 min,特征定量离子为204,特征定性离子为361,217,319;松二糖衍生物1的色谱保留时间为19.479 min,特征定量离子为217,特征定性离子为361,204,319;松二糖衍生物2的色谱保留时间为19.632 min,特征定量离子为217,特征定性离子为361,204,319。麦芽糖衍生物1的色谱保留时间为19.547 min,特征定量离子为217,特征定性离子为361,204,319;麦芽糖衍生物2的色谱保留时间为19.865 min,特征定量离子为217,特征定性离子为361,204,319。异麦芽酮糖衍生物1的色谱保留时间为20.520 min,特征定量离子为204,特征定性离子为217,361,319;异麦芽酮糖衍生物2的色谱保留时间为20.850 min,特征定量离子为204,特征定性离子为217,361,319。异麦芽糖衍生物1的色谱保留时间为21.460 min,特征定量离子为204,特征定性离子为217,361,319;异麦芽糖衍生物2的色谱保留时间为22.034 min,特征定量离子为204,特征定性离子为217,361,319。
75.测定方法:定性测定:通过色谱保留时间与质谱特征定量和定性离子共同定性。
76.低聚糖(蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖和异麦芽糖)湿重含量测定:以内标法定量,配制系列溶剂标准工作液,各标准溶液衍生化处理前均加入20μl的内标物苯基
‑
β
‑
d
‑
葡萄糖苷溶液(0.025 mg/ml),之后进行两步衍生化处理,最后经gc
‑
ms测定(具体步骤参见上文所述)。以低聚糖与内标物苯基
‑
β
‑
d
‑
葡萄糖苷的峰面积比为纵坐标,以低聚糖浓度为横坐标绘制标准曲线。结果在0.07~2.65 μg/ml的测定浓度范围内,蔗糖标准曲线方程为y = 0.4707x 0.0275,r2=0.9984,检出限为0.01 g/100 g,定量
限为0.05 g/100 g;α,β
‑
海藻糖标准曲线方程为y = 1.3105x 0.0115,r2=0.9998,检出限为0.005 g/100 g,定量限为0.01 g/100 g;黑曲霉二糖标准曲线方程为y = 2.1627x
ꢀ‑ꢀ
0.0295,r2=0.9995 检出限为0.005 g/100 g,定量限为0.01 g/100 g;松二糖标准曲线方程为y = 1.4558x 0.0237,r2=0.9999,检出限为0.01 g/100 g,定量限为0.05g/100 g。麦芽糖标准曲线方程为 y = 3.1551x
ꢀ‑ꢀ
0.0017,r2=0.9999,检出限为0.010 g/100 g,定量限为0.02 g/100 g。异麦芽酮糖标准曲线方程为y = 1.9113x
ꢀ‑ꢀ
0.0179,r2=0.9999,检出限为0.010 g/100 g,定量限为0.05 g/100 g。异麦芽糖标准曲线方程为y = 1.955x 0.0034,r2=0.9999,检出限为0.010 g/100 g,定量限为0.05 g/100 g。根据各标准曲线获得待测样品蜂蜜湿重中各低聚糖的含量。
77.低聚糖(蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖)干重含量计算:据蜂蜜湿重中各低聚糖的含量计算获得各低聚糖在蜂蜜干重中含量。
78.本实施例采用上述检测方法,测定并计算酿造1~15天中江苏中蜂枇杷蜂蜜中α,β
‑
海藻糖、黑曲霉二糖、松二糖和蔗糖在蜂蜜干重中的含量,(重复测定3次,结果以平均值
±
标准偏差表示),由不同各低聚糖在蜂蜜干重中平均含量计算t值(参见表5)。由t值可知,酿造1~13天蜂蜜t值为2.28%~4.35%,均不超过5%判定为未成熟蜂蜜;酿造14天和酿造15天蜂蜜t值分别为5.26%和6.85%,均≥5%,判定为成熟蜂蜜。
79.表5 蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖在蜂蜜干重中含量及t值本实施例通过测定不同成熟天数的蜂蜜中糖类物质含量的变化情况,以验证本发明方法的判断准确性。
80.进一步对枇杷蜂蜜干重中的蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖含量,进行单因素方差分析,多重比较结果见表6。
81.表6 蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖在蜂蜜干重中含量(平均值
±
标准偏差)
注:同列含相同字母表示差异不显著(p<0.05)。
82.由表6可看出,随酿造时间延长,蔗糖含量呈降低趋势,α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖呈增加趋势。同时,据方差分析结果显示,酿造14天和酿造15天的蜂蜜中蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、异麦芽酮糖、异麦芽糖含量差异不显著。麦芽糖含量波动上升,在酿造15天蜂蜜中含量上升至最高。由以上结果表明酿造前期蜂蜜中低聚糖含量呈波动性变化,随酿造时间延长大多数低聚糖含量保持稳定,同时多数低聚糖含量酿造14天后低聚糖含量保持稳定,说明其已达到成熟,与本发明的上述方法判断结果一致。
83.实施例3本实验例提供一种本发明的蜂蜜成熟与否鉴别方法。具体如下:以采集于江苏省苏州市的酿造1~15天的枇杷蜂蜜作为样品进行蜂蜜成熟与否的鉴定。蜂蜜样品于
‑
20℃贮存直至分析。依据《进出口蜂蜜检验规程》(sn/t0852
‑
2012),采用阿贝折光仪测定蜂蜜中的水分含量(重复测定3次)。样品采集信息及水分含量见表7。
84.表7c组蜂箱枇杷蜂蜜样品信息表
低聚糖标准贮备液的配制:准确称取苯基
‑
β
‑
d
‑
葡萄糖苷100 mg,用吡啶溶解并定容至5 ml配制成20 mg/ml的标准储备液;准确称取蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖标准品各100 mg,用超纯水溶解并定容至5 ml配制成20 mg/ml的标准储备液,标准储备液均于4 ℃下避光储存,可稳定保存3个月。
85.低聚糖标准工作溶液配制及预处理:吸取适量蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖标准储备液,混匀,配制蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖均为200 μg/ml的混合标准储备液。用80%乙醇稀释混合标准储备液为0.1、0.5、1、5、20 μg/ml系列混合标准工作溶液。吸取各浓度混合标准工作溶液100 μl于1.5 ml离心管,于40 ℃氮吹至干,待衍生。
86.样品前处理方法:称取混合均匀的蜂蜜样品0.30
±
0.005 g,置于50 ml离心管中,加入30 ml 80%的乙醇水溶液,涡旋震荡提取5 min,于8000 r/min,4 ℃离心10 min,吸取上清液1.5 ml,用80%乙醇水溶液稀释10倍(测定蔗糖含量时稀释50倍),吸取稀释液100 μl于1.5 ml离心管,于40 ℃氮吹至干,待衍生。
87.衍生化处理:在上述低聚糖混合标准工作溶液和样品提取物的1.5 ml离心管中分别加入20 μl苯基
‑
β
‑
d
‑
葡萄糖苷溶液(0.025 mg/ml),涡旋混匀,加入350 μl盐酸羟胺吡啶溶液(25 mg/ml),涡旋混匀,75 ℃反应45 min,随后加入350 μl六甲基二硅胺烷和35 μl三氟乙酸酐,涡旋混匀,45 ℃反应45 min,12000 r/min,4 ℃离心10 min,取上清液至进样瓶内,用于gc
‑
ms分析。
88.气相色谱
‑
质谱联用条件:采用日本岛津公司qp 2010 气相色谱
‑
质谱联用仪,色谱柱:db
‑
5(30 m
×
0.25 mm,0.25 μm);载气为氦气,流量0.99 ml/min;进样口温度300 ℃,
不分流进样;升温程序:初始温度180 ℃,保持5 min,以10 ℃/min升温至255 ℃,保持1 min,然后以15 ℃/min升温至265 ℃,保持1 min,然后以1 ℃/min升温至270 ℃,然后以10 ℃/min升温至300 ℃,然后以20 ℃/min升温至320 ℃,保持10 min,进样量1 μl。电子电离源;离子源温度230 ℃;电离能量70 ev;溶剂延迟10 min;扫描类型:选择离子监测(sim)模式。
89.苯基
‑
β
‑
d
‑
葡萄糖苷衍生物的色谱保留时间为12.758 min,特征定量离子为217,特征定性离子为361、204、319;蔗糖衍生物的色谱保留时间为16.294 min,特征定量离子为217,特征定性离子为361、204、319;α,β
‑
海藻糖衍生物的色谱保留时间为18.210 min,特征定量离子为217,特征定性离子为361,204,319;黑曲霉二糖衍生物1的色谱保留时间为19.321 min,特征定量离子为204,特征定性离子为361,217,319;黑曲霉二糖衍生物2的色谱保留时间为19.967 min,特征定量离子为204,特征定性离子为361,217,319;松二糖衍生物1的色谱保留时间为19.390 min,特征定量离子为217,特征定性离子为361,204,319;松二糖衍生物2的色谱保留时间为19.539 min,特征定量离子为217,特征定性离子为361,204,319。麦芽糖衍生物1的色谱保留时间为19.455 min,特征定量离子为217,特征定性离子为361,204,319;麦芽糖衍生物2的色谱保留时间为19.767 min,特征定量离子为217,特征定性离子为361,204,319。异麦芽酮糖衍生物1的色谱保留时间为20.523 min,特征定量离子为204,特征定性离子为217,361,319;异麦芽酮糖衍生物2的色谱保留时间为20.845 min,特征定量离子为204,特征定性离子为217,361,319。异麦芽糖衍生物1的色谱保留时间为21.454 min,特征定量离子为204,特征定性离子为217,361,319;异麦芽糖衍生物2的色谱保留时间为22.023 min,特征定量离子为204,特征定性离子为217,361,319。
90.测定方法:定性测定:通过色谱保留时间与质谱特征定量和定性离子共同定性。
91.图4为本实施例中酿造14天的成熟蜂蜜(图中的a)和酿造1天的未成熟蜂蜜(图中的b)的总离子流色谱图。
92.低聚糖(蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖和异麦芽糖)湿重含量测定:以内标法定量,配制系列溶剂标准工作液,各标准溶液衍生化处理前均加入20μl的内标物苯基
‑
β
‑
d
‑
葡萄糖苷溶液(0.025 mg/ml),之后进行两步衍生化处理,最后经gc
‑
ms测定(具体步骤参见上文所述)。以低聚糖与内标物苯基
‑
β
‑
d
‑
葡萄糖苷的峰面积比为纵坐标,以低聚糖浓度为横坐标绘制标准曲线。结果在0.07~2.65 μg/ml的测定浓度范围内,蔗糖标准曲线方程为y = 0.4535x 0.0063,r2=0.9958,检出限为0.01 g/100 g,定量限为0.05 g/100 g;α,β
‑
海藻糖标准曲线方程为y = 1.1365x 0.0088,r2=0.9999,检出限为0.005 g/100 g,定量限为0.01 g/100 g;黑曲霉二糖标准曲线方程为y = 2.1718x
ꢀ‑ꢀ
0.0491,r2=0.9996 检出限为0.005 g/100 g,定量限为0.01 g/100 g;松二糖标准曲线方程为y = 1.4559x 0.0235,r2=0.9998,检出限为0.01 g/100 g,定量限为0.05g/100 g。麦芽糖标准曲线方程为 y = 2.653x 0.0935,r2=0.9999,检出限为0.010 g/100 g,定量限为0.02 g/100 g。异麦芽酮糖标准曲线方程为y = 1.9039x 0.0248,r2=0.9999,检出限为0.010 g/100 g,定量限为0.05 g/100 g。异麦芽糖标准曲线方程为y = 2.1688x 0.0188,r2=0.9999,检出限为0.010 g/100 g,定量限为0.05 g/100 g。根据各标准曲线获得待测样品蜂蜜湿重中各低聚糖的含量。
93.低聚糖(蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖)干重含量计算:据蜂蜜湿重中各低聚糖的含量计算获得各低聚糖在蜂蜜干重中含量。
94.本实施例采用上述检测方法,测定并计算酿造1~15天中江苏中蜂枇杷蜂蜜中α,β
‑
海藻糖、黑曲霉二糖、松二糖和蔗糖在蜂蜜干重中的含量(重复测定3次,结果以平均值
±
标准偏差表示),由不同各低聚糖在蜂蜜干重中平均含量计算t值(参见表8)。由t值可知,酿造1~13天蜂蜜t值为2.36%~4.71%,均不超过5%判定为未成熟蜂蜜;酿造14天和酿造15天蜂蜜t值分别为6.44%和6.56%,均≥5%,判定为成熟蜂蜜。
95.表8 蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖在蜂蜜干重中含量及t值本实施例通过测定不同成熟天数的蜂蜜中糖类物质含量的变化情况,以验证本发明方法的判断准确性。
96.本实施例进一步对枇杷蜂蜜干重中的蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖含量,进行单因素方差分析,多重比较结果见表9。
97.表9 蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖在蜂蜜干重中含量(平均值
±
标准偏差)
注:同列含相同字母表示差异不显著(p<0.05)。
98.由表9可看出,随酿造时间延长,蔗糖含量呈降低趋势,α,β
‑
海藻糖、黑曲霉二糖、松二糖、麦芽糖、异麦芽酮糖、异麦芽糖呈增加趋势。同时,据方差分析结果显示,酿造14天和酿造15天的蜂蜜中蔗糖、α,β
‑
海藻糖、黑曲霉二糖、松二糖、异麦芽酮糖、异麦芽糖含量差异不显著。由以上结果表明酿造前期蜂蜜中低聚糖含量呈波动性变化,随酿造时间延长大多数低聚糖含量保持稳定,酿造14天后低聚糖含量保持稳定,说明其已达到成熟,与本发明的上述方法判断结果一致。
99.对比例1本对比例提供一种枇杷蜂蜜成熟与否的判定方法。由于α,β
‑
海藻糖、黑曲霉二糖和松二糖随蜂蜜酿造天数增加,其含量增加,因此,以α,β
‑
海藻糖、黑曲霉二糖和松二糖总干重含量判断蜂蜜是否成熟。
100.具体样品、操作过程与实施例1相同,区别仅在于采用如下公式进行判断:。
101.经测定计算得α,β
‑
海藻糖、黑曲霉二糖和松二糖总干重含量结果见表10。
102.表10α,β
‑
海藻糖、黑曲霉二糖和松二糖总干重含量
其与实施例1中通过蜂蜜中各糖类物质含量计算t值所获得的结果不同,在酿造15天的蜂蜜中 α,β
‑
海藻糖、黑曲霉二糖和松二糖总干重含量并非最高,其中酿造7天、14天的蜂蜜中α,β
‑
海藻糖、黑曲霉二糖和松二糖总干重含量高于酿造15天的蜂蜜。因此,无法通过α,β
‑
海藻糖、黑曲霉二糖和松二糖总干重含量准确判断枇杷蜂蜜是否成熟。
103.对比例2本对比例提供一种枇杷蜂蜜成熟与否的判定方法。具体样品、操作过程与实施例1相同,区别仅在于,以实施例1相同的方法额外测定吡喃葡糖基蔗糖的含量,并采用如下公式进行判断:。
104.经测定计算得t1结果见表11。
105.表11 吡喃葡糖基蔗糖在蜂蜜干重中含量及t1结果汇总(平均值
±
标准偏差)
注:同列不同字母表示差异显著(p<0.05)。
106.由表11中蜂蜜干重中吡喃葡糖基蔗糖含量的单因素方差分析结果可知,1~15天中吡喃葡糖基蔗糖含量呈无规则变化。因此,无法通过t1值准确判断蜂蜜是否成熟。
107.虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。