1.本发明涉及医药技术领域,具体涉及一种盐酸拉贝洛尔的制备方法。
背景技术:
2.盐酸拉贝洛尔(labetalol hydrochloride),化学名称为2
‑
羟基
‑5‑
[1
‑
羟基
‑2‑
[(1
‑
甲基
‑3‑
苯基丙基)氨基]乙基]苯甲酰胺盐酸盐(结构式如下),是一种非选择性β受体阻滞作用和选择性突触后α1阻滞作用的抗高血压药,可使周围血管阻力下降,具有膜稳定作用但无内在拟交感活性,可迅速降低血压,适于高血压急症的治疗,如急性主动脉夹层、嗜铬细胞瘤、妊娠高血压综合征。
[0003][0004]
尚芸等(尚芸,王毅军,冯俊才.南京大学学报,1980(1):61
‑
64.)公开了一种盐酸拉贝洛尔的合成方法,以5
‑
溴乙酰基水杨酸甲酯为起始原料与二苄胺缩合,经胺酯交换、氢化脱苄基后与4
‑
苯基
‑2‑
丁酮缩合、还原羰基和成盐制备得到,合成路线如下:
[0005][0006]
国大亮等(国大亮,朱晓薇.化学工业与工程,2008,25(3):276
‑
278)以水杨酰胺为起始原料,经过傅克反应、溴代、二苄胺保护、缩合以及还原得到盐酸拉贝洛尔,合成路线如下:
[0007][0008]
然而,上述合成方法以二苄胺作为氨基源,原料的原子利用率不高。
技术实现要素:
[0009]
有鉴于此,本发明的目的在于提供一种盐酸拉贝洛尔的制备方法,本发明提供的制备方法原料的原子利用率高、产率高且生产成本低。
[0010]
为了实现上述发明目的,本发明提供以下技术方案:
[0011]
本发明提供了一种盐酸拉贝洛尔的制备方法,包括以下步骤:
[0012]
(1)将化合物ii与苄胺进行第一亲核取代反应,得到化合物iii;
[0013]
(2)将所述化合物iii与化合物iv进行第二亲核取代反应,得到化合物v;
[0014]
(3)所述化合物v在催化剂作用下,进行催化氢化反应,得到化合物viii;
[0015]
(4)将所述化合物viii与盐酸溶液混合进行成盐反应,得到具有式i所示结构的盐酸拉贝洛尔;
[0016][0017]
所述化合物ii与化合物iv中x为卤素。
[0018]
优选的,所述步骤(2)替换为:
[0019]
将化合物vi与对甲基苯磺酰氯进行酯化反应,得到化合物vii;
[0020]
将所述化合物vii与化合物iii进行胺酯交换反应,得到化合物v;
[0021][0022]
优选的,所述化合物ii与苄胺的摩尔比为1:0.5~2;
[0023]
所述第一亲核取代反应的温度为
‑
10~35℃,时间为1~5h。
[0024]
优选的,所述化合物iii与化合物iv的摩尔比为1:0.9~1.5;
[0025]
所述第二亲核取代反应的温度为0~100℃。
[0026]
优选的,所述化合物vi与对甲基苯磺酰氯的摩尔比为1:0.5~1.2;
[0027]
所述酯化反应的温度为
‑
10~30℃。
[0028]
优选的,所述化合物iii与化合物vii的摩尔比为1:1~1.2;
[0029]
所述胺酯交换反应的温度为79~83℃。
[0030]
优选的,所述催化剂包括钯碳和/或铂碳。
[0031]
优选的,所述催化氢化反应的氢气压力为0.1~10mpa,温度为25~70℃,时间为4~12h。
[0032]
优选的,所述盐酸溶液包括盐酸水溶液、盐酸醇溶液和盐酸酯溶液中的一种或几种。
[0033]
优选的,所述成盐反应的温度为
‑
10~30℃,时间为30~90min,ph值为1~3。
[0034]
本发明提供了一种盐酸拉贝洛尔的制备方法,包括以下步骤:(1)将化合物ii与苄胺进行第一亲核取代反应,得到化合物iii;(2)将所述化合物iii与化合物iv进行第二亲核取代反应,得到化合物v;(3)将所述化合物v与催化剂混合进行催化氢化反应,得到化合物viii;(4)将所述化合物viii与盐酸溶液混合进行成盐反应,得到具有式i所示结构的盐酸拉贝洛尔;所述步骤(2)替换为:将化合物vi与对甲基苯磺酰氯进行酯化反应,得到化合物vii;将所述化合物vii与化合物iii进行胺酯交换反应,得到化合物v。本发明提供的制备方法,以5
‑
卤代乙酰基水杨酰胺作为起始原料与苄胺进行亲核取代反应,然后与3
‑
卤代丁基苯进行亲核取代反应(或者是与3
‑
羟基丁基苯和对甲基苯磺酰氯酯化反应产物进行胺酯交换反应),再进行催化氢化反应反应后成盐,得到盐酸拉贝洛尔。本发明提供的制备方法,采用苄胺替代二苄胺,原料的原子利用率高、对环境友好,体现了绿色化学的原子经济性。进一步的胺酯交换反应选择性高,所得产物直接用于下一步反应。本发明采用一步法脱除保护剂和还原羰基缩短了工艺路线;制备方法操作简便且稳定性和可控性高,生产成本高,产率高,适宜工业化生产。
附图说明
[0035]
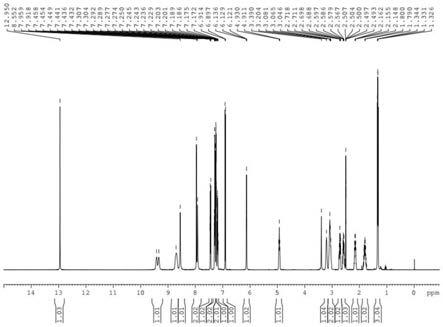
图1为实施例1制备的盐酸拉贝洛尔的氢谱图;
[0036]
图2为实施例1制备的盐酸拉贝洛尔的碳谱图。
具体实施方式
[0037]
本发明提供了一种盐酸拉贝洛尔的制备方法,包括以下步骤:
[0038]
(1)将化合物ii与苄胺进行第一亲核取代反应,得到化合物iii;
[0039]
(2)将所述化合物iii与化合物iv进行第二亲核取代反应,得到化合物v;
[0040]
(3)所述化合物v在催化剂作用下,进行催化氢化反应,得到化合物viii;
[0041]
(4)将所述化合物viii与盐酸溶液混合进行成盐反应,得到具有式i所示结构的盐酸拉贝洛尔;
[0042][0043]
所述化合物ii与化合物iv中x为卤素。
[0044]
在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
[0045]
本发明将化合物ii与苄胺进行第一亲核取代反应,得到化合物iii。在本发明中,所述化合物ii中的x优选包括氯或溴;所述化合物ii优选包括5
‑
溴乙酰基水杨酰胺或5
‑
氯乙酰基水杨酰胺。在本发明中,所述化合物ii与苄胺的摩尔比优选为1:0.5~2,更优选为1:1~1.5。在本发明中,所述第一亲核取代反应优选在碱性条件下进行;所述将化合物ii与苄胺进行第一亲核取代反应优选为将苄胺、碱性试剂和有机溶剂混合后滴加化合物ii溶液混合,进行第一亲核取代反应。在本发明中,所述碱性试剂优选包括有机碱或无机碱,所述无机碱优选包括碳酸钾、氢氧化钠和氢氧化钾中的一种或几种;所述有机碱优选包括三乙胺。在本发明中,所述化合物ii与碱性试剂的摩尔比优选为1:0.8~3,更优选为1:1~2,进一步优选为1:1~1.6。在本发明中,所述有机溶剂优选包括四氢呋喃、醚类溶剂、芳香烃类溶剂和氯代烃溶剂中的一种或几种;所述醚类溶剂优选包括乙醚、乙二醇二甲醚、异丙醚和叔丁醚中的一种或几种;所述芳香烃类溶剂优选包括苯、甲苯和二甲苯中的一种或几种;所述氯代烃溶剂优选包括二氯甲烷;本发明对于所述有机溶剂的用量没有特殊限定,能够使第一亲核取代反应顺利进行即可。在本发明中,所述化合物ii溶液中的溶剂优选与前述有机溶剂相同,在此不再赘述;所述化合物ii溶液的浓度优选为0.1~2g/l,更优选为0.5~1.5g/l,进一步优选为1g/l;所述滴加的温度优选为
‑
10~35℃,更优选为0~25℃;本发明对于所述滴加的速度没有特殊限定,逐滴加入即可。本发明对于所述混合的速度和时间没有特殊限定,能够将原料混合均匀即可。在本发明中,所述第一亲核取代反应的温度优选为
‑
10~35℃,更优选为0~25℃;所述第一亲核取代反应的时间优选为1~5h,更优选为2~3h。在本发明中,所述第一亲核取代反应过程中发生的反应如下:
[0046][0047]
所述第一亲核取代反应后,本发明优选还包括将所述第一亲核取代反应的体系进行后处理,所述后处理包括依次进行水洗和萃取,将所得有机相依次进行饱和碳酸氢钠洗、饱和食盐水洗涤、无水硫酸钠干燥和过滤,将所得滤液浓缩至恒重,得到化合物iii。在本发明中,所述化合物ii的质量和水洗用水的体积之比优选为1g:1~5ml,更优选为1g:2~4ml,进一步优选为1g:3ml,。在本发明中,所述萃取用有机溶剂优选包括酯类溶剂和/或氯代烷烃类溶剂,所述酯类溶剂优选包括乙酸乙酯和/或乙酸异丙酯;所述氯代烷烃类溶剂优选包括二氯甲烷、三氯甲烷和1,2
‑
二氯乙烷中的一种或几种;所述萃取的次数优选为1~3次,更优选为2次;所述化合物ii的质量和单次萃取用有机溶剂的体积之比优选为1g:1~5ml,更优选为1g:2~4ml,进一步优选为1g:3ml。在本发明中,所述浓缩的方式优选为减压蒸馏;本发明对于所述减压蒸馏的条件没有特殊限定,浓缩至恒重即可。在本发明中,所述化合物iii不经纯化直接用于下一步反应。
[0048]
本发明将所述化合物iii与化合物iv进行第二亲核取代反应,得到化合物v。在本发明中,所述第二亲核取代反应优选在碱性条件下进行;所述将所述化合物iii与化合物iv进行第二亲核取代反应优选为将化合物iii、碱性试剂和有机溶剂混合后加入化合物iv进行第二亲核取代反应。在本发明中,所述化合物iii与化合物iv的摩尔比优选为1:0.9~1.5,更优选为1:1~1.2。在本发明中,所述碱性试剂优选包括碳酸钾、三乙胺、氢氧化钠和氢氧化钾中的一种或几种;所述化合物iii与碱性试剂的摩尔比优选为1:0.9~2.5,更优选为1:1~2,进一步优选为1:1.5。在本发明中,所述有机溶剂优选包括n,n
‑
二甲基甲酰胺(dmf)、二氯甲烷、乙腈和四氢呋喃中的一种或几种;本发明对于所述有机溶剂的用量没有特殊限定,能够使第二亲核取代反应顺利进行即可;在本发明的实施例中,所述化合物iii的质量和有机溶剂的体积之比优选为1g:3~8ml,更优选为1g:5~6ml。本发明对于所述混合的速度和时间没有特殊限定,能够将原料混合均匀即可。在本发明中,所述第二亲核取代反应的温度优选为0~100℃,更优选为40~80℃;所述第二亲核取代反应的时间优选以tlc检测至化合物iii完全反应完为准;所述第二亲核取代反应过程中发生的反应如下:
[0049][0050]
所述第二亲核取代反应后,本发明优选还包括将所述第二亲核取代反应的体系进行后处理,所述后处理包括依次进行的恢复至室温、水洗和萃取,将所得有机相无水硫酸钠干燥后过滤,将所得滤液浓缩至恒重,得到化合物v。在本发明中,所述化合物iii的质量和水洗用水的体积之比优选为1g:2~6ml,更优选为1g:4~5ml。在本发明中,所述萃取用有机
溶剂优选包括酯类溶剂和/或氯代烷烃类溶剂,所述酯类溶剂优选包括乙酸乙酯和/或乙酸异丙酯;所述氯代烷烃类溶剂优选包括二氯甲烷、三氯甲烷和1,2
‑
二氯乙烷中的一种或几种;所述萃取的次数优选为1~3次,更优选为2次;所述化合物iii的质量和单次萃取用有机溶剂的体积之比优选为1g:3~8ml,更优选为1g:5~6ml。在本发明中,所述浓缩的方式优选为减压蒸馏;本发明对于所述减压蒸馏的条件没有特殊限定,浓缩至恒重即可。在本发明中,所述化合物v不经纯化直接用于下一步反应。
[0051]
在本发明中,所述化合物v还可以通过以下方法制备得到:
[0052]
将化合物vi与对甲基苯磺酰氯进行酯化反应,得到化合物vii;
[0053]
将所述化合物vii与化合物iii进行胺酯交换反应,得到化合物v;
[0054][0055]
本发明将化合物vi与对甲基苯磺酰氯进行酯化反应,得到化合物vii。在本发明中,所述酯化反应优选在碱性条件下进行;所述将化合物vi(3
‑
羟基丁基苯)与对甲基苯磺酰氯进行酯化反应优选为将化合物vi溶解于有机溶剂中后加入碱性试剂和对甲基苯磺酰氯混合,进行酯化反应。在本发明中,所述化合物vi与对甲基苯磺酰氯的摩尔比优选为1:0.5~1.2,更优选为1:0.8~1。在本发明中,所述有机溶剂优选包括n,n
‑
二甲基甲酰胺(dmf)、二氯甲烷、乙腈和四氢呋喃中的一种或几种;本发明对于所述有机溶剂的用量没有特殊限定,能够使酯化反应顺利进行即可;在本发明的实施例中,所述化合物vi的质量和有机溶剂的体积之比优选为1g:4~9ml,更优选为1g:5~7ml。在本发明中,所述碱性试剂优选包括无机碱和/或有机碱,所述无机碱优选包括碳酸钾、氢氧化钠和氢氧化钾中的一种或几种;所述有机碱优选包括三乙胺。在本发明中,所述化合物vi与碱性试剂的摩尔比优选为1:1~1.5,更优选为1:1.2~1.3。在本发明中,所述碱性试剂和对甲基苯磺酰氯的加入温度优选为
‑
10~30℃,更优选为
‑
5~5℃。本发明对于所述混合的速度和时间没有特殊限定,能够将原料混合均匀即可。在本发明中,所述酯化反应的温度优选为
‑
10~30℃,更优选为0~10℃;所述酯化反应的时间优选以tlc检测至化合物vi完全反应完为准;所述酯化反应过程中发生的反应如下:
[0056][0057]
所述酯化反应后,本发明优选还包括将所述酯化反应的体系进行后处理,所述后处理包括水洗,将所得有机相无水硫酸钠干燥后过滤,将所得滤液浓缩至恒重,得到化合物vii。在本发明中,所述化合物vi的质量和水洗用水的体积之比优选为1g:0.8~2ml,更优选为1g:1~1.5ml。在本发明中,所述浓缩的方式优选为减压蒸馏;本发明对于所述减压蒸馏的条件没有特殊限定,浓缩至恒重即可。在本发明中,所述化合物vii不经纯化直接用于下一步反应。
[0058]
得到化合物vii后,本发明将所述化合物vii与化合物iii进行胺酯交换反应,得到化合物v。在本发明中,所述胺酯交换反应优选在碱性条件下进行;所述将所述化合物vii与化合物iii进行胺酯交换反应优选为将化合物vii、化合物iii、碱性试剂和有机溶剂混合,进行酯交换反应。在本发明中,所述化合物iii与化合物vii的摩尔比优选为1:1~1.2,更优选为1:1.1。在本发明中,所述碱性试剂优选包括碳酸钾、三乙胺、氢氧化钠和氢氧化钾中的一种或几种;所述化合物iii与碱性试剂的摩尔比优选为1:2~3.5,更优选为1:2.5~3。在本发明中,所述有机溶剂优选包括乙腈和/或n,n
‑
二甲基甲酰胺(dmf);本发明对于所述有机溶剂的用量没有特殊限定,能够使胺酯交换反应顺利进行即可;在本发明的实施例中,所述化合物iii的质量和有机溶剂的体积之比优选为1g:5~10ml,更优选为1g:8~9ml。在本发明中,所述胺酯交换反应的温度优选为30~85℃,更优选为50~80℃,进一步优选为60~70℃;所述胺酯交换反应的时间优选以tlc检测至化合物iii完全反应完为准;所述胺酯交换反应过程中发生的反应如下:
[0059][0060]
所述胺酯交换反应后,本发明优选还包括将所述胺酯交换反应的体系进行后处理,所述后处理包括冷却至室温后过滤,将所得滤液浓缩至恒重,得到化合物v。本发明对于所述冷却的方式没有特殊限定,采用本领域技术人员熟知的冷却方式即可。在本发明中,所述浓缩的方式优选为减压蒸馏;本发明对于所述减压蒸馏的条件没有特殊限定,浓缩至恒重即可。在本发明中,所述化合物v不经纯化直接用于下一步反应。
[0061]
得到化合物v后,所述化合物v在催化剂作用下进行催化氢化反应,得到化合物viii。在本发明中,所述化合物v在催化剂作用下进行催化氢化反应优选为将化合物v、催化剂和有机溶剂混合,在氢气存在条件下进行催化氢化反应。在本发明中,所述催化剂优选包括钯碳和/或铂碳;所述钯碳中钯的质量分数优选为10%(10%钯碳);所述铂碳中铂的质量分数优选为10%(10%铂碳);所述化合物v和催化剂的质量比优选为1:0.05~0.5,更优选为1:0.1~0.2。在本发明中,所述有机溶剂优选包括醇类溶剂,更优选包括甲醇、乙醇和异丙醇中的一种或几种;本发明对于所述有机溶剂的用量没有特殊限定,能够使催化氢化反应顺利进行即可;在本发明的实施例中,所述化合物v的质量和有机溶剂的体积之比优选为1g:3~10ml,更优选为1g:5~6ml。在本发明中,所述催化氢化反应的氢气压力优选为0.1~10mpa,更优选为2~5mpa;所述催化氢化反应的温度优选为25~70℃,更优选为40~50℃;所述催化氢化反应的时间优选为6~12h,更优选为8~10h;所述催化氢化反应优选在加氢反应釜中进行;所述催化氢化反应过程中发生的反应如下:
[0062]
[0063]
所述催化氢化反应后,本发明优选还包括将所述催化氢化反应的体系进行后处理,所述后处理包括冷却至室温后过滤以除去催化剂,将所得滤液浓缩至恒重,得到化合物viii。本发明对于所述冷却的方式没有特殊限定,采用本领域技术人员熟知的冷却方式即可。在本发明中,所述浓缩的方式优选为减压蒸馏;本发明对于所述减压蒸馏的条件没有特殊限定,浓缩至恒重即可。在本发明中,所述化合物viii不经纯化直接用于下一步反应。
[0064]
得到化合物viii后,本发明将所述化合物viii与盐酸溶液混合进行成盐反应,得到具有式i所示结构的盐酸拉贝洛尔。在本发明中,所述盐酸溶液优选包括盐酸水溶液、盐酸醇溶液和盐酸酯溶液中的一种或几种;所述盐酸醇溶液优选包括盐酸乙醇溶液、盐酸甲醇溶液和盐酸异丙醇溶液中的一种或几种;所述盐酸酯溶液优选包括盐酸乙酸乙酯溶液;所述盐酸溶液的浓度优选为5~25wt%,更优选为8~20wt%,进一步优选为10~15wt%;本发明对于所述盐酸溶液的用量没有特殊限定,能够保证体系的ph值为1~3即可;所述ph值更优选为2在本发明中,所述成盐反应的温度优选为
‑
10~30℃,更优选为0~10℃;所述成盐反应的时间优选为30~90min,更优选为60~70min;所述成盐反应过程中发生的反应如下:
[0065][0066]
所述成盐反应后,本发明优选还包括将所述成盐反应的体系进行后处理,所述后处理包括固液分离,将所得固体产物进行有机溶剂洗涤后干燥,得到盐酸拉贝洛尔。本发明对于所述固液分离的方式没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如抽滤。在本发明中,所述有机溶剂洗涤用有机溶剂优选包括醇类溶剂,所述醇类溶剂优选包括异丙醇、甲醇、乙醇和正丁醇中的一种或几种;所述有机溶剂洗涤的方式优选为淋洗。在本发明中,所述干燥的温度优选为室温~80℃,更优选为50℃;所述干燥的时间优选为6~24h,更优选为12h。
[0067]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0068]
实施例1
[0069]
(1)2
‑
羟基
‑5‑
{2
‑
[(苯基甲基)氨基]乙酰基}
‑
苯甲酰胺(化合物iii)的制备
[0070]
在四口烧瓶中加入500ml四氢呋喃、42g苄胺和59g三乙胺,降温至
‑
10℃后滴加200ml浓度为0.5g/l的化合物ii(x=br,5
‑
溴乙酰基水杨酰胺)的四氢呋喃溶液,控制温度不超过
‑
5℃,滴加完毕后保温搅拌1h,升至室温后保温第一亲核反应反应2h,加入200ml水洗涤,用乙酸乙酯萃取2次(每次加入300ml),将所得有机层用100ml饱和碳酸氢钠洗涤1次,100ml饱和食盐水洗涤1次,无水硫酸钠干燥,过滤,将所得滤液减压浓缩至恒重,得到化合物iii(100.62g,收率91.0%),不经纯化直接用于下一步反应。
[0071]
(2)5
‑
[n
‑
苄基
‑
n
‑
(4
‑
苯基丁烷
‑2‑
基)甘氨酰基]2
‑
羟基苯甲酰胺(化合物v)的制
备
[0072]
在四口烧瓶中加入300ml乙腈、36.5g碳酸钾和50.0g化合物iii于室温条件下搅拌30min后加入41.05g化合物iv(x=br,3
‑
溴丁基苯),升温至60℃后进行第二亲核取代反应,tlc检测至化合物iii消失后反应完毕,冷却至室温,加入200ml水洗涤,二氯甲烷萃取2次(单次用300ml)将所得有机层进行无水硫酸钠干燥,过滤,将所得滤液减压浓缩至恒重,得到化合物v(62.25g,收率84.98%),不经纯化直接用于下一步反应。
[0073]
(3)拉贝洛尔(化合物viii)的制备
[0074]
在加氢反应釜中加入300ml甲醇,50g化合物v和5.0g 10%钯碳,在50℃、氢气压力为0.5mpa条件下催化加氢反应8h,冷却至室温,过滤除去10%钯碳,将所得滤液减压浓缩至恒重,得到化合物viii(38.11g,收率96.68%)。
[0075]
(4)盐酸拉贝洛尔(具有式i所示结构)的制备
[0076]
在四口烧瓶中加入100ml异丙醇和35g化合物viii,降温至10℃后滴加盐酸异丙醇溶液至ph值为2.0,保温成盐反应1h,抽滤,将所得固体产物异丙醇淋洗后在50℃条件下干燥12h,得到盐酸拉贝洛尔(35.21g,收率90.65%)。
[0077]
本实施例制备的盐酸拉贝洛尔的氢谱图如图1所示,碳谱图如图2所示,由图1~2可知,本发明成功制备得到具有式i所示结构的盐酸拉贝洛尔。
[0078]
实施例2
[0079]
其他制备条件与实施例1相同,与实施例1的区别仅在于:步骤(1)中将式5
‑
溴乙酰基水杨酰胺替换为5
‑
氯乙酰基水杨酰胺(即化合物ii中x=cl),得到化合物iii(93.48g,收率84.54%)。
[0080]
实施例3
[0081]
其他制备条件与实施例1相同,与实施例1的区别仅在于:步骤(2)中将式5
‑
溴乙酰基水杨酰胺替换为5
‑
氯乙酰基水杨酰胺(即化合物iv中x=cl),得到化合物v(61.16g,收率83.49%)。
[0082]
实施例4
[0083]
其他制备条件与实施例1相同,与实施例1的区别仅在于:步骤(1)中化合物ii中x=cl,得到化合物iii(93.48g,收率84.54%);步骤(2)中化合物iv中x=cl,得到化合物v(61.16g,收率83.49%)。
[0084]
实施例5
[0085]
(1)按照实施例1步骤(1)制备化合物iii。
[0086]
(2)4
‑
甲基苯磺酸4
‑
苯基丁
‑2‑
基酯(化合物vii)的制备
[0087]
在四口烧瓶中加入500ml二氯甲烷、80.05g化合物vi(3
‑
羟基丁基苯),降温至
‑
10℃后加入70.16g三乙胺,控制温度不超过5℃,加入106.75g对甲基苯磺酰氯,升至室温后进行酯化反应,tlc检测化合物vi消失后反应完毕,加入100ml水洗涤,将所得有机层无水硫酸钠干燥后过滤,将所得滤液减压浓缩至恒重,得到化合物vii(154.60g,收率95.32%)。
[0088]
(3)在四口烧瓶中加入800ml乙腈、95.67g化合物iii,139.41g碳酸钾和112.64g化合物vii混合均匀,升温至81℃后进行胺酯交换反应,tlc检测至化合物iii消失后反应完毕,冷却至室温后过滤,将所得滤液减压浓缩至恒重,得到化合物v(119.13g,收率85.0%)。
[0089]
(4)按照实施例1步骤(3)制备拉贝洛尔。
[0090]
(5)按照实施例1步骤(4)制备盐酸拉贝洛尔。
[0091]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。