1.本发明涉及一种纳米聚合物载体材料的合成,具体涉及肿瘤靶向性的还原响应型载体材料,属于生物医用材料和纳米药物制剂领域。
背景技术:
2.癌症已经成为人类健康的主要威胁之一。近年来,肿瘤的发生率和死亡率明显呈上升趋势。在过去的几十年里,人们为战胜癌症付出了巨大的努力,使得我们在癌症治疗领域取得了巨大进展。然而,癌症的一线治疗方法,如手术、化疗和放疗等,依然受到明显的副作用和系统毒性所限制。所以,为了对癌症进行更安全、有效的治疗,发展新兴的治疗方式是必不可少的。
3.光热疗法、光动力疗法和化学动力疗法因其可忽略的侵袭性、低毒性、高选择性以及微创性而成为潜在的抗癌方式。其中,光动力疗法(photodynamic therapy,pdt)是通过光动力作用处理癌症的一种新的治疗模式。pdt是使用光敏剂(pi),通过产生活性氧(ros),如单线态氧(1o2)在光照条件下杀死肿瘤细胞。二氢卟吩因其在红光区的高消光系数和高单线态氧量子产率而被频繁用作光敏剂,其中二氢卟吩e6(chlorin e6,ce6)是最常用的光敏剂,它可在650纳米激光照射条件下产生ros,从而被近红外光激活并迅速从体内消除,它可以将光能转化为热能用于肿瘤的消融。然而,张静等人研究虽然发现叶酸修饰光敏剂ce6可以主动靶向作用聚集靶组织,而不会对周围正常组织造成损伤,但是这也会使光敏剂聚集,造成一定的毒性作用(张静,李园,何婷.化学修饰光敏剂的研究进展[j].化学试剂,2014,36(11):983
‑
987 1020.)。李东红等人采用peg作为桥连连接叶酸和ce6,虽然解决了光敏剂聚集的问题,但是peg修饰纳米载体,到达靶点后,存在不容易释放出来的缺陷(李东红,刁俊林,刘建仓,臧家涛.光敏剂叶酸
‑
卟啉对宫颈癌hela细胞的靶向性和光动力活性[j].中国激光医学杂志,2010,19(05):273
‑
277 336.)。
[0004]
peg是最常用的聚合物之一,也是为数不多获得fda批准用于药物和制药应用的聚合物之一。其分子具有亲水性,当与疏水性聚合物结合时,会自组装成两亲性分子。同时聚赖氨酸(poly
‑
l
‑
lysine,pll)也是一种常见的聚合物,它是由几十个赖氨酸连接而成具有氨基官能团的生物大分子,是一种可生物降解的阳离子多肽,它能与细胞相互作用并粘附在细胞上。
[0005]
由于peg和pll属于生物大分子,为了防止大聚集体的形成,传统的方法是通过插入桥基(例如,可旋转的含σ键的苯环)或柔性接头(例如,硫化物键),使分子结构不那么刚性,从而防止长程有序的分子堆积。其中,最常用的柔性接头之一是二硫键(s
‑
s),具有几乎垂直的双键角和单二面角,在分子自组装过程中提高结构柔性和平衡分子间力方面起着重要作用。
[0006]
由于传统的药物治疗在运输方式上存在系统性障碍和细胞障碍,容易导致高毒性,低靶向性等缺点,所以需要制备一种多功能载体材料进行优化。功能性嵌段共聚物因其具有较高的稳定性,低临界胶束浓度,大小可控以及可功能化修饰等优点,常被应用于构建
新型的纳米药物载体,赵顺新等人所制备的叶酸修饰星型端氨基peg
‑
plga的纳米胶束,虽然有着主动靶向肿瘤组织,对肿瘤细胞具有较好的杀伤作用的优势,但是在制备以peg
‑
plga为载体的纳米粒时,所使用的溶剂容易被plga所溶解,对机体可能有免疫抑制或者免疫刺激作用,从而诱导机体产生不同程度的免疫反应(马桂蕾,赵顺新,金旭,陈旼旼,张政朴,宋存先.叶酸修饰星型端氨基peg
‑
plga纳米胶束的制备及肿瘤细胞靶向作用[j].高等学校化学学报,2012,33(08):1854
‑
1859.)。魏露露公开了叶酸修饰一种刺激响应的两亲性嵌段共聚物的制备,所制备的纳米载体材料较为复杂,成本较高(魏路路.叶酸修饰的肿瘤细胞响应性嵌段共聚物的制备及其对dox的控释行为研究[d].石河子大学,2018.)。
[0007]
叶酸(folic acid,fa)是一种小分子维生素。很多肿瘤细胞的细胞膜上都有一种过表达的特异性的蛋白质(叶酸受体),在正常组织中低表达,而在肿瘤细胞中高表达。与其他肿瘤细胞的靶向受体相比,它具有低免疫原性,易于改造,存储稳定性,与各种有机和水溶性试剂兼容以及成本低等优点。因此,叶酸修饰的纳米载体材料表面具有靶向癌细胞递送的特性。
[0008]
查阅相关的文献和专利发现,本发明制备工艺简单可控、条件温和,所用原材料简单,并且目前还未发现使用光敏剂标记叶酸修饰二硫键连接的两嵌段共聚物作为载体材料将光能转化为热能用于肿瘤的消融以及肿瘤细胞的靶向的报道。
技术实现要素:
[0009]
本发明所要解决的技术问题在于:针对现有的技术条件下,提供一种肿瘤靶向性的还原响应型载体材料。
[0010]
本发明解决其技术问题所采用的技术方案是:
[0011]
一种肿瘤靶向性的还原响应型载体材料,由多嵌段聚合物在水中自组装形成的胶束组成,所述多段聚合物为叶酸
‑
聚乙二醇
‑
ss
‑
聚赖氨酸
‑
二氢卟吩ce6(fa
‑
peg
‑
ss
‑
pll(
‑
g
‑
ce6)),其亲水端为peg,疏水端为pll,通过二硫键进行连接,叶酸靶向肿瘤细胞上的叶酸受体。
[0012]
其结构如下式所示:
[0013][0014]
其中n≥2,x≥2;
[0015]
式中所示聚合物叶酸
‑
聚乙二醇
‑
ss
‑
聚赖氨酸
‑
二氢卟吩ce6(fa
‑
peg
‑
ss
‑
pll(
‑
g
‑
ce6)),其亲水端为peg,疏水端为pll,通过二硫键进行连接;所述叶酸靶向受体为一种锚定
在细胞膜中甘油磷脂酰肌醇上的单链糖蛋白,在正常组织中低表达,而在肿瘤细胞表面高表达。
[0016]
所述载体材料自组装形成的胶束是一种壳核结构,两亲性嵌段共聚物含有亲水端peg和疏水端pll,疏水端的“核”用以包裹疏水性药物和ce6,亲水端的“壳”由于其亲水性所以可以使整个聚合物胶束更好的溶解在水中。由于在肿瘤环境中,gsh还原酶含量较高,胶束中的二硫键将会还原成巯基,穿透细胞膜,二硫键的断裂,使得药物的光敏剂释放。
[0017]
一种肿瘤靶向性的还原响应型载体材料的制备方法,具体实验步骤包括:
[0018]
(1)制备叶酸的无水dmso溶液,用dcc和nhs溶液活化2~5h,边搅拌边逐滴滴加聚氧乙烯双胺的无水dmso溶液,反应24~48h后,将反应物透析,最后冷冻干燥得到叶酸修饰的聚氧乙烯双胺固体产物fa
‑
peg
‑
nh2;
[0019][0020]
(2)将fa
‑
peg
‑
nh2和丁二酸酐溶解,逐滴滴加1.5~10倍三乙胺溶液,反应24~48h后,旋蒸除去大部分溶剂,浓缩液再用冰乙醚中沉淀,抽滤后真空常温干燥,得到fa
‑
peg
‑
cooh;
[0021][0022]
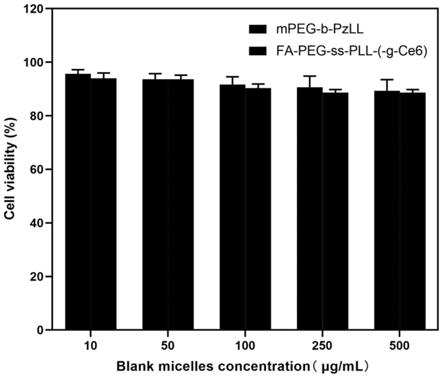
(3)将fa
‑
peg
‑
cooh用dmf溶解,并加入dcc和nhs溶液活化5~6h,将摩尔比为1:5~20的胱胺溶解后,胱胺溶液逐滴滴加到fa
‑
peg
‑
cooh溶液中,室温下反应24~48h,将反应物用冰乙醚沉淀,抽滤后烘干,得到fa
‑
peg
‑
ss
‑
nh2;
[0023][0024]
(4)将步骤(3)所得固体与zll
‑
nca以一定比例溶解于无水dmf中,30~35℃下,反应48~72h,将反应物透析,冷冻干燥后得到fa
‑
peg
‑
ss
‑
pzll;
[0025][0026]
(5)将步骤(4)所得产物用三氟乙酸溶解,并加入冰溴酸溶液,冰水浴,反应1~4h后,将反应物用冰乙醚沉淀,抽滤后冻干,得到fa
‑
peg
‑
ss
‑
pll
‑
nh2;
[0027][0028]
(6)将ce6溶解于无水dmf中,加入edc
·
nhs进行活化2~5h,缓慢加入到fa
‑
peg
‑
ss
‑
pll溶液中,反应24~48h,将反应物透析,冷冻干燥后得到fa
‑
peg
‑
ss
‑
pll(
‑
g
‑
ce6)。
[0029][0030]
步骤(1)中所述的叶酸与nh2‑
peg
‑
nh2的摩尔比为2~6:1;所述的反应需要在n2保护下进行,目的是隔绝空气;所述的透析具体为用透析袋在纯水中透析24h后,将溶液透过微孔滤膜;所述透析袋的分子量截留为500~1500。
[0031]
步骤(2)中所述需要加入三乙胺溶液,三乙胺与fa
‑
peg
‑
nh2摩尔比为1.5~10:1,目的是作为缚酸剂;所述fa
‑
peg
‑
nh2与丁二酸酐的摩尔比为0.5~2:1。
[0032]
步骤(3)中所述加入nhs和dcc溶液,目的是为了活化fa
‑
peg
‑
cooh的羧基端;所述fa
‑
peg
‑
nh2与胱胺的摩尔比为1:5~20。
[0033]
步骤(4)中所述fa
‑
peg
‑
ss
‑
nh2与zll
‑
nca摩尔比为1:15~20;所述的透析袋的分子量截留为3500~5000。
[0034]
步骤(5)中所述加入冰溴酸溶液,目的是为了断裂酰胺键;所述冰溴酸体积约为0.5~5ml。
[0035]
步骤(6)中所述加入nhs和dcc溶液,目的是为了活化光敏剂ce6的羧基端;所述fa
‑
peg
‑
ss
‑
pll与ce6摩尔比为0.5~2:1。
[0036]
所述载体材料用于制备肿瘤靶向性药物,能广泛应用于靶向化疗或靶向光疗体内各种肿瘤部位。
[0037]
本发明制备的一种肿瘤靶向性的还原响应型载体作为一种具有创新性的载体材料,具有以下优点:
[0038]
1.本发明制备的肿瘤靶向性的还原响应型载体材料,原材料叶酸和聚氧乙烯双胺简单易得,制备条件温和,是一种优良抗癌药物靶向载体。
[0039]
2.本发明制备的载体材料由叶酸修饰二硫键连接的两嵌段聚合物,有利于叶酸主动靶向靶组织,并且用还原响应型二硫键连接嵌段共聚物的还原响应有利于载体材料根据肿瘤环境中gsh浓度断裂二硫键,释放药物而不会对周围组织造成损伤。
[0040]
3.本发明制备的肿瘤靶向性的还原响应型载体材料,有利于载体材料上聚赖氨酸的多个氨基接枝多个光敏剂ce6,从而提高胶束的载药量和包封率。
[0041]
4.本发明制备的肿瘤靶向性的还原响应型载体材料具有良好的生物相容性,并不会造成细胞毒性。
[0042]
5.本发明制备的肿瘤靶向性的还原响应型载体材料自组装形成的载药胶束,具有化疗和光动力疗法双重响应的效果。
附图说明
[0043]
图1为本发明制备的中间体fa
‑
peg
‑
nh2的核磁共振氢谱图;
[0044]
图2为本发明制备的中间体fa
‑
peg
‑
ss
‑
pzll的核磁共振氢谱图;
[0045]
图3为本发明制备的fa
‑
peg
‑
ss
‑
pll(
‑
g
‑
ce6)的核磁共振氢谱图;
[0046]
图4为dox标准曲线图;
[0047]
图5为ph7.4 pbs和ph7.4 pbs 10mm dtt缓冲液中不同胶束释放曲线图;
[0048]
图6为两种载体材料的细胞毒性图;
[0049]
图7为不同载体材料在有无激光照射条件下的细胞毒性图;
[0050]
图8为两种不同载体材料的细胞摄图。
具体实施方式
[0051]
下面结合实施例,进一步阐释本发明。应理解,这些实施例仅用于说明本发明,而不用于限制本发明的范围。此外应理解,在阅读本发明讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
[0052]
实施例1
[0053]
(1)fa
‑
peg
‑
nh2合成:称取451.99mg叶酸(fa),1690.26mg二环己基碳二亚胺(dcc)
和647.99mgn
‑
羟基丁二酰亚胺(nhs)溶解于20ml无水二甲基亚砜(dmso)中,在n2保护下,室温避光反应4h,得到活化后的fa溶液;称取5.12g nh2‑
peg
‑
nh2溶解于10ml dmso中,加入3ml无水三乙胺,将活化的fa溶液缓慢滴加到nh2‑
peg
‑
nh2溶液中,n2保护下,室温避光反应48h后,用1000d透析袋透析48h,冷冻干燥后得到产物2.14g fa
‑
peg
‑
nh2。
[0054]
(2)fa
‑
peg
‑
cooh合成:称取1.58g fa
‑
peg
‑
nh2,0.42g丁二酸酐和0.51g4
‑
二甲氨基吡啶(dmap)加入100ml圆底烧瓶中,再加入30.00ml二氧六环溶液,滴加0.58ml三乙胺溶液,磁力搅拌24h后,旋蒸出二氧六环;将残留物各用100ml 0.1m盐酸与乙酸乙酯萃取,有机相弃去,重复萃取3次,水相用100ml二氯甲烷(dcm)萃取3次,合并有机相,采用无水硫酸钠干燥,抽滤,滤液用旋转蒸发仪旋蒸除去大部分dcm,保留约5~10ml dcm,加入150~200ml冰乙醚沉淀12h,抽滤后真空常温干燥得到产物1.20g fa
‑
peg
‑
cooh。
[0055]
(3)fa
‑
peg
‑
ss
‑
nh2合成:称取1.00g fa
‑
peg
‑
cooh,123.00mg dcc,70.00mg nhs和18.30mg dmap溶解于30mln,n
‑
二甲基甲酰胺(dmf)中,n2保护下,冰水浴反应5h后得到活化的fa
‑
peg
‑
cooh溶液;称取867.26mg胱胺溶解于10ml dmf中,将活化后的fa
‑
peg
‑
cooh溶液缓慢的滴加到胱胺溶液中,n2保护下,室温反应24h后,抽滤,滤液用旋转蒸发仪保留约5~10ml,加入150~200ml冰乙醚沉淀12h,抽滤后烘干得到产物1.01g fa
‑
peg
‑
ss
‑
nh2。
[0056]
(4)fa
‑
peg
‑
ss
‑
pzll合成:称取1.01g fa
‑
peg
‑
ss
‑
nh2和2.91g n(ε)
‑
苄氧羰基
‑
l
‑
赖氨酸
‑
n
‑
羧酸酐(zll
‑
nca)溶解于30ml dmf中,35℃下,反应72h后,将反应物用3500d透析袋透析24h,再将溶液过0.45μm滤膜,冷冻干燥后得到产物810.14mg fa
‑
peg
‑
ss
‑
pzll。
[0057]
(5)fa
‑
peg
‑
ss
‑
pll
‑
nh2合成:称取160.00mg fa
‑
peg
‑
ss
‑
pzll溶解于10ml三氟乙酸中,加入0.5ml溴化氢33wt%冰醋酸溶液,冰水浴0℃下,反应1h后,加入150~200ml冰乙醚沉淀12h,将乙醚抽滤,滤渣溶解于10ml dmf中,溶解后放入ph9.0碱性溶液中3500d透析袋透析4~5h后,再用纯水在3500d透析袋中透析24h,冷冻干燥后得到产物131.68mg fa
‑
peg
‑
ss
‑
pll
‑
nh2。
[0058]
(6)fa
‑
peg
‑
ss
‑
pll(
‑
g
‑
ce6)合成:称取28.83mg二氢卟吩e6(ce6),62.10mg edc和46.04mg nhs溶解于10ml无水dmf中,n2保护下,反应4h,得到活化的ce6溶液;称取100.00mg fa
‑
peg
‑
ss
‑
pll溶解于20ml无水dmf中,将活化的ce6溶液缓慢滴加到fa
‑
peg
‑
ss
‑
pll溶液中,n2保护下,室温反应48h后,用3500d透析袋透析48h,冷冻干燥后得到产物65.83mgfa
‑
peg
‑
ss
‑
pll(
‑
g
‑
ce6)。
[0059]
表1实施例1的合成收率
[0060]
步骤(1)(2)(3)(4)(5)(6)理论产量2.50g1.58g1.22g1.01g160mg79.88mg实际产量2.14g1.20g1.01g810.14mg131.68mg65.83mg收率85.60%75.95%82.78%79.97%82.30%82.41%
[0061]
最终产品总收率为29.19%。
[0062]
实施例2
[0063]
(1)fa
‑
peg
‑
nh2合成:称取662.10mg fa,2475.96mg dcc和949.2mgnhs溶解于20ml无水dmso中,在n2保护下,室温避光反应4h,得到活化后的fa溶液;称取7.50g nh2‑
peg
‑
nh2溶解于10ml dmso中,加入3ml无水三乙胺,将活化的fa溶液缓慢滴加到nh2‑
peg
‑
nh2溶液中,n2保护下,室温避光反应48h后,用1000d透析袋透析48h,冷冻干燥后得到产物3.02g fa
‑
peg
‑
nh2。(2)fa
‑
peg
‑
cooh合成:称取2.37g fa
‑
peg
‑
nh2,0.50g丁二酸酐和0.51g dmap加入100ml圆底烧瓶中,再加入30.00ml二氧六环溶液,滴加0.58ml三乙胺溶液,磁力搅拌24h后,旋蒸出二氧六环;将残留物各用100ml 0.1m盐酸与乙酸乙酯萃取,有机相弃去,重复萃取3次,水相用100ml dcm萃取3次,合并有机相,采用无水na2so4干燥,抽滤,滤液用旋转蒸发仪旋蒸除去大部分dcm,保留约5~10ml dcm,加入150~200ml冰乙醚沉淀12h,抽滤后真空常温干燥得到产物2.12g fa
‑
peg
‑
cooh。
[0064]
(3)fa
‑
peg
‑
ss
‑
nh2合成:称取1.50g fa
‑
peg
‑
cooh,185.70mg dcc,103.60mg nhs和19.12mg dmap溶解于30ml dmf中,n2保护下,冰水浴反应5h后得到活化的fa
‑
peg
‑
cooh溶液;称取1324.84mg胱胺溶解于10ml dmf中,将活化后的fa
‑
peg
‑
cooh溶液缓慢的滴加到胱胺溶液中,n2保护下,室温反应24h后,抽滤,滤液用旋转蒸发仪保留约5~10ml,加入150~200ml冰乙醚沉淀12h,抽滤后冷冻干燥得到产物1.29g fa
‑
peg
‑
ss
‑
nh2。
[0065]
(4)fa
‑
peg
‑
ss
‑
pzll合成:称取1.04g fa
‑
peg
‑
ss
‑
nh2和3.00g zll
‑
nca溶解于30ml dmf中,35℃下,反应72h后,将反应物用3500d透析袋透析24h,再将溶液过0.45μm滤膜,冷冻干燥后得到产物875.28mg fa
‑
peg
‑
ss
‑
pzll。
[0066]
(5)fa
‑
peg
‑
ss
‑
pll
‑
nh2合成:称取244.00mg fa
‑
peg
‑
ss
‑
pzll溶解于10ml三氟乙酸中,加入0.5ml溴化氢33wt%冰醋酸溶液,冰水浴0℃下,反应1h后,加入150~200ml冰乙醚沉淀12h,将乙醚抽滤,滤渣溶解于10mldmf中,溶解后放入ph9.0碱性溶液中透析4~5h后,再用纯水3500d透析24h,冷冻干燥后得到产物198.80mg fa
‑
peg
‑
ss
‑
pll
‑
nh2。
[0067]
(6)fa
‑
peg
‑
ss
‑
pll(
‑
g
‑
ce6)合成:称取44.75mg ce6,92.02mg edc和69.05g nhs溶解于10ml无水dmf中,n2保护下,反应4h,得到活化的ce6溶液;称取150.00mg fa
‑
peg
‑
ss
‑
pll溶解于20ml无水dmf中,将活化的ce6溶液缓慢滴加到fa
‑
peg
‑
ss
‑
pll溶液中,n2保护下,室温反应48h后,用3500d透析袋透析48h,冷冻干燥后得到产物112.53mg fa
‑
peg
‑
ss
‑
pll(
‑
g
‑
ce6)。
[0068]
表2实施例2的合成收率
[0069]
步骤(1)(2)(3)(4)(5)(6)理论产量3.66g2.37g1.50g1.04g244.00mg150.00mg实际产量3.02g2.12g1.29g875.28g198.80mg112.53mg收率82.43%89.45%86.00%84.16%81.47%75.02%
[0070]
本发明最终产品总收率为32.62%。
[0071]
合成的中间体及载体材料的核磁共振氢谱图如图1
‑
3所示。
[0072]
图1为fa
‑
peg
‑
nh2的1hnmr分析
[0073]
化学位移3.5ppm处的强信号峰是peg的重复单元(
‑
ch2‑
o
‑
)的质子特征峰;化学位移2.4
‑
2.7ppm处的信号峰是fa上不同位置的
‑
ch2的质子特征峰;化学位移7.7ppm,6.7ppm处的信号峰是fa上苯环的质子特征峰;化学位移8.7ppm处的信号峰是fa上的质子特征峰。
[0074]
图2为fa
‑
peg
‑
ss
‑
pzll的1hnmr分析
[0075]
化学位移3.5ppm的强信号峰是peg的重复单元(
‑
ch2
‑
o
‑
)的质子特征峰;化学位移2.4
‑
2.7ppm处的信号峰是fa上不同位置的
‑
ch2的质子特征峰;化学位移1.3
‑
1.7ppm,3.0ppm,5.0ppm,7.3ppm是pzll重复单元中h的质子特征峰。
[0076]
图3为fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)的1hnmr分析
[0077]
化学位移11.5ppm处的信号峰是ce6上
‑
oh的质子特征峰;化学位移8.0ppm的信号峰是
‑
nh
‑
的质子特征峰;化学位移3.5ppm处的强信号峰peg的重复单元(
‑
ch2
‑
o
‑
)的质子特征峰;化学位移2.4
‑
2.7ppm处的强信号峰是fa上不同位置的
‑
ch2的质子特征峰;化学位移1.3
‑
1.7ppm,2.7
‑
3.1ppm处的信号峰是pzll重复单元中h的质子特征峰。
[0078]
实施例3
[0079]
载药率和包封率实验
[0080]
称取5.00mg mpeg
‑
b
‑
pzll和1.00mg阿霉素(dox)溶解于1ml甲醇溶液中,超声10~15min,搅拌4h后旋蒸。加入10ml纯化水,搅拌过夜形成mpeg
‑
b
‑
pzll载药胶束溶液。
[0081]
称取5.00mg fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)和1.00mg阿霉素(dox)溶解于1ml甲醇溶液中,超声10~15min,避光搅拌4h后旋蒸。加入10ml纯化水,避光搅拌过夜形成fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)载药胶束溶液。
[0082]
(1)色谱条件
[0083]
色谱柱:tianhetm kromasil c18柱(4.6mm
×
250mm,5μm)
[0084]
检测波长:252nm
[0085]
流动相:乙腈:ph 7.4磷酸盐缓冲液(25:75)
[0086]
柱温:35℃
[0087]
流速:1.0ml/min
[0088]
进样量:10μl
[0089]
(2)标准曲线的测定
[0090]
精密称取5mg阿霉素标准品,加入少量甲醇溶液置于25ml容量瓶中利用超声使其溶解,继续滴加甲醇定容至刻度,即得浓度为200μg/ml阿霉素储备液。采用甲醇对阿霉素储备液进行不同浓度梯度的稀释为:200,150,100,50,20,5,1μg/ml的不同浓度的阿霉素储备液。过0.45μm的微孔滤膜后,将样品注入进样小瓶内,待高效液相色谱仪测定。测定结束后,利用色谱分析软件进行积分,按照横轴为阿霉素的浓度,且纵轴为峰面积,绘制阿霉素于甲醇中的标准曲线,如图4所示。
[0091]
(3)载药率和包封率测定
[0092]
将制备的mpeg
‑
b
‑
pzll载药胶束溶液和fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)载药胶束溶液进行超声操作,目的在于破坏胶束的核壳结构,使得所包载的阿霉素从胶束中释放出来。胶束溶液过0.45um微孔滤膜,进行高效液相色谱法(hplc)测定其载药量(drugloadingcapacity,dlc)和包封率(encapsulationefficiency,ee),公式如下:
[0093][0094][0095]
高效液相色谱法测定的结果为:mpeg
‑
b
‑
pzll胶束溶液载药率为5.0%,包封率为73.4%;fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)胶束溶液载药率为15.3%,包封率为95.8%。
[0096]
实施例4
[0097]
ce6接枝量测定实验
[0098]
将制备的fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)胶束溶液,取2ml装入比色皿中,2mlpbs溶液作
为对照装入另一个比色皿中,采用紫外可见分光光度计测定全波长下ce6的吸光度,根据吸光度(660nm)测定ce6的接枝率。计算的ce6接枝率为4.76%。
[0099]
实施例5
[0100]
采用反向动态透析法进行药物的体外释放试验
[0101]
制备的mpeg
‑
b
‑
pzll载药胶束溶液和fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)载药胶束溶液(实施例3制备),将胶束溶液一分为二,转移至一定大小的透析袋中,透析袋两端用棉绳扎紧,放置在分别装有30ml释放介质(pbs或含有10mmdtt的pbs)的离心管中。将整个释放体系置于恒温摇床中(摇床参数为37℃,100rpm),在预定的时间点(0.5,1,2,4,6,8,12,24,48,60,72h)进行取样,每次取1ml释放介质,并补充1ml新鲜介质于离心管中。样品过0.45um微孔滤膜后,注入进样小瓶中,采用hplc进行测定,计算累积释放率,公式如下:
[0102][0103]
如图5所示,相比于游离的阿霉素溶液,其他四种溶液明显存在控释特性。但是,载dox的fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)胶束溶液在肿瘤环境中有着明显的长效缓控释特性。
[0104]
实施例6
[0105]
细胞毒性实验
[0106]
(1)空白胶束的细胞毒性评价
[0107]
称取5.00mg mpeg
‑
b
‑
pzll溶解于1ml甲醇溶液中,超声10~15min,搅拌4h后旋蒸。加入10ml纯化水,搅拌过夜形成mpeg
‑
b
‑
pzll空白胶束溶液。
[0108]
称取5.00mg fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)溶解于1ml甲醇溶液中,超声10~15min,避光搅拌4h后旋蒸。加入10ml纯化水,避光搅拌过夜形成fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)空白胶束溶液。
[0109]
采用mtt法对mpeg
‑
b
‑
pzll和fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)两种空白胶束进行细胞毒性评价。将4t1细胞进行复苏、传代、冻存。在96孔板中接种细胞悬液。将制备的空白胶束(浓度为:500μg/ml)进行不同浓度的稀释,浓度为:10μg/ml、50μg/ml、100μg/ml、250μg/ml、500μg/ml。取100μl不同浓度的空白胶束溶液加入到混有细胞悬液的96孔板中,每组设置3个复孔。加入5mg/ml的预先配置的mtt溶液各20μl进行培养,直至底部出现蓝紫色甲瓒,弃去每孔中的上层清液后,各孔加入适量dmso,采用紫外
‑
可见分光光度计在540nm处测定各组的吸光度,根据吸光度测定各组细胞的存活率(cell survival rate,csr)。
[0110][0111]
如图6所示:两种胶束的存活率均不低于90%,表明其没有细胞毒性。
[0112]
(2)载药胶束的细胞毒性评价
[0113]
采用mtt法进行载药胶束的细胞毒性评价。将稀释并计数后的细胞悬液接种于96孔板后,置于培养箱内继续培养。待贴壁后,弃去每孔中上层清液,分别加入100μl含有药物浓度为0.1μg/ml、1μg/ml、5μg/ml、10μg/ml、50μg/ml的dox&ce6,dox&ce6 激光照射组,dox@fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)和dox@fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6) 激光照射组。同时设置空白、阴性和阳性对照组,每组各3个复孔。孵育细胞24h后,每孔加入预先配置的mtt溶液20μl,继续
在培养箱内孵育,弃去上清液,加入适量dmso,振荡,采用酶标仪检测各组的吸光度,根据吸光度测定各组对4t1细胞的细胞毒性。
[0114]
如图7所示:当激光照射组和无激光照射组相比,对应的激光照射组的4t1细胞的存活率均低于无激光照射组,这说明在激光照射下,化疗药物和光敏剂同时发挥作用对4t1细胞造成的杀伤效果要高于单一的化疗药物。
[0115]
实施例7
[0116]
细胞摄取实验
[0117]
将细胞计数后,接种于共聚焦专用的细胞培养皿中,每孔2ml细胞悬液,继续培养。弃去每孔中上层清液后,用无菌pbs溶液冲洗细胞,分别加入药物浓度均为2μg/ml的游离药物以及两种不同的载药胶束,即dox@mpeg
‑
b
‑
pzll和dox@fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)。在培养箱中孵育细胞4h后,弃去每孔中上层清液,用无菌pbs溶液冲洗细胞。每孔加入适量的多聚甲醛(4%)固定细胞,15min后弃去固定液,再次用无菌pbs溶液冲洗细胞。避光下,各培养皿中加入适量dapi染色,弃去染色剂,使用无菌pbs溶液冲洗细胞,并在激光共聚焦显微镜油镜下观察各组细胞的摄取情况,如图8示:
[0118]
(a)dox@mpeg
‑
b
‑
pzll;(b)dox@fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6);(c)游离dox。当游离dox与4t1细胞共孵育4h后,主要由于游离dox在细胞液中很快被转运到细胞核中与dna结合的缘故;而用dox@mpeg
‑
b
‑
pzll处理后的细胞,细胞质和细胞核的荧光强度相对较弱,表明其进入细胞是由被动扩散所致,它释放dox是一个缓慢的过程;而用dox@fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6)处理后的细胞,荧光强度明显强于dox@mpeg
‑
b
‑
pzll,且细胞核的荧光强度大于细胞质中荧光强度,说明与dox@mpeg
‑
b
‑
pzll相比,dox@fa
‑
peg
‑
ss
‑
pll
‑
(
‑
g
‑
ce6),具有主动靶向作用的fa可以将更多的dox转运至细胞核中,且具有主动靶向肿瘤细胞的作用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。