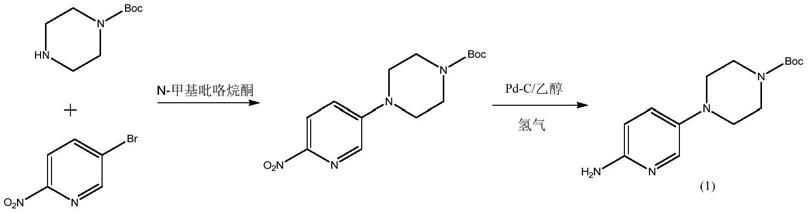
一种4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯的工业化制备方法
技术领域
1.本发明属于化学合成技术领域,具体涉及一种4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯的工业化制备方法。
背景技术:
2.帕布昔利布(palbociclib),化学名为2
‑
[(4
‑
哌啶基)苄基]
‑6‑
乙酰基
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮,由辉瑞公司研制开发,2015年2月在美国率先批准上市,是一种周期蛋白
‑
依赖激酶(cdk)4和6的抑制剂,主要通过调节细胞周期、抑制(cdk)4和6活性来阻止细胞由g1期到s期进而抑制dna的合成,临床主要用于治疗晚期乳腺癌患者。
[0003]4‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式(1))是合成帕布昔利布的关键中间体,cas号为571188
‑
59
‑
5,结构式为:
[0004][0005]
文献报道的4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯的合成方法主要有以下两种:
[0006]
方法一:imaeda,yasuhiro等人(bioorganic&medicinal chemistry,16(6),3125
‑
3140;2008)采用5
‑
溴
‑2‑
硝基吡啶与1
‑
boc
‑
哌嗪在n
‑
甲基吡咯烷酮中反应,得到4
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯,再在乙醇中用钯碳催化氢化还原硝基得到目标产物,反应路线如下:
[0007][0008]
该合成方法的路线短,收率尚可,但因制备1
‑
boc
‑
哌嗪(哌嗪与boc酸酐反应)的收率低,价格较高的boc酸酐利用率低,三废多,不利于环保要求,造成1
‑
boc
‑
哌嗪不仅价格高昂,而且含有大量的双
‑
boc
‑
哌嗪,纯度低,致使上述反应成本高。
[0009]
方法二:jv卡列尼等人在中国专利cn105384741b中报道了5
‑
氯
‑2‑
硝基吡啶首先与哌嗪在正丁醇中通过亲核取代反应得到1
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪盐酸盐,再在四氢呋喃中,以碳酸钾做缚酸剂,与boc酸酐反应,再通过催化还原,得到4
‑
(6
‑
氨基吡啶
‑3‑
基)哌
嗪
‑1‑
羧酸叔丁酯的工艺路线,反应路线如下:
[0010][0011]
该路线制备1
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪盐酸盐的步骤,文献虽然给出了82.3%的收率和熔点范围(mp>230℃),但在碱性条件下,哌嗪两端的氮容易与双分子卤代吡啶发生潜在副反应,生成双分子缩合副产物(tetrahedron letters 39(1998)617
‑
620)。
[0012]
在实际制备过程中,我们观测到2个哌嗪双取代杂质,杂质式(4)和杂质式(5),其反应路线如下:
[0013][0014]
虽然杂质式(4)和杂质式(5),均不与boc哌嗪反应,但其在正丁醇和水中的溶解度均较小,虽然专利cn105384741b中通过步骤2的过滤,杂质式(4)和杂质式(5)均大部分去除,但因其在四氢呋喃中有一定的溶解度,带入4
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯中,通过加氢还原,得到其衍生物杂质式(6)和杂质式(7):
[0015][0016]
杂质式(6)和杂质式(7)在4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯中均难以去除,从而无法得到高纯度的产品。
[0017]
我们在研究钯碳催化还原硝基的过程中,发现微量的杂质式(5),在钯碳还原过程中因发生脱溴反应,生成微量的氢溴酸,进而促使4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯的boc基团水解,生成一系列杂质,其中包含具有潜在基因毒性的杂质,进而影响产品的质量。
[0018][0019]
另外,文献报道的4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯的色泽,均为棕黄色固体,未见白色至类白色固体的报道。但作为以医药原料药及中间体为主的企业,客户给我们提出了浅黄色固体的要求。
[0020]
作为重磅炸弹级新药,帕布昔利布的用量很大,其关键中间体4
‑
(6
‑
氨基
‑3‑
吡啶基)哌嗪
‑1‑
羧酸叔丁酯的用量也会逐年增多。随着医药化工企业对环境保护意识的逐年增强,同时为了满足追求高品质产品客户的需求,开发一种低成本、高收率、高纯度、低毒性、环境友好的4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯的合成方法是当前需要解决的问题。
技术实现要素:
[0021]
针对现有技术的不足,本发明提供了一种4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯的工业化制备方法,该方法采用5
‑
溴
‑2‑
硝基吡啶与哌嗪为起始原料,在正丁醇和水的混合溶剂中,以酸为催化剂通过亲核取代反应制备得到高纯度1
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪,再与boc酸酐在有机溶剂和水的存在下,以弱碱为缚酸剂,制备得到高纯度4
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯,再通过催化氢化,精制脱色,得到高纯度、浅色泽的4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯。该方法操作简便、对环境污染少、收率高、成本低、产品质量好,更适合于工业化生产。
[0022]
本发明的技术方案是:一种4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯的工业化制备方法,其特征是,包括以下步骤:
[0023]
s1:在醇类有机溶剂与水的混合溶剂中,以酸为催化剂,5
‑
溴
‑2‑
硝基吡啶和哌嗪反应生成化合物式(2)化合物的酸式盐,反应结束后,加水并通过共沸蒸馏回收溶剂,得到含有式(2)化合物酸式盐的水溶液,通过过滤除去不溶杂质,用碱调ph10以上,离心,得到高纯度的式(2)化合物湿品;
[0024]
s2:式(2)化合物湿品在有机溶剂与水的混合溶剂中,在缚酸剂的存在下,与boc酸酐反应,反应液后处理得到式(3)化合物;
[0025]
s3:式(3)化合物在极性溶剂中,在催化剂和缚酸剂的存在下,催化氢化,后处理生成式(1)化合物粗品;
[0026]
s4:将式(1)化合物粗品加入水中,再加入酸以使固体溶解,活性炭脱色,用氢氧化钠溶液调ph碱性,经过析晶、离心、洗涤、烘干,得到高纯度(≥99.8%),颜色为白色至浅黄色的4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯。
[0027]
合成路线如下:
[0028][0029]
其中,
[0030]
步骤s1的酸为盐酸、氢溴酸、磷酸、硫酸、醋酸、柠檬酸等的一种或两种混合,优选氢溴酸,所使用酸与哌嗪的摩尔比为0.5~1.5:1,进一步优选为1:1。通过哌嗪成单酸盐的方式,保护哌嗪的氨基的一端,防止双哌嗪取代产物的生成。
[0031]
步骤s1的醇类有机溶剂为正丁醇、正戊醇、异戊醇、叔丁醇、仲丁醇、1
‑
丙醇中的一种,优选正丁醇;其与水的比例为10:0.3~5(v/v),优选10:0.5~2,进一步优选为10:1。
[0032]
步骤s1中的5
‑
溴
‑2‑
硝基吡啶、哌嗪的摩尔比为1:1.2~2,优选1:1.2。哌嗪用量增多,虽有利于哌嗪的单取代产物生成,减少哌嗪双取代产物,但从环保角度考虑,在不影响反应结果的前提下,尽量减少哌嗪的用量,有利于三废处理和环保要求。
[0033]
步骤s1中的反应温度为50~100℃,优选60~65℃。
[0034]
步骤s1中的共沸回收溶剂,通过分批次补充水的方式,共沸蒸馏得到溶剂与水的混合物,通过分液分出有机溶剂和水,有机溶剂直接用于下一批反应,水用于下一批次物料共沸蒸馏,实现溶剂的零排放。
[0035]
步骤s1中通过过滤除去式(4)及式(5)双聚杂质,再用碱调至碱性,得到高纯度的式(2)化合物。
[0036][0037]
反应过程产生的哌嗪双取代杂质式(4)和式(5)两个杂质,在酸性条件下,略溶解于有机溶剂,不溶于水。采用共沸除有机溶剂的方式,将反应液替换成水,中间体式(2)酸式盐溶解于水中,少量的哌嗪双取代杂质直接通过过滤去除,再采用式(2)化合物在强碱中溶解度较小的特点,将反应液调ph值至10以上,析出中间体式(2),通过离心得到中间体式(2)湿品,不用干燥,可直接进行下一步反应。
[0038]
步骤s2中的缚酸剂为碳酸钠、碳酸钾、三乙胺、二异丙基乙胺、醋酸钠、氢氧化钠、氢氧化钾中的一种,优选碳酸钠,式(2)化合物与缚酸剂、boc酸酐的摩尔比为1:1.2~1.8:1.0~1.5。
[0039]
步骤s2中的有机溶剂为二氯甲烷、三氯甲烷、甲苯、四氢呋喃、二氧六环中的一种,优选甲苯和二氯甲烷。
[0040]
步骤s2中的后处理为:升温至固体溶解后,静置分液,分出水层,有机层洗涤,减压浓缩,重结晶。
[0041]
步骤s3中的催化剂为钯碳、铂碳、雷尼镍中的一种,优选铂碳,式(3)化合物与催化剂的质量比为1:0.01~1:0.05,优选1:0.03。
[0042]
步骤s3中的缚酸剂为醋酸钠、醋酸钾、三乙胺、氨水、碳酸钠、碳酸氢钠中的一种,优选醋酸钠。式(3)化合物与缚酸剂的摩尔比为1:0.01~0.1,优选1:0.01~0.05,进一步优选为1:0.02。硝基的催化氢化还原,微量式(5)杂质的脱溴反应,生成的溴化氢溶解于氢化还原产生的水,导致反应液具有一定的酸性,导致boc集团发生微量降解,导致生成多个式(1)化合物中的微量杂质,其中包含具有遗传毒性警示结构的杂质。采用增加少量缚酸剂,中和产生的氢溴酸的方式,以阻止boc基团的降解,有利于提高产品的纯度和质量。
[0043]
步骤s3中的极性溶剂为甲醇、乙醇、异丙醇、正丁醇中的一种,优选甲醇。甲醇更有利于回收处理,经济成本也更低,同时,甲醇对缚酸剂醋酸钠也有一定的溶解性。
[0044]
步骤s3中的反应温度为10~60℃,优选10~30℃。
[0045]
步骤s3的后处理为:过滤,滤液减压浓缩至干,正庚烷重结晶。
[0046]
步骤s4的酸为醋酸、甲酸、柠檬酸、草酸、丙酸中的一种,优选醋酸。
[0047]
本发明的技术特点和优益效果:
[0048]
1、本发明利用5
‑
溴
‑2‑
硝基吡啶与哌嗪,以正丁醇等有机溶剂与水为混合溶剂,以氢溴酸等为催化剂,制备得到1
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪的酸式盐,通过共沸蒸馏,使溶剂实现回收利用,通过过滤去除产生的微量哌嗪双取代,再通过调碱,得到高纯度的1
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪(式(2))。该步骤反应条件温和,后处理简洁,产品无需烘干,质量好,收率高,直接按100%投料。有机溶剂可重复利用,废水为无机盐类,易于处理,更适合工业化生产。
[0049]
2、本发明直接采用1
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪湿品,在两相体系中与boc酸酐反应,通过分液,洗涤去除无机物,减压蒸馏,再结晶得到高纯度的4
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式(3)),蒸出的有机溶剂可以直接回收套用。
[0050]
3、本发明通过加入醋酸钠等缚酸剂的方式,采用更廉价的铂碳等做催化剂进行硝基的氢化还原,制备4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式(1))。缚酸剂的加入阻止了boc基团的降解,阻碍了潜在的基因毒性杂质的产生,提高了产品的安全性,进一步的提高了帕布昔利布的安全性。
[0051]
4、本发明创造性的通过4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯在弱酸性溶解的特点,采用活性炭脱色的方式,去除影响色泽的高分子集团,通过调碱得到高纯度、浅色泽的4
‑
(6
‑
氨基
‑3‑
吡啶基)哌嗪
‑1‑
羧酸叔丁酯。
[0052]
综合以上,本发明为4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯的合成,提供了一条高质量、低成本、对环境友好、高收率(≥85%)、高纯度(纯度可达99.9%)的适合工业化生产的制备方法。
具体实施方式
[0053]
下面结合具体实施例对本发明作更进一步的说明,以便本领域的技术人员更了解本发明,但并不因此限制本发明。
[0054]
实施例1:1
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪(式(2)化合物)的实验室制备
[0055]
2000ml反应瓶中,加入5
‑
溴
‑2‑
硝基吡啶101.5g(0.5mol),无水哌嗪51.6g(0.6mol),正丁醇1000ml,水100ml及氢溴酸(48%)100g,控制60~65℃搅拌反应24h。反应毕,加入300ml水,减压蒸馏,再通过阶段补充的方式,补充1200ml水,蒸馏至馏出液变清。过滤除去不溶固体,母液用30%氢氧化钠调ph12,析出亮黄色固体。过滤,收集固体得1
‑
(6
‑
硝基
‑3‑
吡啶基)哌嗪湿品,以100%计算收率,直接投下一步反应,hplc纯度≥99.5%。
[0056]
实施例2:4
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式(3)化合物)的实验室制备
[0057]
2000ml反应瓶中,加入上步制备的1
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪湿品全部,甲苯1000ml,水500ml,碳酸钠79.5g(0.75mol),控制反应温度10~30℃,缓慢加入boc酸酐131g(0.6mol),加毕继续反应1h,tlc检测至原料消失。反应毕,升温至95℃,固体溶解后,静置分液,分出水层,有机层加入500ml水,保持90~95℃,洗涤一次,减压蒸除约500ml甲苯,降温至0~10℃搅拌析晶,过滤,烘干得亮黄色固体142g,两步反应收率92.2%,纯度99.8%(hplc)。
[0058]
实施例3:1
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪(式(2)化合物)的工业化制备
[0059]
2000l搪玻璃反应釜中,依次投入正丁醇1000l,水100kg,氢溴酸(48%)100kg,5
‑
溴
‑2‑
硝基吡啶101.5kg,无水哌嗪51.6kg,保温60~65℃搅拌反应24h。反应毕,加入300kg纯化水,减压蒸馏,再补充1200kg纯化水,观察馏出液不再浑浊后,停止蒸馏。采用囊式过滤器压滤,50kg水洗涤,滤液用30%氢氧化钠调ph值至12,降温至0
‑
10℃,离心,得亮黄色固体135kg(湿重),hplc纯度99.7%,按100%计直接投下一步反应。
[0060]
实施例4:4
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式(3)化合物)的工业化制备
[0061]
2000l不锈钢反应釜中,依次投入二氯甲烷1000l,水500kg,碳酸钠80kg及上步制备的1
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪湿品135kg,降温至10℃,缓慢加入131kg boc酸酐,加毕继续反应1h,tlc控制反应终点。反应毕,静置分层,有机层用500kg水洗涤一次,常压蒸馏回收二氯甲烷,减压蒸馏残余溶剂。加入500l甲苯,升温至固体溶解,降温至0~10℃搅拌析晶,离心,烘干得145kg亮黄色固体,两步收率94.2%,纯度99.9%(hplc)。
[0062]
实施例5:4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式(1)化合物)的工业化制备
[0063]
2000l高压釜中,投入1000l无水甲醇,145kg 4
‑
(6
‑
硝基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯,醋酸钠0.77kg,通氮气置换3次,将5kg铂碳分散于100l甲醇中,打入高压釜。氮气置换三次,通氢反应3h。压滤,100l甲醇洗涤,收集铂碳回收套用,滤液减压浓缩至干,加入1000l正庚烷,降温至室温,搅拌析晶,离心,正庚烷洗涤,烘干得褐色固体126kg,收率96.3%,纯度99.7%。
[0064]
3000l反应釜中,加入2000kg水,126kg 4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯粗品,30kg醋酸,室温搅拌至固体溶解,加入3kg活性炭(日本白鹭),继续搅拌1h,过滤,100kg水洗涤。母液用10%氢氧化钠调ph值至10,降温至0~10℃析晶2h,离心,水洗,烘干得类白色固体122kg,精制收率96.8%,纯度99.9%(hplc)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。