一种氯代丁二酰亚胺和氨水与1
‑
甲基
‑
哌嗪一步合成1
‑
氨基
‑4‑
甲基哌嗪的方法
技术领域
1.本发明属于医药中间体合成工艺领域,具体涉及一种氯代丁二酰亚胺(ncs)在氢氧化钠作为强碱性试剂,在明胶存在下低温搅拌反应,经醚类溶剂萃取后滴加1
‑
甲基
‑4‑
哌嗪溶液,低温至室温搅拌反应,最后通过减压精馏分离获得目标产物1
‑
氨基
‑4‑
甲基哌嗪的绿色合成方法。
背景技术:
2.利福平是一种很好的抗痨药,认为现在抗痨治疗已进入利福平时代,并认为过去要手术治疗的结核病,有了利福平完全可以不需手术而把病情控制下来。利福平的结构式如下:
[0003][0004]1‑
氨基
‑4‑
甲基哌嗪是一种n
‑
氨基环状化合物,是合成利福平的重要中间体,结构式如下:
[0005][0006]1‑
甲基
‑
哌嗪经亚硝化后再还原是1
‑
氨基
‑4‑
甲基哌嗪主要制备方法。还原步骤最为常用的为锌酸还原,其中使用的最多的为醋酸和锌体系,auelbek ov,s.a.;mirzaabdullaev,a.b.等1985年在pharmaceutical chemistry journ al发表文章“synthesis and antiviral activity of gossypol derivatives”报道了醋酸条件下锌还原1
‑
甲基
‑4‑
亚硝基哌嗪制备1
‑
氨基
‑4‑
甲基哌嗪;2018年wu,yachuang和zhao,yan fang等在european journal of medicinal chemistry发表的文章“synthesis and antibacterial activity evaluation of novel biaryloxa zolidinone analogues containing a hydrazone moiety as promising antibacteri al agents“使用的锌酸还原法合成1
‑
氨基
‑4‑
甲基哌嗪;沈阳药科大学赵燕芳等申请的专利cn107721943“含联芳基腙结构的噁唑烷酮类化合物及基制备方法“中也是采用的锌酸还原法制备1
‑
氨基
‑4‑
甲基哌嗪。此工艺成熟但要生成大量的氧化锌和醋酸锌废渣,对环境不友好。
[0007]
[0008]
我们研究团队于2018年在chemistry of heterocyclic compounds发表文章“selective reduction of n
‑
nitroso aza
‑
aliphatic cyclic compounds to the corresponding n
‑
amino products using zinc dust in co2–
h2o medium”也是首先亚硝化,然后采用锌酸还原法制备1
‑
氨基
‑4‑
甲基哌嗪。其特点是采用碳酸代替醋酸,相对绿色,但也要生成大量碳酸锌和氧化锌废渣,对环境同样不友好。
[0009][0010]
氢化铝锂还原法也是常见的1
‑
甲基
‑4‑
亚硝基哌嗪制备1
‑
氨基
‑4‑
甲基哌嗪的方法。valenta,vladimir和holubek,jiri等在collection of czechoslovak chemical communications发表的文章“synthesis of 4
‑
methoxyphenoxyacetic and 3,4,5
‑
trimethoxyphenoxyacetic acid amides and hydrazides as potential neurotropic and cardiovascular agents”采用醚类溶剂,以氢化铝锂还原1
‑
甲基
‑4‑
亚硝基哌嗪制备1
‑
氨基
‑4‑
甲基哌嗪。氢化铝锂还原同样要生成大量含铝和锂废渣,同时需要无水操作,非常繁琐,不利于工业化生产。
[0011][0012]
我们的专利zl202010393030.6“一种催化加氢合成1
‑
氨基
‑4‑
甲基哌嗪的方法”介绍了一种1
‑
甲基
‑4‑
亚硝基哌嗪在pd/fe3o4‑
feo催化下氢化合成1
‑
氨基
‑4‑
甲基哌嗪的绿色合成方法。它的特征在于1
‑
甲基
‑4‑
亚硝基哌嗪在水、有机溶剂和催化剂三相体系下,一定的温度内进行氢化反应,最后通过减压蒸馏分离获得目标产物1
‑
氨基
‑4‑
甲基哌嗪。其合成路线如下:
[0013][0014]
上述所有方法均需要制备n
‑
亚硝基产物,而n
‑
亚硝基类化合物是一类具有基因毒性的中间体,其残留会大大影响药品的安全性。研究一种由1
‑
甲基
‑
哌嗪一步不经亚硝中间体直接生成1
‑
氨基
‑4‑
甲基哌嗪的方法显得非常必要。
技术实现要素:
[0015]
本发明是一种氯代丁二酰亚胺(ncs)在氢氧化钠作为强碱性试剂,在明胶存在下低温搅拌反应,经醚类溶剂萃取后滴加1
‑
甲基
‑4‑
哌嗪溶液,低温至室温搅拌反应,最后通过减压精馏分离获得目标产物。由1
‑
甲基
‑4‑
哌嗪无需经亚硝化步骤一步反应制备1
‑
氨基
‑4‑
甲基哌嗪。其合成路线如下:
[0016][0017]
本发明涉及一种由1
‑
甲基
‑4‑
哌嗪无需经亚硝化步骤一步反应制备1
‑
氨基
‑4‑
甲基哌嗪的绿色合成方法,该方法包括如下步骤:先向25%氨水的中加入新制明胶溶液催化,
低温0℃温度下,滴加入ncs和氢氧化钠配成的水溶液。滴加完成且继续在0℃温度搅拌反,然后低温下以醚类溶剂萃取两次,然后向合并的醚类溶剂中滴加1
‑
甲基哌嗪的溶液,于0℃温度下搅拌反,自然升温至室温搅拌反应,减压回收溶剂后精馏回收未反应的原料1
‑
甲基哌嗪,再精馏得目标产品1
‑
氨基
‑4‑
甲基哌嗪。
[0018]
进一步详细阐述如下:
[0019]
一种1
‑
甲基
‑4‑
哌嗪无需经亚硝化一步反应制备1
‑
氨基
‑4‑
甲基哌嗪的绿色合成方法,向6l 25%氨水的反应瓶内加入1l的1%新制明胶溶液,开启低温浴,将反应体系冷却至0℃。保持在0℃温度下,向反应瓶内滴加入200g的ncs和180g氢氧化钠配成的1l水溶液。滴加完成且继续在0℃温度搅拌反应15min,然后加入预冷至0℃的2.5l甲基叔丁基醚,搅拌15min后分出有机层,再加入预冷至0℃的2.5l甲基叔丁基醚,搅拌15min后再分出有机层。保持在0℃温度下,向合并的有机层的反应瓶中滴加溶有150g 1
‑
甲基哌嗪的甲基叔丁基醚溶液1l,滴加完成后,于0℃温度下搅拌反应60min,自然升温至室温搅拌反应2h,gc分析。减压回收溶剂得1
‑
甲基哌嗪和1
‑
氨基
‑4‑
甲基哌嗪混合液,将混合液转入带精馏柱的250ml单颈瓶中,水泵减压,控制真空度30mmhg条件下精馏,收集62~70℃馏份88.6g为未反应的原料1
‑
甲基哌嗪,可套用于下次反应,回收率59.0%,同样真空度30mmhg条件下,收集90~96℃馏份59.0g为目标产品1
‑
氨基
‑4‑
甲基哌嗪收率34.3%,gc分析纯度>99%。
[0020]
前述合成方法中,其特征是起始原料氨水与ncs的质量比为100:0.01~100:20,最佳比例为25:1~35:1。
[0021]
前述合成方法中,其特征是是起始原料ncs与碱氢氧化钠的摩尔量比为1:1~1:5,最佳百分比为1:2.5~1:3.5。
[0022]
前述合成方法中,其特征是是起始原料1
‑
甲基哌嗪与ncs的摩尔量比为1:0.5~1:3,最佳比例为1:0.8~1:1.2。
[0023]
前述合成方法中,其特征其特征是醚类溶剂为甲基叔丁醚,四氢呋喃,乙醚,1,4
‑
二氧六环等醚中的一种。
[0024]
前述合成方法中,其特征是碱可以为无机碱氢氧化钠naoh,氢氧化钾koh或有机碱甲醇钠等,以氢氧化钠最优。
[0025]
前述合成方法中,其特征特征原料为1
‑
甲基
‑
哌嗪、其特征目标产物为1
‑
氨基
‑4‑
甲基哌嗪,结构式分别如下所示:
[0026][0027]
前述合成方法中,其特征是使用1
‑
甲基哌嗪为原料,一步不通过亚硝化步骤直接生成目标产物1
‑
氨基
‑4‑
甲基哌嗪,无具有基因毒性的n
‑
亚硝基中间体生成,目标物总产率较高,较传统方法更简洁高效。
附图说明
[0028]
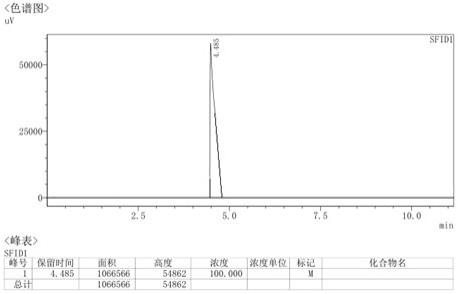
图1为n
‑
甲基哌嗪gc图谱;
[0029]
图2为1
‑
氨基
‑4‑
甲基哌嗪gc图谱;
[0030]
图3为1
‑
氨基
‑4‑
甲基哌嗪的核磁图;
[0031]
图4为1
‑
氨基
‑4‑
甲基哌嗪的质谱图。
具体实施方式
[0032]
以下通过实施例说明本发明的具体工艺步骤,但不受实施例限制。
[0033]
实施例1:四氢呋喃为溶剂1
‑
甲基哌嗪在与碱性条件下的ncs和氨水作用下制备1
‑
氨基
‑4‑
甲基哌嗪步骤如下:
[0034][0035]
向100ml的装有60ml 25%氨水的反应瓶内加入10ml的1%的新制明胶溶液,开启低温浴,将反应体系冷却至0℃。保持在0℃温度下,向反应瓶内滴加入2g(15mmol)的ncs和1.8g(45mmol)氢氧化钠配成的10ml水溶液。滴加完成后继续在0℃温度搅拌反应5min,然后加入预冷至0℃的25ml四氢呋喃,搅拌5min后分出有机层,再加入预冷至0℃的25ml四氢呋喃,搅拌5min后再分出有机层。保持在0℃温度下,向合并的有机层的反应瓶中滴加溶有1.5g(15mmol)1
‑
甲基哌嗪的四氢呋喃溶液10ml,滴加完成后,于0℃温度下搅拌反应30min,自然升温至室温搅拌反应1h,gc分析,减压蒸去溶剂,柱色谱分离得目标产品1
‑
氨基
‑4‑
甲基哌嗪0.56g,收率32.4%。
[0036]
实施例2:甲基叔丁基醚为溶剂1
‑
甲基哌嗪与碱性条件下的ncs和氨水作用下制备1
‑
氨基
‑4‑
甲基哌嗪步骤如下:
[0037][0038]
向100ml的装有60ml 25%氨水的反应瓶内加入10ml的1%的新制明胶溶液,开启低温浴,将反应体系冷却至0℃。保持在0℃温度下,向反应瓶内滴加入2g(15mmol)的ncs和1.8g(45mmol)氢氧化钠配成的10ml水溶液。滴加完成后继续在0℃温度搅拌反应5min,然后加入预冷至0℃的25ml甲基叔丁基醚,搅拌5min后分出有机层,再加入预冷至0℃的25ml甲基叔丁基醚,搅拌5min后再分出有机层。保持在0℃温度下,向合并的有机层的反应瓶中滴加溶有1.5g(15mmol)1
‑
甲基哌嗪的甲基叔丁基醚溶液10ml,滴加完成后,于0℃温度下搅拌反应30min,自然升温至室温搅拌反应1h,gc分析,减压蒸去溶剂,柱色谱分离得目标产品1
‑
氨基
‑4‑
甲基哌嗪0.62g,收率35.9%。
[0039]
实施例3:1
‑
甲基哌嗪一步制备1
‑
氨基
‑4‑
甲基哌嗪放大实验步骤如下:
[0040][0041]
向10l的装有6l 25%氨水的反应瓶内加入1l的1%的新制明胶溶液,开启低温浴,将反应体系冷却至0℃。保持在0℃温度下,向反应瓶内滴加入200g(1.5mol)的ncs和180g(4.5mol)氢氧化钠配成的1l水溶液。滴加完成且继续在0℃温度搅拌反应15min,然后加入预冷至0℃的2.5l甲基叔丁基醚,搅拌15min后分出有机层,再加入预冷至0℃的2.5l甲基叔
丁基醚,搅拌15min后再分出有机层。保持在0℃温度下,向合并的有机层的反应瓶中滴加溶有150g(1.5mol)1
‑
甲基哌嗪的甲基叔丁基醚溶液1l,滴加完成后,于0℃温度下搅拌反应60min,自然升温至室温搅拌反应2h,gc分析。减压回收溶剂得1
‑
甲基哌嗪和1
‑
氨基
‑4‑
甲基哌嗪混合液,将混合液液转入带精馏柱的250ml单颈瓶中,水泵减压,控制真空度30mmhg条件下精馏,收集62~70℃馏份90.2g为未反应的原料1
‑
甲基哌嗪,可用套用下次反应,回收率60.1%,同样控制真空度30mmhg条件下,收集90~96℃馏份59.0g为目标产品1
‑
氨基
‑4‑
甲基哌嗪收率34.2%,gc分析纯度>99%。
[0042]
实施例4:回收套用的1
‑
甲基哌嗪一步制备1
‑
氨基
‑4‑
甲基哌嗪实验步骤如下:
[0043][0044]
向10l的装有6l 25%氨水的反应瓶内加入1l的1%的新制明胶溶液,开启低温浴,将反应体系冷却至0℃。保持在0℃温度下,向反应瓶内滴加入200g(1.5mol)的ncs和180g(4.5mol)氢氧化钠配成的1l水溶液。滴加完成且继续在0℃温度搅拌反应15min,然后加入预冷至0℃的2.5l甲基叔丁基醚,搅拌15min后分出有机层,再加入预冷至0℃的2.5l甲基叔丁基醚,搅拌15min后再分出有机层。保持在0℃温度下,向合并的有机层的反应瓶中滴加溶有150g(1.5mol)1
‑
甲基哌嗪(反应精馏回收套用)的甲基叔丁基醚溶液1l,滴加完成后,于0℃温度下搅拌反应60min,自然升温至室温搅拌反应2h,gc分析。减压回收溶剂得1
‑
甲基哌嗪和1
‑
氨基
‑4‑
甲基哌嗪混合液,将混合液液转入带精馏柱的250ml单颈瓶中,水泵减压,控制真空度30mmhg条件下精馏,收集62~70℃馏份88.6g为未反应的原料1
‑
甲基哌嗪,可用套用下次反应,回收率59.0%,收集90~96℃馏份59.0g为目标产品1
‑
氨基
‑4‑
甲基哌嗪收率34.2%,gc分析纯度>99%。
[0045]1‑
氨基
‑4‑
甲基哌嗪gc分析方法:
[0046]
采用岛津gc
‑
2014c型气相色谱仪,气相色谱柱:wondacap5毛细管色谱柱(柱长:30m,内径:0.25mm,膜厚:0.25um,最高使用温度:325℃)柱温程序升温50
‑
220℃,10℃/min,进样器温度220℃,检测器温度220℃,h2压强0.1mpa、空气0.16mpa、柱前压1.5mpa。含量计算采用面积归一化法,保留时间为n
‑1‑
甲基哌嗪4.48min和1
‑
氨基
‑4‑
甲基哌嗪6.11min。
[0047]
图1为n
‑
甲基哌嗪gc图谱,图2为1
‑
氨基
‑4‑
甲基哌嗪gc图谱。
[0048]1‑
氨基
‑4‑
甲基哌嗪核磁(nmr)和高分辨质谱(hrms)表征:
[0049]
以氘代氯仿为溶剂进行了核磁(nmr),以高分辨质谱(hrms)对目标物结构表征,结果如下。1
‑
氨基
‑4‑
甲基哌嗪1h nmr(300mhz,cdcl3)δ3.11(s,2h),2.99
–
2.29(m,6h),2.25(d,j=2.3hz,3h);1h nmr(300mhz,dmso)δ2.63(d,j=63.5hz,4h),2.13(s,4h),1.96(s,3h);hrms(esi):exact mass calcd for c5h
13
n3[m h]
:116.1175.found:116.1175。
[0050]
图3为1
‑
氨基
‑4‑
甲基哌嗪的核磁图,图4为1
‑
氨基
‑4‑
甲基哌嗪的质谱图。
[0051]
本发明提供的方法,一种氯代丁二酰亚胺(ncs)在氢氧化钠作为强碱性试剂,在明胶存在下低温搅拌反应,经醚类溶剂萃取后滴加1
‑
甲基
‑4‑
哌嗪中,低温至室温搅拌反应,最后通过减压精馏分离获得目标产物1
‑
氨基
‑4‑
甲基哌嗪,其优点主要有如下四点:
[0052]
(1)首次使用一步法由1
‑
甲基
‑
哌嗪直接合成1
‑
氨基
‑4‑
甲基哌嗪。传统工艺均需要制备n
‑
亚硝基产物,而n
‑
亚硝基类化合物是一类具有基因毒性的中间体,其残留会大大
影响药品的安全性。本发明的方法是由1
‑
甲基
‑
哌嗪一步不经亚硝中间体直接生成1
‑
氨基
‑4‑
甲基哌嗪,完全避免了n
‑
亚硝基类化合物基因毒性对药品的安全性影响。
[0053]
(2)使用n
‑
氯代丁二酰亚胺(ncs)与氨水等为原料,原料成本低廉,优势明显。
[0054]
(3)无高压加氢,金属酸还等危险反应,操作简便易于工业化。
[0055]
(4)由1
‑
甲基
‑
哌嗪直接合成1
‑
氨基
‑4‑
甲基哌嗪原料经套用后总的收率高,优势明显。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。