一种硫辛酸衍生物6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸的制备方法
技术领域
[0001]
本发明属于药物化学合成技术领域,具体涉及一种硫辛酸衍生物6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸的制备方法。
背景技术:
[0002]
硫辛酸(α
‑
lipoic acid)是一种具有生物活性的天然产物,1951年由reed首次从猪肝中分离得到。硫辛酸在欧美已广泛地用于临床医学领域,如肝病、阿尔茨海默氏病、糖尿病、癌症、白内障、心脏病、帕金森氏症、艾滋病、牛皮癣、湿疹、风湿病、心脏病、神经性疾病、亚急性坏死脑病、放射性伤害症、重金属中毒等疾病的治疗,被誉为“万能抗氧剂”。随着硫辛酸的药理药效学的深入研究发展,医学界研究人员对各种硫辛酸的衍生物及其它们盐类进行了应用开发,极大的丰富和扩展了硫辛酸系列产品的适应症范围和治疗效果,以满足医药临床和市场需求。
[0003]
专利wo2008131117a1公开了一类硫辛酸衍生物或其盐类,包括开环的硫醚和二硫醚结构(如式ii、iii),该类硫辛酸衍生物可应用于药物制剂的组合物成分,治疗涉上述各种疾病,诸如糖尿病、阿尔茨海默氏病及癌症等。硫辛酸及其衍生物由于其氧化还原电位特性,保持自sh至s
‑
s的氧化还原转换的可用性,以具有所需的抗氧化作用。
[0004][0004]
式中r1、r2可以是相同的或不同的各种取代基,或者其中一个是取代基而另一个是未取代。该专利对列举的若干硫辛酸衍生物硫醚和二硫醚结构系列品种,并无具体实施例公开其制备方法或工艺陈述。
技术实现要素:
[0005]
针对现有技术中存在的不足和限制,本发明的目的是提供一种新的硫辛酸衍生物的制备方法,该方法工艺简洁,制备条件温和,原料价廉易得,满足硫辛酸衍生物的放大生产要求并能体现优异的绿色环保效果。
[0006]
本发明的目的是这样来达到的,一种硫辛酸衍生物6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸的制备方法,包括如下步骤:a) 制备6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸:6
‑
巯基
‑8‑
(三苯甲硫基)辛酸与氯化苄在碱和溶剂体系中进行硫醚化反应,得到6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸;b)制备8
‑
巯基
‑6‑
(苄基硫基)辛酸:
将由步骤a)得到的6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸在三乙基硅烷和酸的条件下进行脱保护反应,得到8
‑
巯基
‑6‑
(苄基硫基)辛酸;c) 制备6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸:将由步骤b)得到的8
‑
巯基
‑6‑
(苄基硫基)辛酸与三甲基氯硅烷在缚酸剂碱和溶剂体系中进行硅烷化反应,得到6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸;d) 制备6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸:将由步骤c)得到的6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸与苯甲醛在溶剂体系中进行加成反应,得到的加成物中间体接着进行酸解脱保护,得到6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸。
[0007]
在进一步是实施例中,步骤a)中所述碱为甲醇钠、乙醇钠或异丙醇钠;所述的溶剂为甲醇、乙醇或异丙醇;所述的6
‑
巯基
‑8‑
(三苯甲硫基)辛酸、氯化苄、碱的摩尔比为1.0∶(1.1~1.3)∶(1.5~2.0)。
[0008]
在进一步是实施例中,步骤a)中所述的硫醚化反应的温度为25~80℃,反应时间为6~12h。
[0009]
在进一步是实施例中,步骤b)中所述的酸为乙酸、三氟乙酸、草酸、甲酸或丙酸;所述的6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸与三乙基硅烷的摩尔比为1.0∶(2.0~2.5)。
[0010]
在进一步是实施例中,步骤b)中所述的脱保护反应的温度为20~35℃,反应时间为30~60min。
[0011]
在进一步是实施例中,步骤c)中所述的缚酸剂碱为三乙胺、吡啶、n,n
‑
二异丙基乙胺、4
‑
二甲氨基吡啶或2,6
‑
二甲基吡啶;所述的溶剂为二氯甲烷、氯仿或1,2
‑
二氯乙烷;所述的8
‑
巯基
‑6‑
(苄基硫基)辛酸、三甲基氯硅烷、缚酸剂碱的摩尔比为1.0∶(1.0~1.5)∶(1.4~2.0)。
[0012]
在进一步是实施例中,步骤c)中所述的硅烷化反应的温度为20~35℃,反应时间为2~6h。
[0013]
在进一步是实施例中,步骤d)中所述的溶剂为甲醇、乙醇、异丙醇、二氯甲烷、1,2
‑
二氯乙烷、氯仿或甲苯;所述的6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸与所述的苯甲醛的摩尔比为1.0∶(1.5~2.0)。
[0014]
在进一步是实施例中,步骤d)中所述的加成反应的温度为30~60℃,反应时间为6~12h。
[0015]
在进一步是实施例中,步骤d)中所述的酸解脱保护,是在20~30℃下用6n盐酸溶液酸化至ph=2,搅拌1h。
[0016]
有益效果:(1)硫醚化反应比较温和,工艺操作简单,避免了使用价格昂贵的原料,降低成本。
[0017]
(2)两次脱保护反应工艺简单,成本较低。
[0018]
(3)整个工艺路线简洁,副反应可控,杂质较少,无污染物产生,体现绿色环保效果。
[0019]
(4)起始原料和所用的试剂易得,可以大量生产来满足原料药的使用需求,适用于工业化生产。
具体实施方式
[0020]
以下结合数个较佳实施例对本发明技术方案作进一步非限制性的详细说明。
[0021]
实施例1a)制备6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸:6
‑
巯基
‑8‑
(三苯甲硫基)辛酸(20.0g,44.4mmol)、氯化苄(6.2g,48.8mmol)溶于甲醇(200ml),滴加甲醇钠的甲醇溶液(甲醇钠66.6mmol,40ml),25℃反应1h,然后升温60℃反应12h,反应完毕,减压旋蒸至干,二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,得到6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸,白色固体(20.5g),收率85%,本步骤所述的硫醚化反应的反应式如下:。
[0022]
b) 制备8
‑
巯基
‑6‑
(苄基硫基)辛酸:6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸(20.0g,37.0mmol)溶于乙酸(200ml),缓慢加三乙基硅烷(8.6g,74.0mmol),保温20℃反应1h,反应完毕,抽滤除去不溶物,收集滤液,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,得到8
‑
巯基
‑6‑
(苄基硫基)辛酸,类白色固体(9.5g),收率86%,本步骤所述的脱保护反应的反应式如下:。
[0023]
c)制备6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸:8
‑
巯基
‑6‑
(苄基硫基)辛酸(9.0g,30.2mmol)、三乙胺(4.3g,42.2mol)溶于二氯甲烷(100ml),降温至10℃,滴加三甲基氯硅烷(3.3g,30.2mol)的二氯甲烷(10ml)溶液,保温20℃反应6h,反应完毕,滴加水淬灭反应液,减压浓缩除去有机溶剂,加入二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,真空干燥,得到6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸(10.0g),收率90%,本步骤所述的硅烷化反应的反应式如下:
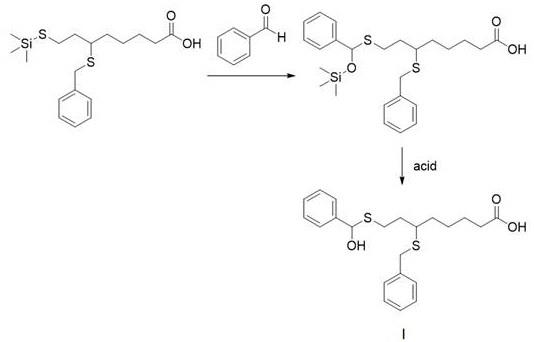
。
[0024]
d)制备6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸:6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸(10.0g,27.0mmol)、苯甲醛(4.3g,40.5mol)溶于乙醇(100ml),保温30℃反应12h,20℃下用6n盐酸溶液酸化至ph=2,搅拌1h,二氯甲烷萃取,分层,有机相用食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,得到6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸,白色固体(9.4g),收率86%,本步骤所述的加成反应及酸解脱保护反应的反应式如下:式中的i为所述硫辛酸衍生物6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸。
[0025]
实施例2a)制备6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸:6
‑
巯基
‑8‑
(三苯甲硫基)辛酸(45.0g,0.10mol)、氯化苄(15.0g,0.12mol)溶于乙醇(500ml),滴加乙醇钠的乙醇溶液(乙醇钠0.18mol,50ml),25℃反应1h,然后升温75℃反应9h,反应完毕,减压旋蒸至干,二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,得到6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸,白色固体(47.5g),收率88%。
[0026]
b)制备8
‑
巯基
‑6‑
(苄基硫基)辛酸:6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸(47.0g,86.9mmol)溶于三氟乙酸(500ml),缓慢加三乙基硅烷(22.0g,0.19mol),保温25℃反应40min,反应完毕,抽滤除去不溶物,收集滤液,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,得到8
‑
巯基
‑6‑
(苄基硫基)
辛酸,类白色固体(23.0g),收率89%。
[0027]
c)制备6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸:8
‑
巯基
‑6‑
(苄基硫基)辛酸(23.0g,77.1mmol)、吡啶(10.0g,0.13mol)溶于氯仿(300ml),降温至10℃,滴加三甲基氯硅烷(10.0g,92.0mmol)的氯仿(300ml)溶液,保温30℃反应4h,反应完毕,滴加水淬灭反应液,减压浓缩除去有机溶剂,加入二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,真空干燥,得到6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸(26.0g),收率91%。
[0028]
d)制备6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸:6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸(26.0g,70.1mmol)、苯甲醛(13.0g,0.12mol)溶于1,2
‑
二氯乙烷(400ml),保温50℃反应8h,25℃下用6n盐酸溶液酸化至ph=2,搅拌1h,二氯甲烷萃取,分层,有机相用食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,得到6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸,白色固体(25.0g),收率88%。
[0029]
本实施例中未涉及的内容均同对实施例1的描述。
[0030]
实施例3a)制备6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸:6
‑
巯基
‑8‑
(三苯甲硫基)辛酸(55.0g,0.12mol)、氯化苄(20.0g,0.16mol)溶于异丙醇(600ml),滴加异丙醇钠的异丙醇溶液(异丙醇钠0.24mol,50ml),25℃反应1h,然后升温80℃反应6h,反应完毕,减压旋蒸至干,二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,得到6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸,白色固体(60.0g),收率91%。
[0031]
b)制备8
‑
巯基
‑6‑
(苄基硫基)辛酸:6
‑
(苄基硫基)
‑8‑
(三苯甲硫基)辛酸(60.0g,0.11mol)溶于草酸(600ml),缓慢加三乙基硅烷(32.0g,0.28mol),保温35℃反应30min,反应完毕,抽滤除去不溶物,收集滤液,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,得到8
‑
巯基
‑6‑
(苄基硫基)辛酸,类白色固体(30.0g),收率91%。
[0032]
c)制备6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸:8
‑
巯基
‑6‑
(苄基硫基)辛酸(30.0g,0.10mol)、n,n
‑
二异丙基乙胺(26.0g,0.20mol)溶于1,2
‑
二氯乙烷(500ml),降温至10℃,滴加三甲基氯硅烷(16.0g,0.15mol)的1,2
‑
二氯乙烷(50ml)溶液,保温35℃反应2h,反应完毕,滴加水淬灭反应液,减压浓缩除去有机溶剂,加入二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,真空干燥,得到6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸(35.0g),收率94%。
[0033]
d)制备6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸:6
‑
(苄基硫基)
‑8‑
[(三甲基硅基)硫基]辛酸(35.0g,94.4mmol)、苯甲醛(20.0g,0.19mmol)溶于甲苯(500ml),保温60℃反应6h,30℃下用6n盐酸溶液酸化至ph=2,搅拌1h,二氯甲烷萃取,分层,有机相用食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯
‑
石油醚混合溶剂重结晶,得到6
‑
(苄基硫基)
‑8‑
[(羟基苯基甲基)硫基]辛酸,白色固体(34.8g),收率91%。
[0034]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种等同变换,这些等同变换均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
![一种硫辛酸衍生物6-(苄基硫基)-8-[(羟基苯基甲基)硫基]辛酸的制备方法与流程](http://img.xjishu.com/img/zl/2021/11/30/tjbc74wro.jpg)