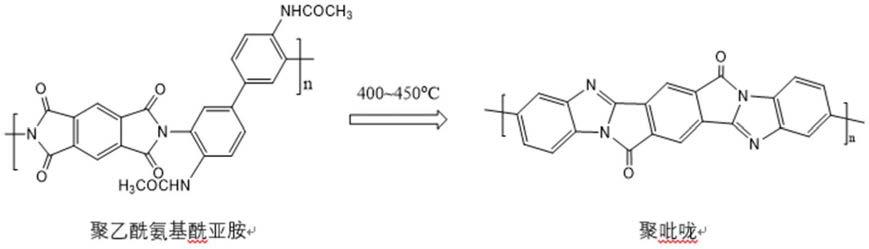
1.本技术涉及聚吡咙材料技术领域,具体涉及到一种高强高模聚吡咙纤维及其制备方法。
背景技术:
2.聚吡咙是一类刚性的梯形或半梯形聚芳杂环大分子聚合物,具有良好的耐高温性和抗氧化性,部分聚吡咙的分解温度超过700℃。这种聚合物纤维不仅是一类耐高温阻燃纤维,还是一类高强度,高模量的高性能纤维。自上世纪六十年代至今,该类聚合物已经得到了广泛的研究,其一般是在多聚磷酸等高沸点溶剂中高温聚合而成。由于聚吡咙既不熔融,也不溶于普通有机溶剂,其可加工性受到了极大的限制,同时对其应用的开发造成极大的阻碍。此外,传统制备聚吡咙粗纤维或薄膜是将其溶解在甲磺酸、氯磺酸等超强质子酸中加工而得,这些溶剂毒性强,沸点高,不仅很难从所制备的纤维或膜材料中去除干净,也容易造成环境污染。因此,非常有必要改变聚吡咙的合成路径,实施一种类似于制备聚酰亚胺的两步合成法,先合成普通有机溶剂可溶的高分子量聚吡咙的前聚体,通过前聚体溶液先制备前聚体纤维(或纳米纤维)或前聚体膜,再在高温下吡咙化,形成聚吡咙纤维(或纳米纤维)或聚吡咙膜。
技术实现要素:
3.针对上述技术问题,本发明的第一方面提供了一种高强高模聚吡咙纤维,所述聚吡咙纤维的聚吡咙化合物具有如下结构:
[0004][0005]
所述聚吡咙纳米纤维的直径为8~15μm。
[0006]
作为本发明的一种优选技术方案,所述聚吡咙纤维的拉伸强度不低于1.8gpa。
[0007]
作为本发明的一种优选技术方案,所述聚吡咙纤维的拉伸模量不低于80gpa。
[0008]
本发明的第二个方面提供了如上所述的高强高模聚吡咙纤维的制备方法,其包括如下步骤:
[0009]
(1)原料单体的制备:将乙酸酐溶解在有机溶剂a中得到反应料a,然后将所述反应料a加入到取代联苯二胺的有机溶剂b溶液中,并在不高于5℃的反应温度下反应2~6小时得到中间粗产物;将所述中间粗产物中加入氧化钙沉淀,过滤,浓缩得到粗产物;然后对所述粗产物进行重结晶得到所述原料单体;
[0010]
(2)中间体聚乙酰氨基酰胺酸的合成:将所述原料单体加入到反应器中在纺丝溶剂中与二元酸酐进行缩合反应,得到聚乙酰氨基酰胺酸溶液;
[0011]
(3)湿法纺丝:将所述聚乙酰氨基酰胺酸溶液在40
‑
60℃下进行化学亚胺化,得到
半化学亚胺化的聚乙酰氨基酰亚胺酰胺酸溶液,经过滤后通过高压经喷丝板挤出进入凝固浴中凝固,洗涤、烘干,得到初生丝;
[0012]
(4)聚吡咙化:将所述初生丝在280℃下热亚胺化和牵伸,然后在450
‑
500℃下加热牵伸聚吡咙化,后处理得到所述高强高模聚吡咙纤维。
[0013]
作为本发明的一种优选技术方案,步骤(3)中在进行化学亚胺化过程中加入所需量的乙酸酐和三乙胺。
[0014]
作为本发明的一种优选技术方案,步骤(3)中所述凝固浴中的凝固剂包含nmp的水溶液。
[0015]
作为本发明的一种优选技术方案,所述凝固剂还包含所述纺丝溶剂,其含量为5~15wt%。
[0016]
作为本发明的一种优选技术方案,步骤(1)中所述取代联苯二胺具有如下结构:
[0017][0018]
其中取代基r1和取代基r2分别独立地为氢原子或乙酰氨基;优选的,所述取代基r1和取代基r2为乙酰氨基。
[0019]
作为本发明的一种优选技术方案,步骤(2)中所述二元酸酐选自联苯四甲酸二酐、均苯四甲酸二酐、萘四甲酸二酐、二苯酮四酸二酐、二苯砜二酐、2,3,3',4'
‑
二苯醚四甲酸二酐、三苯二苯醚二酐中的一种或多种。
[0020]
作为本发明的一种优选技术方案,步骤(3)中所述化学亚胺化过程进行5~7小时;优选的,所述化学亚胺化过程中溶液呈透明状态。
[0021]
有益效果:本方案采用了含两个隐形氨基(乙酰氨基)的四氨基联苯(4,4
’‑
二乙酰氨基
‑
3,3
’‑
二氨基联苯)为单体先合成聚酰亚胺的前聚体(聚乙酰氨基酰胺酸),后者在空气气氛中亚胺化(300
‑
350℃的高温)形成聚乙酰氨基酰亚胺(聚吡咙的前聚体),再在空气气氛中高温吡咙化(450
‑
500℃的高温)形成高性能聚吡咙聚合物。这个方案比直接采用3,3’,4,4
’‑
四氨基联苯作为单体合成聚吡咙的优点就在于:在空气气氛中高温亚胺化和吡咙化,而不会破坏聚合物的化学结构,因为乙酰氨基比氨基更能耐高温和耐空气氧化。如果直接采用3,3’,4,4
’‑
四氨基联苯作单体合成聚氨基酰胺酸(聚酰亚胺的前聚体),并在高温空气中亚胺化,氨基会被氧化成氮
‑
氧化合物,则不能在后续的吡咙化反应中与酰亚胺环上的羰基发生反应而形成含碳氮双键的聚吡咙结构,得不到聚吡咙化合物。此外,聚氨基酰胺酸中的自由氨基很活泼,使得该前聚体聚合物不够稳定,容易产生凝胶,使溶液的流动性受到严重影响,以至于不能顺利纺丝。
具体实施方式
[0022]
下面结合具体实施方式对本发明提供技术方案中的技术特征作进一步清楚、完整的描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0023]
本发明中的词语“优选的”、“优选地”、“更优选的”等是指,在某些情况下可提供某
些有益效果的本发明实施方案。然而,在相同的情况下或其他情况下,其他实施方案也可能是优选的。此外,对一个或多个优选实施方案的表述并不暗示其他实施方案不可用,也并非旨在将其他实施方案排除在本发明的范围之外。
[0024]
应当理解,除了在任何操作实例中,或者以其他方式指出的情况下,表示例如说明书和权利要求中使用的成分的量的所有数字应被理解为在所有情况下被术语“约”修饰。因此,除非相反指出,否则在以下说明书和所附权利要求中阐述的数值参数是根据本发明所要获得的期望性能而变化的近似值。至少并不是试图将等同原则的适用限制在权利要求的范围内,每个数值参数至少应该根据报告的有效数字的个数并通过应用普通舍入技术来解释。
[0025]
本发明的第一方面提供了一种高强高模聚吡咙纤维,所述聚吡咙纤维的聚吡咙化合物具有如下结构:
[0026][0027]
所述聚吡咙纳米纤维的直径为8~15μm,其直径可以为8~10μm、10~12μm、12~15μm、8μm、9μm、10μm、11μm、12μm、13μm、14μm、15μm。
[0028]
在一些实施方式中,所述聚吡咙纤维的拉伸强度不低于1.8gpa;进一步优选的,所述聚吡咙纤维的拉伸强度不低于2.0gpa;进一步优选的,所述聚吡咙纤维的拉伸模量不低于80gpa;进一步优选的,所述聚吡咙纤维的拉伸模量不低于100gpa;进一步优选的聚吡咙纤维的玻璃化转变温度为350~500℃。本技术的所述纤维的拉伸强度和拉伸模量是采用上海中辰超细纤维单丝拉伸测试仪jq03b进行测试得到,可以根据iso 11566
‑
1996标准进行测试。
[0029]
本发明的第二个方面提供了如上所述的高强高模聚吡咙纤维的制备方法,其包括如下步骤:
[0030]
(1)原料单体的制备:将乙酸酐溶解在有机溶剂a中得到反应料a,然后将所述反应料a加入到取代联苯二胺的有机溶剂b溶液中,并在不高于5℃的反应温度下反应2~6小时得到中间粗产物;将所述中间粗产物中加入氧化钙沉淀,过滤,浓缩得到粗产物;然后对所述粗产物进行重结晶得到所述原料单体;
[0031]
(2)中间体聚乙酰氨基酰胺酸的合成:将所述原料单体加入到反应器中在纺丝溶剂中与二元酸酐进行缩合反应,得到聚乙酰氨基酰胺酸溶液;
[0032]
(3)湿法纺丝:将所述聚乙酰氨基酰胺酸溶液在40
‑
60℃下进行化学亚胺化,得到半化学亚胺化的聚乙酰氨基酰亚胺酰胺酸溶液,经过滤后通过高压经喷丝板挤出进入凝固浴中凝固,洗涤、烘干,得到初生丝;
[0033]
(4)聚吡咙化:将所述初生丝在280℃下热亚胺化和牵伸,然后在450
‑
500℃下加热牵伸聚吡咙化,后处理得到所述高强高模聚吡咙纤维。
[0034]
在一些实施方式中,步骤(1)中所述取代联苯二胺具有如下结构:
[0035][0036]
其中取代基r1和取代基r2分别独立地为氢原子或乙酰氨基;优选的,所述取代基r1和取代基r2为乙酰氨基。本发明中的聚乙酰氨基酰亚胺单体中的氨基可以在第2、3、4位中的任一碳原子上,同样的取代基r1和取代基r2也可以在第2、3、4位中的任一碳原子上。优选的,取代基r1和取代基r2分别独立地为氢原子或乙酰氨基。其中取代基r1和取代基r2可以为相同取代基,也可以为不同取代基。
[0037]
进一步的,所述取代基r1和取代基r2相同;优选的,所述取代基r1和取代基r2为乙酰氨基;进一步优选的,所述聚乙酰氨基酰亚胺单体中的氨基取代的是第3位的碳原子,而乙酰氨基取代的是第4位的碳原子,化学名称为3,3
′‑
二氨基
‑
4,4
′‑
二乙酰氨基联苯,其具有如下结构:
[0038][0039]
原料单体的制备:在一些实施方式中,所述3,3
′‑
二氨基
‑
4,4
′‑
二乙酰氨基联苯是通过3,3
′‑
二氨基联苯胺与乙酸酐在低温下反应获得。由于3,3
′‑
二氨基联苯胺结构中含有四个氨基,而每一个氨基的活性不同,因此反应需要再低温下进行,保证乙酸酐只与高活性的4.4
′‑
位的二氨基反应,形成3,3
′‑
二氨基
‑
4,4
′‑
二乙酰氨基联苯。在一些实施方式中,所述反应的温度不高于5摄氏度;优选的,反应的温度不高于0摄氏度;进一步的,反应的温度为
‑
5~0℃。此外,为了避免乙酸酐与3,3
′‑
二氨基联苯胺上的所有氨基发生反应,产生不必要的副产物,进一步控制乙酸酐的添加速度,保证反应过程中体系中的乙酸酐含量保持少于3,3
′‑
二氨基联苯胺含量。
[0040]
在一些实施方式中,上述反应是在液相态下进行;优选的,将3,3
′‑
二氨基联苯胺溶于有机溶剂b中配制成10~20wt%的溶液,同时将乙酸酐采用有机溶剂a配制成5~15wt%的溶液,并将乙酸酐的溶液加入到3,3
′‑
二氨基联苯胺的溶液中进行反应。在一些优选的实施方式中,所述反应料a加入到取代联苯二胺的有机溶剂b溶液中的滴加速度为1~3ml/min。
[0041]
本发明中对所述有机溶剂a和有机溶剂b的具体种类并不做特殊限定,可以选用本领域技术人员所熟知的各类能够溶解乙酸酐的有机溶剂,包括但不限于四氢呋喃、乙二醇二甲醚、碳酸甲酯、二甲基亚砜、dmf、dmac等。在一些优选的实施方式中,所述有机溶剂a和有机溶剂b相同;进一步优选的,所述有机溶剂a和有机溶剂b为四氢呋喃。
[0042]
由于在上述反应中会产生副产物乙酸,因此在反应产物中加入适量的氧化钙,使体系中的乙酸酐形成乙酸钙沉淀,通过过滤除去体系中未反应的乙酸酐,然后将滤液通过旋转蒸发等方式进行浓缩,除去其中的溶剂得到粗产品。本发明中对所述氧化钙的用量并不做特殊限定,可以根据实际情况进行确定,在一些优选的实施方式中,所述氧化钙的含量为所述乙酸酐摩尔量的0.3~0.8倍;进一步优选的,所述氧化钙的含量为所述乙酸酐摩尔量的0.5倍。本发明中对氧化钙沉淀过滤所得的粗产品进行重结晶对其进一步净化。本发明中对所述重结晶步骤并不做特殊限定,可以根据本领域技术人员所熟知的方式进行即可。
在一些优选的实施方式中,所述重结晶采用的溶剂为乙醇和四氢呋喃的混合溶剂;优选的所述乙醇和四氢呋喃的体积比为1:1。
[0043]
本发明中的所述中间体聚乙酰氨基酰胺酸是采用上述步骤1中制备得到的原料单体与二元酸酐进行缩合制备得到。本技术中对与所述聚乙酰氨基酰胺酸单体反应的酸酐的具体种类并不做特殊限定,可以采用本领域技术人员所熟知的各类二元酸酐。在上述反应中的原料单体与二元酸酐的摩尔比例为1:(0.8~1.2),优选摩尔比例为1:1。在一些实施方式中,所述缩合反应温度为5~15℃。在上述反应条件下对反应原料进行搅拌,搅拌速度为150~250r/min,反应4~9小时,得到固含量为8~20wt%(优选10~20wt%)的,粘稠的中间体聚乙酰氨基酰胺酸(paaa)溶液。
[0044]
在一些实施方式中,步骤(2)中所述二元酸酐选自联苯四甲酸二酐、均苯四甲酸二酐、萘四甲酸二酐、二苯酮四酸二酐、二苯砜二酐、2,3,3',4'
‑
二苯醚四甲酸二酐、三苯二苯醚二酐中的一种或多种。
[0045]
本发明中对缩合反应中采用的纺丝溶剂的具体种类并不做特殊限定,可以选用本领域技术人员所熟知的各类溶剂,包括单不限于dmf、dmac。
[0046]
本发明中通过上述步骤制备得到的中间体聚乙酰氨基酰胺酸溶液进一步通过湿法纺丝制备本发明的纤维。
[0047]
在一些实施方式中,步骤(3)中在进行化学亚胺化过程中加入所需量的乙酸酐和三乙胺。本发明中对中间体聚乙酰氨基酰胺酸溶液添加乙酸酐和三乙胺,对其进行部分化学亚胺化处理,形成溶剂可溶的聚乙酰氨基酰亚胺
‑
酰胺酸溶液,然后将聚乙酰氨基酰亚胺
‑
酰胺酸的纺丝溶剂(dmac)溶液进行湿法纺丝。
[0048]
在一些实施方式中,所述乙酸酐的添加量为预聚体(聚乙酰氨基酰胺酸)单元结构摩尔数(或二酐残基摩尔数)的0.5
‑
1.0倍(其理论最高添加量为聚乙酰氨基酰胺酸摩尔量的两倍)。进一步的,所述三乙胺的添加摩尔量为乙酸酐摩尔量的1~1.4倍;进一步的,所述化学亚胺化过程进行5~7小时;优选的,所述化学亚胺化过程中溶液呈透明状态。本发明中所述的透明状态是本领域技术人员通过主观的方式进行判断,通过改变乙酸酐和三乙胺的含量的来调节体系的透明度,如果体系中乙酸或乙酸酐及三乙胺等非溶剂含量过高,会引起溶液分相,变浑浊,影响纺丝的结果。溶液经过部分化学亚胺化之后在挤入凝固浴前进行1~3微米滤网过滤。
[0049]
本发明中经过过滤之后的纺丝液经高压经喷丝板(12k、24k、48k等)挤出进入凝固浴中凝固,洗丝池中对成型的纤维进行洗涤、60
‑
100℃烘干室、300
‑
350℃热牵伸,形成直径在10
‑
15微米的高强、耐高温(聚乙酰氨基酰亚胺纤维。
[0050]
本发明中的纺丝溶液在凝固浴中,在凝固剂的作用下形成溶剂与凝固剂之间的双向扩散,使纺丝溶液中的纺丝溶剂扩散到凝固浴中,并由凝固浴中的凝固剂成分部分替代成型纤维中的纺丝溶剂,并在后续的洗涤、烘干操作中去除得到最终的成型纤维。
[0051]
在一些实施方式中,步骤(3)中所述凝固浴中的凝固剂包含nmp的水溶液。
[0052]
进一步优选的,所述凝固剂还包含所述纺丝溶剂,其含量为5~15wt%;优选为15wt%。
[0053]
进一步优选的,所述凝固剂由10~20wt%纺丝溶剂、8~12wt%nmp(n
‑
甲基吡咯烷酮),0.05~0.15wt%表面活性剂和余量的水组成。
[0054]
本发明中对所述表面活性剂的具体种类并不做特殊限定,可以选用本领域技术人员所熟知的各类表面活性剂,包括但不限于非离子表面活性剂、阴离子表面活性剂包括但不限于360渗透剂。
[0055]
在一些实施方式中,步骤(3)中所述化学亚胺化过程进行5~7小时;优选的,所述化学亚胺化过程中溶液呈透明状态。
[0056]
本发明中对在空气气氛中亚胺化(300
‑
330℃的高温)形成聚乙酰氨基酰亚胺(聚吡咙的前聚体),再在空气气氛中高温吡咙化(430
‑
300℃的高温),是聚合物结构中的乙酰氨基中的氮元素在高温张力下与酰亚胺环中羰基作用,形成碳氮双键,脱去乙酸分子,形成高性能聚吡咙聚合物;进一步的在450~500℃下退火15
‑
30分钟。
[0057][0058]
本技术中采用了含两个隐形氨基(乙酰氨基)的四氨基联苯(4,4
’‑
二乙酰氨基
‑
3,3
’‑
二氨基联苯)为单体先合成聚酰亚胺的前聚体(聚乙酰氨基酰胺酸),后者在空气气氛中亚胺化(300
‑
330℃的高温)形成聚乙酰氨基酰亚胺(聚吡咙的前聚体),再在空气气氛中高温吡咙化(430
‑
300℃的高温)形成高性能聚吡咙聚合物。这个方案比直接采用3,3’,4,4
’‑
四氨基联苯作为单体合成聚吡咙的优点就在于:在空气气氛中高温亚胺化和吡咙化,而不会破坏聚合物的化学结构,因为乙酰氨基比氨基更能耐高温和耐空气氧化。如果直接采用3,3’,4,4
’‑
四氨基联苯作单体合成聚氨基酰胺酸(聚酰亚胺的前聚体),并在高温空气中亚胺化,氨基会被氧化成氮
‑
氧化合物,则不能在后续的吡咙化反应中与酰亚胺环上的羰基发生反应而形成含碳氮双键的聚吡咙结构,得不到聚吡咙化合物。此外,聚氨基酰胺酸中的自由氨基很活泼,使得该前聚体聚合物不够稳定,容易产生凝胶,使溶液的流动性受到严重影响,以至于不能顺利纺丝。
[0059]
下面通过实施例对本发明进行具体描述。有必要在此指出的是,以下实施例只用于对本发明作进一步说明,不能理解为对本发明保护范围的限制,该领域的专业技术人员根据上述本发明的内容做出的一些非本质的改进和调整,仍属于本发明的保护范围。
[0060]
其中的反应方程式参见如下示意结构:
[0061][0062]
实施例1:
[0063]
(1)原料的制备:将100克(466.7mmol)3,3
’‑
二氨基联苯胺置于反应器中加入567克thf溶解配制成浓度为15%的溶液,然后在上述3,3
’‑
二氨基联苯胺(m=214.27)的thf溶液中以3
‑
5g/min的速度滴加10%乙酸酐的thf溶液952.9克(2*466.7mmol乙酸酐(m=102.09),重量95.29克),控制反应温度在
‑
5~0℃范围内反应4小时,然后加入干燥氧化钙(m=56.077)22.6g(466.7mmol),与乙酸反应形成乙酸钙沉淀,从thf溶液中析出;过滤去除体系中的乙酸钙,并将滤液旋蒸浓缩得到3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯(m=298.27)的粗产品136克(理论产量为139.2克);在上述3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯粗产品中加入体积比为1/1的乙醇/thf溶剂,进行重结晶得到纯度为99%的3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯产品131克,最终收率为94%。
[0064]
(2)中间体聚乙酰氨基酰胺酸的合成:以3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯和均苯四甲酸二酐单体为原料进行缩合聚合反应,以dmac为溶剂,控制二胺和二酐的摩尔比为1:1、溶液的固含量为15wt.%,在10
‑
15℃的温度范围内进行缩合聚合8小时,形成粘稠的聚乙酰氨基酰胺酸溶液。
[0065]
(3)上述聚乙酰氨基酰胺酸中加入其单元结构摩尔量的乙酐和1.2倍摩尔量的三乙胺在50℃下进行化学亚胺化6小时,形成半化学亚胺化的聚乙酰氨基酰亚胺
‑
酰胺酸溶液,将此溶液经过2微米滤网过滤后,通过高压经喷丝板(12k)挤出进入凝固浴(凝固剂为10%nmp,15%dmac,0.1%表面活性剂(360渗透剂)和75%的去离子水)、洗丝池(去离子水洗)、80℃烘干室、280℃下热亚胺化和牵伸,再在480℃加热牵伸,乙酰氨基中的氮元素在高温张力下与酰亚胺环中羰基作用,形成碳氮双键,脱去乙酸分子,形成具有聚吡咙结构高性
能纤维。
[0066]
实施例2:
[0067]
(1)原料的制备:将100克(466.7mmol)3,3
’‑
二氨基联苯胺置于反应器中加入567克thf溶解配制成浓度为15%的溶液,然后在上述3,3
’‑
二氨基联苯胺(m=214.27)的thf溶液中以3
‑
5g/min的速度滴加10%乙酸酐的thf溶液952.9克(2*466.7mmol乙酸酐(m=102.09),重量95.29克),控制反应温度在
‑
5~0℃范围内反应4小时,然后加入干燥氧化钙(m=56.077)22.6g(466.7mmol),与乙酸反应形成乙酸钙沉淀,从thf溶液中析出;过滤去除体系中的乙酸钙,并将滤液旋蒸浓缩得到3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯(m=298.27)的粗产品136克(理论产量为139.2克);在上述3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯粗产品中加入体积比为1/1的乙醇/thf溶剂,进行重结晶得到纯度为99%的3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯产品131克,最终收率为94%。
[0068]
(2)中间体聚乙酰氨基酰胺酸的合成:以3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯和联苯四甲酸二酐单体为原料进行缩合聚合反应,以dmac为溶剂,控制二胺和二酐的摩尔比为1:1、溶液的固含量为15wt.%,在10
‑
15℃的温度范围内进行缩合聚合8小时,形成粘稠的聚乙酰氨基酰胺酸溶液。
[0069]
(3)上述聚乙酰氨基酰胺酸中加入其单元结构摩尔量的乙酐和1.2倍摩尔量的三乙胺在50℃下进行化学亚胺化6小时,形成半化学亚胺化的聚乙酰氨基酰亚胺
‑
酰胺酸溶液,将此溶液经过2微米滤网过滤后,通过高压经喷丝板(24k)挤出进入凝固浴(凝固剂为10%nmp,15%dmac,0.1%表面活性剂(360渗透剂)和75%的去离子水)、洗丝池(去离子水洗)、80℃烘干室、280℃下热亚胺化和牵伸,再在480℃加热牵伸,乙酰氨基中的氮元素在高温张力下与酰亚胺环中羰基作用,形成碳氮双键,脱去乙酸分子,形成具有聚吡咙结构高性能纤维。
[0070]
实施例3:
[0071]
(1)原料的制备:将100克(466.7mmol)3,3
’‑
二氨基联苯胺置于反应器中加入567克thf溶解配制成浓度为15%的溶液,然后在上述3,3
’‑
二氨基联苯胺(m=214.27)的thf溶液中以3
‑
5g/min的速度滴加10%乙酸酐的thf溶液952.9克(2*466.7mmol乙酸酐(m=102.09),重量95.29克),控制反应温度在
‑
5~0℃范围内反应4小时,然后加入干燥氧化钙(m=56.077)22.6g(466.7mmol),与乙酸反应形成乙酸钙沉淀,从thf溶液中析出;过滤去除体系中的乙酸钙,并将滤液旋蒸浓缩得到3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯(m=298.27)的粗产品136克(理论产量为139.2克);在上述3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯粗产品中加入体积比为1/1的乙醇/thf溶剂,进行重结晶得到纯度为99%的3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯产品131克,最终收率为94%。
[0072]
(2)中间体聚乙酰氨基酰胺酸的合成:以3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯和二苯酮四酸二酐单体为原料进行缩合聚合反应,以dmac为溶剂,控制二胺和二酐的摩尔比为1:1、溶液的固含量为15wt.%,在10
‑
15℃的温度范围内进行缩合聚合8小时,形成粘稠的聚乙酰氨基酰胺酸溶液。
[0073]
(3)上述聚乙酰氨基酰胺酸中加入其单元结构摩尔量的乙酐和1.2倍摩尔量的三乙胺在50℃下进行化学亚胺化6小时,形成半化学亚胺化的聚乙酰氨基酰亚胺
‑
酰胺酸溶液,将此溶液经过2微米滤网过滤后,通过高压经喷丝板(48k)挤出进入凝固浴(凝固剂为
10%nmp,15%dmac,0.1%表面活性剂(360渗透剂)和75%的去离子水)、洗丝池(去离子水洗)、80℃烘干室、280℃下热亚胺化和牵伸,再在480℃加热牵伸,乙酰氨基中的氮元素在高温张力下与酰亚胺环中羰基作用,形成碳氮双键,脱去乙酸分子,形成具有聚吡咙结构高性能纤维。
[0074]
实施例4:
[0075]
(1)原料的制备:将100克(466.7mmol)3,3
’‑
二氨基联苯胺置于反应器中加入567克thf溶解配制成浓度为15%的溶液,然后在上述3,3
’‑
二氨基联苯胺(m=214.27)的thf溶液中以3
‑
5g/min的速度滴加10%乙酸酐的thf溶液952.9克(2*466.7mmol乙酸酐(m=102.09),重量95.29克),控制反应温度在
‑
5~0℃范围内反应4小时,然后加入干燥氧化钙(m=56.077)22.6g(466.7mmol),与乙酸反应形成乙酸钙沉淀,从thf溶液中析出;过滤去除体系中的乙酸钙,并将滤液旋蒸浓缩得到3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯(m=298.27)的粗产品136克(理论产量为139.2克);在上述3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯粗产品中加入体积比为1/1的乙醇/thf溶剂,进行重结晶得到纯度为99%的3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯产品131克,最终收率为94%。
[0076]
(2)中间体聚乙酰氨基酰胺酸的合成:以3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯和2,3,3',4'
‑
二苯醚四甲酸二酐单体为原料进行缩合聚合反应,以dmac为溶剂,控制二胺和二酐的摩尔比为1:1、溶液的固含量为15wt.%,在10
‑
15℃的温度范围内进行缩合聚合8小时,形成粘稠的聚乙酰氨基酰胺酸溶液。
[0077]
(3)上述聚乙酰氨基酰胺酸中加入其单元结构摩尔量的乙酐和1.2倍摩尔量的三乙胺在50℃下进行化学亚胺化6小时,形成半化学亚胺化的聚乙酰氨基酰亚胺
‑
酰胺酸溶液,将此溶液经过2微米滤网过滤后,通过高压经喷丝板(12k)挤出进入凝固浴(凝固剂为10%nmp,15%dmac,0.1%表面活性剂(360渗透剂)和75%的去离子水)、洗丝池(去离子水洗)、80℃烘干室、280℃下热亚胺化和牵伸,再在480℃加热牵伸,乙酰氨基中的氮元素在高温张力下与酰亚胺环中羰基作用,形成碳氮双键,脱去乙酸分子,形成具有聚吡咙结构高性能纤维。
[0078]
实施例5:
[0079]
(1)原料的制备:将100克(466.7mmol)3,3
’‑
二氨基联苯胺置于反应器中加入567克thf溶解配制成浓度为15%的溶液,然后在上述3,3
’‑
二氨基联苯胺(m=214.27)的thf溶液中以3
‑
5g/min的速度滴加10%乙酸酐的thf溶液952.9克(2*466.7mmol乙酸酐(m=102.09),重量95.29克),控制反应温度在
‑
5~0℃范围内反应4小时,然后加入干燥氧化钙(m=56.077)22.6g(466.7mmol),与乙酸反应形成乙酸钙沉淀,从thf溶液中析出;过滤去除体系中的乙酸钙,并将滤液旋蒸浓缩得到3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯(m=298.27)的粗产品136克(理论产量为139.2克);在上述3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯粗产品中加入体积比为1/1的乙醇/thf溶剂,进行重结晶得到纯度为99%的3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯产品131克,最终收率为94%。
[0080]
(2)中间体聚乙酰氨基酰胺酸的合成:以3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯和萘四甲酸二酐单体为原料进行缩合聚合反应,以dmac为溶剂,控制二胺和二酐的摩尔比为1:1、溶液的固含量为15wt.%,在10
‑
15℃的温度范围内进行缩合聚合8小时,形成粘稠的聚乙酰氨基酰胺酸溶液。
[0081]
(3)上述聚乙酰氨基酰胺酸中加入其单元结构摩尔量的乙酐和1.2倍摩尔量的三乙胺在50℃下进行化学亚胺化6小时,形成半化学亚胺化的聚乙酰氨基酰亚胺
‑
酰胺酸溶液,将此溶液经过2微米滤网过滤后,通过高压经喷丝板(12k)挤出进入凝固浴(凝固剂为10%nmp,15%dmac,0.1%表面活性剂(360渗透剂)和75%的去离子水)、洗丝池(去离子水洗)、80℃烘干室、280℃下热亚胺化和牵伸,再在480℃加热牵伸,乙酰氨基中的氮元素在高温张力下与酰亚胺环中羰基作用,形成碳氮双键,脱去乙酸分子,形成具有聚吡咙结构高性能纤维。
[0082]
实施例6:
[0083]
(1)原料的制备:将100克(466.7mmol)3,3
’‑
二氨基联苯胺置于反应器中加入567克thf溶解配制成浓度为15%的溶液,然后在上述3,3
’‑
二氨基联苯胺(m=214.27)的thf溶液中以3
‑
5g/min的速度滴加10%乙酸酐的thf溶液952.9克(2*466.7mmol乙酸酐(m=102.09),重量95.29克),控制反应温度在
‑
5~0℃范围内反应4小时,然后加入干燥氧化钙(m=56.077)22.6g(466.7mmol),与乙酸反应形成乙酸钙沉淀,从thf溶液中析出;过滤去除体系中的乙酸钙,并将滤液旋蒸浓缩得到3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯(m=298.27)的粗产品136克(理论产量为139.2克);在上述3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯粗产品中加入体积比为1/1的乙醇/thf溶剂,进行重结晶得到纯度为99%的3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯产品131克,最终收率为94%。
[0084]
(2)中间体聚乙酰氨基酰胺酸的合成:以3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯和三苯二苯醚二酐单体为原料进行缩合聚合反应,以dmac为溶剂,控制二胺和二酐的摩尔比为1:1、溶液的固含量为15wt.%,在10
‑
15℃的温度范围内进行缩合聚合8小时,形成粘稠的聚乙酰氨基酰胺酸溶液。
[0085]
(3)上述聚乙酰氨基酰胺酸中加入其单元结构摩尔量的乙酐和1.2倍摩尔量的三乙胺在50℃下进行化学亚胺化6小时,形成半化学亚胺化的聚乙酰氨基酰亚胺
‑
酰胺酸溶液,将此溶液经过2微米滤网过滤后,通过高压经喷丝板(12k)挤出进入凝固浴(凝固剂为10%nmp,15%dmac,0.1%表面活性剂(360渗透剂)和75%的去离子水)、洗丝池(去离子水洗)、80℃烘干室、280℃下热亚胺化和牵伸,再在480℃加热牵伸,乙酰氨基中的氮元素在高温张力下与酰亚胺环中羰基作用,形成碳氮双键,脱去乙酸分子,形成具有聚吡咙结构高性能纤维。
[0086]
实施例7:
[0087]
(1)原料的制备:将100克(466.7mmol)3,3
’‑
二氨基联苯胺置于反应器中加入567克thf溶解配制成浓度为15%的溶液,然后在上述3,3
’‑
二氨基联苯胺(m=214.27)的thf溶液中以3
‑
5g/min的速度滴加10%乙酸酐的thf溶液952.9克(2*466.7mmol乙酸酐(m=102.09),重量95.29克),控制反应温度在
‑
5~0℃范围内反应4小时,然后加入干燥氧化钙(m=56.077)22.6g(466.7mmol),与乙酸反应形成乙酸钙沉淀,从thf溶液中析出;过滤去除体系中的乙酸钙,并将滤液旋蒸浓缩得到3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯(m=298.27)的粗产品136克(理论产量为139.2克);在上述3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯粗产品中加入体积比为1/1的乙醇/thf溶剂,进行重结晶得到纯度为99%的3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯产品131克,最终收率为94%。
[0088]
(2)中间体聚乙酰氨基酰胺酸的合成:以3,3
’‑
二氨基
‑
4,4
’‑
二乙酰氨基联苯和二
苯砜二酐单体为原料进行缩合聚合反应,以dmac为溶剂,控制二胺和二酐的摩尔比为1:1、溶液的固含量为15wt.%,在10
‑
15℃的温度范围内进行缩合聚合8小时,形成粘稠的聚乙酰氨基酰胺酸溶液。
[0089]
(3)上述聚乙酰氨基酰胺酸中加入其单元结构摩尔量的乙酐和1.2倍摩尔量的三乙胺在50℃下进行化学亚胺化6小时,形成半化学亚胺化的聚乙酰氨基酰亚胺
‑
酰胺酸溶液,将此溶液经过2微米滤网过滤后,通过高压经喷丝板(12k)挤出进入凝固浴(凝固剂为10%nmp,15%dmac,0.1%表面活性剂(360渗透剂)和75%的去离子水)、洗丝池(去离子水洗)、80℃烘干室、280℃下热亚胺化和牵伸,再在480℃加热牵伸,乙酰氨基中的氮元素在高温张力下与酰亚胺环中羰基作用,形成碳氮双键,脱去乙酸分子,形成具有聚吡咙结构高性能纤维。
[0090]
性能测试
[0091]
申请人对上述实施例中的实验样品进行了纤维直径、力学强度等测试,具体如下:
[0092]
1、纤维直径测试:主要在显微镜下测量多组样品中随机截取的纤维平均直径(μm)。
[0093]
2、玻璃化转变温度测试:采用dma测试上述实施例中样品的玻璃化转变温度t
g
。
[0094]
3、分解温度测试:采用tga测试上述实施例中样品的热分解温度,其中的分解温度是指热失重达到5wt%时的温度t
d
/
5%
。
[0095]
4、拉伸强度:采用上海中辰超细纤维单丝拉伸测试仪jq03b进行纤维拉伸强度的测试,得到拉伸强度(gpa)、模量(gpa)以及伸长率(%)等参数。
[0096]
上述性能测试结果参见如下表1。
[0097]
表1
[0098] 纤维直径tg/℃td/℃拉伸强度伸长率模量实施例112
‑
15/700
‑
7202.0
‑
2.42.0
‑
4.0220
‑
250实施例210
‑
12480
‑
500715
‑
7352.5
‑
304.0
‑
6.0150
‑
180实施例310
‑
12400
‑
430680
‑
7001.8
‑
2.24.0
‑
6.0110
‑
150实施例48
‑
10380
‑
400660
‑
6802.0
‑
2.56.0
‑
8.0100
‑
130实施例512
‑
15/730
‑
7503.0
‑
3.53.0
‑
5.0250
‑
280实施例68
‑
10350
‑
370630
‑
6502.1
‑
2.78.0
‑
10.080
‑
100实施例710
‑
12410
‑
430650
‑
6701.8
‑
2.44.0
‑
6.0160
‑
180
[0099]
对比案例
[0100]
将14.8615克(69.36mmol)3,3
’‑
二氨基联苯胺置于反应器中加入180克dmac溶解配制成浓度为~7.5%的溶液,并将反应器置于冷浴中,控制反应体系温度在
‑
5~0℃之间,然后将15.1385g(69.36mmol)均苯四酸二酐(pmda)粉末加入到该反应体系中,在机械搅拌下进行反应4小时,形成15%的聚氨基酰胺酸的dmac溶液。然后在该溶液中加入14.1622g(138.72mmol)乙酸酐,继续搅拌4小时后,将该溶液温度升至50℃,再加入7.0811g(69.36mmol)乙酸酐和8.4219g(83.23mmol)三乙胺,搅拌反应6小时,结果溶液呈现部分凝胶状态,有一定的流动性,但没有粘滞性,或粘滞性低。当将此溶液经过2微米滤网过滤时,发现这种有一定流动性,低粘滞性的凝胶状半固体液几乎完全不能透过2微米滤网,无法通过过滤方法获取用于湿法喷丝板纺丝所需的具有流动性的洁净的高粘滞纺丝液。失败原因
可归因为用于与聚氨基酰胺酸中自由氨基反应的乙酸酐并没有完全反应,有可能参与了后面的亚胺化反应,使亚胺化程度提高到50%以上,导致聚合物难溶;另一方面,未反应的自由氨基也可能参与了与相邻聚合物分子上酰胺基之间的交换反应,导致部分交联结构的形成,致使该聚乙酰氨基酰亚胺或酰胺酸溶液凝胶化或部分凝胶化,而导致过滤网堵塞,不能过滤。
[0101]
以上所述仅是本发明的较佳实施例而已,并非是对发明作其他形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或更改为等同变化的等效实施例,但凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改,等同变化与改型,仍属于本发明技术方案的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。