1.本发明涉及基因工程技术领域,具体涉及一种色氨酸脱羧酶基因原核表达载体及其应用。
背景技术:
2.色氨酸脱羧酶(tryptophan decarboxylase,tdc)是色氨酸脱羧反应的脱羧酶,在植物生长素合成、生物碱类以及次生代谢物合成等众多植物代谢过程中发挥重要作用。研究表明tdc能够将色氨酸和酪氨酸催化脱羧,生成色胺和酪胺。色氨途径是植物细胞内生长素合成的重要途径之一。tdc是色氨途径iaa合成的关键酶。tdc基因是个多基因家族,在烟草、水稻、喜树等多种植物中已被克隆。过量表达tdc基因提高了植物体内iaa含量,表明tdc和生长素生物合成之间的密切联系。
3.利用原核表达系统对外源基因进行表达,具有周期短、成本低、操作简便以及表达量高等优点。目前已有利用原核表达系统生产色氨酸脱羧酶的报道,例如:专利cn111849950a将川桑色氨酸脱羧酶基因tdc连入pcold
‑
tf质粒kpni和sali酶切位点,获得重组表达载体pcold
‑
tf
‑
tdc,再将获得的重组表达载体转化大肠杆菌,诱导表达,获得重组川桑色氨酸脱羧酶。专利cn104232664a将易变山羊草tdc全长序列连接至克隆载体peasy
‑
t1,获得aetdc双元植物表达载体。谢全亮等将棉花tdc基因连入pet32a。构建得到pet32a
‑
ghtdc原核表达载体。但是,由于不同植物中的tdc基因的同源性存在差异,即使是同一种植物中也会存在多条色氨酸脱羧酶基因,因此,需要根据具体的色氨酸编码基因序列选择相应的表达载体;若表达载体选择不当,很可能会导致目的蛋白不能正常可溶表达。
技术实现要素:
4.针对上述现有技术,本发明的目的是提供一种色氨酸脱羧酶基因原核表达载体及其应用。
5.为实现上述目的,本发明采用如下技术方案:
6.本发明的第一方面,提供一种色氨酸脱羧酶基因原核表达载体,其由如下方法构建而成:
7.(1)以pegx
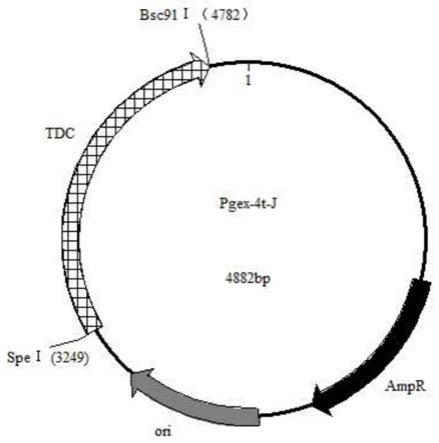
‑
4t
‑
1为出发质粒,将pegx
‑
4t
‑
1利用pflmⅰ和btgⅰ酶切之后连入长度为100bp的人造链,构建得到质粒pegx
‑
4t
‑
j;
8.(2)将编码水稻色氨酸脱羧酶的基因进行密码子优化,优化后的水稻色氨酸脱羧酶的编码基因序列如seq id no.1所示;
9.(3)采用speⅰ和bsc91ⅰ对质粒pegx
‑
4t
‑
j进行双酶切处理,通过dna连接酶将seq id no.1所示的核苷酸片段连入双酶切后的质粒pegx
‑
4t
‑
j上,即构建得到色氨酸脱羧酶基因原核表达载体。
10.优选的,步骤(1)中,先采用pflmⅰ在pegx
‑
4t
‑
1第3250位点处进行单酶切,再采用btgⅰ在pegx
‑
4t
‑
1第4869位点处进行单酶切。
11.优选的,步骤(1)中,质粒pegx
‑
4t
‑
j的长度为3449bp,所构建的质粒pegx
‑
4t
‑
j仅在第3249位点处存在speⅰ酶切位点,仅在第3349位点处存在bsc91ⅰ酶切位点。
12.本发明的第二方面,提供含有上述色氨酸脱羧酶基因原核表达载体的重组菌。
13.本发明的第三方面,提供上述色氨酸脱羧酶基因原核表达载体或者重组菌在色氨酸脱羧酶生产中的应用。
14.本发明的有益效果:
15.(1)本发明从营养匮乏的水稻叶片中克隆得到色氨酸脱羧酶的编码基因。为了使编码基因能够更适用于原核表达系统,本发明对编码基因进行了密码子优化,采用优化后的色氨酸脱羧酶编码基因进行原核表达,目的蛋白(色氨酸脱羧酶)的表达量显著提高。
16.(2)本发明以pgex
‑
4t
‑
1为出发质粒,pgex
‑
4t
‑
1为诱导型质粒,需要加入诱导物诱导后才能表达目的基因,因此可以控制目的基因的表达时间。本发明进一步对pgex
‑
4t
‑
1进行了改造,将质粒pegx
‑
4t
‑
1在第3250位点和第4869位点处进行双酶切之后又连接一段100bp的碱基,这样改造处理的好处是减小质粒大小,原质粒4969bp,经过改造后只有3449bp。而且在改造后的质粒第3249和3349位点处分别设有speⅰ和bsc91ⅰ两个酶切位点,这两个酶切位点与优化后的色氨酸脱羧酶的编码基因相呼应,编码基因的编码区不存在上述两个酶切位点,这样既方便目的基因的插入,又能防止目的基因被误剪。
附图说明
17.图1:优化前的密码子相对适应度图。
18.图2:优化后的密码子相对适应度图。
19.图3:表达载体pgex
‑
4t
‑
j的结构示意图。
20.图4:色氨酸脱羧酶基因原核表达载体的结构示意图。
21.图5:sds
‑
page测定结果;图中,泳道1:空载体(pgex
‑
4t
‑
j)未诱导;泳道2:空载体(pgex
‑
4t
‑
j) iptg诱导;泳道3:pegx
‑
已优化(pegx
‑
tdc
‑
2)未诱导;泳道4:pegx
‑
未优化(pegx
‑
tdc
‑
1)诱导2h;泳道5:pegx
‑
未优化(pegx
‑
tdc
‑
1)诱导4h;泳道6:pegx
‑
已优化(pegx
‑
tdc
‑
2)诱导2h。
具体实施方式
22.应该指出,以下详细说明都是例示性的,旨在对本技术提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本技术所属技术领域的普通技术人员通常理解的相同含义。
23.为了使得本领域技术人员能够更加清楚地了解本技术的技术方案,以下将结合具体的实施例详细说明本技术的技术方案。
24.本发明实施例中所用的试验材料均为本领域常规的试验材料,均可通过商业渠道购买得到。
25.实施例1:水稻色氨酸脱羧酶(tdc)基因的克隆
26.选取营养匮乏水稻的叶片,通过ctab法提取材料的总rna,根据从genbank est数据库(https://www.ncbi.nlm.nih.gov/genbank/genbank)中所获得的完整cdna orf序列信息设计引物(tdcsense和tdcantisense),以反转录合成的cdna为模板,进行pcr扩增全长
开放读码框。
27.tdcsense:5
’‑
gtaaggtatagtatcatc
‑3’
;(seq id no.4)
28.tdcantisense:5
’‑
gtgagttagctaggttggtgtg
‑3’
。(seq id no.5)
29.水稻色氨酸脱羧酶(tdc)基因的cdna全长包含完整开放读码框为1533bp,核苷酸序列如seq id no.3所示;编码507个氨基酸的多肽,理论分子质量为56.21ku。
30.实施例2:水稻色氨酸脱羧酶(tdc)基因的密码子优化
31.为了使水稻色氨酸脱羧酶的编码基因能更好的适应于原核表达系统,本发明进一步对水稻色氨酸脱羧酶的编码基因进行了密码子优化。经密码子优化后的水稻色氨酸脱羧酶的编码基因的核苷酸序列如seq id no.1所示;实际编码507个氨基酸的多肽,其氨基酸序列如seq id no.2所示。
32.优化前的密码子相对适应度图如图1所示;优化后的密码子相对适应度图如图2所示。
33.实施例2:表达载体pgex
‑
4t
‑
j的构建:
34.以质粒pgex
‑
4t
‑
1为出发质粒,先采用pflmⅰ在3250位点处进行单酶切,3’末端再切2个,5’末端切6个,3’末端加a,5’末端加ctagt;再保护;再用btgⅰ在4869位点处进行单酶切,5’末端切4个再加aaga,去掉所有的保护;再加上5
’‑
ctagt
……
g
‑3’
的全长100bp的人造链,人造链中间省略的序列,其只要不包含speⅰ和bsc91ⅰ两个酶切位点,中间省略的序列可任意选择。
35.用speⅰ和bsc91ⅰ对质粒pgex
‑
4t
‑
j进行双酶切并鉴定,双酶切后pegx
‑
4t
‑
j出现3349和100两条,则证明表达载体pgex
‑
4t
‑
j构建成功(图3)。
36.密码子优化后的目的基因没额外增加酶切位点,在质粒上增加speⅰ和bsc91ⅰ两个酶切位点才能将目的基因整合到质粒上。
37.实施例3:色氨酸脱羧酶基因原核表达载体的构建
38.将实施例1扩增得到的pcr产物用speⅰ和bsc91ⅰ进行双酶切,回收得到水稻色氨酸脱羧酶的编码基因tdc;同时用speⅰ和bsc91ⅰ双酶切实施例3构建的表达载体pgex
‑
4t
‑
j;通过dna连接酶将基因tdc(seq id no.3)整合到双酶切后的表达载体pgex
‑
4t
‑
j上,得到色氨酸脱羧酶基因原核表达载体pegx
‑
tdc
‑
1。
39.对构建的色氨酸脱羧酶基因原核表达载体pegx
‑
tdc
‑
1进行单酶切和双酶切(speⅰ和bsc91ⅰ)验证,单酶切后pegx
‑
tdc
‑
1为4782bp,双酶切出现1533和3349两条带,证明目的基因已经整合到质粒上。
40.将实施例2优化后的tdc基因进行pcr扩增,同样使用speⅰ和bsc91ⅰ对质粒进行双酶切,再通过dna连接酶将优化后的tdc基因(seq id no.1)整合到双酶切后的表达载体pgex
‑
4t
‑
j上,得到色氨酸脱羧酶基因原核表达载体pegx
‑
tdc
‑
2。
41.将上述构建原核表达载体pegx
‑
tdc
‑
1和pegx
‑
tdc
‑
2导入大肠杆菌b21(de3),首先在氨苄霉素选择培养基(所述氨苄霉素选择培养基是在lb固体培养基加60μg/ml的氨苄霉素,目的是为了筛选高拷贝质粒的菌株,注意一点,氨苄霉素需在培养基温度降至55℃后再通过滤膜加进去)上筛选,后通过sds
‑
page测定(图5),结果表明tdc基因在工程菌e.coli内表达;经密码子优化后的tdc基因的表达水平明显提高。
42.对比例1:
43.将实施例2优化后的tdc基因通过现有基因工程的方法连接到表达载体pgex
‑
4t
‑
1上,得到色氨酸脱羧酶基因原核表达载体pegx
‑
tdc
‑
3。
44.对比例2:
45.将实施例2优化后的tdc基因通过现有基因工程的方法连接到表达载体pet32a上,得到色氨酸脱羧酶基因原核表达载体pet32a
‑
tdc。
46.对比例3:
47.将实施例2优化后的tdc基因通过现有基因工程的方法连接到表达载体pcold
‑
tf上,得到色氨酸脱羧酶基因原核表达载体pcold
‑
tf
‑
tdc。
48.将本发明实施例构建的色氨酸脱羧酶基因原核表达载体pegx
‑
tdc
‑
1和pegx
‑
tdc
‑
2,以及对比例1
‑
对比例3制备的色氨酸脱羧酶基因原核表达载体导入大肠杆菌b21(de3),分别获得重组菌;将重组菌接入到同样的lb液体培养基中进行诱导培养,诱导培养条件相同(iptg终浓度均为1mm,25℃诱导培养6h),诱导培养后超声破菌,分离上清液,检测上清液中目的蛋白(色氨酸脱羧酶)的表达情况以及活力。其中:
49.目的蛋白的表达情况是利用植物色氨酸脱羧酶(tdc)检测试剂盒(购自于上海臻科生物科技有限公司)测定色氨酸脱羧酶(tdc)的浓度进行表征,其检测原理为:用纯化的色氨酸脱羧酶(tdc)抗体包被微孔板,制成固相抗体,往包被单抗的微孔中依次加入色氨酸脱羧酶(tdc),再与hrp标记的色氨酸脱羧酶(tdc)抗体结合,形成抗体
‑
抗原
‑
酶标抗体复合物,经过彻底洗涤后加底物tmb显色。tmb在hrp酶的催化下转化成蓝色,并在酸的作用下转化成最终的黄色。颜色的深浅和样品中的色氨酸脱羧酶(tdc)呈正相关。用酶标仪在450nm波长下测定吸光度(od值),通过标准曲线计算样品中色氨酸脱羧酶(tdc)浓度。检测方法按检测试剂盒说明书操作。
50.色氨酸脱羧酶的酶活的计算方法如下:
51.通过液相测定色胺生成量计算酶活,取1g l
‑
色氨酸 0.01g辅酶plp,加0.900g色氨酸脱羧酶(通过不同原核表达载体制备得到),溶解,用稀硫酸和naoh溶液调ph为6.0,定容100ml,25℃水浴。通过液相色谱测定反应开始后5
‑
30min的色胺并绘制曲线,最终计算出酶活力。
52.结果见表1。
53.表1:
54.表达载体色氨酸脱羧酶(tdc)浓度色氨酸脱羧酶(tdc)酶活pegx
‑
tdc
‑
10.010μmol/l0.95
‑
1.28upegx
‑
tdc
‑
20.014μmol/l1.03
‑
1.35upegx
‑
tdc
‑
36nmol/l0.90
‑
1.10upet32a
‑
tdc3nmol/l0.85
‑
0.95upcold
‑
tf
‑
tdc4nmol/l0.75
‑
0.90u
55.以上所述仅为本技术的优选实施例而已,并不用于限制本技术,对于本领域的技术人员来说,本技术可以有各种更改和变化。凡在本技术的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。