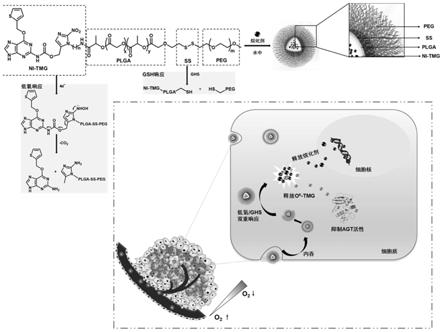
1.本发明属于生物医用高分子材料技术领域,涉及一种多功能纳米载体及其制备方法和应用。具体的说是一种兼有抗耐药性和低氧/谷胱甘肽双重响应性的多功能纳米载体及其制备方法和作为药用载体的应用。
背景技术:
2.自氮芥用于治疗恶性淋巴瘤以来,烷化剂相关的基础研究和临床应用迅速发展,烷化剂已成为癌症化疗中的一类重要药物。烷化剂可以产生带正电的碳离子中间体,与细胞中dna形成共价键,导致dna损伤,进而干扰 dna的正常复制或在dna复制时引起碱基错配,造成dna的结构和功能破坏,最终导致细胞死亡。然而,多数抗癌烷化剂存在水溶性差、稳定性差、毒副作用大等缺点,限制其在临床上的应用。此外,dna损伤修复机制中涉及的多种蛋白能够修复烷化剂引起的dna损伤,使肿瘤细胞产生耐药性,严重限制烷化剂的临床应用。其中,o6‑
烷基鸟嘌呤
‑
dna烷基转移酶(agt) 在多种肿瘤细胞中高表达,可以通过修复dna鸟嘌呤o6位上的烷化损伤来修复烷化剂造成的肿瘤细胞dna损伤,从而使肿瘤细胞对抗癌烷化剂产生耐药性。因此,agt是氯乙基亚硝基脲(cenus)、替莫唑胺、顺铂等烷化剂耐药性主要原因。
3.为了克服agt介导的耐药性,临床上常辅以agt抑制剂来提高上述烷化剂类药物的治疗效果。然而,直接将agt抑制剂与烷化剂联用,药物通过血液循环到达肿瘤组织的剂量有限;并且agt抑制剂在抑制肿瘤组织中 agt活性的同时也抑制了正常组织的agt活性,使正常组织的dna损伤不能被修复,导致抗癌烷化剂对正常细胞的毒性增强,进而带来严重的副作用。因此,迫切需要开发具有肿瘤靶向性的药物递送系统,将本身不具备肿瘤靶向性的抗癌烷化剂和agt抑制剂靶向递送至肿瘤组织,二者同时发挥抗癌和抗耐药的作用,以达到高效低毒的治疗效果。近年来纳米颗粒、脂质体、胶束等药物递送系统在临床前和临床试验中得到了广泛的研究,并已被证明其有效性。其中,两亲性嵌段共聚物,在水溶液中进行自组装,可得到具有典型核壳结构的纳米级聚合物胶束。这些纳米胶束的内核空腔包载水溶性差、化学性质不稳定的小分子化疗药物,其亲水外壳可躲避网状内皮系统的识别吞噬,延长体内循环时间,同时解决疏水药物的给药问题。因此,开发具有集成智能功能的聚合物胶束纳米载体,将烷化剂包封于纳米胶束中,不仅可以克服药物水溶性差、稳定性差的问题,而且能够有效解决其不具备肿瘤靶向性的缺陷,从而提高其药效并减小其毒副作用。
4.为了实现药物的靶向递送,基于肿瘤组织生物学特征(如低ph、低氧及酶的特异性表达),开发刺激响应型的纳米载体是有效策略。肿瘤组织中存在低氧微环境,是区别于正常组织的重要特征;同时,肿瘤细胞中还原物质谷胱甘肽(gsh)的表达水平显著高于正常细胞。因此,基于肿瘤组织低氧和 gsh高表达的特征,设计一种兼有低氧和gsh双重刺激响应性并同时具有抗耐药性的纳米药物载体,用于抗癌烷化剂的靶向递送,将有助于实现药物靶向作用于肿瘤部位,从而发挥高效低毒的抗肿瘤作用。
技术实现要素:
5.本发明的目的是提供一种兼有抗耐药性和低氧/gsh双重响应性的多功能纳米载体及其制备方法和应用。本发明的纳米递送系统载体是一种两亲性嵌段共聚物,能够通过分子间作用力包载疏水性化疗药物,形成纳米胶束。在低氧和gsh高表达的肿瘤组织中,该纳米载体上的硝基能够被还原,使得硝基咪唑与o6‑
噻吩甲基鸟嘌呤(o6‑
tmg)之间的偶联断裂,同时二硫键被谷胱甘肽还原也发生断裂,从而导致纳米胶束碎裂。胶束碎裂后释放出内部包载的烷化剂和能够抑制agt活性的o6‑
tmg,从而发挥靶向抗肿瘤作用,同时特异性的提高肿瘤细胞对烷化剂的敏感性。
6.所述低氧响应基团为2
‑
硝基咪唑(ni),gsh响应基团为二硫键(ss)。
7.所述agt抑制剂为o6‑
tmg。
8.所述两亲性嵌段共聚物由具有gsh响应性的二硫键作为连接基团,一端连接亲水端聚乙二醇(peg),另一端连接疏水端聚乳酸
‑
羟基乙酸共聚物 (plga)。将o6‑
tmg与低氧响应基团ni连接后,再共价连接到上述两亲性嵌段共聚物plga
‑
ss
‑
peg上,形成具有低氧/gsh双重响应性的偶联物ni
‑ꢀ
tmg
‑
plga
‑
ss
‑
peg,通过自组装反应形成纳米胶束用于递送烷化剂类抗肿瘤药物。
9.本发明提供“一种兼有抗耐药性和低氧/gsh双重响应性的多功能纳米载体”的制备方法及包括的步骤:
10.1)低氧/gsh双靶向性agt抑制剂两亲性嵌段共聚物偶联物ni
‑
tmg
‑ꢀ
plga
‑
ss
‑
peg的合成方法:
11.本发明提供低氧/gsh双靶向性偶联物ni
‑
tmg
‑
plga
‑
ss
‑
peg的结构式为:
[0012][0013]
称取o6‑
tmg溶于二氯甲烷中,得到浓度为8
‑
20mg/ml的o6‑
tmg二氯甲烷溶液。再称取三光气,在0
‑
10℃条件下,将o6‑
tmg溶液滴加到三光气中,滴加完毕后在n2保护、有机碱作为催化剂的条件下,于25℃反应4
‑ꢀ
10h。再加入(1
‑
(氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基)甲醇,于25℃继续反应4
‑ꢀ
12h。其中o6‑
tmg、有机碱、三光气、(1
‑
(氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基) 甲醇的投料摩尔比为1:(6
‑
12):(1
‑
6):(2
‑
10)。将反应液经35℃减压旋蒸除去溶剂后,采用柱层析法进行分离提纯,固定相为硅胶,流动相为石油醚和乙酸乙酯,以石油醚/乙酸乙酯(v/v)为1:1
‑
1:5的比例进行洗脱,洗脱液经35℃减压旋蒸后得到产物(1
‑
(2
‑
氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基)甲基(6
‑
(噻吩
‑2‑
基甲氧基)
‑
9h
‑
嘌呤
‑2‑
基)氨基甲酸酯(ni
‑
tmg)。
[0014]
称取plga
‑
ss
‑
peg溶于无水吡啶中,得到浓度为20
‑
60mg/ml的 plga
‑
ss
‑
peg溶液,向其中加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐 (edc
·
hcl)、n
‑
羟基琥珀酰亚胺(nhs),其中plga
‑
ss
‑
peg、edc
·
hcl、 nhs的投料摩尔比为1:(1
‑
8):(1
‑
8),于25℃下反应2
‑
6h得到反应液a;再称取ni
‑
tmg,按plga
‑
ss
‑
peg与ni
‑
tmg的投料摩尔比为1:(1
‑
10)的比例
加入到反应液a中,于25℃下继续反应8
‑
12h,得到含有ni
‑
tmg
‑
plga
‑ꢀ
ss
‑
peg的混合液b。将混合液b在50℃下进行减压旋蒸,然后将浓缩液置于透析袋中,透析袋截留分子量为3000
‑
4000da,使用无水乙醇/蒸馏水 (v/v)=3:1
‑
1:1透析,透析时间为24
‑
72h。将透析液真空冷冻干燥,即得浅褐色粉末ni
‑
tmg
‑
plga
‑
ss
‑
peg偶联物。
[0015]
2)一种兼有抗耐药性和低氧/gsh双重响应性的多功能纳米载体ni
‑ꢀ
tmg
‑
plga
‑
ss
‑
peg nps的制备:
[0016]
称取ni
‑
tmg
‑
plga
‑
ss
‑
peg溶于蒸馏水中得到浓度为20
‑
60mg/ml的偶联物水溶液,0
‑
4℃下超声处理10
‑
20min,超声功率设为300
‑
600w。将上述反应液用混合纤维素滤膜针式过滤器(0.45μm)过滤,真空冷冻干燥24
‑ꢀ
72h,即得纳米载体ni
‑
tmg
‑
plga
‑
ss
‑
peg nps。
[0017]
3)载化疗药物的一种兼有抗耐药性和低氧/gsh双重响应性的多功能抗肿瘤纳米药物ni
‑
tmg
‑
plga
‑
ss
‑
peg/drug nps的制备:
[0018]
将疏水性烷化剂类化疗药物溶于有机溶剂中得到浓度为20
‑
120mg/ml 的药物溶液,将ni
‑
tmg
‑
plga
‑
ss
‑
peg溶于蒸馏水中得到浓度为20
‑
55 mg/ml的ni
‑
tmg
‑
plga
‑
ss
‑
peg水溶液,将药物溶液用注射器均匀地注入 ni
‑
tmg
‑
plga
‑
ss
‑
peg水溶液中,继续搅拌10
‑
20min。然后在0
‑
4℃下超声处理10
‑
35min,超声功率设为300
‑
600w。接下来,将上述超声后的反应液转移至透析袋,中,透析袋截留分子量为2500da,透析液为ph=7.4的 pbs,透析4
‑
10h以除去未包封的药物。将透析袋中后的液体真空冷冻干燥,即得包载化疗药物的抗耐药性和低氧/gsh双重响应性的多功能抗肿瘤纳米药物ni
‑
tmg
‑
plga
‑
ss
‑
peg/drug nps。
[0019]
优选地,所述步骤1)中o6‑
tmg二氯甲烷溶液的浓度优选为8.2
‑
16 mg/ml;有机碱优选为吡啶或三乙胺;o6‑
tmg、有机碱、三光气、(1
‑
(氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基)甲醇的投料摩尔比优选为1:(6
‑
10):(1
‑
3):(2
‑
8); plga
‑
ss
‑
peg吡啶溶液的浓度优选为30
‑
60mg/ml;所述plga
‑
ss
‑
peg的分子量优选为4000da;plga
‑
ss
‑
peg、edc
·
hcl、nhs的投料摩尔比优选为1:(1
‑
5):(1
‑
5),,plga
‑
ss
‑
peg与ni
‑
tmg的摩尔比优选为1:(5
‑
10),透析时间优选为48
‑
72h。
[0020]
优选地,步骤2)中ni
‑
tmg
‑
plga
‑
ss
‑
peg水溶液浓度优选为25
‑
30 mg/ml;超声处理的时间优选为10
‑
15min;超声功率优选为300
‑
400w;真空冷冻干燥时间优选为24
‑
48h。
[0021]
优选地,步骤3)所述的疏水性化疗药物为dna烷化剂类药物,优选为氯乙基亚硝基脲类抗癌烷化剂及替莫唑胺(tmz);进一步优选为卡莫司汀 (bcnu)、牛磺莫斯汀(tcnu)和tmz。所述有机溶剂为二甲基亚砜(dmso)、乙醇、甲醇、四氢呋喃中的一种或多种,优选为乙醇或dmso;药物溶液的浓度优选为30
‑
60mg/ml;ni
‑
tmg
‑
plga
‑
ss
‑
peg水溶液的浓度优选为20
‑ꢀ
35mg/ml;超声处理时间优选为5
‑
20min,超声功率优选为300
‑
400w;透析时间优选为6
‑
10h。
[0022]
本发明所述肿瘤为脑胶质瘤、脑瘤、骨髓瘤、淋巴瘤、恶性黑色素瘤、乳腺癌、肺癌、胃癌、前列腺癌、结肠癌、淋巴癌、白血病的一种或多种;为脑胶质瘤、脑瘤、前列腺癌、肺癌、肝癌的一种或多种。
[0023]
本发明具有的实质性特点:
[0024]
1)本发明制备功能性纳米载体具有抗耐药性和低氧/gsh双重响应性。在低氧和gsh高表达的肿瘤微环境中,载体上的2
‑
硝基咪唑被还原,释放 agt抑制剂o6‑
tmg,发挥抗耐药作用,同时二硫键被gsh还原,进一步释放纳米胶束核心所包载的抗肿瘤药物,从而实
现抗肿瘤药物的靶向递送和可控释放,发挥增效减毒的作用。
[0025]
2)本发明所用的两亲性嵌段共聚物材料具有良好的生物相容性、生物可降解性,能在水溶液中自组装形成粒径均匀和分散度好的球形纳米载体。
[0026]
3)本发明所设计的功能性纳米载体能够包载疏水性化疗药物,克服了烷化剂类化疗药物体内不稳定、易被清除以及疏水性药物难溶性带来的生物利用度低等缺点。
[0027]
4)本发明所制备的功能性纳米载体是具有壳核结构的球形纳米颗粒,其平均粒径为100
‑
200nm,在肿瘤组织高渗透长滞留效应下,有效地富集在肿瘤部位,通过胞吞进入细胞。
[0028]
5)本发明制备方法简单,经济成本低廉,具有很广阔的应用前景。
附图说明
[0029]
附图是用来对本发明的进一步阐述,与下面具体实施方式共同用于解释本发明,但并不构成对本发明的限制。附图中:
[0030]
图1为本发明的“一种兼有抗耐药性和低氧/gsh双重响应性的多功能纳米载体”的制备路线及在肿瘤组织中药物递送示意图;
[0031]
图2为本发明实施例1中ni
‑
tmg
‑
plga
‑
ss
‑
peg nps的粒径分布和透射电镜形貌图;
[0032]
图3为本发明实施例3中ni
‑
tmg
‑
plga
‑
ss
‑
peg/bcnu nps的粒径分布和透射电镜形貌图;
[0033]
图4为本发明实施例5中ni
‑
tmg
‑
plga
‑
ss
‑
peg/tcnu nps的粒径分布和透射电镜形貌图;
[0034]
图5为本发明实施例7中ni
‑
tmg
‑
plga
‑
ss
‑
peg/tmz nps的粒径分布和透射电镜形貌图;
[0035]
图6为实验例2中各功能性载药载体在h2o、pbs和含10%血清(fbs) 的mem培养基中稳定性;
[0036]
图7为实验例3中各功能性载药载体在不加gsh条件下的低氧还原敏感性(图a、c、e为常氧条件下的测定结果,图b、d、f为低氧条件下的测定结果);
[0037]
图8为实验例4中各功能性载药载体在常氧条件下的gsh响应性(图a、 c、e为不加gsh条件下的测定结果,图b、d、f为加入gsh条件下的测定结果);
[0038]
图9为实验例5中在不加gsh的条件下,常氧和低氧反应体系中各游离药物与功能性纳米载体的药物累积释放曲线;
[0039]
图10为实验例5中在加入gsh的条件下,常氧和低氧反应体系中各游离药物与功能性纳米载体的药物累积释放曲线;
具体实施方式
[0040]
为了更清楚地阐述本发明,以下申请人将按照本发明技术方案的实施例对本发明作更进一步的详细说明。
[0041]
实施例1一种兼有抗耐药性和低氧/gsh双重响应的多功能纳米载体的 (ni
‑
tmg
‑
plga
‑
ss
‑
peg nps)制备:
[0042]
(1)ni
‑
tmg
‑
plga
‑
ss
‑
peg的合成
[0043]
称取0.12g(0.5mmol)o6‑
tmg溶于10ml二氯甲烷中,得到浓度为12 mg/ml的o6‑
tmg二氯甲烷溶液,再称取0.15g(0.5mmol)三光气,在0
‑
10℃条件下,将o6‑
tmg溶液滴加到三光气中,滴加完毕后在n2保护、0.25ml (3mmol)吡啶作为催化剂的条件下,于25℃反应4h。再加入0.17g(1mmol) (1
‑
(氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基)甲醇,于25℃继续反应6h。将上述反应液经35℃减压旋蒸除去溶剂后,采用柱层析法进行分离提纯,固定相为硅胶,流动相为石油醚和乙酸乙酯,以石油醚/乙酸乙酯(v/v)为1:1
‑
1:5的比例进行洗脱,洗脱液经35℃减压旋蒸后得到浅褐色固体ni
‑
tmg。
[0044]
称取0.32g plga
‑
ss
‑
peg溶于10ml无水吡啶中,得到浓度为32 mg/ml的plga
‑
ss
‑
peg溶液,向其中加入0.02g(0.1mmol)edc
·
hcl、0.01 g(0.1mmol)nhs,于25℃下反应2h得到反应液a;再称取0.2g(0.4mmol) ni
‑
tmg加入到反应液a中,于25℃下继续反应8h,得到含有ni
‑
tmg
‑ꢀ
plga
‑
ss
‑
peg的混合液b。将混合液b在50℃下进行减压旋蒸,然后将浓缩液置于透析袋中,透析袋截留分子量为3500da,使用无水乙醇/蒸馏水 (v/v)=3:1
‑
1:1透析,透析时间为48h。将透析液真空冷冻干燥,即得ni
‑ꢀ
tmg
‑
plga
‑
ss
‑
peg偶联物。
[0045]
ir(kbr压片)υ/cm
‑1:3412.8(n
‑
h);2983.6(c
‑
h);1790.8(c=o);1685.6 (c=n);1605.3(c=c);1531.3(n=o);1142.6(c
‑
o
‑
c);1075.2(c
‑
o);629.1(c
‑
s); 542.6(s
‑
s);
[0046]1h nmr(400mhz,dmso)δ:1.42(d,3h,ch3);5.03(s,2h,ch2‑
o6);5.32(s, 2h,ch2);5.72(s,2h,ch2‑
n);6.93
‑
7.47(m,4h,ch);8.08(s,h,nh);10.32(s,h, nh);13.86(s,h,nh);
[0047]
(2)ni
‑
tmg
‑
plga
‑
ss
‑
peg np的制备
[0048]
称取250mg步骤(1)中所制备的ni
‑
tmg
‑
plga
‑
ss
‑
peg溶于10ml蒸馏水中得到浓度为25mg/ml的偶联物水溶液,0
‑
4℃下超声处理10min,超声功率设为300w。将上述反应液用混合纤维素滤膜针式过滤器(0.45μm)过滤,真空冷冻干燥36h,即得纳米载体ni
‑
tmg
‑
plga
‑
ss
‑
peg nps。
[0049]
实施例2一种兼有抗耐药性和低氧/gsh双重响应的多功能纳米载体的 (ni
‑
tmg
‑
plga
‑
ss
‑
peg nps)制备:
[0050]
(1)ni
‑
tmg
‑
plga
‑
ss
‑
peg的合成
[0051]
称取0.2g(0.8mmol)o6‑
tmg溶于15ml二氯甲烷中,得到浓度为13 mg/ml的o6‑
tmg二氯甲烷溶液,再称取0.3g(1mmol)三光气,在0
‑
10℃条件下,将o6‑
tmg溶液滴加到三光气中,滴加完毕后在n2保护、0.72ml (5.6mmol)三乙胺作为催化剂的条件下,于25℃反应6h。再加入0.3g(1.8 mmol)(1
‑
(氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基)甲醇,于25℃继续反应8h。将上述反应液经35℃减压旋蒸除去溶剂后,采用柱层析法进行分离提纯,固定相为硅胶,流动相为石油醚和乙酸乙酯,以石油醚/乙酸乙酯(v/v)为1:1
‑
1:5 的比例进行洗脱,洗脱液经35℃减压旋蒸后得到浅褐色固体ni
‑
tmg。
[0052]
称取0.4g plga
‑
ss
‑
peg溶于12ml无水吡啶中,得到浓度为33 mg/ml的plga
‑
ss
‑
peg溶液,向其中加入0.038g(0.2mmol)edc
·
hcl、 0.023g(0.2mmol)nhs,于25℃下反应3h得到反应液a;再称取0.25g (0.55mmol)ni
‑
tmg加入到反应液a中,于25℃下继续反应9h,得到含有 ni
‑
tmg
‑
plga
‑
ss
‑
peg的混合液b。将混合液b在50℃下进行减压旋蒸,然后将浓缩液置于透析袋中,透析袋截留分子量为4000da,使用无水乙醇/ 蒸馏水(v/v)=3:1
‑
1:1透析,透析时间为54h。将透析液真空冷冻干燥,即得 ni
‑
tmg
‑
plga
‑
ss
‑
peg偶联物。
[0053]
ir(kbr压片)υ/cm
‑1:3396.8(n
‑
h);2973.7(c
‑
h);1783.1(c=o);1643.6 (c=n);1587.4(c=c);1519.8(n=o);1132.7(c
‑
o
‑
c);1022.5(c
‑
o);644.2(c
‑
s); 566.7(s
‑
s);
[0054]1h nmr(400mhz,dmso)δ:1.65(d,3h,ch3);5.22(s,2h,ch2‑
o6);5.63(s, 2h,ch2);6.14(s,2h,ch2‑
n);7.05
‑
7.78(m,4h,ch);8.45(s,h,nh);10.74(s,h, nh);13.97(s,h,nh);
[0055]
(2)ni
‑
tmg
‑
plga
‑
ss
‑
peg nps的制备
[0056]
称取300mg步骤(1)中所制备的ni
‑
tmg
‑
plga
‑
ss
‑
peg溶于10ml蒸馏水中得到浓度为30mg/ml的偶联物水溶液,0
‑
4℃下超声处理15min,超声功率设为400w。将上述反应液用混合纤维素滤膜针式过滤器(0.45μm)过滤,真空冷冻干燥48h,即得纳米载体ni
‑
tmg
‑
plga
‑
ss
‑
peg nps。
[0057]
实施例3载卡莫司汀的一种兼有抗耐药性和低氧/gsh双重响应的多功能纳米载体的(ni
‑
tmg
‑
plga
‑
ss
‑
peg/bcnu nps)制备
[0058]
(1)ni
‑
tmg
‑
plga
‑
ss
‑
peg的合成
[0059]
称取0.25g(1mmol)o6‑
tmg溶于18ml二氯甲烷中,得到浓度为13 mg/ml的o6‑
tmg二氯甲烷溶液,再称取0.45g(1.5mmol)三光气,在0
‑
10℃条件下,将o6‑
tmg溶液滴加到三光气中,滴加完毕后在n2保护、0.7ml(8 mmol)吡啶作为催化剂的条件下,于25℃反应8h。然后加入0.43g(2.5 mmol)(1
‑
(氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基)甲醇,于25℃继续反应6h。将上述反应液经35℃减压旋蒸除去溶剂后,采用柱层析法进行分离提纯,固定相为硅胶,流动相为石油醚和乙酸乙酯,以石油醚/乙酸乙酯(v/v)为1:1
‑
1:5 的比例进行洗脱,洗脱液经35℃减压旋蒸后得到浅褐色固体ni
‑
tmg。
[0060]
称取0.8g plga
‑
ss
‑
peg溶于20ml无水吡啶中,得到浓度为40 mg/ml的plga
‑
ss
‑
peg溶液,向其中加入0.095g(0.5mmol)edc
·
hcl、 0.058g(0.5mmol)nhs,于25℃下反应4h得到反应液a;再称取0.55g (1.2mmol)ni
‑
tmg加入到反应液a中,于25℃下继续反应10h,得到含有 ni
‑
tmg
‑
plga
‑
ss
‑
peg的混合液b。将混合液b在50℃下进行减压旋蒸,然后将浓缩液置于透析袋中,透析袋截留分子量为3500da,使用无水乙醇/ 蒸馏水(v/v)=3:1
‑
1:1透析,透析时间为54h。将透析液真空冷冻干燥,即得 ni
‑
tmg
‑
plga
‑
ss
‑
peg偶联物。
[0061]
ir(kbr压片)υ/cm
‑1:3422.4(n
‑
h);2983.2(c
‑
h);1765.3(c=o);1623.5 (c=n);1546.7(c=c);1502.9(n=o);1121.4(c
‑
o
‑
c);1056.3(c
‑
o);683.1(c
‑
s); 556.7(s
‑
s);
[0062]1h nmr(400mhz,dmso)δ:1.57(d,3h,ch3);5.34(s,2h,ch2‑
o6);5.76(s, 2h,ch2);6.45(s,2h,ch2‑
n);7.25
‑
7.86(m,4h,ch);8.65(s,h,nh);12.06(s,h, nh);13.65(s,h,nh);
[0063]
(2)ni
‑
tmg
‑
plga
‑
ss
‑
peg/bcnu nps的制备
[0064]
称取3mg bcnu溶于100μl无水乙醇中得到浓度为30mg/ml的 bcnu乙醇溶液,称取40mg ni
‑
tmg
‑
plga
‑
ss
‑
peg溶于2ml去离子水中得到浓度为20mg/ml的偶联物水溶液,将上述bcnu溶液用注射器均匀地滴加至偶联物水溶液中,继续搅拌反应10min。然后在冰浴条件下超声处理反应液5min,超声功率设为300w。接下来,将上述超声后的反应液加至透析袋中,透析袋截留分子量为2500da,透析液为ph=7.4的pbs,透析时间为6h以除去未包封的药物。最后,将透析袋中液体冻干,即得ni
‑
tmg
‑ꢀ
plga
‑
ss
‑
peg/bcnu nps。
[0065]
实施例4载卡莫司汀的一种兼有抗耐药性和低氧/gsh双重响应的多功能纳米载体
的(ni
‑
tmg
‑
plga
‑
ss
‑
peg/bcnu nps)制备
[0066]
(1)ni
‑
tmg
‑
plga
‑
ss
‑
peg的合成
[0067]
称取0.3g(1.2mmol)o6‑
tmg溶于20ml二氯甲烷中,得到浓度为15 mg/ml的o6‑
tmg二氯甲烷溶液,再称取0.65g(2.16mmol)三光气,在0
‑ꢀ
10℃条件下,将o6‑
tmg溶液滴加到三光气中,滴加完毕后在n2保护、 1.39ml(10mmol)三乙胺作为催化剂的条件下,于25℃反应8h。然后加入 0.62g(3.6mmol)(1
‑
(氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基)甲醇,于25℃继续反应 6h。将上述反应液经35℃减压旋蒸除去溶剂后,采用柱层析法进行分离提纯,固定相为硅胶,流动相为石油醚和乙酸乙酯,以石油醚/乙酸乙酯(v/v)为 1:1
‑
1:5的比例进行洗脱,洗脱液经35℃减压旋蒸后得到浅褐色固体ni
‑ꢀ
tmg。
[0068]
称取1.2g plga
‑
ss
‑
peg溶于25ml无水吡啶中,得到浓度为48 mg/ml的plga
‑
ss
‑
peg溶液,向其中加入0.16g(0.83mmol)edc
·
hcl、0.094g(0.83mmol)nhs,于25℃下反应5h得到反应液a;再称取0.83g (1.8mmol)ni
‑
tmg加入到反应液a中,于25℃下继续反应12h,得到含有 ni
‑
tmg
‑
plga
‑
ss
‑
peg的混合液b。将混合液b在50℃下进行减压旋蒸,然后将浓缩液置于透析袋中,透析袋截留分子量为4000da,使用无水乙醇/ 蒸馏水(v/v)=3:1
‑
1:1透析,透析时间为60h。将透析液真空冷冻干燥,即得 ni
‑
tmg
‑
plga
‑
ss
‑
peg偶联物。
[0069]
ir(kbr压片)υ/cm
‑1:3376.2(n
‑
h);2862.9(c
‑
h);1715.3(c=o);1605.2 (c=n);1542.4(c=c);1482.5(n=o);1152.3(c
‑
o
‑
c);937.2(c
‑
o);638.9(c
‑
s); 543.5(s
‑
s);
[0070]1h nmr(400mhz,dmso)δ:1.46(d,3h,ch3);5.32(s,2h,ch2‑
o6);5.43 (s,2h,ch2);6.64(s,2h,ch2‑
n);7.37
‑
7.93(m,4h,ch);8.56(s,h,nh);11.23 (s,h,nh);12.43(s,h,nh);
[0071]
(2)ni
‑
tmg
‑
plga
‑
ss
‑
peg/bcnu nps的制备
[0072]
称取6mg bcnu溶于100μldmso中得到浓度为60mg/ml的bcnudmso溶液,称取60mg ni
‑
tmg
‑
plga
‑
ss
‑
peg溶于2ml去离子水中得到浓度为30mg/ml的偶联物水溶液,将上述bcnu溶液用注射器均匀地滴加至偶联物水溶液中,继续搅拌反应15min。然后在冰浴条件下超声处理反应液15min,超声功率设为350w。接下来,将上述超声后的反应液加至透析袋中,透析袋截留分子量为2500da,透析液为ph=7.4的pbs,透析时间为 8h以除去未包封的药物。最后,将透析袋中液体冻干,即得ni
‑
tmg
‑ꢀ
plga
‑
ss
‑
peg/bcnu nps。
[0073]
实施例5载牛磺莫斯汀的一种兼有抗耐药性和低氧/gsh双重响应的多功能纳米载体的(ni
‑
tmg
‑
plga
‑
ss
‑
peg/tcnu nps)制备
[0074]
(1)ni
‑
tmg
‑
plga
‑
ss
‑
peg的合成
[0075]
称取0.4g(1.6mmol)o6‑
tmg溶于25ml二氯甲烷中,得到浓度为16 mg/ml的o6‑
tmg二氯甲烷溶液,再称取0.96g(3.2mmol)三光气,在0
‑ꢀ
10℃条件下,将o6‑
tmg溶液滴加到三光气中,滴加完毕后在n2保护、1.1 ml(13.6mmol)吡啶作为催化剂的条件下,于25℃反应6h。然后加入(1
‑
(氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基)甲醇0.96g(5.6mmol),于25℃继续反应6h。将上述反应液经35℃减压旋蒸除去溶剂后,采用柱层析法进行分离提纯,固定相为硅胶,流动相为石油醚和乙酸乙酯,以石油醚/乙酸乙酯(v/v)为1:1
‑ꢀ
1:5的比例进行洗脱,洗脱液经35℃减压旋蒸后得到浅褐色固体ni
‑
tmg。
[0076]
称取1.6g plga
‑
ss
‑
peg溶于30ml无水吡啶中,得到浓度为53 mg/ml的plga
‑
ss
‑
peg溶液,向其中加入0.23g(1.2mmol)edc
·
hcl、0.14 g(1.2mmol)nhs,于25℃下反应6h得
ni
‑
tmg
‑
plga
‑
ss
‑
peg溶于2ml去离子水中得到浓度为35mg/ml的偶联物水溶液,将上述tcnu溶液用注射器均匀地滴加至偶联物水溶液中,继续搅拌反应20min。然后在冰浴条件下超声处理反应液20min,超声功率设为400w。接下来,将上述超声后的反应液加至透析袋中,透析袋截留分子量为2500da,透析液为ph=7.4的pbs,透析时间为 8h以除去未包封的药物。最后,将透析袋中液体冻干,即得ni
‑
tmg
‑ꢀ
plga
‑
ss
‑
peg/tcnu nps。
[0089]
实施例7载替莫唑胺的一种兼有抗耐药性和低氧/gsh双重响应的多功能纳米载体的(ni
‑
tmg
‑
plga
‑
ss
‑
peg/tmz nps)制备
[0090]
(1)ni
‑
tmg
‑
plga
‑
ss
‑
peg的合成
[0091]
称取0.49g(2mmol)o6‑
tmg溶于35ml二氯甲烷中,得到浓度为14 mg/ml的o6‑
tmg二氯甲烷溶液,再称取1.5g(5mmol)三光气,在0
‑
10℃条件下,将o6‑
tmg溶液滴加到三光气中,滴加完毕后在n2保护、1.35ml (16.2mmol)吡啶作为催化剂的条件下,于25℃反应6h。再加入1.72g(10 mmol)(1
‑
(氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基)甲醇,于25℃继续反应6h。将上述反应液经35℃减压旋蒸除去溶剂后,采用柱层析法进行分离提纯,固定相为硅胶,流动相为石油醚和乙酸乙酯,以石油醚/乙酸乙酯(v/v)为1:1
‑
1:5 的比例进行洗脱,洗脱液经35℃减压旋蒸后得到浅褐色固体ni
‑
tmg)。
[0092]
称取2.4g plga
‑
ss
‑
peg溶于50ml无水吡啶中,得到浓度为48 mg/ml的plga
‑
ss
‑
peg溶液,向其中加入0.52g(2.7mmol)edc
·
hcl、0.31 g(2.7mmol)nhs,于25℃下反应4h得到反应液a;再称取2.48g(5.4 mmol)ni
‑
tmg加入到反应液a中,于25℃下继续反应12h,得到含有ni
‑ꢀ
tmg
‑
plga
‑
ss
‑
peg的混合液b。将混合液b在50℃下进行减压旋蒸,然后将浓缩液置于透析袋中,透析袋截留分子量为3500da,使用无水乙醇/蒸馏水(v/v)=3:1
‑
1:1透析,透析时间为66h。将透析液真空冷冻干燥,即得ni
‑ꢀ
tmg
‑
plga
‑
ss
‑
peg偶联物。
[0093]
ir(kbr压片)υ/cm
‑1:3476.8(n
‑
h);2893.6(c
‑
h);1754.3(c=o);1667.7 (c=n);1523.5(c=c);1433.4(n=o);1152.8(c
‑
o
‑
c);1067.3(c
‑
o);674.9(c
‑
s); 535.5(s
‑
s);
[0094]1h nmr(400mhz,dmso)δ:1.64(d,3h,ch3);5.36(s,2h,ch2‑
o6);5.83 (s,2h,ch2);6.46(s,2h,ch2‑
n);7.23
‑
7.45(m,4h,ch);8.89(s,h,nh);11.65 (s,h,nh);13.86(s,h,nh);
[0095]
(2)ni
‑
tmg
‑
plga
‑
ss
‑
peg/tmz nps的制备
[0096]
称取4mg tmz溶于100μl无水乙醇中得到浓度为40mg/ml的tmz 乙醇溶液,称取40mg ni
‑
tmg
‑
plga
‑
ss
‑
peg溶于2ml去离子水中得到浓度为20mg/ml的偶联物水溶液,将上述tmz溶液用注射器均匀地滴加至偶联物水溶液中,继续搅拌反应10min。然后在冰浴条件下超声处理反应液10 min,超声功率设为350w。接下来,将上述超声后的反应液加至透析袋中,透析袋截留分子量为2500da,透析液为ph=7.4的pbs,透析时间为8h以除去未包封的药物。最后,将透析袋中液体冻干,即得ni
‑
tmg
‑
plga
‑
ss
‑ꢀ
peg/tmz nps。
[0097]
实施例8载替莫唑胺的一种兼有抗耐药性和低氧/gsh双重响应的多功能纳米载体的(ni
‑
tmg
‑
plga
‑
ss
‑
peg/tmz nps)制备
[0098]
(1)ni
‑
tmg
‑
plga
‑
ss
‑
peg的合成
[0099]
称取0.62g(2.5mmol)o6‑
tmg溶于40ml二氯甲烷中,得到浓度为15 mg/ml的o6‑
tmg二氯甲烷溶液,再称取2.25g(7.5mmol)三光气,在0
‑
10℃条件下,将o6‑
tmg溶液滴加到三光气中,滴加完毕后在n2保护、3.13ml (22.5mmol)三乙胺作为催化剂的条件下,于25℃反应
6h。然后加入2.6g (15mmol)(1
‑
(氨基乙基)
‑2‑
硝基
‑
1h
‑
咪唑
‑5‑
基)甲醇,于25℃继续反应6h。将上述反应液经35℃减压旋蒸除去溶剂后,采用柱层析法进行分离提纯,固定相为硅胶,流动相为石油醚和乙酸乙酯,以石油醚/乙酸乙酯(v/v)为1:1
‑ꢀ
1:5的比例进行洗脱,洗脱液经35℃减压旋蒸后得到浅褐色固体ni
‑
tmg。
[0100]
称取3.2g plga
‑
ss
‑
peg溶于60ml无水吡啶中,得到浓度为53 mg/ml的plga
‑
ss
‑
peg溶液,向其中加入0.77g(4mmol)edc
·
hcl、0.46g (4mmol)nhs,于25℃下反应6h得到反应液a;再称取3.67g(8mmol) ni
‑
tmg加入到反应液a中,于25℃下继续反应10h,得到含有ni
‑
tmg
‑ꢀ
plga
‑
ss
‑
peg的混合液b。将混合液b在50℃下进行减压旋蒸,然后将浓缩液置于透析袋中,透析袋截留分子量为4000da,使用无水乙醇/蒸馏水 (v/v)=3:1
‑
1:1透析,透析时间为72h。将透析液真空冷冻干燥,即得ni
‑ꢀ
tmg
‑
plga
‑
ss
‑
peg偶联物。
[0101]
ir(kbr压片)υ/cm
‑1:3434.6(n
‑
h);2817.8(c
‑
h);1725.6(c=o);1634.1 (c=n);1543.6(c=c);1399.0(n=o);1134.1(c
‑
o
‑
c);1045.4(c
‑
o);668.4(c
‑
s); 548.4(s
‑
s);
[0102]1h nmr(400mhz,dmso)δ:1.49(d,3h,ch3);5.37(s,2h,ch2‑
o6);5.92 (s,2h,ch2);6.87(s,2h,ch2‑
n);7.43
‑
7.86(m,4h,ch);8.79(s,h,nh);11.78 (s,h,nh);13.64(s,h,nh);
[0103]
(2)ni
‑
tmg
‑
plga
‑
ss
‑
peg/tmz的制备
[0104]
称取5mg tmz溶于100μl dmso中得到浓度为50mg/ml的tmzdmso溶液,称取60mg ni
‑
tmg
‑
plga
‑
ss
‑
peg溶于2ml去离子水中得到浓度为30mg/ml的偶联物水溶液,将上述tmz溶液用注射器均匀地滴加至偶联物水溶液中,继续搅拌反应10min。然后在冰浴条件下超声处理反应液 15min,超声功率设为400w。接下来,将上述超声后的反应液加至透析袋中,透析袋截留分子量为2500da,透析液为ph=7.4的pbs,透析时间为10h以除去未包封的药物。最后,将透析袋中液体冻干,即得ni
‑
tmg
‑ꢀ
plga
‑
ss
‑
peg/tmz nps。
[0105]
实验例1:各载药的功能性纳米载体包封率和载药量的测试
[0106]
1.实验材料
[0107]
将上述透析前各载药功能性纳米载体的反应液置于超滤管(3kd)中, 1000rpm离心8min,收集滤液,并利用有机溶剂破坏法取适量甲醇滴入反应液中,经0.22μm水膜过滤,高效液相色谱法(hplc)测定药物含量并计算包封率与载药量。
[0108][0109][0110]
2.实验结果:见表1。
[0111]
表1各载药功能性纳米载体包封率和载药量
[0112]
[0113]
由表1中数据可知,制备的一种兼有抗耐药性和低氧/gsh双重响应性的多功能纳米载体对各疏水性药物的包封率均在70%以上,载药量均在6%以上,说明该载体可高效包封各疏水性药物,且具有较高载药量。
[0114]
实验例2:各载药的功能性纳米载体的稳定性测试
[0115]
取适量实施例3
‑
8中所制备的载药功能性纳米载体冻干粉末,溶于蒸馏水、pbs、或含10%血清(fbs)的mem培养基中,在0、12、24、48、72、 96h测定三种载药功能性纳米载体的粒径变化,结果如图6所示。由图6可知,三种载药功能性纳米颗粒在各溶剂中的平均粒径均无明显变化,说明该各载药纳米颗粒具有良好的稳定性。
[0116]
实验例3:各载药的功能性纳米载体的低氧还原敏感性测试
[0117]
利用na2s2o4法模拟低氧还原条件,称取8mg实施例3
‑
8中制备的载药的功能性纳米载体冻干粉末于离心管中,溶于3ml蒸馏水中,并向体系中加入1ml浓度为5μm的na2s2o4溶液,封闭离心管管口,于37℃水浴锅中孵育30min,利用马尔文粒度仪检测其平均粒径变化情况,结果如图7所示。加入na2s2o4后,各载药的功能性纳米载体的平均粒径变化明显,平均粒径分布较广,说明在低氧条件下各纳米药物载体中的硝基咪唑被还原,导致纳米药物载体发生裂解,颗粒散裂;而在常氧条件下,三种载药的功能性纳米载体的平均粒径均无明显变化,说明在常氧条件下硝基咪唑不会被还原,各纳米药物载体不能发生裂解,颗粒保持稳定,具有良好的低氧选择性。
[0118]
实验例4:各载药的功能性纳米载体的gsh响应性测试
[0119]
称取8mg实施例3
‑
8中制备的载药的功能性纳米载体冻干粉末于离心管中,溶于2ml pbs中,加入1ml浓度为150u/ml的gsh溶液,于37℃水浴条件下振荡孵育10min。利用马尔文粒度仪检测其平均粒径变化情况,结果如图8所示。在加入gsh模拟肿瘤微环境的反应体系中,各纳米药物的平均粒径变化明显,与未加入gsh组相比,加入gsh后溶液中平均粒径分布变广,溶液中粒径分布不均匀。而在未加入gsh的纳米药物平均粒径分布较为集中,说明该纳米药物具有良好的gsh响应性。
[0120]
实验例5:各载药的功能性纳米载体中药物释放行为测试
[0121]
将实施例3
‑
8中制备的载药的功能性纳米载体和游离的化疗药物(bcnu、 tcnu和tmz)分别置于再生纤维透析袋中(透析袋截留分子量为1000
‑
2000 da),将透析袋置于含有大鼠肝微粒体、烟酰胺腺嘌呤二核苷酸磷酸(nadph) 的pbs工作液中,通过向体系内充入氮气和添加150u/ml的gsh以分别模拟低氧和gsh高表达的肿瘤微环境,各组样品在以上条件下进行透析。在5、 10、20、30、60、90、120、180、240min时取0.5ml透析袋外的pbs,用高效液相色谱法测定溶液中bcnu、tcnu和tmz的含量,从而得到三种药物的累计释放量。在不加gsh的条件下,常氧和低氧体系中各游离药物与功能性纳米载体的累积释药量如图9所示,三种游离药物组在常氧和低氧条件下,药物均快速释放,累积释药量均高于80%。三种载药的功能性纳米载体组在常氧和低氧条件下则表现出不同的释药能力:在常氧条件下,各组累积释药量均低于10%,表明在常氧条件下,各纳米颗粒保持完整,不会发生碎裂,因此包载的药物不能被释放出来;而在低氧条件下,各组累积释药量均明显上升,这是由于低氧条件可以触发ni
‑
tmg
‑
plga
‑
ss
‑
peg载体中的硝基咪唑基团发生还原,进而使纳米载体降解,纳米核心中的药物被有效释放。在加入gsh的条件下,常氧和低氧体系中各游离药物与功能性纳米载体的累积释药量变化情况如图10所示,三种游离药物组在常氧和低氧条件下,药物均
快速释放,累积释药量均高于80%。三种载药的功能性纳米载体组在常氧和低氧条件下表现出不同的释药能力,而且与不加gsh的纳米载体组表现出明显差异:在常氧条件下,各组累积释药量明显高于不加gsh 的纳米载体组,这说明gsh可以使纳米载体的二硫键断裂,进而导致载体碎裂,使药物从纳米核心释放出来;而且,在低氧且加入gsh的条件下,各纳米药物组的累积释药量达到最高,不但高于加入gsh的常氧组,而且高于不加gsh的低氧组,这表明在低氧和gsh的协同作用下,纳米载体上硝基咪唑被还原并伴随着二硫键的断裂,更加促使纳米核心的药物充分被释放出来。该实验结果表明,ni
‑
tmg
‑
plga
‑
ss
‑
peg nps在正常组织中可保持良好的稳定性,有利于延长药物的血液循环时间,防止药物提前释放;而在到达低氧与gsh高表达的肿瘤微环境中,ni
‑
tmg
‑
plga
‑
ss
‑
peg nps能迅速降解,高效释放药物,从而发挥良好的靶向抗肿瘤作用。
[0122]
实验例6:各载药的功能性纳米载体的细胞毒性实验
[0123]
1、实验材料
[0124]
供试化合物:游离的bcnu、tcnu、tmz、实施例3
‑
8中制备的ni
‑ꢀ
tmg
‑
plga
‑
ss
‑
peg/bcnu nps、ni
‑
tmg
‑
plga
‑
ss
‑
peg/tcnu nps和ni
‑ꢀ
tmg
‑
plga
‑
ss
‑
peg/tmz nps。
[0125]
细胞系:人神经胶质瘤sf763、sf126细胞,人肝癌hepg2细胞,人前列腺癌du145细胞,人乳腺癌mcf
‑
7细胞。
[0126]
2、实验方法
[0127]
5种细胞均以1000个/孔接种于96孔板,在37℃、5%co2培养24h后,以浓度为20μm、50μm、100μm、200μm、500μm、800μm、1000μm和 2000μm的各载药的功能性纳米载体和游离药物进行处理,每组6个复孔,并设置对照组。设置常氧/不加gsh、低氧/不加gsh、常氧/加gsh和低氧/ 加gsh组;向体系内充入氮气及加入150u/ml gsh以模拟低氧/gsh高表达的肿瘤微环境。然后每孔加入10μl cck
‑
8溶液,作用4h。最后,在450 nm波长下检测吸光度值,按以下公式计算细胞存活率,并计算药物的半数抑制率ic
50
。
[0128]
肿瘤细胞存活率(%)=(a
药物处理组
‑
a
空白组
)/(a
对照组
‑
a
空白组
)
×
100%
[0129]
a
药物处理组
为具有培养基、肿瘤细胞、药物溶液和cck
‑
8溶液的孔的吸光度值;
[0130]
a
空白组
为具有培养基和cck
‑
8溶液,但没有肿瘤细胞和药物的孔的吸光度值;
[0131]
a
对照组
为具有培养基、肿瘤细胞、cck
‑
8溶液,但没有药物溶液的孔的吸光度值。
[0132]
3、实验结果:见表2。
[0133]
表2肿瘤细胞的半数抑制率(ic
50
,μm)
[0134][0135][0136]
由表2中数据可知,在常氧/不加gsh条件下,功能性纳米载体包载各化疗药物的
ic
50
值比游离的化疗药物均显著提高。这是由于纳米载体ni
‑ꢀ
tmg
‑
plga
‑
ss
‑
peg nps对各化疗药物进行了有效的包封,且在常氧/不加 gsh条件下不能发生裂解,药物不能被释放,因此不能发挥对肿瘤细胞的抑制作用。这说明在正常组织中ni
‑
tmg
‑
plga
‑
ss
‑
peg nps保持稳定,不能碎裂且不能释放纳米核心的药物,因而能够有效降低药物对正常组织的毒副作用。
[0137]
在低氧/不加gsh条件下,各功能性纳米载体包载各化疗药物的ic
50
值比游离的化疗药物显著降低。这表明ni
‑
tmg
‑
plga
‑
ss
‑
peg nps具有良好的低氧响应性,其中的硝基咪唑在低氧条件下能够有效地被还原,从而使纳米载体碎裂,释放出纳米核心的药物和agt抑制剂o6‑
tmg。o6‑
abg能有效抑制agt的活性,阻断了dna烷化损伤的修复,增加了肿瘤细胞对药物的敏感性。
[0138]
在常氧/加gsh条件下,功能性纳米载体包载各化疗药物的ic
50
值与游离化疗药物的ic
50
值相比无明显差异。这表明所合成的ni
‑
tmg
‑
plga
‑
ss
‑ꢀ
peg nps具有良好的gsh响应性,其中的二硫键在gsh的作用下发生断裂,从而使纳米载体碎裂,释放包载药物;但是,由于常氧环境不能使硝基咪唑还原,所以不能释放agt抑制剂o6‑
tmg,因此各纳米组在常氧/加gsh条件下的抗肿瘤效果与游离药物相似。
[0139]
在低氧/加gsh条件下,各功能性纳米载体包载各化疗药物的比游离的化疗药物和其它各种条件下的纳米组均显著降低。导致这一结果的原因是,低氧环境促使硝基咪唑发生还原,从而使纳米载体碎裂并释放出agt抑制剂o6‑
tmg;同时,二硫键在gsh的作用下发生断裂,纳米载体进一步碎裂,使得所包载的抗肿瘤药物得以充分释放。所以,相比于低氧/不加gsh条件下的各组,该条件下纳米载体的二硫键断裂,能够更加充分的释放药物;相比于常氧/加gsh条件下的各组,该条件下纳米载体的硝基咪唑能够有效的被还原,释放o6‑
tmg的同时,也使得纳米壳碎裂,从而进一步释放包载的药物。可见,在低氧/加gsh条件下,纳米载体核内包载的药物释放更加充分,且伴随o6‑
tmg的释放,因此使得ic
50
值显著降低。
[0140]
以上结果表明,本发明所合成的功能性纳米载体能够发挥良好的抗耐药性和低氧/gsh双重响应性。在低氧和gsh高表达的肿瘤微环境中,该载体上低氧靶向基团硝基咪唑和gsh响应基团二硫键都能够被特异性地还原,使得纳米载体碎裂,释放壳结构中的agt抑制剂与核中的抗癌药物。本发明在靶向递送抗肿瘤药物的同时,发挥抗耐药的作用,提高了化疗药物的生物利用度,降低了游离药物的全身毒性。此外,功能性纳米载体ni
‑
tmg
‑ꢀ
plga
‑
ss
‑
peg nps对抗癌药物进行包载,显著提高了药物的稳定性以及血液循环时间,从而有利于药物更好地发挥抗肿瘤作用。
[0141]
最后本发明的实施例仅为较易的实施方案,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。