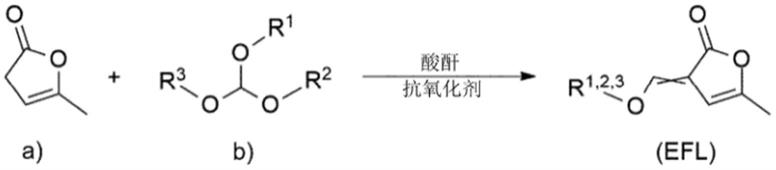
官能化的
α
‑
当归内酯单体和由其获得的聚合物
技术领域
1.本发明涉及官能化的α
‑
当归内酯化合物。更具体地,本发明涉及由环外烷氧基亚甲基官能化的α
‑
当归内酯,由所述官能化的α
‑
当归内酯化合物获得的均聚物和共聚物,以及基于所述均聚物或共聚物的组合物。
背景技术:
2.可再生资源,包括源自生物来源(生物质)的材料,是可用于许多应用,特别是用作化学和聚合物合成中的原材料的石油化工产品的重要替代品。再生资源的开发提供了一种克服有关石油化工产品供应、成本、环境影响和可持续性的问题的手段。此外,生物衍生材料通常可以是生物相容的、生物可吸收的和/或生物可降解的。
3.乙酰丙酸(la)是可以衍生自可再生资源的最高增值化学品之一,正如bozell et al.technology development for the production of biobased products from biorefinery carbohydrides
‑
the us department of energy’s“top 10”revisited,green chem.2010,12,539
‑
554所认可的。它在广泛选择的具有潜在工业应用的分子的合成中的应用使乙酰丙酸成为从碳水化合物中获得的最有希望的构筑单元之一,正如werpy et al.top value added chemicals for bio
‑
mass.volume i
–
results of screening for potential candidates from sugars and synthesis gas,us department of energy(2004)所讨论的。
4.就乙酰丙酸衍生物而言,当归内酯在过去几年受到越来越多的关注:例如,最近在lima et al.angelica lactones:from biomass
‑
derived plat
‑
form chemicals to value
‑
added products,chemsuschem 2018,11,25
‑
47中总结了它们的合成和应用。该参考文献广泛报道了α
‑
当归内酯(α
‑
al)向γ
‑
戊内酯和其他增值化学品的转化,以及α
‑
和β
‑
当归内酯在共轭加成反应中的广泛应用。
5.在asaoka et al.new synthetic method of 5
‑
and 2,5
‑
substituted 3
‑
furoates and 1,4
‑
dicarbonyl compounds from unsaturated lactones,chemistry letters,pp.171
‑
174,1977chemical society of japan中,标题化合物由通过4
‑
取代的2
‑
或3
‑
丁烯
‑4‑
内酯与原酸酯和乙酸酐反应获得的4
‑
取代的2
‑
(1'
‑
乙氧基亚烷基)
‑3‑
丁烯
‑4‑
内酯3来合成。
6.就当归内酯的成功聚合而言,文献中仅存在非常有限数量的报道:本发明人知道的此类报道之一是tarabanko et al.new biodegradable polymers based onα
‑
angelica lactone.chem.sustainable develop(2010),18,pages321
‑
328。缺乏文献的原因可能在于五元内酯对聚合过程的反应性通常较差。特别地,用于合成在其重复单元中保留内酯结构的聚合物的当归内酯的乙烯基聚合似乎只是一个出版物的目标,具体是marvel et al.the structure of vinyl polymers.iii.the polymer fromα
‑
angelica lactone(1939)1682
‑
1684。在这项陈旧(age
‑
worn)的工作中,作者描述了当α
‑
当归内酯与二硫化碳中催化量的三氟化硼反应时,以深红色固体形式获得的低分子量聚合物(mw=800
‑
900da)的形成。
7.将侧链(pendant)内酯部分结合到聚合物链中被认为有可能提供具有有用特性的材料,特别是具有高玻璃化转变温度(tg)的材料。然而,迄今为止,仅对于通过官能化γ
‑
戊内酯和丁内酯化合物反应获得的聚合物才实现这些特性。在这方面,有经验的读者的注意力可能被引导到:akkapeddi poly(a
‑
methylene
‑
y
‑
butyrolactone)synthesis,configurational structure,and properties,macromolecules(1979)546
‑
551;manzer catalytic synthesis ofα
‑
methylene
‑
γ
‑
valerolactone:a biomass
‑
derived acrylic monomer,applied catalysis a:general(2004)272,249
‑
256;vobecka et al.poly(a
‑
methylene
‑
g
‑
valerolactone):sustainable monomer synthesis and radical polymerization studies,polymer(2015)74,262
‑
271;和gowda et al.sustainable polymers from biomass
‑
derivedα
‑
methylene
‑
γ
‑
butyrolactones,in encyclopedia of polymer science and technology,4
th ed.,mark,h.f.,ed.;wiley:hoboken,nj,(2014)8,235
‑
271。
8.就本发明人的知识所及,使用α
‑
当归内酯获得具有侧链内酯基团的高分子量聚合物从未在文献中报道过。因此,本发明着眼于本领域中的这一缺陷以提供基于官能化的α
‑
当归内酯的聚合物,该聚合物在其重复单元中保留了内酯结构。
技术实现要素:
9.根据本发明的第一方面,提供了一种用于链增长聚合、优选用于阴离子聚合的单体,所述单体具有通式(efl)
[0010][0011]
其中:r
a
是c1‑
c
30
烷基、c3‑
c
30
环烷基、c6‑
c
18
芳基或c2‑
c
12
烯基。
[0012]
符号在本文中用于表示所有立体异构体都落入该通式(efl)的范围内。因此,所述单体可具有以下结构:
[0013][0014]
其中,例如,两种所述立体异构体的取代基r
a
可以是c1‑
c
18
烷基、c3‑
c
18
环烷基、c6‑
c
18
芳基或c2‑
c
10
烯基。例如,本发明提供一种单体(efl),其中r
a
是乙基(et),并且其在室温下以结晶固体的形式分离:
[0015][0016]
在单体(efl)的一个重要实施方案中,r
a
是c1‑
c
12
烷基或c2‑
c8烯基,特别是c1‑
c6烷基或c2‑
c4烯基。在不旨在与上述实施方案相互排斥的另一个实施方案中,所述单体(efl)的特征在于r
a
是c3‑
c6烷基或c2‑
c4烯基。
[0017]
根据本发明的第二个方面,提供了一种合成如上文和所附权利要求中定义的单体(efl)的方法,所述方法包括在酸酐和抗氧化剂存在下使以下物质反应的步骤:
[0018]
a)α
‑
当归内酯;和
[0019]
b)具有通式(1)的原酸酯
[0020][0021]
其中:r1、r2和r3独立地选自c1‑
c
30
烷基、c3‑
c
30
环烷基、c6‑
c
18
芳基和c2‑
c
12
烯基。
[0022]
该合成方法在实施方案中非常有效地进行,在该实施方案中酸酐是乙酸酐、丙酸酐、丁酸酐或琥珀酸酐中的一种,并且其中相对于反应物(a)、b))的总摩尔数,所述酸酐以摩尔过量存在。同样,已经获得了良好的结果,其中基于反应物(a)、b))的总重量,所述抗氧化剂以至多10重量%的量存在,并且优选地包含至少一种空间位阻酚或由至少一种空间位阻酚组成。
[0023]
根据本发明的第三方面,提供了一种用于如上文和所附权利要求中所定义的至少一种单体(efl)的链增长聚合的方法。特别地,提供了一种用于如上文和所附权利要求中所定义的至少一种单体(efl)的阴离子聚合的方法,其中所述阴离子聚合在选自以下的引发剂存在下进行:碱金属有机化合物;碱金属醇盐;碱金属硫醇盐;碱金属氨化物(alkali metal amides);以及元素周期表第3a族元素的化合物。为了获得在其重复单元中具有侧链内酯官能团的均聚物或共聚物(p
‑
efl)的合适产率,优选的是该阴离子聚合过程在50℃至150℃范围内的温度下进行。
[0024]
本发明还提供了一种通过链增长聚合、特别是通过如上和所附权利要求中定义的阴离子聚合的方法可获得的均聚物或共聚物(p
‑
efl),该聚合物(p
‑
efl)具有以下通式:
[0025][0026]
其中:r
a
是c1‑
c
30
烷基、c3‑
c
30
环烷基、c6‑
c
18
芳基或c2‑
c
12
烯基,并且
[0027]
n为至少20的整数。
[0028]
通过上文和所附权利要求中定义的阴离子聚合方法可获得或获得的在其重复单元中具有侧链内酯官能团的聚合物(p
‑
efl)应具有以下至少一个特征:i)至少2500g/mol、优选10000g/mol至150000g/mol的数均分子量(mn),如通过凝胶渗透色谱法(gpc)在四氢呋喃中使用聚苯乙烯标样(polystyrenestandard)测定;ii)50℃
‑
200℃,优选100℃
‑
200℃的玻璃化转变温度(tg);iii)1.1至2.0、优选1.10至1.80的多分散指数(pdi)。
[0029]
在其重复单元中具有侧链内酯官能团的所述聚合物(p
‑
efl)在选自以下的至少一种其他单体的开环聚合中可用作大分子单体:环状碳酸酯;环状酸酐;草酸盐;和具有5
‑
、6
‑
和/或7
‑
元环的环酯。可选地,具有侧链内酯官能团的所述聚合物(p
‑
efl)可用作与至少两种能够形成酯键的共聚单体的酯化反应中的大分子单体。
e.carraher,jr.,第159
‑
161页的工作中定义的机理。链增长聚合也称为加成聚合,并且基于其中单个引发物质引起聚合物链的增长的自由基、阳离子、阴离子和配位反应。在链增长聚合中,活性物质(诸如引发剂)添加一个单体分子以产生新的活性中心,其然后再添加另一个单体分子以产生另一个活性中心,依此类推,从而使链增长以化学链反应的形式进行。
[0043]
本文所用的术语“阴离子聚合”是指“advanced organic chemistry”,第三版,jerry march,第151
‑
161页的工作中定义的机理。具体而言,它是指其中动力链载体(kinetic chain carrier)为阴离子的离子聚合。因此,阴离子聚合反应是一种链反应,其中聚合物链的增长通过单体和聚合物链上的反应位点之间的反应进行,反应位点在每个增长步骤结束时再生。在本文中,阴离子聚合用于由含有碳
‑
碳双键的单体生产大分子。通过亲核加成到单体的双键来引发聚合,其中引发剂包含阴离子,诸如氢氧化物、醇盐、氰化物或负碳离子。
[0044]
本文所用的术语“开环聚合”表示其中在合适的催化剂存在下环状化合物(单体)开环以形成线性聚合物的聚合。反应体系趋向于在所需的所得高分子化合物、环状化合物的混合物和/或线性低聚物之间达到平衡,该平衡的实现在很大程度上取决于环状单体的性质和量、所用的催化剂以及反应温度。不建议在聚合中使用溶剂和/或乳液,因为一旦反应完成,它们的去除可能很复杂。除此之外,可以在inter alia nuyken et al.,ring
‑
opening polymerization—an introductory review polymers 2013,5,361
‑
403中找到开环聚合的指导性公开。
[0045]
本文所用的术语“原酸酯”涉及包含连接至三个烷氧基的碳原子的化合物。
[0046]
名称α
‑
当归内酯(cas 591
‑
12
‑
8)与5
‑
甲基
‑
3h
‑
呋喃
‑2‑
酮同义使用。
[0047]
术语“外(exo)”根据其在本领域的标准定义使用。
[0048]
本文所用的“c1‑
c
n
烷基”基团是指含有1至n个碳原子的一价基团,其是烷烃的基团并且包括直链和支链的有机基团。同样地,“c1‑
c
30
烷基”基团是指含有1至30个碳原子的一价基团,其是烷烃的基团并且包括直链和支链的有机基团。烷基的实例包括但不限于:甲基;乙基;丙基;异丙基;正丁基;异丁基;仲丁基;叔丁基;正戊基;正己基;正庚基;和2
‑
乙基己基。在本发明中,此类烷基基团可以是未取代的或可以被一个或多个取代基取代,诸如卤素、硝基、氰基、酰胺基、氨基、磺酰基、亚磺酰基、硫烷基(sulfanyl)、次硫酸基(sulfoxy)、脲、硫脲、胺磺酰基、磺酰胺和羟基。上面列出的示例性烃基的卤化衍生物尤其可以作为合适的取代烷基的实例提及。然而,通常应注意优选含有1
‑
18个碳原子的未取代烷基(c1‑
c
18
烷基),例如含有1
‑
12个碳原子的未取代烷基(c1‑
c
12
烷基)。
[0049]
术语“c3‑
c
30
环烷基”应理解为表示具有3至30个碳原子的饱和的、单
‑
、双
‑
或三环烃基。通常,应注意优选含有3
‑
18个碳原子的环烷基(c3‑
c
18
环烷基)。环烷基的实例包括:环丙基;环丁基;环戊基;环己基;环庚基;环辛基;金刚烷;和降莰烷。
[0050]
如本文所用,单独使用或作为较大部分的一部分使用的“c6‑
c
18
芳基”基团——如在“芳烷基”中一样——是指任选取代的单环、双环和三环体系,其中单环体系是芳族的,或双环或三环体系中的环中的至少一个环是芳族的。双环和三环体系包括苯并稠合的2
‑
3元碳环。示例性芳基包括:苯基;茚基;萘基、四氢萘基、四氢茚基;四氢蒽基;和蒽基。并且可以注意优选苯基。
[0051]
本文所用的“c2‑
c
12
烯基”是指具有2至12个碳原子和至少一个烯属不饱和单元的
烃基。烯基可以是直链、支链或环状的,并且可以任选地被取代。如本领域普通技术人员所理解的,术语“烯基”还包括具有“顺式(cis)”和“反式(trans)”构型,或者“e”和“z”构型的基团。然而,通常应注意优选含有2至10个(c2‑
10
)或2至8个(c2‑8)碳原子的未取代烯基。所述c2‑
c
12
烯基基团的实例包括但不限于:
‑
ch
═
ch2;
‑
ch
═
chch3;
‑
ch2ch
═
ch2;
‑
c(
═
ch2)(ch3);
‑
ch
═
chch2ch3;
‑
ch2ch
═
chch3;
‑
ch2ch2ch
═
ch2;
‑
ch
═
c(ch3)2;
‑
ch2c(
═
ch2)(ch3);
‑
c(
═
ch2)ch2ch3;
‑
c(ch3)
═
chch3;
‑
c(ch3)ch
═
ch2;
‑
ch
═
chch2ch2ch3;
‑
ch2ch
═
chch2ch3;
‑
ch2ch2ch
═
chch3;
‑
ch2ch2ch2ch
═
ch2;
‑
c(
═
ch2)ch2ch2ch3;
‑
c(ch3)
═
chch2ch3;
‑
ch(ch3)ch
═
chch;
‑
ch(ch3)ch2ch
═
ch2;
‑
ch2ch
═
c(ch3)2;1
‑
环戊
‑1‑
烯基;1
‑
环戊
‑2‑
烯基;1
‑
环戊
‑3‑
烯基;1
‑
环己
‑1‑
烯基;1
‑
环己
‑2‑
烯基;和1
‑
环己基
‑3‑
烯基。
[0052]
本文所用的“烷基芳基”是指烷基取代的芳基,而“取代的烷基芳基”是指进一步带有一个或多个上述取代基的烷基芳基。
[0053]
本文所用的术语“杂”是指含有一个或多个杂原子诸如n、o、si和s的基团或部分。因此,例如“杂环”是指具有例如n、o、si或s作为环结构的一部分的环状基团。“杂烷基”和“杂环烷基”部分分别是包含n、o、si或s作为其结构的一部分的如上文所定义的烷基和环烷基。
[0054]
本文所用的术语“催化量”是指相对于反应物,亚化学计量量的催化剂,除非另有明确说明。
[0055]
本文使用的术语“路易斯酸”表示能够通过与来自第二分子或离子的两个电子形成共价键而与另一分子或离子组合的任何分子或离子——通常称为亲电体:因此路易斯酸是电子受体。
[0056]
本说明书中提及的分子量可以通过凝胶渗透色谱法(gpc)使用聚苯乙烯校准标准物进行测量,诸如根据astm 3536所进行的。
[0057]
本文所用的术语“多元醇”应包括二醇和更高官能度的羟基化合物。
[0058]
本文所用的“结晶”是指具有高度规则的化学结构的固体。特别地,结晶化合物可以以其一种或多种单一结晶形式——多晶型物或假多晶型物——生产。结晶固体的颗粒可以以单晶、聚集体和附聚物的任意组合存在。
[0059]
在提到的地方,聚合物或共聚物的计算玻璃化转变温度(“tg”)是可以通过使用fox方程计算的温度(t.g.fox,bull.am.physics soc.,volume 1,issue no.3,page 123(1956))。某些均聚物的玻璃化转变温度可以在已公开的文献中找到,诸如在interscience publishers的由j.brandrup和e.h.immergut编辑的“polymer handbook”中。
[0060]
聚合物的实际玻璃化转变温度(tg)可以通过差示扫描量热法(dsc)确定。使用dsc来确定t
g
在本领域中是众所周知的,并且被b.cassel and m.p.divito in"use of dsc to obtain accurate thermodynamic and kinetic data",american laboratory,january 1994,pp 14
‑
19和b.wunderlich in thermal analysis,academic press,inc.,1990所描述。本专利申请中具体测量的玻璃化转变温度(t
g
)根据deutsches institut f
ü
r normung(din)11357的方法进行测量。
[0061]
术语“无水”在本文中旨在表示基于混合物或组分的重量,适用的反应混合物或组分包含小于0.25重量%的水。术语“基本上不含溶剂”应类似地解释为表示相关组合物包含小于0.25重量%的溶剂。
[0062]
发明详述
[0063]
官能化的α
‑
当归内酯(efl)的合成
[0064]
官能化的α
‑
当归内酯(efl)的合成最广泛地以以下反应方案为特征:
[0065][0066]
没有特别意图限制获得反应物α
‑
当归内酯(a))的方式:除了所述化合物可商购获得外,它还可以通过技术人员已知的多种合成路线来合成。在这方面可以参考http://www.molbase.com/en/synthesis_591
‑
12
‑8‑
moldata
‑
4778.html。当基于所采用的合成路线方便时,可以使用本领域已知的方法分离和纯化α
‑
当归内酯。在这方面可以提及萃取、蒸发、蒸馏和色谱法作为合适的技术。
[0067]
可用于上述反应方案的原酸酯反应物具有以下通式(b)):
[0068][0069]
其中:r1、r2和r3独立地选自c1‑
c
30
烷基、c3‑
c
30
环烷基、c6‑
c
18
芳基和c2‑
c
12
烯基。
[0070]
在式(b))的原酸酯的优选实施方案中,r1、r2和r3独立地选自c1‑
c
18
烷基和c2‑
c
12
烯基;r1、r2和r3可以例如独立地选自c1‑
c
12
烷基和c2–
c8烯基或独立地选自c1‑
c6烷基或c2‑
c4烯基。作为上述实施方案的替代或补充,优选地,式(1)中的r1、r2和r3中的至少两个是相同的。
[0071]
用于本发明的合适的原酸酯(b))的实例包括但不限于:原甲酸三乙酯(r1=r2=r3=et);原甲酸三甲酯(r1=r2=r3=me);原甲酸三丁酯(r1=r2=r3=bu);原甲酸三丙氧酯(tripropoxy orthoformate)(r1=r2=r3=npr);二乙基乙烯基原甲酸酯(r1=r2=et,r3=ch2=ch2);原甲酸三
‑
十八烷基酯(r1=r2=r3=c
18
h
37
);和原甲酸三戊酯(r1=r2=r3=c5h
11
)。
[0072]
如上述方案所述,反应在酸酐的存在下进行。通常,所述酸酐是乙酸酐、丙酸酐、丁酸酐或琥珀酸酐中的一种。应注意优选乙酸酐。除此之外,酸酐应该以催化量存在,在这点上,可以包括相对于反应物(a)、b))的总摩尔数,亚化学计量量的所述酸酐,但不排除酸酐相对于反应物(a)、b))的总摩尔数以摩尔过量(例如至多20%摩尔过量)存在。
[0073]
该反应也在合适的抗氧化剂存在下进行,基于反应物(a)、b))的总重量,该抗氧化剂通常占至多10重量%或至多5重量%。本文优选的是使用一种或多种空间位阻酚——包括但不限于2,6
‑
二叔丁基
‑4‑
甲基苯酚(bht)和/或丁基化的羟基茴香醚(bha)。
[0074]
虽然不需要存在助催化剂(co
‑
catalyst),但也不排除助催化剂的存在。在一个实施方案中,原酸酯和α
‑
当归内酯之间的反应可以在催化量的强质子酸下进行,所述强质子酸选自h2so4、hno3、hcl、hbr、hi、三氟乙酸(tfa)、h3po4、对甲苯磺酸(p
‑
tsa)和甲磺酸(msa)。
[0075]
上述反应应在无水条件下进行。通过为反应容器提供惰性、干燥的气态覆盖层,可以避免暴露于大气水分。虽然可以使用干燥的氮气、氦气和氩气作为覆盖气体,但在使用普
通氮气作为覆盖层时应采取预防措施,因为此类氮气由于其对水分夹带的敏感性而可能不够干燥;在这里使用之前,氮气可能需要额外的干燥步骤。
[0076]
上述反应可以在溶剂存在下进行。优选惰性溶剂作为溶剂;这些不含与起始化合物反应的反应性基团。惰性、极性、非质子溶剂是特别优选的。如此命名的是例如环醚化合物,特别是四氢呋喃(thf)。
[0077]
反应温度通常为至少40℃,优选至少60℃。虽然反应温度可以为200℃或更高,但优选温度不超过190℃或甚至180℃,以便尤其:保持能工作的的反应器压力;并且,在适用的情况下,在不使催化剂失活或分解的情况下保持足够的催化剂活性。由于反应通常是放热的,因此在进行时可能需要进行一些冷却。
[0078]
工艺压力并不关键:照此,反应可以在低于大气压、大气压或高于大气压的压力下进行,但处于大气压或略高于大气压的压力是优选的。在这方面可以提及100至500mpa或100至200mpa的压力。
[0079]
上述反应的进程可以通过已知技术监测。例如,可以从反应容器中取出样品并使用带有火焰离子化检测(fid)的气相色谱法(gc)进行测试。
[0080]
可以使用本领域已知的方法分离和纯化反应产物。虽然在这方面可以提及萃取、过滤、蒸发、蒸馏和色谱法作为合适的技术,但最方便的是通过蒸馏掉溶剂和任何未反应的起始材料来分离反应产物。
[0081]
外
‑
亚甲基官能化聚合物的形成
[0082]
本发明的第二方面提供了上述定义的单体化合物(efl)的聚合。广义上,聚合通过链增长聚合进行,但特别是可以在阴离子条件下进行:技术人员将选择合适的条件,使得聚合的乙烯基加成途径优于竞争性开环聚合途径。因此,所得均聚物或共聚物(p
‑
efl)在其重复单元中保留了内酯结构。
[0083]
共聚单体
[0084]
如前所述,上述单体(efl)可以被结合到共聚物(p
‑
efl)中。最广泛地,可行的共聚单体是在合适、实用的阴离子聚合条件下提供合理聚合反应速率的那些。
[0085]
在本发明的非限制性和说明性的实施方案中,提供了一种共聚物(p
‑
efl),其包含:
[0086]
a)15重量%至75重量%、优选15重量%至60重量%的至少一种如上文的式(efl)中定义的单体;和
[0087]
b)25重量%至85重量%、优选40重量%至85重量%的至少一种共聚单体。
[0088]
在不旨在与上述说明性实施方案相互排斥的另一个示例性实施方案中,共聚物衍生自以上定义的单体(efl)和至少一种另外的单体,其中所述至少一种另外的单体是选自以下的不提供羰基的烯属不饱和单体:(甲基)丙烯腈;(甲基)丙烯酸烷基酯;(甲基)丙烯酸;乙烯基酯;和乙烯基单体。
[0089]
合适的乙烯基单体包括:1,3
‑
丁二烯;异戊二烯;苯乙烯;二乙烯基苯;杂环乙烯基化合物;和乙烯基卤化物,例如氯丁二烯。优选地,乙烯基单体包括乙烯、苯乙烯、丁二烯和异戊二烯。
[0090]
合适的乙烯基酯包括乙酸乙烯酯、丙酸乙烯酯、叔碳酸乙烯酯和月桂酸乙烯酯。
[0091]
丙烯酸和甲基丙烯酸的合适烷基酯是衍生自c1至c
14
醇的那些并且因此作为非限
制性实例包括:丙烯酸甲酯;甲基丙烯酸甲酯;丙烯酸乙酯;甲基丙烯酸乙酯;丙烯酸正丁酯;甲基丙烯酸正丁酯;丙烯酸2
‑
乙基己酯;甲基丙烯酸2
‑
乙基己酯;丙烯酸异丙酯;甲基丙烯酸羟乙酯;甲基丙烯酸羟丙酯;甲基丙烯酸异丙酯;丙烯酸正丙酯;甲基丙烯酸正丙酯;以及烷烃二醇的二(甲基)丙烯酸酯,诸如1,6
‑
己二醇二丙烯酸酯。
[0092]
聚合方法
[0093]
所存在的单体(efl)和任何共聚单体的阴离子聚合在选自以下的引发剂存在下进行:碱金属有机化合物;碱金属醇盐;碱金属硫醇盐;碱金属氨化物;以及元素周期表第3a族元素的化合物,优选有机铝或有机硼。
[0094]
可以使用的碱金属有机化合物是单
‑
、双
‑
或多官能碱金属烷基、芳基或芳烷基化合物。使用有机锂化合物是有利的,包括但不限于:乙基锂;丙基锂;异丙基锂;正丁基锂;仲丁基锂;叔丁基锂;苯基锂;二苯基己基锂;六亚甲基二锂;丁二烯基锂;异戊二烯基锂;聚苯乙烯基锂;1,4
‑
二锂丁烷(1,4
‑
dilithiobutane);1,4
‑
二锂
‑2‑
丁烯;和1,4
‑
二锂苯(1,4
‑
dilithiobenzene)。
[0095]
可单独或混合使用的碱金属醇盐是锂、钠或钾的脂族、芳族或芳脂族醇盐。实例是锂、钠或钾的甲醇盐、乙醇盐、正丙醇盐、异丙醇盐、正丁醇盐、仲丁醇盐、叔丁醇盐、正戊醇盐、异戊醇盐、己醇盐、戊醇盐、3,7
‑
二甲基
‑3‑
辛醇盐、苯酚盐(phenoxide)、2,4
‑
二叔丁基苯酚盐、2,6
‑
二叔丁基苯酚盐、3,5
‑
二叔丁基苯酚盐、2,4
‑
二叔丁基
‑4‑
甲基苯酚盐和三甲基硅烷酸盐。优选使用脂族醇盐,特别是钠、钾或锂的甲醇盐、乙醇盐、正丙醇盐、异丙醇盐、正丁醇盐、仲丁醇盐和叔丁醇盐。
[0096]
可单独或混合使用的碱金属硫醇盐是锂、钠或钾的脂族、芳族或芳脂族硫醇盐。实例是锂、钠或钾的甲基硫化物、乙基硫化物、丁基硫化物、己基硫化物、癸基硫化物、十二烷基硫化物、硬脂基硫化物、噻吩氧化物、甲苯基硫化物、环己基硫化物,或1,2
‑
二巯基乙烷二锂。优选在烷基链中具有8至18个碳原子的脂族硫醇盐。
[0097]
可以单独或混合使用的碱金属氨化物是氨或者具有脂族、芳族或芳脂族取代基的伯胺或仲胺的锂盐、钠盐或钾盐。合适的氨化物的实例是氨基锂、n
‑
甲基氨基锂、n
‑
乙基氨基锂、n
‑
丙基氨基锂、n
‑
丁基氨基锂、n
‑
戊基氨基锂、n
‑
苯基氨基锂或相应的钠盐或钾盐;n
‑
二甲基氨基锂、n
‑
二乙基氨基锂、n
‑
二丙基氨基锂、n
‑
二丁基氨基锂、n
‑
二戊基氨基锂、n
‑
锂
‑
(n,n
‑
双
‑
三甲基甲硅烷基)氨化物、n
‑
二环己基氨基锂、n
‑
锂
‑
n
‑
甲基苯胺、n
‑
锂
‑
n
‑
乙基苯胺、n
‑
吗啉锂、n
‑
二苯基氨基锂、n
‑
哌啶锂或n
‑
咪唑锂。特别优选仲脂族胺的盐,非常特别优选n
‑
二异丙基氨基锂。
[0098]
可以使用的有机铝或有机硼是式r3al或r3b的那些,其中基团r各自彼此独立地为氢、卤素、c1
‑
c18
‑
烷基或c6
‑
c18
‑
芳基。优选的有机铝是三烷基铝,诸如三甲基铝、三乙基铝、三异丁基铝、三正丁基铝、三异丙基铝、三正己基铝、氢化二乙基铝、氢化二异丁基铝或异戊二烯基铝。特别优选使用三异丁基铝。
[0099]
设想可以使用通过烷基
‑
或芳基铝化合物或与醇盐、硫醇盐、硫化物、氨化物(amides)、酰亚胺、氮化物或磷化物络合的那些的部分或完全水解、醇解、氨解、硫解、膦解或氧化而形成的有机铝。这种带有杂取代基的化合物的实例包括但不限于:二乙基n,n
‑
二丁基氨基铝;二乙基乙醇铝;二异丁基乙醇铝;二异丁基
‑
(2,6
‑
二叔丁基
‑4‑
甲基
‑
苯氧基)铝;甲基铝氧烷;异丁基化甲基铝氧烷;异丁基铝氧烷;四异丁基二铝氧烷;双(二异丁基)氧
化铝;二乙基甲氧基硼;三甲基环三硼氧烷;和2
‑
苯基
‑
1,3,2
‑
二氧杂硼烷。
[0100]
合适引发剂的其他实例包括:烷醇铝,诸如三甲醇铝、三乙醇铝、三丙醇铝和三丁醇铝;以及硼酸三烷基酯。优选使用铝化合物,尤其是具有氧代基团或醇盐基团的那些。非常特别优选使用二乙基乙醇铝、二异丁基乙醇铝、甲基铝氧烷、丙醇铝和三仲丁醇铝。
[0101]
所用引发剂的量没有特别限制,但其基于100重量份的单体,通常为0.0001至5重量份,优选0.05至1重量份。
[0102]
此外,聚合可以在溶液中或在没有溶剂的熔体中进行。当使用时,用于聚合的合适溶剂应该是能够在25℃下溶解至少1重量%、优选超过10重量%的聚合物的非反应性有机液体。二氯甲烷和四氢呋喃(thf)可作为示例性溶剂提及。
[0103]
在某些实施方案中,阴离子聚合过程在路易斯酸的存在下进行。用于本发明的聚合过程的优选路易斯酸的特征是“非质子”:它们是不能作为质子(h )源起作用的路易斯酸。用于本发明目的的特别优选的路易斯酸包括选自铝、锰、铁、钴、硼、铁、钛、锡、铬、镁、钒、铪、锆和锌的元素的卤化物。
[0104]
在本发明的均聚和共聚过程中,(非质子)路易斯酸的量应调节为使得催化剂的活性(如在给定温度下每单位时间反应的单体的重量所测量的)与在相同条件下不存在路易斯酸的情况下的催化剂活性相比,降低不超过20%:在这方面,使用路易斯酸:催化剂重量比在0.1至1.0范围内是有利的。
[0105]
虽然当然无意排除如美国专利号5,777,177和5,689,012中描述的聚合的间歇或连续进行,但聚合反应最适合以半间歇方法进行。
[0106]
聚合反应可以在适合于下述压力和温度的任何类型的容器中进行。在优选的半间歇方法中,容器应具有在反应期间通过其引入单体的一个或多个入口。在不太理想的连续方法中,反应器容器应包含通过其可以排出部分聚合的反应混合物的一部分的至少一个出口。除此之外,用于连续或半间歇操作的示例性容器包括但不限于:管式反应器;环管反应器;和连续搅拌釜反应器(ctsr)。当然,任何反应器都应该配备有提供或移除热量使得聚合混合物的温度可以保持在所需的范围内的装置:无意限制这种构件,但实例包括用于热流体的包壳和内部和/或外部加热器。
[0107]
在聚合过程开始时,将引发剂和任选存在的路易斯酸加入反应容器中。在优选的半间歇方法中,引发剂可以在不存在单体的情况下,在50℃至220℃,例如75℃至180℃的温度下进行初步加热步骤。该初步加热步骤在惰性气氛中进行,并且通常但不一定在低于大气压的压力下进行。此外,初步加热通常进行至少10分钟的时间段:为了说明的目的,可以提及10至30分钟的时间段。
[0108]
单体(efl)的均聚、满足通式(efl)的两种或多种单体的共聚以及单体(efl)与共聚单体的共聚应在无水条件下和不存在任何具有活性氢原子的化合物的情况下(除了起始化合物的有意加入之外)进行。通过为反应容器提供惰性、干燥的气态覆盖层,可以避免暴露于大气水分。虽然可以使用干燥的氮气、氦气和氩气作为覆盖气体,但在使用普通氮气作为覆盖层时应采取预防措施,因为此类氮气由于其对水分夹带的敏感性而可能不够干燥;在这里使用之前,氮气可能需要额外的干燥步骤。
[0109]
聚合温度通常为至少25℃,优选至少50℃。虽然反应温度可以为200℃或更高,但优选温度不超过200℃、175℃或甚至150℃,以便尤其:保持能工作的反应器压力;使聚合物
降解的速度和挥发性杂质或其他副产物的伴随形成最小化;以及如果适用,在不使催化剂失活或分解的情况下保持足够的催化剂活性。在50℃至150℃的典型所需聚合温度范围内,溶剂类型、搅拌速率和压力将决定反应时间,但1至100小时的时间是标准的。
[0110]
工艺压力并不关键:因此,聚合反应可以在低于大气压、大气压或高于大气压的压力下进行,但处于大气压或略高于大气压的压力是优选的。在这方面可以提及100至500mpa或100至200mpa的压力。
[0111]
可以使用本领域已知的方法分离和纯化反应产物。虽然在该上下文中可以提及萃取、蒸发、蒸馏和色谱法作为合适的技术,但最方便的是通过在减压下蒸馏掉溶剂和任何未反应的起始材料来分离反应产物。
[0112]
如果打算在生产时储存(任选纯化的)反应产物,则聚合物应置于具有气密和防潮密封件的容器中。
[0113]
在上述聚合过程中衍生的均聚物或共聚物(p
‑
efl)可以具有:至少2500g/mol,例如10000g/mol至150000g/mol、优选10000至100000g/mol的数均分子量(mn),如通过凝胶渗透色谱法(gpc)在四氢呋喃中使用聚苯乙烯标样测定;ii)50℃至200℃,例如100℃至200℃的玻璃化转变温度(tg);和iii)1.1至2.0,例如1.10至1.90,优选1.10至1.80的多分散指数(pdi)。
[0114]
均聚物和共聚物(p
‑
efl)的聚合物衍生物
[0115]
i)开环聚合
[0116]
本发明的聚合物(p
‑
efl)中的内酯官能团可用于调节选自以下的至少一种单体的开环聚合:环状碳酸酯;环状酸酐;草酸盐;和具有5
‑
、6
‑
和/或7
‑
元环的环酯。特别地,聚合物(p
‑
efl)可以在与至少一种选自以下的单体的开环聚合反应中作为反应物大分子单体存在:丙交酯;乙交酯;ε
‑
己内酯;对二氧环己酮;三亚甲基碳酸酯;1,4
‑
二氧杂环庚烷
‑2‑
酮;1,5
‑
二氧杂环庚烷
‑2‑
酮;γ
‑
丁内酯;α
‑
亚甲基
‑
γ
‑
丁内酯;γ
‑
甲基
‑
α
‑
亚甲基
‑
γ
‑
丁内酯;α
‑
溴
‑
γ
‑
丁内酯;α
‑
羟基
‑
γ
‑
丁内酯;α
‑
乙酰基
‑
γ
‑
丁内酯;螺环
‑
γ
‑
丁内酯;γ
‑
戊内酯;α
‑
当归内酯;和β
‑
当归内酯。衍生的共聚物可以是嵌段共聚(酯)或无规共聚(酯)。
[0117]
虽然没有具体意图限制本发明中采用的开环聚合的机理,并且虽然因此不严格排除通过阴离子途径、经由碱性催化剂进行的环状单体的开环聚合,但在本文中优选通过阳离子途径、经由酸催化进行所述聚合。概括地说,任何合适的酸性开环聚合催化剂都可以在本文中使用,并且同样地,可以使用催化剂的混合物。在该上下文中路易斯酸和布朗斯台德酸可以适用,但后者是优选的,因为它们往往在低于150℃的温度下有效,并且通常在50℃至100℃的温度下有效。
[0118]
合适的路易斯酸的实例包括但不限于:bf3;alcl3;t
‑
bucl/et2alcl;cl2/bcl3;albr3;albr3.ticl4;i2;sbcl5;wcl6;alet2cl;pf5;vcl4;aletcl2;bf3et2o;pcl5;pcl3;pocl3;ticl6;和sncl4。
[0119]
可任选地布置在固体无机载体上的布朗斯台德酸或质子酸型催化剂的实例包括但不限于:hcl;hbr;hi;h2so4;hclo4;对甲苯磺酸;三氟乙酸;以及全氟烷烃磺酸诸如三氟甲磺酸(或三氟甲磺酸(triflic acid),cf3so3h)、c2f5so3h、c4f9so3h、c5f
11
so3h、c6f
13
so3h和c8f
17
so3h。这些强酸中最优选的是三氟甲磺酸(三氟甲磺酸(triflic acid),cf3so3h)。
[0120]
基于要聚合的单体的总重量,用于所述开环聚合的催化剂通常可以以按重量计1
至1000ppm的浓度使用。优选使用按重量计5至150ppm,最优选5至50ppm。当单体和催化剂接触的温度升高时,催化量可能减少。
[0121]
开环聚合可方便地在10℃至150℃范围内的温度下进行。然而,优选地,温度范围为20℃或50℃至100℃,因为避免高温可限制挥发性单体由于其较低沸点而从反应混合物中损失。
[0122]
工艺压力并不关键。因此,聚合反应可以在低于大气压、大气压或高于大气压的压力下进行,但处于大气压或高于大气压的压力是优选的。
[0123]
重要的是,该反应应当在无水条件下和不存在任何具有活性氢原子的化合物的情况下进行。通过为反应容器提供惰性、干燥的气态覆盖层,可以避免暴露于大气水分。虽然可以使用干燥的氮气、氦气和氩气作为覆盖气体,但在使用普通氮气作为覆盖层时应采取预防措施,因为此类氮气由于其对水分夹带的敏感性而可能不够干燥;在这里使用之前,氮气可能需要额外的干燥步骤。
[0124]
反应的持续时间取决于体系达到平衡所需的时间。然而,同样地,可以理解,可以通过恰好在所需的时间停止平衡来获得所需的产物:例如,可以通过分析随时间的粘度或通过使用气相色谱分析单体转化率来监测反应,并且在达到所需的粘度或单体转化率时停止反应。撇开这些考虑不谈,聚合反应通常发生0.5至72小时,更常见的是1至30或1至20小时。聚合反应结束时存在于反应混合物中的酸催化剂可以容易地中和以稳定反应产物。
[0125]
聚合完成后,可以通过例如过滤、错流过滤或离心除去任何固体悬浮化合物。此外,可以使用本领域已知的方法对聚合的输出物进行后处理,以分离和纯化羟基官能化的聚酯。在这方面可以提及萃取、蒸发、蒸馏和色谱法作为合适的技术。分离后,已发现羟基官能化的聚酯的典型产率为至少40%,通常为至少60%。
[0126]
如通过凝胶渗透色谱法(gpc)在四氢呋喃中使用聚苯乙烯标样测量,由该开环聚合方法衍生的聚酯可具有至少5000g/mol,优选10000g/mol至200000g/mol的分子量(mn)。此外,聚合物的特征可以在于1.0至2.5,优选1.0至2.0范围内的多分散指数。
[0127]
ii)聚酯形成
[0128]
在第二示例性实施方案中,本发明的聚合物(p
‑
efl)可用作酯化中的大分子单体,其中所得共聚物包含衍生自能够形成酯键的至少两种共聚单体的非乳酰基单元。更特别地,那些共聚单体包括:i)至少一种二醇;(ii)至少一种二元羧酸或其成酯衍生物。
[0129]
用于上下文中的合适二醇(i)包括饱和和不饱和脂族和脂环族二羟基化合物以及芳族二羟基化合物。这些二醇优选具有250道尔顿或更小的分子量。当在本文中使用时,术语“二醇”应被解释为包括其等效的成酯衍生物,但前提是分子量要求仅涉及二醇而不涉及其衍生物。示例性成酯衍生物包括二醇的乙酸酯,以及例如对于乙二醇而言环氧乙烷或者碳酸亚乙酯。
[0130]
优选的二醇是具有2至10个碳原子的那些。作为这些二醇的实例,可以提及:乙二醇;丙二醇;1,3
‑
丙二醇;1,2
‑
丁二醇;2
‑
甲基丙二醇;1,3
‑
丁二醇;1,4
‑
丁二醇;2,3
‑
丁二醇;新戊二醇;己二醇;癸二醇;己二醇;环己烷二甲醇;间苯二酚;和对苯二酚。可以使用这些二醇的混合物,但在这方面,通常优选的是至少约60摩尔%、优选至少80摩尔%(基于总二醇含量)是相同的二醇。
[0131]
在优选的实施方案中,二醇选自:乙二醇;丙二醇;1,3
‑
丙二醇;1,2
‑
丁二醇;1,3
‑
丁二醇;1,4
‑
丁二醇;2,3
‑
丁二醇;新戊二醇;己二醇;环己烷二甲醇;以及它们的混合物。最优选地,二醇是乙二醇或新戊二醇。
[0132]
适用于上述上下文的二羧酸(ii)包括脂族、脂环族和/或芳族二元羧酸。这些酸应优选具有小于300道尔顿的分子量。本文所用的术语“二元羧酸”包括具有两个官能羧基的二元羧酸的等价物,其在与二醇类和二醇反应形成聚酯时的表现基本上类似于二元羧酸。这些等价物包括酯和成酯反应性衍生物诸如酸性卤化物和酸酐,但前提是上述分子量优选涉及酸而不是其等价的酯或成酯衍生物。因此,包括分子量大于300道尔顿的二羧酸的酯或分子量大于300道尔顿的二元羧酸的酸等价物,前提是该酸具有低于300道尔顿的分子量。此外,二元羧酸可含有基本上不干扰聚合物形成和本发明聚合物的使用的任何取代基或组合。
[0133]
优选的二元羧酸是选自具有总计2至16个碳原子的烷基二元羧酸和具有总计8至16个碳原子的芳基二羧酸的那些。代表性的烷基二元羧酸包括:戊二酸;己二酸;庚二酸;琥珀酸;癸二酸;壬二酸;和丙二酸。这里可以提及优选己二酸。代表性的芳基二元羧酸包括:对苯二甲酸;邻苯二甲酸;间苯二甲酸;所述酸的二甲基衍生物;以及它们的混合物。
[0134]
含有本发明的均聚物和共聚物(p
‑
efl)的组合物
[0135]
本发明的聚合物(p
‑
efl)被认为是通用的,因此具有多种用途。例如,含内酯的聚合物可用于制备与具有阳离子部分的试剂(包括治疗剂诸如肽)的离子复合物。这些聚合物中存在的内酯环也可以被碱金属类氢氧化物打开以形成相应羟基羧酸的碱金属盐。此外,含有内酯基团的聚合物可以通过可与内酯反应的多官能化合物进行交联。在这方面,特别需要多官能胺。
[0136]
预期本发明的官能化聚合物本身可用作涂层组合物、密封剂组合物或粘合剂组合物的可固化、可交联或其他反应性组分。
[0137]
在本发明的一个重要实施方案中,提供了一种具有两种单独的反应性组分的组合物,当它们混合在一起时形成经历固化或硬化的反应性混合物,所述双组分组合物包含:
[0138]
i)在第一组分中的所述聚合物(p
‑
efl);和
[0139]
ii)在第二组分中的未取代的或羟基取代的单烷基胺、二烷基胺或三烷基胺。
[0140]
优选地,烷基胺是伯胺和仲胺中的至少一种。更优选地,烷基胺是伯胺。独立地或另外地,优选的是所述第二组分包含未取代的或羟基取代的单
‑
、二
‑
或三
‑
(c1‑
c
12
)烷基胺。此外,同样独立于或附加于这些优选条件,所述组合物的特征可以在于组分(i)中的内酯基团与组分(ii)中的胺基团的摩尔比在0.8:1至2.5:1的范围内。
[0141]
组分(ii)中的烷基胺的合适实例包括但不限于:甲基胺、二甲基胺或三甲基胺;乙胺、二乙胺或三乙胺;乙醇胺、二乙醇胺或三乙醇胺;三
‑
(羟甲基)
‑
甲胺;2
‑
羟基
‑
叔丁胺;n,n
‑
二甲基
‑
n
‑
(2
‑
羟乙基)
‑
胺;n
‑
甲基
‑
d
‑
葡糖胺;二异丙基乙胺;和乙基二异丙胺。
[0142]
本发明中获得的包含均聚物或共聚物(p
‑
efl)的所述组合物(诸如涂层组合物、密封剂组合物或粘合剂组合物)通常将进一步包含可赋予这些组合物改进性能的佐剂和添加剂。例如,佐剂和添加剂可以赋予以下一种或多种:改善的弹性;改善的弹性恢复;更长可能的处理时间;更快的固化时间;以及更低的残余粘性。此类佐剂和添加剂之中包括催化剂、增塑剂、稳定剂、抗氧化剂、填料、反应性稀释剂、干燥剂、粘合促进剂和uv稳定剂、杀真菌剂、阻燃剂、流变佐剂、彩色颜料或色浆,和/或任选地在小程度上还有溶剂。
[0143]
用于本发明目的的“增塑剂”是降低组合物粘度并因此促进其可加工性的物质。基于组合物的总重量,在本文中增塑剂可以占至多40重量%或至多20重量%,并且优选地选自:聚二甲基硅氧烷(pdms);二氨基甲酸酯(diurethanes);单官能、直链或支链c4
‑
c16醇的醚,诸如cetiol oe(可从cognis deutschland gmbh,d
ü
sseldorf获得);松香酸酯、丁酸酯、硫代丁酸酯、乙酸酯、丙酸酯和柠檬酸酯;基于硝基纤维素和聚乙酸乙烯酯的酯;脂肪酸酯;二羧酸酯;带oh基团的或环氧化的脂肪酸的酯;乙醇酸酯;苯甲酸酯;磷酸酯;磺酸酯;偏苯三酸酯;环氧化增塑剂;聚醚增塑剂,诸如封端的聚乙二醇或聚丙二醇;聚苯乙烯;烃类增塑剂;氯化石蜡;以及它们的混合物。注意,原则上,邻苯二甲酸酯可用作增塑剂,但由于它们的毒理学潜力,这些不是优选的。优选地,增塑剂包含一种或多种聚二甲基硅氧烷(pdms)或由其组成。
[0144]
用于本发明目的的“稳定剂”应理解为抗氧化剂、uv稳定剂或水解稳定剂。在本文中,基于组合物的总重量,稳定剂可以占至多10重量%或至多5重量%。适用于本文的稳定剂的标准市售实例包括:空间位阻酚;硫醚;苯并三唑;二苯甲酮;苯甲酸酯;氰基丙烯酸酯;丙烯酸酯;受阻胺光稳定剂(hals)型胺类;磷;硫;以及它们的混合物。
[0145]
如上所述,根据本发明的组合物可以另外包含填料。此处合适的是例如白垩、石灰粉、沉淀和/或热解硅酸、沸石、膨润土、碳酸镁、硅藻土、氧化铝、粘土、滑石、氧化钛、铁氧化物、氧化锌、沙子、石英、燧石、云母、玻璃粉和其他磨碎的矿物质。也可以使用有机填料,特别是炭黑、石墨、木纤维、木粉、锯末、纤维素、棉花、纸浆、棉花、木屑、切碎的稻草、谷壳、磨碎的胡桃壳和其他切碎的纤维。也可以加入短纤维,诸如玻璃纤维、玻璃长丝、聚丙烯腈、碳纤维、kevlar纤维或聚乙烯纤维。铝粉同样适合作为填料。
[0146]
热解和/或沉淀硅酸有利地具有10m2/g至90m2/g的bet表面积。当使用它们时,它们不会引起根据本发明的组合物的粘度的任何额外增加,但确实有助于增强固化的组合物。
[0147]
同样可以想到使用具有较高bet表面积,有利地为100m2/g至250m2/g,特别是110m2/g至170m2/g的热解和/或沉淀硅酸作为填料:由于较大的bet表面积,用较小重量比例的硅酸实现了强化固化组合物的效果。
[0148]
也适合作为填料的是具有矿物壳或塑料壳的中空球。例如,这些可以是中空玻璃球,其可以商品名glass商购获得。可以使用基于塑料的中空球,诸如或并描述于ep 0 520 426b1中:它们由无机或有机物质组成,并且各自具有1mm或更小,优选500μm或更小的直径。
[0149]
赋予组合物触变性的填料对于许多应用可能是优选的:此类填料也被描述为流变佐剂,例如氢化蓖麻油、脂肪酸酰胺或可溶胀塑料诸如pvc。
[0150]
基于组合物的总重量,存在于本发明组合物中的填料的总量优选为1至80重量%,更优选为5至60重量%。可固化组合物的所需粘度通常由添加的填料总量决定,并且认为为了易于从合适的分配设备(诸如管)中挤出,可固化组合物应具有3000至150,000mpas,优选40,000至80,000mpas,或甚至50,000至60,000mpas的粘度。
[0151]
合适的颜料的实例是二氧化钛、铁氧化物或炭黑。
[0152]
为了更进一步延长保存期限,通常建议通过使用干燥剂使本发明的组合物相对于水分渗透进一步稳定。有时还需要通过使用反应性稀释剂来降低用于特定应用的根据本发明的粘合剂或密封剂组合物的粘度。基于组合物的总重量,存在的反应性稀释剂的总量通
常为至多15重量%、优选为1至5重量%。
[0153]
以下实施例是对本发明的说明,并不以任何方式限制本发明的范围。
实施例
[0154]
实施例中使用了以下材料:
[0155]
α
‑
当归内酯:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
购自sigma aldrich的4
‑
羟基
‑3‑
戊烯酸γ
‑
内酯
[0156]
原甲酸三异丙酯:
ꢀꢀꢀꢀꢀꢀꢀ
cas号4447
‑
60
‑
3,购自sigma aldrich
[0157]
原甲酸三乙酯:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
cas号122
‑
51
‑
0,购自sigma aldrich
[0158]
原甲酸三甲酯:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
cas号149
‑
73
‑
5,购自sigma aldrich
[0159]
原甲酸三丁酯:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
cas号588
‑
43
‑
2,购自sigma aldrich
[0160]
原甲酸三丙氧酯:
ꢀꢀꢀꢀꢀꢀꢀ
cas号621
‑
76
‑
1,购自sigma aldrich
[0161]
二乙基乙烯基原甲酸酯: cas号34712
‑
46
‑
4,购自sigma aldrich
[0162]
原甲酸三
‑
十八烷基酯:
ꢀꢀ
cas号17671
‑
28
‑
2,购自sigma aldrich
[0163]
原甲酸三戊酯:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
cas号637
‑
42
‑
3,购自sigma aldrich
[0164]
实施例1:2
‑
乙氧基亚甲基
‑
α
‑
当归内酯(etomal)的合成
[0165]
在氩气气氛和搅拌下,首先将179ml原甲酸三乙酯(1.07mol)加入到在1l圆底烧瓶中的203ml乙酸酐(2.15mol)中。然后在氩气气氛下将64.4mlα
‑
当归内酯(0.72mol)和150mg 2,6
‑
二叔丁基对甲酚(bht)添加到烧瓶中。将混合物在回流条件(t=110℃
‑
130℃)和氩气气氛下搅拌约7小时。
[0166]
在反应期间分别在0、60、120、260和410分钟时取出五个样品。将100μl和250μl之间的这些样品加入含有用作内标的30μl十二烷的色谱(gc)小瓶中;将1.6ml甲苯加入所有gc小瓶中,并且通过gc
‑
fid分析所得溶液。α
‑
当归内酯的相对量通过相对于内标和相对于用于gc分析的反应混合物的量归一化的积分面积之间的比率来计算。
[0167]
反应完成后,将混合物升至室温并在氩气气氛下储存过夜。随后在真空下蒸馏混合物。
[0168]
在约120℃的温度和0.4毫巴的压力下收集黄色液体形式的77.25g含有所需产物etomal的馏分。gc
‑
fid分析表明该馏分含有约90%的etomal(收率=61%)。在氩气气氛下将25ml二乙醚和20ml己烷加入到收集的馏分中。将混合物浸入乙醇浴中,通过向浴中加入干冰来缓慢降低温度。沉淀出白色结晶材料,将后者在大气条件下过滤,并用戊烷洗涤数次。最后,将产物在真空下干燥过夜并在惰性条件下储存。总收率为约40%。
[0169]
产物通过核磁共振(nmr)表征如下:
[0170][0171]1h nmr(300mhz,cd2cl2,δ):=7.18(q,6j=0.75hz,h6,1h),5.80(m,h3,1h),4.16(q,3j=7.05hz,h7,2h),2.04(dd,4j=1.44hz,6j=0.75hz,h5,3h),1.34(t,3j=7.07hz,h8,3h)ppm。
[0172]
13
c nmr(100mhz,cd2cl2,δ):171.06(c1),154.17(c6),151.54(c4),107.86(c2),
99.22(c3),71.93(c7),15.42(c8),14.31(c5)ppm。
[0173]
实施例2:etomal的阴离子聚合
[0174][0175]
方案:用阴离子引发剂的etomal的聚合
[0176]
实施例2.1:使用异丙醇钠(naipro)作为催化剂的聚
‑
etomal的合成和表征
[0177]
2.1.1合成
[0178]
在惰性条件下,将800mg etomal溶解在1ml甲苯中。在惰性气氛下,在60℃下将溶液加入4mg naipro和1ml甲苯的混合物中。使用额外的甲苯(1ml)将etomal定量转移到反应混合物中。
[0179]
添加etomal后不久,凝胶形式的固体材料开始沉淀。将反应在60℃下放置2小时。然后将约1ml的hcl(0.1m)水溶液加入反应schlenk容器中。然后将固体用甲苯洗涤数次。然后将洗涤过的固体溶解在二氯甲烷中并通过sio2短柱过滤。最后在真空下干燥过夜。总收率通常高于90%。
[0180]
2.1.2聚
‑
etomal的nmr表征
[0181]
产物通过核磁共振(nmr)表征如下:
[0182][0183]1h nmr(300mhz,cd2cl2,δ):=7.43(bs,h3,1h),4.29(bs,h6,1h),3.43(bs,h7,2h),1.45(bs,h5,3h),1.13(bs,h8,3h)ppm。
[0184]
13
c nmr(75mhz,cd2cl2,δ):171.35(c1),156.22(c3),131.78(c2),88.17(c4),76.80(c6),66.77(c7),20.79(c5),15.27(c8)ppm。
[0185]
2.1.3聚
‑
etomal的分子量表征
[0186]
所附的图1说明了当使用聚苯乙烯校准标准物通过凝胶渗透色谱法(gpc)在四氢呋喃(thf)中分析本实施例的聚合物的分子量时获得的双峰分子量分布。
[0187]
实施例2.2:使用异丙醇铝[al[ipro)3]作为催化剂的聚
‑
etomal的合成
[0188]
在惰性条件下,将900mg etomal溶解在1ml甲苯中。在惰性气氛下,在60℃下将溶液加入在1ml甲苯中的al(ipro)3(10摩尔%)中。使用额外的甲苯(1
‑
3ml)将etomal定量转移到反应混合物中。
[0189]
添加etomal后不久,凝胶形式的固体材料开始沉淀。将反应在60℃下放置2小时。然后使用hcl水溶液(0.1m)淬灭反应。将固体在bruckner过滤器上用甲苯、二氯甲烷和二乙醚洗涤数次。最后在真空下干燥过夜。总收率通常高于80%。通过nmr分析鉴定聚合物形成。
[0190]
实施例2.3:通过使用丁基锂作为催化剂来合成聚
‑
etomal
[0191]
在惰性条件下,将170mg etomal溶解在2ml thf中。在惰性气氛下将溶液加热至60℃。然后在搅拌下将一定体积的1.6m buli在己烷中的溶液(相当于1摩尔%对etomal)加入单体溶液中。1小时后用hcl水溶液(1m)猝灭反应。然后将二氯甲烷加入混合物中,并通过硅藻土过滤所得溶液。最后在真空下干燥过夜。通过nmr分析鉴定聚合物形成。
[0192]
实施例3:2
‑
异丙氧基亚甲基
‑
α
‑
当归内酯(
i
promal)的合成
[0193]
在乙酸酐(65.8g,645mmol)和2,6
‑
二叔丁基对甲酚(bht,0.060g,0.27mmol)存在下,将原甲酸三异丙酯(61.1g,321mmol)与α
‑
当归内酯(21.2g,216mmol)在回流条件下反应7小时。
[0194][0195]
在反应期间取出反应混合物的五个部分样品,并加入含有用作内标的30μl十二烷的气相色谱(gc)小瓶中;将1.6ml甲苯加入所有gc小瓶中,并且通过gc
‑
fid分析所得溶液。α
‑
当归内酯的相对量通过相对于内标和相对于用于gc分析的反应混合物的量归一化的积分面积之间的比率来计算。
[0196]
反应完成后,在减压下蒸馏反应混合物,以获得浅黄色油状物形式的
i
promal(12.1g,33.3%收率),该产物表征如下:
[0197]1h nmr(400mhz,cdcl3)δ7.22(s,1h),5.78(s,1h),4.28(sep,j=4hz,1h),2.03(s,3h),1.31(d,6h)ppm。
[0198]
13
c nmr(100mhz,cdcl3)δ171.5,153.3,151.2,107.9,33.5,79.5,22.1,14.5ppm。
[0199]
实施例4:其他合成
[0200]
使用以下原甲酸酯((xo)3ch)以≥25%的收率可行地重复进行上述合成2
‑
异丙氧基亚甲基
‑
α
‑
当归内酯(
i
promal)的方法:原甲酸三甲酯(r1=r2=r3=me);原甲酸三丁酯(r1=r2=r3=bu);原甲酸三丙酯(tripropylorthoformate)(r1=r2=r3=npr);二乙基乙烯基原甲酸酯(r1=r2=et,r3=ch2=ch2);原甲酸三
‑
十八烷基酯(r1=r2=r3=c
18
h
37
);和原甲酸三戊酯(r1=r2=r3=c5h
11
)。
[0201]
鉴于前述描述和实施例,本领域技术人员将明白在不脱离权利要求的范围的情况下可以对其进行等效修改。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。