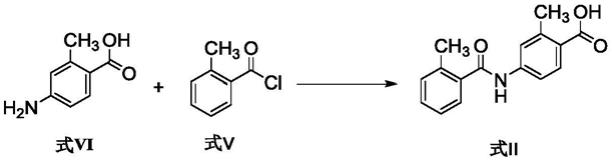
一种升压素v2受体拮抗剂的中间体的制备方法
技术领域
1.本发明属于药物化学领域,具体涉及一种升压素v2受体拮抗剂的中间体的制备方法。
背景技术:
2.托伐普坦是世界上首个口服普坦类药物,由日本otsuka pharm制药公司开发的口服剂型选择性升压素v2受体拮抗剂,于2009年5月19日获美国fda批准,既是目前唯一获准治疗低钠血症的药物,也是中国市场第一个和唯一的v2受体拮抗剂。通过拮抗加压素的作用,使尿液中水排泄量增加,提高了自由水的清除率,降低尿液渗透压,从而增加血钠值,但同时又不改变尿液钠钾分泌,不影响血钾值。可用于治疗由充血性心衰、肝硬化等多种疾病导致的液体潴留、水肿、低钠血症等。此外,托伐普坦能够减少肾脏多发性囊泡的发生和生长,从而减缓多囊肾的病程进展,是首个针对多囊肾发病机制的治疗药物。
3.托伐普坦(tolvaptan),商品名samsca,化学名n-[4-[(5r)-7-氯-5-羟基-2,3,4,5-四氢-1-苯并氮杂卓-1-甲酰基]-3-甲基苯基]-2-甲基苯甲酰胺,其结构如下式所示:
[0004][0005]
2-甲基-4-(2-甲基苯酰胺基)苯甲酸是合成托伐普坦的中间体。专利文献 cn2013000368444.6、cn201711349574.x公开了该中间体的合成方法,采用的是2-甲基-4
-ꢀ
氨基苯甲酸与邻甲基苯甲酰氯在碱催化下于四氢呋喃、1,4-二氧六环、丙酮、氯仿或乙醚等溶剂中发生缩合反应制备。
技术实现要素:
[0006]
本发明要解决的技术问题是克服现有技术的缺陷,提供一种制备2-甲基-4-(2-甲基苯甲酰胺基)苯甲酸的方法,所述方法能够高收率、高纯度地制备得到目标产物,而且反应不需要加入催化剂碱性物质,减少了试剂的使用,节约生产成本,适合工业化应用。
[0007]
本发明提供了一种如下式ii所示的化合物的制备方法,路线如下:
[0008][0009]
式vi化合物与式v化合物在有机溶剂中反应制备式ii化合物,其中所述有机溶剂
选自 n-甲基吡咯烷酮或2-吡咯烷酮。
[0010]
在一些实施方案中,式vi化合物与有机溶剂的质量比为1:2~1:8;在一些典型的实施方案中,式vi化合物与有机溶剂的质量比为1:3~1:6;在一些更典型的实施方案中,式 vi化合物与有机溶剂的质量比为1:3~1:4。
[0011]
在一些实施方案中,所述有机溶剂为n-甲基吡咯烷酮。
[0012]
在一些实施方案中,所述反应不加入催化剂。
[0013]
在一些实施方案中,所述反应的反应温度为20~60℃;优选25~35℃。
[0014]
在一些实施方案中,所述反应的后处理包括:将反应液用碱性溶液调节ph至10以上;加入二氯甲烷或乙酸乙酯萃取;分液;水相用酸性溶液调节ph至7以下;搅拌析晶,过滤,干燥即得。
[0015]
在一些实施方案中,所述后处理步骤中的碱性溶液为氢氧化钠水溶液或氢氧化钾水溶液。任何浓度的碱性溶液均可用于本发明,优选5~30%,更优选21%。
[0016]
在一些实施方案中,所述后处理步骤中的酸性溶液选自盐酸水溶液、氢溴酸水溶液、磷酸水溶液或乙酸水溶液;优选盐酸水溶液或氢溴酸水溶液;更优选盐酸水溶液。任何浓度的酸性溶液均可用于本发明,优选1~10mol/l;更优选2~8mol/l;更优选4mol/l。
[0017]
在一些实施方案中,所述式v化合物通过以下方法制备:将邻甲基苯甲酸与二氯亚砜在有机溶剂或不加溶剂的条件下反应,反应温度为50~70℃,优选55~65℃,反应完毕后减压浓缩即得;所述有机溶剂选自甲苯或二甲苯。
[0018]
在一些典型的实施方案中,所述式v化合物通过以下方法制备:将邻甲基苯甲酸与二氯亚砜在不加溶剂的条件下反应,反应温度为50~70℃,优选55~65℃,反应完毕后减压浓缩即得。
[0019]
在一些更典型的实施方案中,本发明提供了式ii所示化合物的制备方法,具体包括:将邻甲基苯甲酸与二氯亚砜在有机溶剂或不加溶剂的条件下反应制备式v化合物,优选不加溶剂,反应温度为50~70℃,优选55~65℃,反应完毕后减压浓缩即得;将式v化合物加至式 vi化合物的n-甲基吡咯烷酮或2-吡咯烷酮溶液中,优选n-甲基吡咯烷酮,20~60℃下反应,优选25~35℃,反应完全后将反应液用碱性溶液调节ph至10以上;加入二氯甲烷或乙酸乙酯萃取;分液;水相用酸性溶液调节ph至7以下;搅拌析晶,过滤,干燥即得。
[0020]
本发明的有益效果在于本发明提供的式ii化合物的制备方法可以高收率、高纯度地制备得到目标化合物,而且反应时间短、反应体系不需要加入催化剂,减少了试剂的使用,适合工业化应用。
[0021]
相关说明
[0022]
当浓度为“%”表示时,是指质量百分比浓度。
[0023]
n:mol/l
[0024]
说明书附图
[0025]
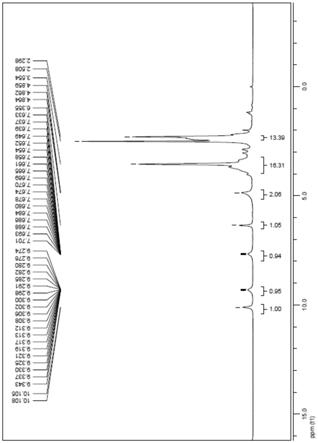
图1为实施例2制得产品的hplc图谱
[0026]
图2为对比例1制得产品的hplc图谱
具体实施方式
[0027]
下面结合具体实施例进一步说明本发明的技术方案,但并不限定本发明。
[0028]
hplc检测方法:
[0029]
色谱条件
[0030]
色谱柱:色谱柱:kromasil c18(4.6mm
×
250mm,5μm);
[0031]
柱温:30℃;
[0032]
流速:1.0ml/min;
[0033]
检测波长:254mn;
[0034]
流动相:a相-0.1%磷酸溶液
[0035]
b相-乙腈
[0036]
梯度洗脱如下:
[0037]
时间(min)流动相a(%)流动相b(%)0802025307033307033.18020408020
[0038]
实施例1
[0039]
将邻甲基苯甲酸466g、氯化亚砜816g加入反应瓶中,55~65℃反应至终点,减压浓缩,后加至4-氨基-2-甲基苯甲酸466g的n-甲基吡咯烷酮(1.92kg)的悬浮液中。加毕,保温25~35℃反应,反应1h后,用21%氢氧化钠水溶液调节ph至10以上,加入二氯甲烷萃取。分液,水相用4n盐酸溶液调节ph至7以下,搅拌析晶,离心,过滤,干燥,得式ii化合物,收率 95.8%,hplc纯度99.3%。
[0040]
实施例2
[0041]
将邻甲基苯甲酸7.50kg、氯化亚砜13.10kg加入反应瓶中,55~65℃反应至终点,减压浓缩,后加至4-氨基-2-甲基苯甲酸7.50kg的n-甲基吡咯烷酮(27.80kg)的悬浮液中。加毕,保温25~35℃反应,反应1h后,用21%氢氧化钠水溶液调节ph至10以上,加入二氯甲烷萃取。分液,水相用4n盐酸溶液调节ph至7以下,搅拌析晶,离心,过滤,干燥,得式ii 化合物,收率98.0%,hplc纯度99.53%,hplc附图见图1。
[0042]
对比例1
[0043]
将邻甲基苯甲酸440g、氯化亚砜760g加入反应瓶中,55~65℃反应至终点,减压浓缩,后加至4-氨基-2-甲基苯甲酸(440g)和氢氧化钠(190g)的n-甲基吡咯烷酮(1.8kg)的悬浮液中。加毕,保温25~35℃反应,反应1h后,用21%的氢氧化钠溶液调节ph至10以上,加入二氯甲烷萃取。分液,水相用4n盐酸溶液调节ph至7以下,搅拌析晶,离心,过滤,干燥,得式ii化合物,收率53.7%,hplc纯度86.5%,hplc附图见图2。
[0044]
对比例2
[0045]
将邻甲基苯甲酸2.5g、氯化亚砜4.3g加入反应瓶中,55~65℃反应至终点,减压浓缩,后加至4-氨基-2-甲基苯甲酸2.78g的1,4二氧六环(20ml)的悬浮液中。加毕,保温25~35℃反应,反应1h后,用21%的氢氧化钠溶液调节ph至10以上,加入二氯甲烷萃取。分液,水相用4n盐酸溶液调节ph至7以下,搅拌析晶,离心,过滤,干燥,得式ii化合物,收率 79%,hplc纯度95.3%。
[0046]
对比例3
[0047]
将邻甲基苯甲酸2.5g、氯化亚砜4.3g加入反应瓶中,55~65℃反应至终点,减压浓缩,后加至4-氨基-2-甲基苯甲酸2.78g的1,4二氧六环/乙酸乙酯(6ml/14ml)的悬浮液中。加毕,保温25~35℃反应,反应1h后,用21%的氢氧化钠溶液调节ph至10以上,加入二氯甲烷萃取。分液,水相用4n盐酸溶液调节ph至7以下,搅拌析晶,离心,过滤,干燥,得式ii 化合物,收率79%,hplc纯度93.6%。
[0048]
对比例4
[0049]
将邻甲基苯甲酸2.5g、氯化亚砜4.3g加入反应瓶中,55~65℃反应至终点,减压浓缩,后加至4-氨基-2-甲基苯甲酸2.78g的n-甲基吡咯烷酮/二氯甲烷(6ml/14ml)的悬浮液中。加毕,保温25~35℃反应,反应1h后,用21%的氢氧化钠溶液调节ph至10以上,加入二氯甲烷萃取。分液,水相用4n盐酸溶液调节ph至7以下,搅拌析晶,离心,过滤,干燥,得式ii 化合物,收率78%,hplc纯度92.8%。
[0050]
对比例5
[0051]
将邻甲基苯甲酸2.5g、氯化亚砜4.3g加入反应瓶中,55~65℃反应至终点,减压浓缩,后加至4-氨基-2-甲基苯甲酸2.78g的四氢呋喃(20ml)的悬浮液中。加毕,保温25~35℃反应,反应1h后,用21%的氢氧化钠溶液调节ph至10以上,加入二氯甲烷萃取。分液,水相用 4n盐酸溶液调节ph至7以下,搅拌析晶,离心,过滤,干燥,得式ii化合物,收率73%, hplc纯度87.9%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。