1.本发明涉及药物制剂技术领域,更具体的说是涉及一种多晶型厄贝沙坦纳米混悬液及其制备方法和应用。
背景技术:
2.厄贝沙坦是一种血管紧张素ii受体拮抗剂,用于治疗高血压、心功能不全、心律失常等心血管疾病。它属于bscⅱ类,是一种难溶性药物。研究表明厄贝沙坦的可吸收剂量为26mg,远低于市售的厄贝沙坦片的最低规格75mg。这是因为其较低的溶解度限制了生物利用度。厄贝沙坦还是一种多晶型药物,具有a和b晶型以及一种无定形。多晶型通常被定义为特定物质的晶格具有两种或多种不同分子排列或构象的能力,在制药领域中,它是值得关注的关键因素。因为在不同的晶型中它们的物理化学性质有显著差异,例如稳定性和溶解度。晶型的这些性质也会影响药物的临床疗效。例如,已上市的厄贝沙坦和氢氯噻嗪的复合片因水溶性较低的b晶型的存在导致其生物利用度降低而被紧急召回。为了改善厄贝沙坦的口服生物利用度,已有研究者将其制备为其它剂型,如固体分散体,自纳米乳化片剂等。中国发明专利cn103497178a和cn110452156a分别公开了使用有机溶剂挥发法制备的厄贝沙坦瑞格列奈共无定型物和厄贝沙坦多奈哌齐共晶物。虽然这些剂型都可有效的提高厄贝沙坦的溶出度,但是这些剂型的制备过程中会引入有机溶剂而产生一定的毒副作用,或加入大量的辅料等而不适于高剂量药物的生产。
3.纳米混悬液是通过加入合适的聚合物和/或表面活性剂稳定剂而制备成的一种粒径小于1000nm的稳定胶体系统,也是一种改善难溶性药物溶解度的有前景的策略。它具有一些明显的优势,如适用于不同物理化学性质的分子,高载药量,改善剂量
‑
生物利用度比例,减少毒副作用,提高对生物膜和组织的黏附性,提高患者的依从性等。纳米混悬液从开发到工业化规模取得了最快的突破。第一个基于纳米悬液的产品在1990年专利申请后仅10年左右就已上市。在制备纳米混悬液的方法中,湿介质研磨法适用于工业化生产,使用该方法制备的部分产品也已投放市场,例如因此通过湿研磨法将多晶型厄贝沙坦制备为纳米混悬液,有助于工业大生产并改善其生物利用度上的一些限制。另外,一种可以从电子结构分析到宏观性能预测的跨尺度计算方法可以用于筛选稳定剂,纳米混悬液中的厄贝沙坦晶型转化情况的监控也是必须的。但是,目前使用计算手段来设计纳米混悬液的处方以及使用湿研磨法制备多晶型厄贝沙坦纳米混悬液的产品较少。
4.因此,如何使用计算方法来设计优化纳米混悬液处方,并提供一种性能稳定的,且能够显著提高溶出度的多晶型厄贝沙坦纳米混悬液及其制备方法是本领域技术人员亟需解决的问题。
技术实现要素:
5.有鉴于此,本发明提供了一种性能稳定的,且能够显著提高溶出度的多晶型厄贝
沙坦纳米混悬液。
6.为了实现上述目的,本发明采用如下技术方案:一种多晶型厄贝沙坦纳米混悬液,所述多晶型厄贝沙坦纳米混悬液由聚合物稳定剂和厄贝沙坦经纳米化处理制成;
7.所述厄贝沙坦的结构包括晶型a,晶型b或无定型,所述聚合物稳定剂为聚维酮va64、大豆磷脂、壳聚糖、聚乙二醇6000(peg6000)、十二烷基硫酸钠(sds)、聚乙烯己内酰胺
‑
聚乙酸乙烯酯
‑
聚乙二醇接枝共聚物(soluplus)、泊洛沙姆407(p407)、维生素e聚乙二醇琥珀酸酯(tpgs)和羟丙基甲基纤维素(hpmce5)中的任一种或几种的组合;
8.所述厄贝沙坦与所述聚合物稳定剂的质量比为10:(1
‑
5)。
9.本发明的有益效果:本发明中的聚合物稳定剂和多晶型厄贝沙坦间产生的氢键或疏水力使得焓减和/或熵增,从而有助于形成粒径变化小且分布均匀的稳定纳米体系。
10.优选地,所述多晶型厄贝沙坦纳米混悬液中厄贝沙坦的粒径为100
‑
500nm,所述多晶型厄贝沙坦纳米混悬液的载药量为12%
‑
35%,所述多晶型厄贝沙坦纳米混悬液zeta电位的绝对值为2
‑
12mv;
11.所述多晶型厄贝沙坦,其溶出性均具有ph依赖性,分别在ph 1.2、4.5和6.8时,多晶型厄贝沙坦纳米混悬液在5~10min的溶出度分别比厄贝沙坦原料药提高9~18、15~25和20~45倍。
12.优选地,所述聚合物稳定剂为聚乙烯己内酰胺
‑
聚乙酸乙烯酯
‑
聚乙二醇接枝共聚物和泊洛沙姆407的组合、维生素e聚乙二醇琥珀酸酯和羟丙基甲基纤维素的组合。
13.优选地,所述聚合物稳定剂经过筛选得到,应用计算机模拟软件通过空间构象和能量匹配原理辅助筛选聚合物稳定剂;
14.所述聚合物稳定剂和多晶型厄贝沙坦在三维空间可进行结构匹配,且产生的氢键或疏水力使得焓减和/或熵增而使它们具有能量自发结合的趋势,有助于形成粒径变化小且分布均匀的稳定纳米体系。
15.具体的筛选方法为:采用schrodinger软件的molecular docking模块和materials studio的blends模块通过空间构象和能量匹配原理辅助筛选聚合物稳定剂。
16.优选地,所述泊洛沙姆407的促晶作用使无定型在聚乙烯己内酰胺
‑
聚乙酸乙烯酯
‑
聚乙二醇接枝共聚物、泊洛沙姆407和无定型的纳米混悬液中转化为a型,羟丙基甲基纤维素的抑晶作用使晶型a在维生素e聚乙二醇琥珀酸酯、羟丙基甲基纤维素和a型的纳米混悬液中转化为无定型。
17.优选地,所述b型的厄贝沙坦在聚乙烯己内酰胺
‑
聚乙酸乙烯酯
‑
聚乙二醇接枝共聚物、泊洛沙姆407和b型的纳米混悬液的研磨和冻干过程中转化为a型和无定型;
18.所述b型的厄贝沙坦在维生素e聚乙二醇琥珀酸酯、羟丙基甲基纤维素和b型的纳米混悬液的研磨过程中转化为a型,在冻干和研磨的共同作用下转化为无定型;
19.所述聚乙烯己内酰胺
‑
聚乙酸乙烯酯
‑
聚乙二醇接枝共聚物与泊洛沙姆407的组合和维生素e聚乙二醇琥珀酸酯与羟丙基甲基纤维素的组合中的羟基基团的“水溶剂”效应诱导b型在与它们制备的物理混合物中发生固态质子转移,转化为a型。
20.本发明中还提供了一种多晶型厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
21.(1)所述的多晶型厄贝沙坦纳米混悬液,称取各原料;
22.(2)将聚合物稳定剂溶于分散介质水中,得到混合液;
23.(3)在混合液中加入厄贝沙坦a型、b型或无定型原料药,搅拌均匀,得粗混悬液;
24.(4)将粗混悬液进行研磨,得到a型和/或无定型厄贝沙坦纳米混悬液。
25.优选地,步骤(1)中,所述的分散介质水为50ml的纯化水。
26.优选地,步骤(2)中,所述搅拌采用磁力搅拌,所述搅拌时间为10
‑
30min。
27.优选地,步骤(4)中,所述研磨过程中加入研磨珠,所述研磨珠为钇稳定的氧化锆研磨珠,所述研磨珠的粒度为0.2
‑
0.8mm。
28.优选地,步骤(4)中,所述研磨时间为15min,速度为2500rpm,温度为10
‑
40℃。
29.优选地,还包括冻干的步骤:多晶型厄贝沙坦纳米混悬液的冻干方法:将所述的多晶型厄贝沙坦纳米混悬液放置于90mm的培养皿中并在
‑
80℃的冰箱中冷冻12
‑
24h,然后使用6l冷冻干燥机(labconco corporation,kansas city,mo,usa)在
‑
40℃~
‑
80℃以下冻干12
‑
24h。
30.本发明中还提供了一种多晶型厄贝沙坦纳米混悬液作为药物制剂中间体的应用,所述药物制剂包括片剂、颗粒剂、胶囊剂、缓释剂。
31.经由上述的技术方案可知,与现有技术相比,本发明公开提供了一种多晶型厄贝沙坦纳米混悬液及其制备方法和应用,具有如下有益效果:
32.(1)本发明不使用有机溶剂的条件下,使用少量的辅料将厄贝沙坦的不同晶型和无定型制备为纳米混悬液,改善药物的溶出度。
33.(2)本发明通过聚合物稳定剂的促晶或抑晶作用使a晶型或无定型发生转化,并通过聚合物稳定剂的羟基基团的“水溶剂”效应诱导b晶型在与它们制备的物理混合物中发生固态质子转移。
34.(3)本发明中的操作过程简单,高效,安全且适用于工业大生产的纳米混悬液制备方法。
附图说明
35.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
36.图1为通过sitemap获得组合的聚合物稳定剂的最佳结合位点;其中,图1
‑
(a)中的聚合物稳定剂为soluplus
‑
p407,图1
‑
(b)tpgs
‑
hpmce5;红色区域表示氢键受体区域,紫色区域表示氢键给体区域,黄色表示疏水单体区域;
37.图2为通过分子对接的相互作用预测;其中,soluplus
‑
p407和tpgs
‑
hpmce5与厄贝沙坦a型/无定型分子的空间对接如图2
‑
(a)和2
‑
(b)所示;图2
‑
(c)和2
‑
(d)表示厄贝沙坦b型分子与soluplus
‑
p407和tpgs
‑
hpmce5的空间对接;黄色虚线表示氢键,蓝色虚线表示芳香氢键,绿色虚线表示pi相互作用;
38.图3为聚合物稳定剂为soluplus
‑
p407(a)和tpgs
‑
hpmce5(b)的红外光谱图;;(a
‑
c)曲线为原料药,(d
‑
f)曲线为物理混合物,(g
‑
i)曲线为纳米混悬液;图3
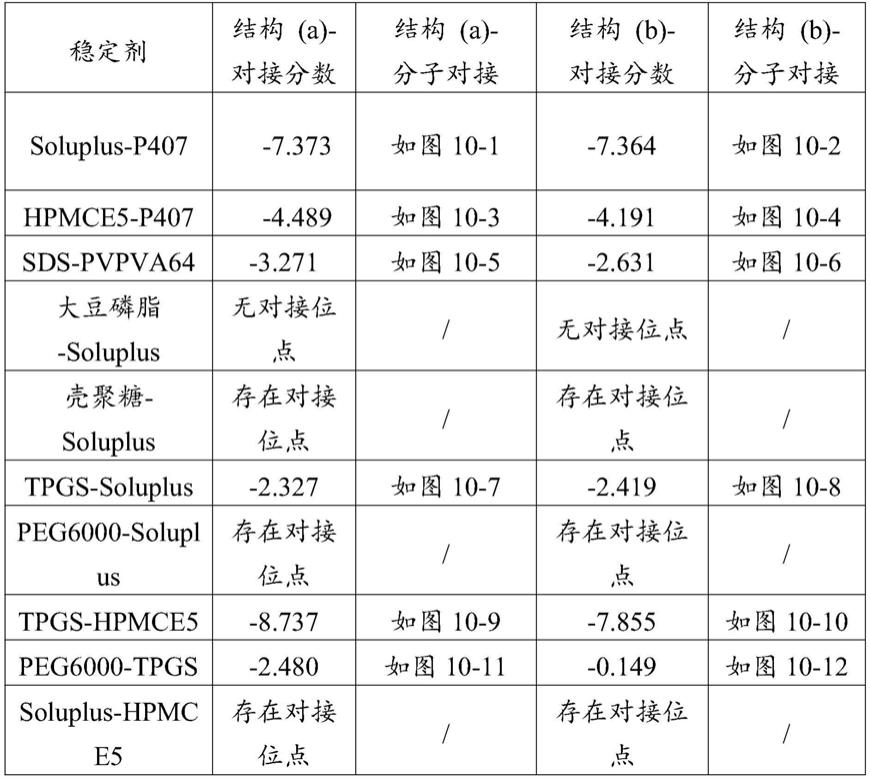
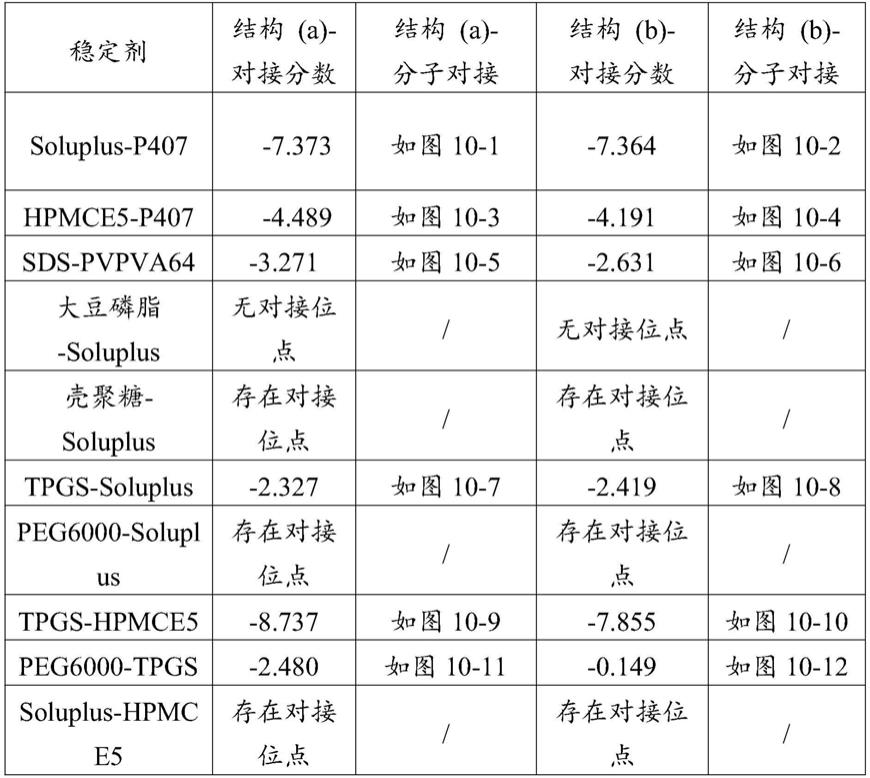
‑
a中的(j)曲线为soluplus,(k)曲线为p407,图3
‑
b中的(j)曲线为tpgs,(k)曲线为hpmce5;
39.鲜红色的线代表厄贝沙坦a型,蓝黑色的线代表厄贝沙坦b型,橙色的线代表厄贝
沙坦无定形;
40.图4为聚合物稳定剂soluplus
‑
p407(a)和tpgs
‑
hpmce5(b)的拉曼光谱图;(a
‑
c)曲线为原料药,(d
‑
f)曲线为物理混合物,(g
‑
i)纳米混悬液;图4
‑
a中的(j)曲线为soluplus,(k)曲线为p407,图4
‑
b中的(j)曲线为tpgs,(k)曲线为hpmce5;鲜红色的线代表厄贝沙坦a型,蓝黑色的线代表厄贝沙坦b型,橙色的线代表厄贝沙坦无定形;
41.图5为不同ph介质中厄贝沙坦的溶出曲线图;其中,(a)、(b)和(c)分别代表了原料药和soluplus
‑
p407纳米混悬液在ph值为1.2、4.5和6.8时的溶出曲线,(d)、(e)和(f)分别代表原料药和tpgs
‑
hpmce5纳米混悬液在ph 1.2、4.5和6.8培养基中的溶出曲线;
42.图6为聚合物稳定剂soluplus
‑
p407(a)和tpgs
‑
hpmce5(b)的xrd图;其中,(a
‑
c)曲线为原料药,(d
‑
f)曲线为物理混合物(药物:聚合物稳定剂=10:4),(g
‑
i)纳米混悬液(药物:聚合物稳定剂=10:4),(l)冻干微悬液;图6
‑
a中的(j)曲线为soluplus,(k)曲线为p407,图6
‑
b中的(j)曲线为tpgs,(k)曲线为hpmce5。鲜红色的线代表厄贝沙坦a型,蓝黑色的线代表厄贝沙坦b型,橙色的线代表厄贝沙坦无定型;
43.图7为聚合物稳定剂soluplus
‑
p407(a)和tpgs
‑
hpmce5(b)的dsc图;其中,(a
‑
c)曲线为原料药,(d
‑
f)曲线为物理混合物(药物:聚合物稳定剂=10:4),(g
‑
i)纳米混悬液(药物:聚合物稳定剂=10:4),(l)冻干微悬液,图7
‑
a中的(j)曲线为soluplus/tpgs,(k)曲线为p407/hpmce5,图7
‑
b中(j)曲线为tpgs,(k)曲线为hpmce5;鲜红色的线代表厄贝沙坦a型,蓝黑色的线代表厄贝沙坦b型,橙色的线代表厄贝沙坦无定型;
44.图8为厄贝沙坦的sem和tem图;其中,8
‑
(1、4、7)为a型原料药,8
‑
(2、5、8)为soluplus
‑
p407物理混合物(药物(b型):聚合物稳定剂=10:4),8
‑
(3、6、9)为tpgs
‑
hpmce5物理混合物(药物(无定型):聚合物稳定剂=10:4),图8
‑
(10
‑
12)为soluplus
‑
p407纳米混悬液(药物:聚合物稳定剂=10:4),(e)tpgs
‑
hpmce5纳米混悬液(药物:聚合物稳定剂=10:4);tpgs
‑
hpmce5
‑
b纳米混悬液(药物:聚合物稳定剂=10:1)和sds
‑
b纳米混悬液(药物:聚合物稳定剂=10:4)分别显示在(b
‑
f
‑
1)和(b
‑
f
‑
2)。
45.图9为厄贝沙坦b晶型处方的xrd图(a)和dsc图(b);其中,(a
‑
c)曲线为原料药,(d)和(f)曲线为物理混合物(药物:稳定剂=10:2),(e)和(g)曲线为物理混合物(药物:稳定剂=10:1),(h)曲线为物理混合物(药物:稳定剂=10:4),(i)曲线为纳米混悬液(药物:稳定剂=10:1),(j)曲线为纳米混悬液(药物:稳定剂=10:4),(k)曲线为冻干微悬液(药物:稳定剂=10:1),(l)曲线为冻干微悬液(药物:稳定剂=10:4);亮红色线代表厄贝沙坦a型,蓝黑色线代表厄贝沙坦b型,橙色线代表厄贝沙坦无定型,绿色线代表soluplus
‑
p407组,粉红色线代表tpgs
‑
hpmce5组,水蓝色线代表sds组。
46.图10为irb结构(a)和(b)与不同组合稳定剂的分子对接附图。
具体实施方式
47.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
48.本发明联用schrodinger软件的molecular docking模块和materials studio的
blends模块通过药物与聚合物稳定剂间的空间构象和能量匹配原理辅助筛选联用聚合物稳定剂。
49.实施例1
50.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
51.(1)称取0.1g的soluplus和0.3g的p407加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
52.(2)在混合液中加入1g的厄贝沙坦a型于磁力搅拌器上搅拌10min,得到粗混悬液;
53.(3)向纳米研磨机的研磨室中依次加入粒度为0.2
‑
0.8mm的钇稳定的氧化锆研磨珠和粗混悬液,在温度为10
‑
40℃、速度为2500rpm条件下研磨15min,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释10倍之后测得的粒径为221.1,pdi为0.271。
54.实施例2
55.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
56.(1)称取0.1g的hpmce5和0.3g的p407加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
57.(2)在混合液中加入1g的厄贝沙坦a型于磁力搅拌器上搅拌10min,得到粗混悬液;
58.(3)向纳米研磨机的研磨室中依次加入粒度为0.2
‑
0.8mm的钇稳定的氧化锆研磨珠和粗混悬液,在温度为10
‑
40℃、速度为2500rpm条件下研磨15min,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释5倍之后测得的粒径为224.5,pdi为0.349。
59.实施例3
60.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
61.(1)称取0.1g的sds和0.3g的pvpva64加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
62.(2)在混合液中加入1g的厄贝沙坦a型于磁力搅拌器上搅拌10min,得到粗混悬液;
63.(3)向纳米研磨机的研磨室中依次加入粒度为0.2
‑
0.8mm的钇稳定的氧化锆研磨珠和粗混悬液,在温度为10
‑
40℃、速度为2500rpm条件下研磨15min,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释10倍之后测得的粒径为240.3,pdi为0.373。
64.实施例4
65.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
66.(1)称取0.1g的soluplus和0.3g的壳聚糖加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
67.(2)在混合液中加入1g的厄贝沙坦a型于磁力搅拌器上搅拌10min,得到粗混悬液;
68.(3)向纳米研磨机的研磨室中依次加入粒度为0.2
‑
0.8mm的钇稳定的氧化锆研磨珠和粗混悬液,在温度为10
‑
40℃、速度为2500rpm条件下研磨15min,得到厄贝沙坦纳米混悬液。所得混悬液用纯化水稀释10倍之后测得的粒径为240.3,pdi为0.373;使用此方法无法制得纳米混悬液。
69.实施例5
70.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
71.(1)称取0.1g的soluplus和0.3g的大豆磷脂加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
72.(2)在混合液中加入1g的厄贝沙坦a型于磁力搅拌器上搅拌10min,得到粗混悬液;
73.(3)向纳米研磨机的研磨室中依次加入粒度为0.2
‑
0.8mm的钇稳定的氧化锆研磨珠和粗混悬液,在温度为10
‑
40℃、速度为2500rpm条件下研磨15min,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释2.5倍之后测得的粒径为891.1,pdi为0.343。
74.实施例6
75.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
76.(1)称取0.1g的tpgs和0.3g的soluplus加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
77.(2)在混合液中加入1g的厄贝沙坦a型于磁力搅拌器上搅拌10min,得到粗混悬液;
78.(3)向纳米研磨机的研磨室中依次加入粒度为0.2
‑
0.8mm的钇稳定的氧化锆研磨珠和粗混悬液,在温度为10
‑
40℃、速度为2500rpm条件下研磨15min,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释5倍之后测得的粒径为230,pdi为0.498。
79.实施例7
80.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
81.(1)称取0.1g的0.1g的soluplus和0.3g的peg6000加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
82.(2)在混合液中加入1g的厄贝沙坦a型于磁力搅拌器上搅拌10min,得到粗混悬液;
83.(3)向纳米研磨机的研磨室中依次加入粒度为0.2
‑
0.8mm的钇稳定的氧化锆研磨珠和粗混悬液,在温度为10
‑
40℃、速度为2500rpm条件下研磨15min,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释10倍之后测得的粒径为1246.1,pdi为0.132。
84.实施例8
85.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
86.(1)称取0.1g的tpgs和0.3g的hpmce5加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
87.(2)在混合液中加入1g的厄贝沙坦a型于磁力搅拌器上搅拌10min,得到粗混悬液;
88.(3)向纳米研磨机的研磨室中依次加入粒度为0.2
‑
0.8mm的钇稳定的氧化锆研磨珠和粗混悬液,在温度为10
‑
40℃、速度为2500rpm条件下研磨15min,研磨,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释6倍之后测得的粒径为134.5,pdi为0.273。
89.实施例9
90.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
91.(1)称取0.1g的tpgs和0.3g的peg6000加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
92.(2)在混合液中加入1g的厄贝沙坦a型于磁力搅拌器上搅拌10min,得到粗混悬液;
93.(3)向纳米研磨机的研磨室中依次加入粒度为0.2
‑
0.8mm的钇稳定的氧化锆研磨珠和粗混悬液,在温度为10
‑
40℃、速度为2500rpm条件下研磨15min,研磨,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释5倍之后测得的粒径为453.2,pdi为0.241。
94.实施例10
95.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
96.(1)称取0.1g的soluplus和0.3g的hpmce5加入到50ml蒸馏水中,于磁力搅拌器上
搅拌10min,得到混合液;
97.(2)在混合液中加入1g的厄贝沙坦a型于磁力搅拌器上搅拌10min,得到粗混悬液;
98.(3)向纳米研磨机的研磨室中依次加入粒度为0.2
‑
0.8mm的钇稳定的氧化锆研磨珠和粗混悬液,在温度为10
‑
40℃、速度为2500rpm条件下研磨15min,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释5倍之后测得的粒径为224.5,pdi为0.471。
99.实施例1
‑
10中,可以得出,采用soluplus
‑
p407
‑
a和tpgs
‑
hpmce5
‑
a复合物所制备的纳米混悬液的粒径和pdi相对较低;peg6000
‑
tpgs
‑
a、granulesten
‑
soluplus
‑
a和soluplus
‑
hpmce5
‑
a复合物的结合能较低,对接分数较高,所制备的纳米混悬液的粒径较高;hpmce5
‑
p407
‑
a的对接分数较低,结合能较高,纳米混悬液的pdi相对较高;预选出的组合型的聚合物稳定剂soluplus
‑
p407/tpgs
‑
hpmce5,与a晶型所制备的纳米混悬液粒径和pdi都符合要求。
100.实施例11
101.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
102.(1)称取0.1g的soluplus和0.3g的p407加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
103.(2)在混合液中加入1g的厄贝沙坦b型于磁力搅拌器上搅拌10min,得到粗混悬液;
104.(3)向纳米研磨机的研磨室中依次加入研磨珠和粗混悬液,研磨,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释10倍之后测得的粒径为187.9,pdi为0.194。
105.实施例12
106.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
107.(1)称取0.1g的tpgs和0.3g的hpmce5的p407加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
108.(2)在混合液中加入1g的厄贝沙坦b型于磁力搅拌器上搅拌10min,得到粗混悬液;
109.(3)向纳米研磨机的研磨室中依次加入研磨珠和粗混悬液,研磨,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释6倍之后测得的粒径为231.1,pdi为0.138。
110.实施例13
111.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
112.(1)称取0.1g的soluplus和0.3g的p407加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
113.(2)在混合液中加入1g的厄贝沙坦b型于磁力搅拌器上搅拌10min,得到粗混悬液;
114.(3)向纳米研磨机的研磨室中依次加入研磨珠和粗混悬液,研磨,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释6倍之后测得的粒径为305.6,pdi为0.374。
115.实施例14
116.厄贝沙坦纳米混悬液的制备方法,包括以下步骤:
117.(1)称取0.1g的tpgs和0.3g的hpmce5加入到50ml蒸馏水中,于磁力搅拌器上搅拌10min,得到混合液;
118.(2)在混合液中加入1g的厄贝沙坦b型于磁力搅拌器上搅拌10min,得到粗混悬液;
119.(3)向纳米研磨机的研磨室中依次加入研磨珠和粗混悬液,研磨,得到厄贝沙坦纳米混悬液;所得混悬液用纯化水稀释6倍之后测得的粒径为320.4,pdi为0.321。
120.效果验证
121.一、组合的聚合物稳定剂的筛选
122.1、空间构象分析筛选:使用schrodinger suite 2017
‑
2版进行了分子对接研究。在蛋白质制备模块的默认参数下,对每个聚合物的结构进行了优化。然后使用binding site detect模块确定组合稳定剂的结合位点,并通过比较sitescore的值获得最佳结合位点。然后利用ligpre模块对默认参数下的药物分子结构进行优化。在前期准备的基础上,通过配体对接模块对不同组合稳定剂与药物分子进行空间结合,并从对接评分中对稳定剂进行评价筛选。对接分数越低代表空间结合越强。
123.表1.irb结构(a)和(b)与不同组合稳定剂的分子对接结果。
[0124][0125]
由表1可知大豆磷脂
‑
soluplus不能产生合适的结合位点,可能是因为组件构建的联用稳定剂链太直,无法形成空洞结构/结合位点并完成对接。结合位点存在于壳聚糖
‑
soluplus、peg6000
‑
soluplus和soluplus
‑
hpmce5中,但未能与药物结合,这可能是由于相互作用太弱,无法与结合。此外,tpgs
‑
hpmce5、soluplus
‑
p407和hpmce5
‑
p407的对接分数在所有组中都是最低的,这可能是适合irb的较优稳定剂组合。
[0126]
2、热力学能量分析筛选:使用materials studio软件计算结合能。三维晶型和无定型结构可以分别使用构建晶体模块和非晶细胞模块建立。然后,通过导入这些结构,从共混模块获得晶型/无定型与稳定剂间的热力学性质。该模块的参数采用了干燥力场和qeq电
荷。base项和screen 1/2项对应于药物和稳定剂的1/2。最终可获得三种能量,即:稳定剂间的能量(es1s2)、药物与稳定剂1间的能量(ebs1)、药物与稳定剂2间的能量(ebs2)。然后通过比较这三种能量在不同反应式中的关系来筛选稳定剂。多晶型irb与稳定剂之间的结合强于稳定剂之间的结合,表明稳定剂的稳定性能更强,稳定剂与药物间可自发结合。结合能越低表示药物与稳定剂间结合越强。
[0127]
表2.irb a晶型与不同联用稳定剂间的结合能(kj/mol)。
[0128][0129]
由表2可知不同联用稳定剂与irb a晶型的结合在能量上有很大差异。tpgs
‑
soluplus、tpgs
‑
hpmce5、soluplus
‑
p407、peg6000
‑
tpgs和大豆磷脂
‑
soluplus的e(4)值均低于其他稳定剂组合,表明这些组合稳定剂与a型结合较强。tpgs
‑
soluplus的e(5)值为正,说明联用稳定剂之间的结合强度可能大于此联用稳定剂与irb a形式之间的结合强度,因此tpgs
‑
soluplus不适合作为irb的稳定剂。因此,soluplus
‑
p407、peg6000
‑
tpgs、大豆磷脂
‑
soluplus和tpgs
‑
hpmce5,是适合厄贝沙坦的组合型聚合物稳定剂。
[0130]
综上所述,空间构象和能量匹配模拟表明,soluplus
‑
p407和tpgs
‑
hpmce5具有相对优势,是预筛选出的联用稳定剂。
[0131]
二、纳米混悬液的稳定性的试验
[0132]
1、实验过程:
[0133]
(1)稳定性考察:将纳米混悬液于4℃冰箱中放置30天,考察其稳定性。分别在第1、3、5、7、15和30天测定其平均粒径和pdi。
[0134]
(2)计算辅助:使用schrodinger suite 2017
‑
2版预测稳定剂提供的作用力。在蛋白质制备模块的默认参数下,对每个聚合物稳定剂的结构进行了优化,然后使用binding site detect模块确定组合稳定剂的结合位点,再根据结合位点图而进一步确定;如图1
‑
2;
[0135]
(3)红外光谱:采用傅里叶变换红外光谱(tensor27,bruker optics,karlsruhe,
germany)研究分子间的相互作用;样品包括原料药irb、tpgs/hpmce5、soluplus/p407、物理混合物和固化纳米混悬液,以1:150的比例与溴化钾混合,磨细并压成片剂。波数范围和分辨率分别选择在400
‑
4000cm
‑1和4cm
‑1;如图3;
[0136]
(4)拉曼光谱:利用拉曼光谱(thermo fisher,waltham,usa)对ftir获得的相互作用进行验证;样品包括原料药irb、tpgs/hpmce5、soluplus/p407、物理混合物和固化纳米混悬液,均置于铝板上。采用785nm二极管激光器作为光源的拉曼光谱系统,所有光谱记录在200
‑
1800cm
‑1的光谱范围内,分辨率为6cm
‑1,采集时间相同(10s);如图4。
[0137]
2、实验结果如下表4:
[0138]
表4.irb多晶型纳米混悬液的粒径和pdi
[0139][0140][0141]
由表4可知,这些纳米混悬液相对稳定。而soluplus
‑
p407
‑
无定形/tpgs
‑
hpmce5
‑
无定形纳米混悬液的粒径和pdi高于a型和b型纳米混悬液。这可能是因为无定型具有较高的表面自由能和化学势,导致纳米粒子间相互吸引而造成聚集。
[0142]
由图1可知,soluplus
‑
p407具有氢键供体和受体的空间位置,在tpgs
‑
hpmce5中,具有氢键供体和疏水单体的空间构象。
[0143]
由图2所示,具体的,irb的芳基h和羰基可以与soluplus
‑
p407的醚基的o和
‑
oh形成氢键;irb的芳基h可以与tpgs
‑
hpmce5的羰基形成氢键。
[0144]
由图3所示,irb中
‑
nh伸缩振动峰、
‑
c=o伸缩振动峰、四唑环伸缩振动峰
‑
联苯桥弯曲振动峰分别为~3440cm
‑1、~1740cm
‑1和~1230cm
‑1;聚合物稳定剂的
‑
oh弯曲振动峰与irb的
‑
nh拉伸振动峰重叠;与原料药相比,各纳米混悬液组
‑
nh和
‑
c=o峰均增强;四唑环
‑
联苯桥的峰强度在soluplus
‑
p407纳米混悬液组中增加,而在tpgs
‑
hpmce5纳米混悬液组中
降低。
[0145]
由图4所示,c=o和四唑环
‑
联苯桥的拉曼吸收峰分别位于~1730cm
‑1和~1240cm
‑1;这些峰的变化与傅里叶变换红外光谱相似。
[0146]
这些结果表明,irb多态性与tpgs
‑
hpmce5之间可能同时形成氢键和疏水相互作用,在soluplus
‑
p407 ns中只形成氢键。在形成疏水力的情况下,水分子在空间结合位点的排出导致熵增加,当稳定剂与药物形成氢键时,焓可能降低。熵增和/或焓减的过程可能会使得整个系统的吉布斯自由能降低,产生稳定的纳米混悬液。
[0147]
三、纳米混悬液体外溶出考察
[0148]
1.实验过程
[0149]
(1)溶出实验:根据美国药典usp32
‑
nf27,选择桨法。使用hanson vision g2 elite8溶出仪(hanson research co.,ltd,america)研究原料药和纳米混悬液在三种不同介质(ph=1.2,4.5和6.8)中的溶出度;首先制备溶出介质900ml,加热至37.0
±
0.5℃,然后加入75mg原料药或相当于原料药质量的纳米混悬液;然后在75rpm的转速下进行溶出实验。分别于5、10、15、15、20、30、45、60min时取样10ml样品,同时补充10ml的相同的等温溶出介质。
[0150]
含量测定:用0.45μm滤膜过滤后,采用岛津lc
‑
2030c 3d高效液相色谱仪(shimadzu research laboratory co.,ltd,japan)测定含量;色谱柱为diamonsii c18250 mm 4.6mm,5μm。流动相为磷酸缓冲液,使用三乙胺调节ph至3.0,乙腈(60%:40%),进样量10μl,流速为1.0ml/min,检测波长为245nm。
[0151]
2.实验结果
[0152]
测得的溶出曲线图如图5所示;
[0153]
总的来说,纳米混悬液组的溶出行为优于原料药,且在ph为6.8和1.2时的溶出行为要优于在ph为4.5时的,可能是电解平衡影响其溶出行为,由于irb在ph 4介质中存在质子化和非质子化形式,难以溶解;当ph接近ph 7时,非离子和阴离子形式同时存在,irb在该ph溶液中更容易溶解;ph为4.5时,从高到低的原料药溶出行为为a型、无定形和b型,而从高到低的纳米混悬液溶出行为为无定形、a型和b型;原料药和多态irb在ph 4.5下溶出行为的差异与原料药本身的溶出/溶解度特性有关。无定型的堆积能更低,更容易破坏,其释放受到这种溶解的限制;相反,晶体的释放行为受其溶解度的限制。
[0154]
四、多晶型irb纳米混悬液的晶型转化研究
[0155]
1.实验过程
[0156]
(1)x射线衍射法:采用x射线衍射仪(bruker co.,ltd.,karlsruhe,germany)测定了irb多晶型原料药、tpgs/hpmce5、soluplus/p407、物理混合物、固化纳米混悬液和固化微悬液的晶型状态。采用镍滤光片cu
‑
kα(管电压40mv,管电流40ma)作为光源。扫描范围(2θ)和速度分别为3
°
~40
°
和2
°
/min,扫描步长0.02
°
。
[0157]
(2)差示扫描量热法:采用差示扫描量热仪(mettler toledo co.,ltd.,zurich,switzerland)测定了irb多晶型原料药、tpgs/hpmce5、soluplus/p407、物理混合物、固化纳米混悬液和固化微悬液的热性能。将这些样品(5~10mg)置于铝坩埚中,用铝片覆盖进行测定。在n2保护下,从40℃到220℃的升温速率为10℃/min。
[0158]
(3)扫描电镜:采用s
‑
3400扫描电子显微镜(hitachi high
‑
technologies co.,
ltd.,tokyo,japan)对irb、tpgs/hpmce5、soluplus/p407、物理混合物和固化纳米混悬液的形貌进行了表征。将样品放置在由铝头固定的双面导电胶带上,并镀金至5纳米厚。
[0159]
(4)透射电镜:采用透射电子显微镜(hitachi high
‑
technologies co.,ltd.,tokyo,japan)表征了纳米混悬液的形貌。将样品稀释3倍后放置在铜网格上,在室温下干燥,然后观察。
[0160]
2.实验结果
[0161]
由图6可知,irb多晶型的衍射峰位置不同。b型的特征衍射峰分别在8.04
°
和16.04
°
处,a型的特征衍射峰分别在12.35
°
处。在固化的soluplus
‑
p407
‑
a/b和tpgs
‑
hpmce5
‑
a/b纳米混悬液中,a和b形态的峰值强度显著降低,与无定型的峰值强度相似。固化的soluplus
‑
p407
‑
无定型纳米混悬液在12.35
°
处有一个小峰,与a型的衍射峰一致,而固化的tpgs
‑
hpmce5
‑
无定型纳米混悬液的的衍射峰在12.60
°
处。这表明,在固化的soluplus
‑
p407
‑
a/b和tpgs
‑
hpmce5
‑
a/b纳米混悬液中,a和b形态可能转化为无定型,而在soluplus
‑
p407
‑
无定型中,a形态可能部分转化为无定型。
[0162]
由图7可知,a、b和无定形的吸热峰分别位于187.31℃、193.12℃和181.50℃。soluplus
‑
p407
‑
b和tpgs
‑
hpmce5
‑
b物理混合物中b型的吸热峰转移到187.31℃,固化后soluplus
‑
p407
‑
b纳米混悬液中的b型的峰值又从187.31℃转至181.50℃。另外,固化后soluplus
‑
p407
‑
amorphous纳米混悬液的吸热峰变为183.28℃,tpgs
‑
hpmce5
‑
a/b/无定型的固化纳米混悬液组的吸热峰均变为181.50℃。这说明在固化的tpgs
‑
hpmce5
‑
a/b纳米混悬液中,b型和a型均有可能转变为无定型。在soluplus
‑
p407
‑
b固化纳米混悬液中,b型可能转变为无定型。由于吸热峰的微小变化,在soluplus
‑
p407
‑
无定形固化纳米混悬液中可能发生一些转变。
[0163]
由图8可知,扫描电镜图显示原料药a型为不规则形状,b型为四方棱柱型,无定型为针型;在soluplus
‑
p407
‑
b和tpgs
‑
hpmce5
‑
b物理混合物中,存在a型的形貌。透射电镜图可知,在soluplus
‑
p407
‑
amorphous/tpgs
‑
hpmce5
‑
a纳米混悬液中显示了a型/无定型的形貌;soluplus
‑
p407
‑
b纳米混悬液的部分形貌类似于a型的小颗粒聚集,以及继续生长的无定型形态;tpgs
‑
hpmce5
‑
b纳米混悬液中存在b型和a型形态;
[0164]
其中,图8
‑
10为soluplus
‑
p407 a型纳米混悬液,8
‑
11为b型纳米混悬液,8
‑
13为无定型纳米混悬液。
[0165]
由图9可知,经xrd分析,所得物理混合物的衍射峰均与b型相一致;在dsc中,只有tpgs
‑
hpmce5
‑
b(药物:稳定剂=10:1)和sds
‑
b物理混合物的吸热峰与b型相一致。这表明,如果联合使用的稳定剂中没有
‑
oh基团,b型在物理混合物中可能不会转化。加入少量tpgs
‑
hpmce5也不能转化b型。这可能是由于与soluplus
‑
p407相比,tpgs
‑
hpmce5中含有少量的
‑
oh基团。
[0166]
这些结果表明,hpmce5能吸附在药物颗粒表面,使a型转变为无定形;p407通过溶解和吸附在无定型表面而降低了无定型的表面自由能,从而降低了结晶能,促进了无定型结晶;另外,soluplus
‑
p407和tpgs
‑
hpmce5的oh通过“水溶剂”效应而俘获了irb四唑环的
‑
nh基团上的质子。
[0167]
本说明书中各个实施例采用递进的方式描述,每个实施例重点说明的都是与其他实施例的不同之处,各个实施例之间相同相似部分互相参见即可。对于实施例公开的装置
而言,由于其与实施例公开的方法相对应,所以描述的比较简单,相关之处参见方法部分说明即可。
[0168]
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。