基于宏基因组测序的微生物对比结果校正的方法和系统
1.相关申请的交叉引用
2.本发明属于申请日为2020.12.14,申请号为202011474395.0,发明名称为“一种检测样本中微生物和耐药基因的方法和系统”的中国发明专利申请的分案申请,其全部内容通过引用合并于此。
技术领域
3.本发明属于宏基因组分析技术领域,具体地,涉及一种基于宏基因组测序的微生物对比结果校正的方法和系统。
背景技术:
4.由病原微生物导致的感染性疾病,尤其是重症肺炎、脓毒血症和脑炎脑膜炎为主的疑难危重感染性疾病的病原鉴定困难,一直是感染病临床医师面临的一个关键问题,故精确的病原微生物鉴定是精准感染治疗至关重要的步骤。
5.传统的病原微生物检测技术主要分为两类:基因细胞培养的方法和基于特异性引物或抗体(分子检测)的方法如pcr检测、多重pcr检测和抗原抗体杂交反应等。目前临床常规病原检测方法以细菌/真菌培养、病毒pcr与感染免疫标志物为主,但分离培养阳性率偏低(15%
‑
20%)且周期过长(3
‑
5天),而分子检测与血清学检测虽然可扩大病原体的检测范围,但存在检测目标固定单一的局限,其难以应对因个体差异、混合感染、罕见感染和国际旅行等因素引起的疑难/复杂感染。
6.而以高通量测序为基础的病原宏基因组学技术可在一定程度上提升时效性、敏感性和鉴定精确度。相比传统技术,高通量测序直接测序可以快速一次性检测包括细菌、真菌、病毒和寄生虫等上千种病原体,且特别适用于罕见、新发和非典型的复杂传染病的病原检测。同时可检测细菌的耐药性、毒力性进行分析。病原宏基因组学由于其高灵敏度性、高效性、覆盖范围广和成本效益,有望部分取代传统检测方法,成为感染病诊治与传染病防控的革新性方法。
7.然而,目前利用宏基因组测序检测样本中微生物的方法存在下机数据分析时间长、微生物比对准确度低、未能有效过滤背景和污染微生物、未能有效区分背定植或感染微生物、未能有效鉴定多重耐药细菌及混合感染、未有效对检测的耐药基因与微生物建立关联、未能有效鉴定新发病原体等不足。
技术实现要素:
8.为了解决上述技术问题中的至少一个,本发明建立了一种检测样本中微生物和/或耐药基因的系统和方法,具体采用的技术方案如下:
9.本发明第一方面提供一种检测样本中微生物的方法,包括以下步骤:
10.s1,获得所述样本的宏基因组测序数据;
11.s2,对所述宏基因组测序数据进行物种分析:
12.s21,利用微生物比对数据库,基于k
‑
mer算法对所述宏基因组测序数据进行比对分析,获得微生物比对结果,
13.s22,利用微生物注释数据库,对所述微生物比对结果进行注释,获得微生物注释结果,
14.s23,对所述微生物注释结果进行初步过滤:a)过滤比对置信度小于第一预设阈值的序列;b)过滤丰度低于第二预设阈值的微生物;c)结合样本类型信息,过滤背景或污染微生物以及定植微生物,从而获得候选微生物信息,
15.s24,利用微生物代表基因组数据库,比对获得的所述候选微生物信息的覆盖率,过滤覆盖率小于第三预设阈值或非连续的比对区域数小于第四预设阈值的微生物,从而获得所述样本中的微生物信息。
16.在本发明中,所述样本为任意可包含微生物的生物样本,优选地是,为被感染的人的生物样本,更优选地,为人的体液样本。
17.在本发明中,所述微生物包括但不限于细菌、真菌、病毒、寄生虫和古菌。
18.在本发明的一些实施方案中,由于微生物的多样性,需要对由于所述样本的核酸提取分别进行。对于不包含rna病毒的生物样本,仅需提取dna并制备测序文库。对于包含rna病毒的生物样本,需要同时提取rna样本,进行反转录后制备测序文库,与dna测序文库一起上机测序。
19.在本发明的一些实施方案中,所述样本的宏基因组测序数据可以由任意二代测序平台或三代测序平台获得。
20.在本发明中,所述方法适用于短读长测序、长读长测序数据,支持单端、双端测序数据,也可支持组装草图序列数据。
21.进一步地,在步骤s2之前,进一步包括对宏基因组测序数据进行前处理(质控和质检)和去除宿主序列的步骤:
22.(1)前处理:a)质控:包括去除接头序列、低质量及重复序列等,得到高质量测序数据;b)根据质控分析的统计信息进行质检,如符合质量标准,则执行后续分析。
23.(2)去除宿主核酸序列:将通过质控和质检的高质量测序数据比对到样本宿主的参考基因组上(来源于ncbi中refseq数据库),过滤宿主核酸序列,得到clean data。
24.在本发明的一些实施方案中,所述微生物比对数据库包括但不限于:ncbi nt库、refseq和genbank库、病毒参考序列库img_vr、真菌和寄生虫参考序列库eupathdb。在本发明的一些优选实施方案中,所述微生物比对数据库还可以包括自建数据库,所述自建数据库是基于大量微生物样本建立的。
25.在本发明的一些实施方案中,所述微生物注释数据库根据微生物类型按照细菌、真菌、古菌、寄生虫和病毒进行归类,数据库中包括但不限于科、属、种、基因型、血清型、微生物中文名、拉丁文名、定植部位、感染部位、致病性、关联疾病、传播途径、革兰氏类型、核酸类型、微生物简介和参考文献信息。
26.在本发明的一些实施方案中,所述微生物代表基因组数据库是在微生物比对数据库的基础上,优先选取refseq数据库的序列,其次是从genbank和nt数据库选取完整地、高质量序列。在本发明的一些具体实施方案中,首先以种为单位,统计各菌株的序列数量和总序列长度,并计算总序列长度的中位数(记为l
median
);然后对不同类别的微生物设置序列数
量的阈值筛选菌株,即细菌序列数量低于300,真菌序列数量低于1000、古菌序列数量低于300、病毒序列数量低于10、寄生虫序列数量低于2000;最后挑选序列数量最少,且总序列长不低于0.9
×
l
median
,且不高于1.1
×
l
median
的菌株作为物种代表序列,如符合标准的菌株存在多株,则任意选取一株作为物种代表序列,如无符合筛选标准的菌株,则表示该物种无代表基因组。
27.在本发明的一些实施方案中,在所述步骤s21中,所述微生物对比结果包括但不限于种拉丁名、种水平序列数、种水平相对丰度、属拉丁名、属水平序列数、属水平相对丰度、属内各个种的序列占比、科拉丁名、科水平序列数、科水平相对丰度、科内各个属的序列占比和分类学谱系信息。
28.进一步地,在步骤s22之前,还包括对s21获得的微生物比对结果进行校正的步骤:
29.s2101,以科为单元进行检索,若注释到各个属的序列总和占该科序列总数小于50%,且该科的相对丰度大于15%,则:
30.a)将注释到该科的reads提取并组装成contig;
31.b)将reads比对到contig,记录read与contig的对应关系;
32.c)将contig比对到微生物比对数据库,获得比对的物种拉丁名和contig覆盖度;
33.d)结合b)和c)的结果,更新属水平序列数、属相对丰度和科内各个属的序列占比,
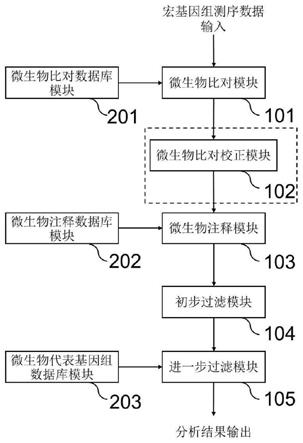
34.s2102,以属为单元进行检索,若注释到各个种的序列总和占该属序列总数小于50%,且该属的相对丰度大于15%,则:
35.a)将注释到该属的read提取并组装成contig;
36.b)将read比对到contig,记录read与contig的对应关系;
37.c)将contigs比对到微生物比对数据库,获得比对的物种拉丁名和contig覆盖度;
38.d)结合b和c的结果,更新种水平序列数、种相对丰度和属内各个种的序列占比,
39.由此,得到校正后的微生物比对结果。该步骤针对微生物初步比对结果中可能存在的局部异常结果运用序列组装后再进行序列比对的方法,保证分析速度的同时提高微生物比对的准确度。
40.在本发明的一些实施方案中,如出现同属内的多个菌,则可参考属内占比,通常属内占比大于80%的物种,其物种注释可信度高,可降低病原检出的假阳性。
41.在本发明的一些实施方案中,将校正结果中contigs覆盖度小于70%作为阈值,用于识别鉴定新微生物变异种。具体地,如contigs覆盖度小于70%,表示该物种为新微生物变异种。
42.在本发明的一些实施方案中,在所述步骤s22中,所述微生物注释结果包括但不限于微生物名称、微生物类别、定植部位、感染部位、致病性、关联疾病和传播途径。
43.在本发明的一些实施方案中,在所述步骤s23中,所述第一预设阈值为0.1
‑
0.3,优选地为0.1。在本发明的一些具体实施方案中,所述过滤比对置信度小于0.1的序列是指过滤比对上该物种的kmer数量占总kmer数量的比例低于0.1的序列。
44.在本发明的一些实施方案中,在所述步骤s23中,所述过滤丰度低于第二预设阈值的微生物,按不同的微生物类别进行归类,并分别按丰度从高到低进行排序,对不同的微生物类别设置不同的丰度阈值,过滤低于丰度阈值的微生物。在本发明的一些优选的实施方案中,不同的微生物类别的过滤第二预设阈值为:细菌read数不大于30,真菌read数不大于
30、古菌read数不大于30、病毒read数不大于3、寄生虫read数不大于100。
45.在本发明的一些实施方案中,在所述步骤s24中,对步骤s23中获得的候选微生物对应的序列集比对到微生物代表基因组数据库中相应的物种参考基因组上,快速、精确地比对后,计算各物种的覆盖率、非连续的比对区域数、平均覆盖深度,并作为微生物鉴定的比对可靠性、真实性的验证指标。
46.在本发明的一些实施方案中,在所述步骤s24中,所述第三阈值为1%
‑
5%,优选地为1%;所述第四阈值为3
‑
8,优选地为3。在本发明的一些具体实施方案中,过滤覆盖率小于1%或非连续的比对区域数小于3的微生物,从而获得所述样本中的微生物信息。
47.进一步地,所述方法还包括步骤s3,对所述宏基因组测序数据进行耐药基因分析:
48.s31,利用耐药基因数据库,对所述宏基因组测序数据进行比对分析,获得预测的耐药基因信息;
49.s32,对所述预测的耐药基因信息进行过滤,过滤掉覆盖率小于10%的耐药基因,从而得到最终的耐药基因信息。
50.本发明的一些实施方案中,使用srst2 v0.2.0将所述样本的宏基因组测序数据比对到细菌耐药基因数据库,获得耐药基因预测结果后,统计比对结果的耐药基因名称、耐药基因read数、覆盖率和覆盖深度。
51.在本发明的一些实施方案中,通过检索细菌耐药基因注释数据库补充耐药基因类别和关联细菌信息,从而用于辅助筛选潜在致病微生物。
52.更进一步地,所述方法还包括将步骤s24中获得的微生物信息和步骤s32中获得的耐药基因信息进行关联分析,得到潜在的致病微生物信息的步骤。
53.本发明的第二方面提供一种检测样本中微生物的系统,包括:
54.微生物分析模块组,包括:微生物比对模块101、微生物注释模块103、初步过滤模块104和进一步过滤模块105;和
55.微生物数据库模块组,包括:微生物比对数据库201、微生物注释数据库202和微生物代表基因组数据库203,
56.其中,
57.所述微生物比对模块101与所述微生物比对数据库201连接,用于利用微生物比对数据库,基于k
‑
mer算法对所述样本的宏基因组测序数据进行比对分析,获得微生物比对结果;
58.所述微生物注释模块103与所述微生物比对模块101和所述微生物注释数据库模块202连接,用于利用微生物注释数据库,对所述微生物比对结果进行注释,获得微生物注释结果;
59.所述初步过滤模块104与所述微生物注释模块103连接,用于对所述微生物注释结果进行初步过滤:a)过滤比对置信度小于第一预设阈值的序列;b)过滤丰度低于第二预设阈值的微生物;c)结合样本类型信息,过滤背景或污染微生物以及定植微生物,从而获得候选微生物信息;
60.所述进一步过滤模块105与所述初步过滤模块104和所述微生物代表基因组数据库模块203连接,用于利用微生物代表基因组数据库,比对获得的所述候选微生物信息的覆盖率,过滤覆盖率小于第三预设阈值或非连续的比对区域数小于第四预设阈值的微生物,
从而获得所述样本中的微生物信息。
61.进一步地,所述系统进一步包括宏基因组测序数据前处理(质控和质检)模块和去除宿主序列模块,其中
62.(1)前处理模块包括:
63.a)质控子模块,用于去除接头序列、低质量及重复序列等,得到高质量测序数据;和
64.b)质检子模块,用于根据质控分析的统计信息进行质检,如符合质量标准,则将高质量数据输入到下一模块。
65.(2)去除宿主序列模块,与质检子模块连接,用于将通过质控和质检的高质量测序数据比对到样本宿主的参考基因组上(来源于ncbi中refseq数据库),过滤宿主核酸序列,得到clean data,并进一步与所述微生物比对模块101连接,用于将clean data输入至所述微生物比对模块101。
66.进一步地,所述微生物分析模块组进一步包括微生物比对校正模块组102,位于所述微生物比对模块101和所述微生物注释模块之间,用于基于本发明第一方面所述的步骤对s21获得的微生物比对结果进行校正。
67.更进一步地,所述系统还包括:
68.耐药基因分析模块组,包括:耐药基因比对模块301和耐药基因过滤模块302;
69.耐药基因数据库模块组:包括:耐药基因比对数据库模块401和耐药基因注释数据库402,
70.其中,
71.所述耐药基因比对模块301与所述耐药基因比对数据库模块401连接,用于利用耐药基因数据库,对所述样本的宏基因组测序数据进行比对分析,获得预测的耐药基因信息;
72.所述耐药基因过滤模块302分别与耐药基因比对模块301和耐药基因注释数据库402连接,用于对所述预测的耐药基因信息进行过滤,过滤掉覆盖率小于10%的耐药基因,从而得到最终的耐药基因信息。
73.更进一步地,所述系统还包括关联模块105,用于对进一步过滤模块105和获得的微生物信息和耐药基因过滤模块302中获得的耐药基因信息进行关联分析,得到潜在的致病微生物信息。
74.本发明的有益效果
75.本发明相对于现有技术,具有以下有益效果:
76.本发明的方法和系统适用范围广,可覆盖人体不同感染部位,能检测多种类型微生物,兼容多种高通量测序平台,并支持短读长测序、长读长测序数据,支持单端、双端测序数据,也可支持组装草图序列。
77.本发明的方法和系统可从测序数据中准确分析样本内的微生物物种及其丰度、覆盖度,并可有效过滤背景/污染微生物,有效区分定植/致病微生物,有效降低假阳性,有效鉴定多重耐药菌及混合感染,可快速、准确、全面且灵敏地检测样本内的微生物。
78.本发明的方法和系统可从测序数据中准确分析样本内的耐药基因,并可有效提示关联细菌信息,为感染精准诊疗提供技术支持。
79.本发明的方法和系统可从测序数据中准确分析潜在的新发病原微生物,为新发传
染病的预警提供技术支持。
附图说明
80.图1示出了本发明实施例1和实施例2检测样本中微生物的系统的示意图。
81.图2示出了本发明实施例3和实施例4检测样本中微生物和耐药基因的系统的示意图。
82.图3示出了本发明实施例5检测样本中微生物及耐药基因的方法和装置的流程示意图。
83.图4示出了实施例6样本微生物组成分布图。
84.图5示出了实施例6样本中鲍曼不动杆菌的基因组覆盖度图。
85.图6示出了实施例7样本微生物组成分布图。
86.图7示出了实施例7样本中斯氏假单胞菌的基因组覆盖度图。
87.图8示出了实施例7样本中猪肉绦虫的基因组覆盖度图。
具体实施方式
88.为了使本发明所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。
89.实施例
90.以下例子在此用于示范本发明的优选实施方案。本领域内的技术人员会明白,下述例子中披露的技术代表发明人发现的可以用于实施本发明的技术,因此可以视为实施本发明的优选方案。但是本领域内的技术人员根据本说明书应该明白,这里所公开的特定实施例可以做很多修改,仍然能得到相同的或者类似的结果,而非背离本发明的精神或范围。
91.除非另有定义,所有在此使用的技术和科学的术语,和本发明所属领域内的技术人员所通常理解的意思相同,在此公开引用及他们引用的材料都将以引用的方式被并入。
92.那些本领域内的技术人员将意识到或者通过常规试验就能了解许多这里所描述的发明的特定实施方案的许多等同技术。这些等同将被包含在权利要求书中。
93.下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的仪器设备,如无特殊说明,均为实验室常规仪器设备;下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。
94.实施例1检测样本中微生物的系统及方法
95.本实施例提供一种检测样本中微生物的系统,如图1所示,包括:
96.微生物分析模块组,包括:微生物比对模块101、微生物注释模块103、初步过滤模块104和进一步过滤模块105;和
97.微生物数据库模块组,包括:微生物比对数据库201、微生物注释数据库202和微生物代表基因组数据库203,
98.其中,
99.微生物比对模块101与微生物比对数据库201连接,用于利用微生物比对数据库,基于k
‑
mer算法对样本的宏基因组测序数据进行比对分析,获得微生物比对结果;
100.微生物注释模块103与微生物比对模块101和微生物注释数据库模块202连接,用
于利用微生物注释数据库,对微生物比对结果进行注释,获得微生物注释结果;
101.初步过滤模块104与微生物注释模块103连接,用于对微生物注释结果进行初步过滤:a)过滤比对置信度小于0.1的序列;b)过滤低丰度微生物;c)结合样本类型信息,过滤背景或污染微生物以及定植微生物,从而获得候选微生物信息;
102.进一步过滤模块105与初步过滤模块104和微生物代表基因组数据库模块203连接,用于利用微生物代表基因组数据库,比对获得的候选微生物信息的覆盖率,过滤覆盖率小于1%或非连续的比对区域数小于3的微生物,从而获得样本中的微生物信息。
103.使用时,包括以下步骤:
104.s1,获得样本的宏基因组测序数据;
105.s2,对宏基因组测序数据进行物种分析:
106.s21,利用微生物比对数据库,基于k
‑
mer算法对宏基因组测序数据进行比对分析,获得微生物比对结果,
107.s22,利用微生物注释数据库,对微生物比对结果进行注释,获得微生物注释结果,
108.s23,对微生物注释结果进行初步过滤:a)过滤比对置信度小于0.1的序列;b)过滤低丰度微生物;c)结合样本类型信息,过滤背景或污染微生物以及定植微生物,从而获得候选微生物信息,
109.s24,利用微生物代表基因组数据库,比对获得的候选微生物信息的覆盖率,过滤覆盖率小于1%或非连续的比对区域数小于3的微生物,从而获得样本中的微生物信息。
110.实施例2检测样本中微生物的系统及方法
111.本实施例对实施例1中检测样本中微生物的系统进行改进,改进的点在于,微生物分析模块组进一步包括微生物比对校正模块组102(如图1虚线框内),位于所述微生物比对模块101和所述微生物注释模块之间,用于基于以下步骤对s21获得的微生物比对结果进行校正。
112.以科为单元进行检索,若注释到各个属的序列总和占该科序列总数小于50%,且该科的相对丰度大于15%,则:
113.a)将注释到该科的reads提取并组装成contig;
114.b)将reads比对到contig,记录read与contig的对应关系;
115.c)将contig比对到微生物比对数据库,获得比对的物种拉丁名和contig覆盖度;
116.d)结合b)和c)的结果,更新属水平序列数、属相对丰度和科内各个属的序列占比,
117.以属为单元进行检索,若注释到各个种的序列总和占该属序列总数小于50%,且该属的相对丰度大于15%,则:
118.a)将注释到该属的read提取并组装成contig;
119.b)将read比对到contig,记录read与contig的对应关系;
120.c)将contigs比对到微生物比对数据库,获得比对的物种拉丁名和contig覆盖度;
121.d)结合b和c的结果,更新种水平序列数、种相对丰度和属内各个种的序列占比,
122.由此,得到校正后的微生物比对结果。
123.实施例3检测样本中微生物和耐药基因的系统及方法
124.本实施例对实施例1或实施例2的用于检测样本中微生物的系统进行改进,如图2所示,改进的点在于,进一步包括:
125.耐药基因分析模块组,包括:耐药基因比对模块301和耐药基因过滤模块302;
126.耐药基因数据库模块组:包括:耐药基因比对数据库模块401和耐药基因注释数据库402,
127.其中,
128.耐药基因比对模块301与耐药基因比对数据库模块401连接,用于利用耐药基因数据库,对样本的宏基因组测序数据进行比对分析,获得预测的耐药基因信息;
129.耐药基因过滤模块302分别与耐药基因比对模块301和耐药基因注释数据库402连接,用于对预测的耐药基因信息进行过滤,过滤掉覆盖率小于10%的耐药基因,从而得到最终的耐药基因信息。
130.使用方法相应地,包括以下步骤:s3,对宏基因组测序数据进行耐药基因分析:
131.s31,利用耐药基因数据库,对宏基因组测序数据进行比对分析,获得预测的耐药基因信息;
132.s32,对预测的耐药基因信息进行过滤,过滤掉覆盖率小于10%的耐药基因,从而得到最终的耐药基因信息。
133.实施例4检测样本中微生物和耐药基因的系统及方法
134.本实施例对实施例3的系统进行改进,改进点的在于,进一步包括关联模块105,如图2中下方虚线处。用于对进一步过滤模块105和获得的微生物信息和耐药基因过滤模块302中获得的耐药基因信息进行关联分析,得到潜在的致病微生物信息。
135.相应地,使用方法中,进一步包括将步骤s24中获得的微生物信息和步骤s32中获得的耐药基因信息进行关联分析,得到潜在的致病微生物信息的步骤。
136.实施例5检测临床感染样本中致病微生物及耐药基因的方法的建立
137.本实施例提供实施例4一种系统的详细建立步骤及使用方法:
138.一、数据库的构建
139.数据库的构建包括微生物参考数据库构建和细菌耐药基因数据库构建:
140.1、微生物参考数据库的构建
141.微生物参考数据库包括微生物比对数据库、微生物注释数据库和微生物代表基因组数据库。
142.(1)微生物比对数据库:该数据库包含的参考基因组序列用于宏基因组测序的微生物分析比对。参考基因组序列包括但不限于从ncbi nt库(ftp://ftp.ncbi.nlm.nih.gov/blast/db/fasta/nt.gz)、refseq和genbank库(ftp://ftp.ncbi.nlm.nih.gov/genomes)、病毒参考序列库(https://genome.jgi.doe.gov/portal/img_vr)、真菌和寄生虫参考序列库(https://eupathdb.org/eupathdb/)等权威数据库中获取的数据库,并可以根据数据库的增加或更新进行完善。
143.微生物比对数据库包含全面完整的细菌、真菌、病毒、寄生虫、古菌及其他人体定植微生物的基因组序列,并去除冗余重复及可信度较低的基因组序列,共包含19616种微生物。微生物比对数据库确保微生物序列的完整性、准确性和全面性,且减少序列冗余,提高了分析比对速度。
144.(2)微生物注释数据库:该数据库用于对所鉴定微生物物种进行注释。该数据库根据微生物类型按照细菌、真菌、古菌、寄生虫和病毒进行归类,数据库包含科、属、种、基因
型、血清型、微生物中文名、拉丁文名、定植部位、感染部位、致病性、关联疾病、传播途径、革兰氏类型、核酸类型、微生物简介和参考文献等信息。数据库共包含3013种微生物,包括人体定植/致病微生物,以及常见的背景/污染微生物。微生物注释数据库的示例如表1,即肺炎衣原体的注释信息。
145.表1微生物注释数据库示例表
[0146][0147][0148]
(3)微生物代表基因组数据库:该数据库包含的物种代表基因组序列用于对候选病原的精确比对分析,并计算覆盖率、覆盖深度和非连续的比对区域数。该数据库的每个物种,是在微生物比对数据库的基础上,优先选取refseq数据库的序列作为物种代表序列;如refseq不存在物种序列,则从genbank和nt数据库中选取完整地、高质量序列作为物种代表序列,优选规则如下:首先以种为单位,统计各菌株的序列数量和总序列长度,并计算总序列长度的中位数(记为l
median
);然后对不同类别的微生物设置序列数量的阈值筛选菌株,即细菌序列数量低于300,真菌序列数量低于1000、古菌序列数量低于300、病毒序列数量低于10、寄生虫序列数量低于2000;最后挑选序列数量最少,且总序列长不低于0.9
×
l
median
,且不高于1.1
×
l
median
的菌株作为物种代表序列,如符合标准的菌株存在多株,则随机选取一
株作为物种代表序列,如无符合筛选标准的菌株,则表示该物种无代表基因组。最终得到12816种微生物的代表基因组。微生物代表基因组数据库对每个物种所对应的基因组序列精挑细选,确保精简且准确,提高了候选致病微生物的比对分析的速度、准确性和权威性。
[0149]
2、细菌耐药基因数据库的构建
[0150]
细菌耐药基因数据库包括细菌耐药基因比对数据库和细菌耐药基因注释数据库。
[0151]
(1)细菌耐药基因比对数据库:该数据库用于对宏基因组测序的细菌耐药基因分析比对。耐药基因参考序列主要从card(https://card.mcmaster.ca)和arg
‑
annot(https://github.com/katholt/srst2/blob/master/data/argannot_r3.fasta)数据库获取。包含全面完整的细菌的耐药基因序列,并去除冗余重复及可信度较低的基因序列,确保耐药基因序列的完整性和准确性,且减少序列冗余,提高了分析比对速度。
[0152]
(2)细菌耐药基因注释数据库:该数据库用于对所鉴定的耐药基因进行注释。细菌耐药基因注释数据库整理了所有耐药基因的注释信息,包括耐药基因名、耐药类别名称和关联菌列表等,并标记关联菌列表中的临床高重要性的细菌病原体,标记对应的耐药基因。
[0153]
二、检测临床感染样本中致病微生物及耐药基因的方法
[0154]
该方法包括两大步骤:样本测序(湿实验)和测序分析(干实验),如图3所示。
[0155]
1、样本测序(湿实验)
[0156]
如图3中湿实验部分所示,详细步骤如下:
[0157]
1)使用qiagen rneasy kit对样品提取rna,使用qiaamp dna mini kit(qiagen)提取dna。
[0158]
核酸提取后,使用agilent 2100bioanalyzer(agilent)进行核酸浓度的测定和质量检测;
[0159]
2)用covaris e210(covaris)将rna片段化,并进行反转录合成cdna;
[0160]
3)用qia quick pcr extraction kit(qiagen)对cdna和dna片段进行纯化和洗脱,并进行片段末端修复;
[0161]
4)分别对rna和dna文库进行加接头和纯化扩增,扩增完成后合并rna和dna文库;
[0162]
5)使用illumina miseq平台进行se75的宏基因组测序,测序下机后对原始bam文件进行数据格式转换和barcode拆分,得到样本的测序原始数据(fastq格式)。
[0163]
2、测序数据分析
[0164]
如图3中干实验部分所示,分析模式分为快速分析模式和综合分析模式,其区别在于如使用快速分析模式,则跳过第4步(微生物比对校正步骤,图中未包含)。详细步骤如下:
[0165]
(1)测序数据前处理:
[0166]
a)质控:使用fastp v0.20对测序原始数据进行质控,参数设置为
‑
n 2
ꢀ‑
q 15
ꢀ‑
x
ꢀ‑5ꢀ‑3ꢀ‑
y。首先过滤含有2个以上n碱基的read,对保留下来的序列,在序列首尾两端使用滑动窗口检测碱基质量,切除质量值低于q15的短片段序列,同时检测序列尾端的polyx序列并去除。此外,还过滤低复杂度序列,接头序列。上述所有过滤步骤通过后,过滤长度小于15bp的序列;
[0167]
b)质检:统计质控后的序列质量,以q20(单条序列的平均错误率<=1%)作为序列质量评价标准。若符合q20的序列高于80%,则本次测序数据通过质检;
[0168]
c)序列去冗余:使用soapnuke v1.5.6的默认参数对上一步数据进行序列去冗余,
得到非冗余序列。
[0169]
(2)去除人源宿主:使用bwa v0.7.17的默认参数将上述数据比对到人参考基因组序列(hg19)和人转录本数据(refmrna),并用samtools v1.9提取未比对上的序列,得到非人序列。
[0170]
(3)微生物比对:使用kraken2 v2.0.8(基于k
‑
mer的算法)对上述数据进行快速的比对注释,所用的数据库为微生物比对数据库。统计比对结果的种拉丁名、种水平序列数、种水平相对丰度(种水平read数占总序列的百分比)、属拉丁名、属水平序列数、属水平相对丰度、属内各个种的序列占比、科拉丁名、科水平序列数、科水平相对丰度、科内各个属的序列占比和分类学谱系信息。
[0171]
(4)微生物比对校正(综合分析模式选用步骤):如启用综合模式,则对于上述的比对结果中,首先会以科为单元进行检索遍历,若注释到各个属的序列总和占该科序列总数小于50%,且该科的相对丰度大于15%,则:
[0172]
a)将注释到该科的reads提取后,使用idba_hybrid v1.1.3以de brujin graph算法组装成contigs;
[0173]
b)使用blastn将reads比对到contigs,记录read与contig的对应关系;
[0174]
c)然后使用blastn v2.2.26将contigs比对到微生物比对数据库,并获得比对的物种拉丁名、contig覆盖度;
[0175]
d)结合b)和c)的结果,更新属水平序列数、属相对丰度和科内各个属的序列占比。
[0176]
然后以属为单元进行检索遍历,若注释到各个种的序列总和占该属序列总数小于50%,且该属的相对丰度大于15%,则:
[0177]
a)将注释到该属的reads提取后,使用idba_hybrid v1.1.3以de brujin graph算法组装成contigs;
[0178]
b)使用blastn将reads比对到contigs,记录read与contig的对应关系;
[0179]
c)然后使用blastn将contigs比对到微生物比对数据库,并获得比对的物种拉丁名、contigs覆盖度;
[0180]
d)结合b和c的结果,更新种水平序列数、种相对丰度和属内各个种的序列占比。最后得到校正后的微生物比对结果。
[0181]
(5)微生物注释:将微生物比对结果与微生物注释数据库进行关联,补充注释信息,包括微生物中文名、微生物类别、定植部位、感染部位、致病性、关联疾病和传播途径等。
[0182]
(6)微生物注释结果的初步过滤:
[0183]
a)过滤比对置信度小于0.1的序列,过滤标准:比对上该物种的kmer数量占总kmer数量的比例低于0.1;
[0184]
b)过滤低丰度微生物:按不同的微生物类别进行归类,按细菌read数大于30,真菌read数大于30、古菌read数大于30、病毒read数大于3、寄生虫read数大于100,分别过滤不同类别的低丰度微生物;
[0185]
c)结合样本类型信息及微生物注释信息,过滤常见背景/污染菌,区分定植或感染微生物,得到候选致病微生物注释结果。
[0186]
(7)候选致病微生物的覆盖率计算:使用megablast v2.2.26对上述候选致病微生物列表对应的序列集比对到相应的物种参考基因组上,所用的数据库为微生物代表基因组
数据库。
[0187]
快速、精确的比对后计算各物种的覆盖率、非连续的比对区域数、平均覆盖深度,并作为微生物鉴定的比对可靠性、真实性的验证指标。
[0188]
(8)候选致病微生物注释结果的进一步过滤:过滤覆盖率小于1%或非连续的比对区域数小于3的微生物。
[0189]
(9)耐药基因鉴定:使用srst2 v0.2.0将非人序列(第2步得到的数据集)比对到细菌耐药基因数据库。获得耐药基因预测结果后,统计比对结果的耐药基因名称、耐药基因read数、覆盖率和覆盖深度,过滤覆盖率小于10%的耐药基因结果。此外通过检索细菌耐药基因注释数据库补充耐药基因类别和关联细菌信息。
[0190]
(10)结果整合:
[0191]
a)若核酸提取的质检未能通过质量标准,则该样本实验处理不合格,重新进行样本实验处理环节。
[0192]
b)若核酸提取的质检符合质量标准,但测序下机数据质检未能通过质量标准,则该检测实验不合格,仅展示质检报告。
[0193]
c)若核酸提取的质检和测序下机数据的质检均符合质量标准,则进行完整的测序数据分析和结果过滤,得到候选致病微生物鉴定结果和耐药基因鉴定结果。最后,
[0194]
1)根据耐药基因鉴定结果的关联菌提示信息,辅助筛选潜在致病微生物;
[0195]
2)如出现同属内的多个菌,则可参考属内占比,通常属内占比大于80%的物种,其物种注释可信度高,故可降低病原检出的假阳性;
[0196]
3)将微生物比对校正结果中contigs覆盖度小于70%,作为新微生物变异种的阈值。
[0197]
综上,经过上述步骤,最终展示潜在致病微生物、耐药基因结果及潜在的新微生物变异种。
[0198]
实施例6利用实施例1建立的方法对肺泡灌洗液进行dna和rna测序并分析
[0199]
1、样本实验处理
[0200]
按照实施例5的湿实验方法对1例肺泡灌洗液样本dna和rna提取并进行宏基因组测序。
[0201]
2、测序数据分析
[0202]
该实施例采用快速模式对测序数据进行分析。
[0203]
根据表2可知,测序数据量约为19.9m,而序列的q20比例达到85.5%,数据有效率达到99.2%,本实验测序质量通过标准,可进行后续分析。
[0204]
表2样本数据质控信息
[0205][0206][0207]
表2说明:
[0208]
raw read:原始下机数据的序列总数;
[0209]
filter reads:低质量序列数;
[0210]
filter(%):低质量序列数占总序列数的百分比;
[0211]
clean reads:质控后剩余序列数;
[0212]
clean q20(%):质控后序列中,平均错误率小于1%的比例;
[0213]
effective(%):质控后得到的clean reads数占raw reads数的比例。
[0214]
经过去除人源序列后余下的序列用于微生物比对注释,约0.206m reads注释到微生物(表3)。
[0215]
表3样本宿主及微生物注释统计表
[0216][0217]
其中丰度最高的前9种微生物的组成见图4,由图4可知,前9种微生物分别是鲍曼不动杆菌(acinetobacter baumannii)、痤疮丙酸杆菌(propionibacterium acnes)、溶血葡萄球菌(staphylococcus haemolyticus)、金黄色葡萄球菌(staphylococcus aureus)、山羊葡萄球菌(staphylococcus caprae)、人葡萄球菌(staphylococcus hominis)、皮特不动杆菌(acinetobacter pittii)、医院不动杆菌(acinetobacter nosocomialis)、约氏不动杆菌(acinetobacter johnsonii)。相对丰度分别为:90.1%、6.66%、0.12%、0.09%、0.05%、0.05%、0.04%、0.03%、0.02%。经过分析且最终筛选得到的微生物注释结果见表4。
[0218]
表4样本部分微生物的注释信息表
[0219][0220][0221]
其中鲍曼不动杆菌(acinetobacter baumannii)检出186465条序列,占总微生物序列数90.7%,覆盖率92.98%,非连续的比对区域数79(均通过检出阈值),对应的鲍曼不动杆菌基因组覆盖图见图5。此外,相比皮特不动杆菌(acinetobacter pittii),鲍曼不动杆菌在不动杆菌属(acinetobacter)内的占比高达91.8%,故比对注释可信度高。此外,由于本实施例采用的是高通量测序的短读长测序模式,短序列存在部分高度同源序列难区分的问题。但本实施例中检测结果中皮特不动杆菌的属内占比低于0.1%(比例极低),表明本发明能提供更加灵敏而准确的微生物注释比对分析。
[0222]
此外,表4列出的痤疮丙酸杆菌(propionibacterium acnes)是常见污染菌,结合统计信息,综合判断后不予检出。而溶血葡萄球菌(staphylococcus haemolyticus)、金黄
色葡萄球菌(staphylococcus aureus)是皮肤定植菌,而本实验是肺泡关系液样本,本次实验认为是实验操作引入的污染菌,故不予检出。
[0223]
此外,本实验分析得到的耐药基因结果如表5所示。
[0224]
表5样本耐药基因结果信息表
[0225][0226]
结果表明,该样本存在多种耐药基因,对β
‑
内酰胺酶类、氨基糖苷类和四环素类等药物耐受,且预测结果进一步表明携带这些耐药基因的微生物是鲍曼不动杆菌。
[0227]
最后给予检出的感染微生物是鲍曼不动杆菌(acinetobacter baumannii),且是多重耐药鲍曼不动杆菌。
[0228]
实施例7利用实施例1建立的方法对脑脊液进行dna测序并分析
[0229]
1、样本实验处理
[0230]
按照实施例1的湿实验方法对1例脑脊液样本进行dna提取和文库构建,并进行宏基因组测序。
[0231]
2、测序数据分析
[0232]
该实施例采用快速模式对测序数据进行分析。
[0233]
根据表6可知,测序数据量约为14.7m,而序列的q20比例达到82.62%,且数据有效率达到98.7%,本实验测序质量好,可进行后续分析。
[0234]
表6样本数据质控信息
[0235][0236]
表6说明:
[0237]
raw read:原始下机数据的序列总数;
[0238]
filter reads:低质量序列数;
[0239]
filter(%):低质量序列数占总序列数的百分比;
[0240]
clean reads:质控后剩余序列数;
[0241]
clean q20(%):质控后序列中,平均错误率小于1%的比例;
[0242]
effective(%):质控后得到的clean reads数占raw reads数的比例。
[0243]
经过去除人源序列后余下的序列用于微生物注释,约0.33m reads注释到微生物(表7)。
[0244]
表7样本宿主及微生物注释统计表
[0245][0246]
其中丰度最高的前9种微生物的组成见图6,由图6可知,前9种微生物分别是斯氏假单胞菌(pseudomonas stutzeri)、猪肉绦虫(taenia solium)、痤疮丙酸杆菌(cutibacterium acnes)、门多萨假单胞菌(pseudomonas mendocina)、约氏不动杆菌(acinetobacter johnsonii)、奥斯陆莫拉菌(moraxella osloensis)、荧光假单胞菌(pseudomonas fluorescens)、洋葱伯克霍尔德菌(burkholderia cepacia),多噬伯克霍尔德氏菌(burkholderia multivorans)。相对丰度分别为:50.6%、14.5%、8.47%、6.21%、1.73%、1.04%、0.77%、0.75%、0.72%。
[0247]
经过分析且最终筛选得到的微生物注释结果见表8。
[0248]
表8样本部分微生物的注释信息表
[0249][0250]
其中痤疮丙酸杆菌(cutibacterium acnes)和约氏不动杆菌(acinetobacter johnsonii)是常见污染菌,综合统计信息,判断后予以剔除。而斯氏假单胞菌(pseudomonas stutzeri)检出165451条序列,占总微生物序列数50.61%,对应的基因组覆盖图见图7。
[0251]
猪肉绦虫(taenia solium)检出48047条序列,占总微生物序列数的14.52%,对应的猪肉绦虫基因组覆盖图见图8。
[0252]
此外,本实验未预测到细菌耐药基因。
[0253]
结合根据表8的注释信息,斯氏假单胞菌(pseudomonas stutzeri)和猪肉绦虫(taenia solium)均可引起脑部感染,结合样本类型和统计信息综合判定是混合感染,感染微生物是斯氏假单胞菌(pseudomonas stutzeri)和猪肉绦虫(taenia solium)。
[0254]
实施例8方法的时效性、计算资源消耗检测
[0255]
1、实验数据
[0256]
为了测试本发明实施例5的测序分析方法运算时间和计算资源消耗,使用5个se150,reads数为50mb的宏基因组数据进行测试,比较实施例5建立的分析方法与常规的宏
基因组测序基因检测方法[clarke el,taylor lj,zhao c,et al.sunbeam:an extensible pipeline for analyzing metagenomic sequencing experiments.microbiome.2019 mar 22;7(1):46.doi:10.1186/s40168
‑
019
‑
0658
‑
x.]在分析用时和计算资源消耗方面的差异。
[0257]
2、实验结果
[0258]
从表9可知,同样的cpu核数分析约50mb数据量的样本,常规的宏基因组测序数据分析流程比实施例5建立的快速模式方法耗时多4倍,服务器内存资源消耗多2倍多。而实施例5建立的方法即使使用综合模式,在不增加内存峰值情况下,其耗时也比常规方法少3倍。
[0259]
表9实施例5建立的方法与常规宏基因组测序数据分析方法的时效性、计算资源消耗对比表
[0260][0261]
结果表明,实施例5建立的方法通过设计多数据库系统,特别是精简、完整且准确的微生物比对数据库,加上精心设计的分析策略和参数优化节省了分析时长、降低了内存消耗,达到快速且准确检测宏基因组测序数据中微生物的效果。
[0262]
实施例9方法的准确度检测
[0263]
1、实验数据
[0264]
埃希氏菌属和志贺氏菌属是临床常见感染病原,但其属间基因组相似度高,基于read的序列比对的准确率较低。为了评价本发明实施例5建立的方法的准确度,以大肠埃希菌、志贺氏菌和人的参考基因组为实验对象,按不同物种设置不同的序列占比,使用pirs v2.0.0软件生成5份模拟的se150测序数据,每份数据共10m reads,具体的数据情况见表10。
[0265]
表10模拟测序数据表
[0266][0267]
2、实验结果
[0268]
模拟数据集按实施例5建立的方法进行微生物检测分析,并同时也用常规的宏基因组测序数据分析流程[clarke el,taylor lj,zhao c,et al.sunbeam:an extensible pipeline for analyzing metagenomic sequencing experiments.microbiome.2019 mar 22;7(1):46.doi:10.1186/s40168
‑
019
‑
0658
‑
x.]进行分析,得到的微生物注释统计见表11。
[0269]
表11所有数据集的不同分析方法得到的微生物注释结果对比
[0270][0271]
从表11结果可知,实施例5建立的方法在快速模式下,注释到目标微生物的序列数量是常规方法的约1.4倍,而综合模式是常规方法的4倍以上。
[0272]
进一步地,发明人统计了所有模拟数据集的大肠埃希菌、志贺氏菌的平均检出率,结果如表12所示。
[0273]
表12实施例5建立的方法与常规宏基因组测序数据分析方法的2种同源物种的检出率对比
[0274][0275]
从表12结果可知:实施例5建立的分析方法在快速模式下的分析效果优于常规方法,真阳性率相对较高(~20%vs~15%),其得益于实施例5建立的准确度极高的微生物比对数据库。
[0276]
更为重要的是,实施例5建立的分析方法在综合模式下的分析效果明显优于在快速模式分析和常规方法,真阳性率高达64%左右,相比常规方法提高了4倍。此外,在综合模式下分析的假阳性率低至7%,相比普通方法降低了2.5倍。
[0277]
实施例5建立的方法能根据微生物间的同源性高低,选择合适的分析模式,并整合分析结果,保证分析速度的同时,准确地鉴定基因组相似高的属或种,保证真阳性率,又能降低假阳性率。为鉴定样本内微生物,尤其是对临床影响偏高的病原微生物,提供快速、准确的分析结果。
[0278]
实施例10实施例5建立的方法用于新发病原体检测
[0279]
1、实验数据
[0280]
为了评价本发明实施例5建立的方法对新发病原体的检测性能,发明人采集1例疑似2019新冠病毒(sars
‑
cov
‑
2)感染患者的肺泡灌洗液,按照实施例5的方法对该例样本进行宏基因组测序。并且将微生物参考数据库的2019新冠病毒参考序列剔除。
[0281]
2、测序数据分析
[0282]
按照实施例5建立的方法的综合分析模式进行微生物检测分析,首先得到微生物鉴定结果,如表13所示。
[0283]
表13样本的微生物鉴定信息表
[0284][0285]
表13结果显示,鉴定到高丰度的sars冠状病毒,对应乙型冠状病毒属(~1mreads),但该种的属内占比仅有34%(小于50%),且属相对丰度是75%(高于15%)。故触发了微生物的比对校正分析,将乙型冠状病毒属序列集提取后进行组装,共获得26个contig,总长度24561bp,组装后比对结果如表14所示。
[0286]
表14样本中乙型冠状病毒属序列的组装后再比对的结果
[0287][0288]
表14结果表明,绝大部分contig均比对到sars冠状病毒,但其对应的contigs(22891bp)只有37.63%的区域与sars冠状病毒可比对上,远远低于鉴定新微生物变异种的阈值70%,表明是一个新的冠状病毒种。
[0289]
为验证上述假设,将微生物参考数据库纳入sars
‑
cov
‑
2参考基因组,重新分析,结果如表15所示。
[0290]
表15样本中的微生物重鉴定信息表
[0291][0292]
表15结果显示,确实鉴定到了sars
‑
cov
‑
2,且属内占比及覆盖度都极高(>98%)。
[0293]
以上结果表明,实施例1建立的方法具有优异的新发病原的检测性能,可以为新发传染病的预警提供技术支持。
[0294]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。