一种非对称苊基
α
‑
二亚胺配体及其钯配合物和该配合物的制备与应用
技术领域
1.本发明属于乙烯聚合催化技术领域,具体涉及一种非对称苊基α
‑
二亚胺配体及其钯配合物和该配合物的制备与应用。
技术背景
2.聚烯烃是目前市场需求量最大的聚合物材料,在现代社会中起着举足轻重的作用。聚烯烃有超过300个不同的商业产品等级,年产量已超过1.8亿吨,其生产仍处于快速增长阶段。先进的烯烃聚合催化剂的开发一直是全球科研工作者和工业界关注的焦点。鉴于非功能化的聚乙烯(pe)和聚丙烯(pp)等聚烯烃的优异性能,以现有聚烯烃为基础制备功能化的聚烯烃备受关注。然而,聚烯烃材料的非极性特性,使其粘附性和相容性差,这些都极大地限制了它在许多重要领域的应用。在聚烯烃中引入极性官能团可以有效地改善黏附,韧性,涂装性,混溶性,流变性和染色等性质,拓宽聚烯烃的功能,增加聚烯烃的价值。实现非极性烯烃与极性乙烯基单体有效共聚合并非易事,主要原因是非极性烯烃在自由基聚合过程中活性较低,需要高温和高压等苛刻的条件才能功进行聚合,获得的聚烯烃高度支化。而高温导致极性乙烯基单体在自由基过程中的活性太高,容易阻碍烯烃双键与金属中心形成π配位,进而导致链增长不足,甚至导致聚合反应终止。
3.此外,工业上目前广泛研究与应用的前过渡金属催化剂不适合催化烯烃与极性单体的共聚,主要是因为极性官能团容易毒化金属中心,如氧,氮,膦等杂原子,使催化剂失活,不能够催化烯烃与极性单体共聚。并且除了精确控制组分和分子量分布,大多数前过渡金属催化剂制备的聚烯烃都是线性结构。
4.上世纪90年代发明的brookhart型a
‑
二亚胺钯催化剂具有良好极性官能团耐受性,在催化烯烃
‑
极性单体共聚领域实现里程碑式的发展。该类型钯催化剂的链行走或者链异构化会在聚合物链上引入短支链,长支链甚至支链带支链的结构。由于其独特的链行走机理,通过对相应均聚物做很小的改变即可大幅度改变聚烯烃的性质,实现功能化。极性基团随机分布在聚合物链上,可破坏非功能化聚烯烃的晶体结构,而官能团的类型、数量以及沿着非极性链结构的分布位置,对非极性烯烃的性能产生有影响。但在pd(ii)体系中,聚合活性通常比较温和,聚乙烯的分枝密度依然保持不变,经典的pd(ii)体系只能够获得分枝密度在20
‑
40/1000c左右的分支低密度聚乙烯,而由于条件严苛恶劣,共聚物的共聚活性和分子量都很低。
技术实现要素:
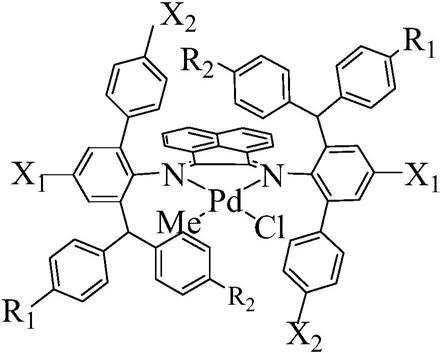
5.为了解决上述问题,本发明的目的之一在于提供一种非对称苊基α
‑
二亚胺配体,该非对称苊基α
‑
二亚胺配体的结构式如式(ⅰ)所示:
[0006][0007]
其中,x1为甲基
‑
ch3、乙基
‑
ch2ch3、丁基
‑
bu或叔丁基
‑
tbu中任意一种;x2为氢原子
‑
h、甲基
‑
ch3中任意一种;r1为甲基
‑
ch3、氟
‑
f、氯
‑
cl、氢原子
‑
h、甲氧基
‑
ome或叔丁基
‑
tbu中任意一种;r2为甲基
‑
ch3、氟
‑
f、氯
‑
cl、氢原子
‑
h、甲氧基
‑
ome中任意一种。
[0008]
本发明还提供上述一种非对称苊基α
‑
二亚胺配体的制备方法,包括以下步骤:
[0009]
s1.在圆底烧瓶中放入0.5
‑
4n zncl2、0.5
‑
3n苊醌和3
‑
20ml冰醋酸的悬浮液,加入0.8
‑
6n的2
‑
(x2基
‑
苯基)
‑4‑
(x1基)
‑6‑
[1,1
‑
(r1‑
苯基)
‑
(r2‑
苯基)]苯胺同类物,加热搅拌回流0.5
‑
5h,冷却至室温;n为当量值;
[0010]
s2.s1中溶液冷却至室温,沉淀出橘红色固体,过滤并分离所述橘红色固体,依次使用乙酸和乙醚洗涤后,在真空下干燥得到橘红色提纯固体;
[0011]
s3.将所述橘红色提纯固体放入盛有5
‑
50ml二氯甲烷的圆底烧瓶中,加入0.5
‑
5n草酸钾水溶液1
‑
20ml,并以500
‑
2000r/min速度搅拌20min;
[0012]
s4.搅拌后溶液进行两相分离,水洗有机溶剂层,并用mgso4干燥除去有机溶剂层水,再减压过滤;
[0013]
s5.过滤后在真空干燥箱中去除溶剂,得到产品为橙色粉末,即为本技术所需的非对称苊基α
‑
二亚胺配体。
[0014]
该制备方法的反应过程为:
[0015][0016]
优选的,所述2
‑
(x2基
‑
苯基)
‑4‑
(x1基)
‑6‑
[1,1
‑
(r1‑
苯基)
‑
(r2‑
苯基)]苯胺同类物的制备方法,具体步骤为:
[0017]
s11.氮气气氛下,将5
‑
15n的4
‑
x2‑
苯基硼酸、5
‑
20n的2
‑
溴
‑4‑
x1‑
苯胺、1
‑
2n的pd(pph3)4和20
‑
100n的k
2 co3的混合物加入四氢呋喃与水的混合物液中,再将混合物液加热至65
‑
80℃,搅拌18
‑
36h;四氢呋喃与水以任意比例混合均可;
[0018]
s12.将反应后的混合物液转移到旋转蒸发器中,去除溶剂,并用二氯甲烷提取残渣,继续转移到分液漏斗,加入水,震荡后静置分离出下层有机相,并用mgso4干燥所述有机相;
[0019]
s13过滤除去mgso4固体,将有机相转移到旋转蒸发器中浓缩,浓缩液通过硅胶柱层析(硅胶:pe/dcm=1:1)分离,分离得到的第二个组分即为2
‑
(x2基
‑
苯基)
‑4‑
(x1基)
‑
苯胺白色固体;
[0020]
s14.将1,1
‑
(r1‑
苯基)
‑
(r2‑
苯基)
‑
甲醇4
‑
16n和s13制备的2
‑
(x2基
‑
苯基)
‑4‑
(x1基)
‑
苯胺4
‑
16n的混合物加热到100
‑
150℃后,向混合物中加入2.2n无水氧化锌和浓盐酸0.5
‑
2ml溶液,继续升温至160
‑
220℃;
[0021]
s15.在160
‑
220℃条件下反应20
‑
80min后,冷却至室温,产物溶于二氯甲烷中,并进行两相分离;
[0022]
s16.分离后,使用100ml水水洗有机溶剂层三遍,并用mgso4干燥除去有机溶剂层水,干燥后溶液在真空干燥箱中浓缩;
[0023]
s17.再用100ml甲醇洗涤浓缩后产物三次,即得到最终产物为2
‑
(x2基
‑
苯基)
‑4‑
(x1基)
‑6‑
[1,1
‑
(r1‑
苯基)
‑
(r2‑
苯基)]苯胺同类物。
[0024]
本发明的目的之二在于提供一种非对称苊基α
‑
二亚胺钯配合物,所述配合物由上述的非对称苊基α
‑
二亚胺配体与钯化合物形成,所述钯化合物为(cod)pdmecl,所述配合物的结构式如式(ⅱ)所示:
[0025][0026]
其中,x1为甲基
‑
ch3、乙基
‑
ch2ch3、丁基
‑
bu或叔丁基
‑
tbu中任意一种;x2为氢原子
‑
h、甲基
‑
ch3中任意一种;r1为甲基
‑
ch3、氟
‑
f、氯
‑
cl、氢原子
‑
h、甲氧基
‑
ome或叔丁基
‑
tbu中任意一种;r2为甲基
‑
ch3、氟
‑
f、氯
‑
cl、氢原子
‑
h、甲氧基
‑
ome中任意一种。
[0027]
上述的非对称苊基α
‑
二亚胺钯配合物的制备方法为:在氮气氛围下,将非对称苊基α
‑
二亚胺配体和(cod)pdmecl按摩尔比1:0.5
‑
2混合并溶解在惰性溶剂中,室温下搅拌反应1
‑
5天;搅拌结束后,过滤得到固体产物,并将所述固体产物溶解在惰性溶剂中,用柱层析法分离出第二组分层,旋转蒸发去除第二分层中溶剂,得到所需的非对称苊基α
‑
二亚胺钯配合物。
[0028]
该反应的过程如下:
[0029][0030]
优选的,所述惰性溶剂为二氯甲烷或三氯甲烷。
[0031]
本发明的目的之三在于提供上述非对称苊基α
‑
二亚胺钯配合物的应用,所述非对称苊基α
‑
二亚胺钯配合物用于烯烃聚合反应的催化剂。
[0032]
优选的,所述应用具体为:所述非对称苊基α
‑
二亚胺钯配合物对乙烯、丙烯或α
‑
烯烃中任意一种或多种进行催化聚合,得到数均分子量mn在11000
‑
30000,分支密度在90
‑
130/1000c的聚烯烃。
[0033]
优选的,所述应用具体为:所述非对称苊基α
‑
二亚胺钯配合物对烯烃与丙烯酸酯类化合物进行催化共聚,获得插入比大于10mol%的烯烃
‑
丙烯酸酯类共聚物。
[0034]
优选的,所述丙烯酸酯类化合物为丙烯酸甲酯、丙烯酸乙酯和丙烯酸正丁酯中任意一种或多种。
[0035]
本发明的有益效果在于:
[0036]
1.本发明提供的非对称苊基α
‑
二亚胺配体为含2
‑
(x2基
‑
苯基)
‑4‑
(x1基)
‑6‑
[1,1
‑
(r1‑
苯基)
‑
(r2‑
苯基)]的非对称苊基α
‑
二亚胺配体,并基于其制备了非对称苊基α
‑
二亚胺pd(ii)配合物,通过引入2
‑
(x2基
‑
苯基)
‑4‑
(x1基)
‑6‑
[1,1
‑
(r1‑
苯基)
‑
(r2‑
苯基)]甲烯胺基取代基,可以有效抑制基于“杂化”苯胺的基于“杂化”苯胺的不对称苊烯基α
‑
二亚胺pd(ii)催化剂体系中的链转移相对于链增长的速度,进而能够在提高所得聚烯烃的支化度的同时,获得较高分子量的无定型聚烯烃。
[0037]
2.本发明提供的非对称苊烯基α
‑
二亚胺钯配合物可以作为烯烃聚合反应的催化剂,由于其独特的环烷结构屏蔽了轴向结合位点,在与丙烯酸酯的结合中表现出了很高的效率,能有效地促进烯烃与丙烯酸酯类的共聚反应,得到高度支化的较高插入比的极性功能化烯烃
‑
丙烯酸酯类共聚物。另外现有技术中对调节聚乙烯分支密度较难,本发明提供的非对称苊烯基α
‑
二亚胺钯配合物与现有的pd(ii)催化剂相比,可产生更高的支链聚乙烯和e
‑
ma共聚物。
附图说明
[0038]
图1a为非对称苊基α
‑
二亚胺配体l1的核磁氢谱;图1b为非对称苊基α
‑
二亚胺配体l1的质谱。
[0039]
图2a为非对称苊基α
‑
二亚胺配体l2的核磁氢谱;图2b为非对称苊基α
‑
二亚胺配体l2的质谱。
[0040]
图3a为非对称苊基α
‑
二亚胺配体l3的核磁氢谱;图3b为非对称苊基α
‑
二亚胺配体l3的质谱。
[0041]
图4a为非对称苊烯基α
‑
二亚胺钯配合物pd1的核磁氢谱;图4b为非对称苊烯基α
‑
二亚胺钯配合物pd1的质谱。
[0042]
图5a为非对称苊烯基α
‑
二亚胺钯配合物pd2的核磁氢谱;图5b为非对称苊烯基α
‑
二亚胺钯配合物pd2的质谱。
[0043]
图6a为非对称苊烯基α
‑
二亚胺钯配合物pd3的核磁氢谱;图6b为非对称苊烯基α
‑
二亚胺钯配合物pd3的质谱。
[0044]
上述核磁共振所用的氘代溶剂在使用前经过干燥和蒸馏,氢谱由jnm
‑
ecz600r光谱仪在室温下记录。
[0045]
上述通过(esi)lcms
‑
2010a来完成非对称苊基α
‑
二亚胺配体l1、l2和l3的质谱分析,通过atouflex speed maldi
‑
tof ms来完成苊基α
‑
二亚胺配体钯配合物pd1、pd1和pd3的质谱分析。
具体实施方式
[0046]
除非另有说明,本文中所使用的术语均具有本领域技术人员常规理解的含义。
[0047]
下面结合实施例对本发明的技术方案做出更为具体的说明:
[0048]
实施例1:
[0049]
一种非对称苊基α
‑
二亚胺配体l1,其结构式如下所示:
[0050][0051]
s1.在圆底烧瓶中放入zncl2(0.17g,1.25mmol)、苊醌(0.18g,1mmol)和5ml冰醋酸的悬浮液,加入2mmol的2
‑
苯基和6
‑
二芳基苯胺,加热搅拌回流0.5h;
[0052]
s2.s1中溶液冷却至室温,沉淀出明亮的橘红色固体,过滤并分离该橘红色固体,依次使用5ml乙酸洗涤三遍、5ml乙醚洗涤5遍,去除残留的乙酸后,在真空下干燥得到明亮的橘红色提纯固体;
[0053]
s3.将上述橘红色提纯固体放入盛有15ml二氯甲烷的圆底烧瓶中,加入草酸钾(0.14g,1.25mmol)水溶液2ml,并快速搅拌20min;
[0054]
s4.搅拌后溶液进行两相分离,5ml水洗有机溶剂层三遍,并用mgso4干燥;
[0055]
s5.过滤后在真空去除溶剂,得到产品为橙色粉末,在高真空下干燥,即得到目标产物非对称苊基α
‑
二亚胺配体l1。
[0056]
本技术制备l1所得质量0.70g,产率73%。l1的氢谱和质谱如下:
[0057]1h nmr(600mhz,cdcl3)δ7.66(d,j=7.6hz,4h,ar
‑
h),7.41(d,j=8.2hz,2h,ar
‑
h),7.14(t,j=7.7hz,4h,ar
‑
h),7.06(dd,j=12.9,4.7hz,6h,ar
‑
h),6.97
–
6.89(m,6h,ar
‑
h),6.83(d,j=8.6hz,4h,ar
‑
h),6.09(d,j=7.2hz,2h,ar
‑
h),5.95(d,j=8.4hz,4h,ar
‑
h),5.36(s,2h,char2),5.28(d,j=8.4hz,4h,ar
‑
h),3.81(d,j=14.9hz,6h,och3),2.95(d,j=14.8hz,6h,och3),2.37(d,j=14.8hz,6h,ch3).
13
c nmr(151mhz,cdcl3)δ162.76(c=n),157.75,156.02,145.57,140.52,139.89,136.37,134.15,133.29,131.34,130.82,130.41,129.60,129.56,129.39,129.31,129.12,128.19,127.28,127.19,126.69,122.78,113.37,112.42,55.29(och3),54.23(och3),50.33(char2),21.35(ch3).
[0058]
apci
‑
ms(m/z):calcd for c
68
h
57
n2o
4
:965.4313,found,965.4340,[m h]
.
[0059]
实施例2:
[0060]
一种非对称苊基α
‑
二亚胺配体l2,其结构式如下所示:
[0061][0062]
s1.在圆底烧瓶中放入zncl2(0.17g,1.25mmol)、苊醌(0.18g,1mmol)和5ml冰醋酸的悬浮液,加入2mmol的2
‑
苯基和6
‑
二芳基苯胺,加热搅拌回流0.5h;
[0063]
s2.s1中溶液冷却至室温,沉淀出明亮的橘红色固体,过滤并分离该橘红色固体,
依次使用5ml乙酸洗涤三遍、5ml乙醚洗涤5遍,去除残留的乙酸后,在真空下干燥得到明亮的橘红色提纯固体;
[0064]
s3.将上述橘红色提纯固体放入盛有15ml二氯甲烷的圆底烧瓶中,加入草酸钾(0.14g,1.25mmol)水溶液2ml,并快速搅拌20min;
[0065]
s4.搅拌后溶液进行两相分离,5ml水洗有机溶剂层三遍,并用mgso4干燥;
[0066]
s5.过滤后在真空去除溶剂,得到产品为橙色粉末,在高真空下干燥,即得到目标产物非对称苊基α
‑
二亚胺配体l2。
[0067]
本技术制备l2所得质量0.62g,产率69%。l2的氢谱和质谱如下:
[0068]1h nmr(600mhz,cdcl3)δ7.66(d,j=7.5hz,4h,ar
‑
h),7.41(d,j=8.2hz,2h,ar
‑
h),7.14(t,j=7.7hz,4h,ar
‑
h),7.09(d,j=8.2hz,6h,ar
‑
h),7.03(d,j=8.0hz,4h,ar
‑
h),6.95
–
6.87(m,6h,ar
‑
h),6.10(d,j=7.1hz,2h,ar
‑
h),5.93(d,j=7.8hz,4h,ar
‑
h),5.56(d,j=7.7hz,4h,ar
‑
h),5.37(s,2h,char2),2.39(s,6h,ch3),2.36(s,6h,ch3),1.36(s,6h,ch3).
13
c nmr(151mhz,cdcl3)δ162.81(c=n),145.68,141.08,140.45,140.03,138.00,135.21,133.94,133.74,133.18,131.32,129.76,129.55,129.50,129.42,129.34,129.10,128.71,128.20,127.97,127.14,126.71,122.74,51.17(char2),21.35(ch3),21.13(ch3),20.18(ch3).
[0069]
apci
‑
ms(m/z):calcd for c
68
h
57
n
2
:901.4516,found,901.4512,[m h]
.
[0070]
实施例3:
[0071]
一种非对称苊基α
‑
二亚胺配体l3,其结构式如下所示:
[0072][0073]
s1.在圆底烧瓶中放入zncl2(0.17g,1.25mmol)、苊醌(0.18g,1mmol)和5ml冰醋酸的悬浮液,加入2mmol的2
‑
苯基和6
‑
二芳基苯胺,加热搅拌回流0.5h;
[0074]
s2.s1中溶液冷却至室温,沉淀出明亮的橘红色固体,过滤并分离该橘红色固体,依次使用5ml乙酸洗涤三遍、5ml乙醚洗涤5遍,去除残留的乙酸后,在真空下干燥得到明亮的橘红色提纯固体;
[0075]
s3.将上述橘红色提纯固体放入盛有15ml二氯甲烷的圆底烧瓶中,加入草酸钾(0.14g,1.25mmol)水溶液2ml,并快速搅拌20min;
[0076]
s4.搅拌后溶液进行两相分离,5ml水洗有机溶剂层三遍,并用mgso4干燥;
[0077]
s5.过滤后在真空去除溶剂,得到产品为橙色粉末,在高真空下干燥,即得到目标产物非对称苊基α
‑
二亚胺配体l3。
[0078]
本技术制备l3所得质量0.53g,产率58%。l3的氢谱和质谱如下:
[0079]1h nmr(400mhz,cdcl3)δ7.65(d,j=7.5hz,4h,ar
‑
h),7.51(d,j=8.2hz,2h,ar
‑
h),7.13(dd,j=14.4,6.7hz,6h,ar
‑
h),6.99(ddt,j=25.1,16.9,8.4hz,12h,ar
‑
h),6.85(s,2h,ar
‑
h),6.12(d,j=7.1hz,2h,ar
‑
h),6.05
–
5.92(m,4h,ar
‑
h),5.49(t,j=8.4hz,4h,ar
‑
h),5.41(s,2h,char2),2.40(s,6h,ch3).
13
c nmr(101mhz,cdcl3)δ162.71(c=n),162.56,160.97,160.13,158.54,145.41,140.25,139.72,139.27,136.42,136.39,133.63,133.17,131.49,131.17,131.09,130.73,130.66,129.94,129.50,129.44,129.27,128.67,128.13,128.05,127.16,126.70,122.64,114.95,114.74,114.03,113.82,50.28(char2),21.24(ch3).
19
f nmr(376mhz,cdcl3)δ
‑
117.29(s),
‑
118.01(s).
[0080]
apci
‑
ms(m/z):calcd for c
64
h
45
f4n
2
:917.3513,found,917.3517,[m h]
.
[0081]
实施例4
[0082]
利用实施例1
‑
3中制备的非对称苊基α
‑
二亚胺配体l1、l2和l3分别与钯化合物pd(cod)mecl反应,制备得到非对称苊烯基α
‑
二亚胺钯配合物pd1、pd2和pd3。
[0083]
制备方法为:在氮气氛围下,将0.5mmol非对称苊烯基α
‑
二亚胺配体(l1、l2或l3)和pd(cod)mecl(133mg,0.5mmol)溶解在10ml二氯甲烷中,在室温下搅拌3天,随着搅拌进行,溶液的颜色逐渐加深。搅拌结束后,用柱层析法分离出所需的非对称苊烯基α
‑
二亚胺钯配合物(pd1、pd2或pd3)。该配合物为橙色或红色的固体。
[0084]
其中,pd1的结构式为:
[0085][0086]
本实施例制备的pd1质量为0.47g,产率84%。pd1的氢谱和质谱如下:
[0087]1h nmr(600mhz,cdcl3)δ8.13(d,j=7.1hz,2h,ar
‑
h),7.95(d,j=7.5hz,2h,ar
‑
h),7.59(d,j=8.3hz,1h,ar
‑
h),7.55(d,j=8.3hz,1h,ar
‑
h),7.35(d,j=8.5hz,2h,ar
‑
h),7.21(td,j=7.9,3.4hz,4h,ar
‑
h),7.13
–
7.09(m,3h,ar
‑
h),7.04(td,j=8.1,2.5hz,5h,ar
‑
h),6.97(t,j=7.4hz,1h,ar
‑
h),6.91(s,1h,ar
‑
h),6.86(dd,j=8.6,4.8hz,4h,ar
‑
h),6.09(d,j=7.1hz,1h,ar
‑
h),6.05(d,j=7.2hz,1h,ar
‑
h),6.02(d,j=8.3hz,2h,ar
‑
h),5.92(s,1h,char2),5.85
–
5.76(m,3h,ar
‑
h,char2),5.22(dd,j=8.0,6.1hz,4h,ar
‑
h),3.82(s,3h,och3),3.81(s,3h,och3),2.89(s,6h,och3),2.40(s,3h,ch3),2.36(s,3h,ch3),1.38(s,3h,pd
‑
ch3).
13
c nmr(151mhz,cdcl3)δ173.24(c=n),169.06(c=n),158.16,157.93,156.18,156.11,143.18,140.18,139.83,139.44,139.00,137.44,136.76,136.24,135.44,134.63,134.61,133.77,133.21,132.76,130.94,130.80,130.73,130.36,130.11,130.08,129.74,129.69,129.36,128.87,128.70,128.52,128.30,128.25,127.96,127.38,127.27,126.15,125.66,124.30,124.09,113.64,113.49,112.92,112.72,55.35(och3),55.30(och3),54.19(och3),50.57(char2),50.27(char2),21.49(ch3),4.32(pd
‑
ch3).
[0088]
esi
‑
ms(m/z):calcd for c
69
h
59
cln2o4pd
:1123.32,found,1123.27,[m]
.anal.calcd for c
69
h
59
cln2o4pd:c,73.86;h,5.30;n,2.50;found,c,73.65;h,5.24;n,2.61.
[0089]
pd2的结构式为:
[0090][0091]
本实施例制备的pd2质量为0.41g,产率77%。pd2的氢谱和质谱如下:
[0092]1h nmr(600mhz,cdcl3)δ8.15(d,j=7.1hz,2h,ar
‑
h),7.97(d,j=7.6hz,2h,ar
‑
h),7.58(d,j=8.2hz,1h,ar
‑
h),7.54(d,j=8.3hz,1h,ar
‑
h),7.32(d,j=7.9hz,2h,ar
‑
h),7.21(q,j=8.2hz,4h,ar
‑
h),7.15
–
6.90(m,14h,ar
‑
h),6.08(d,j=7.1hz,1h,ar
‑
h),6.05(d,j=7.1hz,1h,ar
‑
h),5.97(d,j=7.8hz,2h,ar
‑
h),5.90(s,1h,char2),5.80(s,1h,char2),5.76(d,j=7.8hz,2h,ar
‑
h),5.49(dd,j=7.4,4.2hz,4h,ar
‑
h),2.40(s,3h,ch3),2.37(s,3h,ch3),2.36(s,3h,ch3),2.34(s,3h,ch3),1.37(s,3h,pd
‑
ch3),1.26(s,6h,ch3).
13
c nmr(151mhz,cdcl3)δ173.24(c=n),169.07(c=n),143.28,140.29,140.00,139.76,139.53,139.30,138.92,138.03,137.62,137.30,137.16,136.50,136.12,135.91,135.39,134.63,134.20,133.91,133.80,130.75,130.09,129.87,129.82,129.67,129.43,129.36,129.30,128.98,128.82,128.71,128.54,128.51,128.32,128.26,128.20,127.91,127.44,127.31,126.15,125.64,124.25,124.08,51.43(char2),51.13(char2),21.51(ch3),21.48(ch3),21.21(ch3),21.14(ch3),20.12(ch3),4.29(pd
‑
ch3).
[0093]
esi
‑
ms(m/z):calcd for c
69
h
59
cln2pd
:1059.34,found,1059.26,[m]
.anal.calcd for c
69
h
59
cln2pd:c,78.32;h,5.62;n,2.65;found,c,78.41;h,5.74;n,2.59.
[0094]
pd3的结构式为:
[0095][0096]
本实施例制备的pd3质量为0.43g,产率80%。pd3的氢谱和质谱如下:
[0097]1h nmr(600mhz,cdcl3)δ8.11(d,j=5.6hz,2h,ar
‑
h),7.93(d,j=7.5hz,2h,ar
‑
h),7.69(d,j=8.3hz,1h,ar
‑
h),7.65(d,j=8.3hz,1h,ar
‑
h),7.42
–
7.30(m,2h,ar
‑
h),7.30
–
7.17(m,4h,ar
‑
h),7.14(s,1h,ar
‑
h),7.04(ddq,j=44.3,18.8,9.3,8.7hz,11h,ar
‑
h),6.85(s,1h,ar
‑
h),6.12(d,j=7.1hz,1h,ar
‑
h),6.13
–
6.03(m,4h,ar
‑
h),5.97(s,1h,char2),5.85(d,j=8.9hz,3h,ar
‑
h,char2),5.45(dt,j=13.5,8.4hz,4h,ar
‑
h),2.42(s,3h,ch3),2.38(s,3h,ch3),1.37(s,3h,pd
‑
ch3).
13
c nmr(151mhz,cdcl3)δ173.12(c=n),168.98(c=n),162.54,162.48,160.91,160.87,160.65,159.02,143.14,140.02,139.63,139.35,138.76,138.31,137.86,137.62,136.67,136.57,136.47,136.01,135.98,134.83,133.94,131.38,131.33,131.28,131.24,131.19,131.14,130.77,130.72,130.57,130.34,130.09,129.86,129.57,129.27,128.73,128.54,128.32,128.04,127.56,127.43,125.79,125.24,124.27,124.09,115.35,115.21,115.08,114.94,114.69,114.55,114.46,114.32,50.59(char2),50.24(char2),21.49(ch3),4.54(pd
‑
ch3).
19
f nmr(565mhz,cdcl3)δ
‑
116.12(q,j=10.3,9.0hz),
‑
116.65
–‑
116.81(m),
‑
116.96,
‑
117.22
–‑
117.36(m).
[0098]
esi
‑
ms(m/z):calcd for c
65
h
47
clf4n2pd
:1077.25,found,1077.31,[m]
.anal.calcd for c
65
h
47
clf4n2pd:c,72.69;h,4.41;n,2.61;found,c,72.84;h,4.31;n,2.74.
[0099]
实施例5
[0100]
利用实施例4制备的非对称苊烯基α
‑
二亚胺钯配合物作为催化剂进行乙烯均聚的一般方法为:
[0101]
首先在真空90℃下干燥一台连接高压气体管路的350ml不锈钢压力反应器,干燥时间至少为1h;然后将反应器调整到所需的聚合温度(本实验实施温度为30℃和50℃)。在氮气气氛下向反应器中加入18ml甲苯以及所需量的nabarf,然后通过注射器将含所需苊烯基α
‑
二亚胺钯催化剂的2ml二氯甲烷溶液注入聚合体系中,本实施例中催化剂加入量为2μmol。快速搅拌后,反应器加压并保持在4atm的乙烯状态。在所需的时间后,将压力反应器放空,将聚合物在真空下干燥过夜。
[0102]
上述催化反应中,pd1、pd2和pd3分别在30℃、50℃条件下进行1次,则试验分别得到pd1、pd2和pd3三份催化剂的6份催化聚合物产物,测得聚聚合物产物的各项数据如下表1所示。表中pd0项为对照组数据,对照数据使用含二亚胺配合体pd催化剂,具体为具有吸电子取代基pd的催化剂pd4、pd5,其结构式分别表示如下:
[0103][0104]
对照数据使用低温(20℃)聚合,以便获得较高分子量,其他反应条件同试验组,在最优化聚合条件下进行。
[0105]
聚合物的分子量和分子量分布均通过凝胶渗透色谱法(gpc),以四氢呋喃溶剂在40℃的条件下测定,并使用聚苯乙烯为标准进行校准。
[0106]
表1 pd1、pd2和pd3催化聚合产物比较
[0107][0108]
高支化的聚乙烯,pdi范围小。催化活性,和产物分子量以及聚合物的分子量成正相关,在需求的分子量范围内,支化度越高表明催化效果越好。
[0109]
从表1中可以看出,本技术制备的pd1、pd2和pd3的催化活性act.
b
随温度升高而增加,但聚乙烯的分子量m
nc
降低,说明生成了分子量适中的高支链聚乙烯。对照组生成的聚乙烯对比pd1、pd2和pd3,具有更高的活性和更高的分子量,其得到的聚乙烯分枝密度受聚合温度影响较小,这主要是因为在α
‑
二亚胺pd(ii)体系中,链的行走速度远远大于链的生长速度。此外,对照组催化剂生成9
‑
40/1000c低分支密度的聚乙烯,所获得的聚乙烯pdi分布在2.3
‑
3.3之间,而pd1、pd2和pd3生成了95
‑
102/1000c的高支化聚乙烯,其分子量分布范围pdi=m
w
/m
n
仅在1.5
‑
1.8之间。表明本技术的“杂化”α
‑
二亚胺pd(ii)体系可以显著地影响聚合物的微观结构。产生这种现象的主要原因可能是由于催化剂金属中心附近的空间结构发生了变化,杂化取代基提供了更开放的空间结构。含me取代基的配合物pd2制备的聚乙烯分枝密度b
d
最高。所以可以精确调控催化剂中杂化苯胺基团的电子效应和空间位阻,延缓链转移相对于链增长速度,获得高分子量,高支化程度的聚合物。
[0110]
实施例6
[0111]
利用实施例4制备的非对称苊烯基α
‑
二亚胺钯配合物作为催化剂将丙烯酸甲酯与乙烯共聚,试验方法为:
[0112]
首先在真空90℃下干燥一台连接高压气体管路的300ml不锈钢压力反应器,干燥时间至少为1h;然后将反应器调整到所需的聚合温度(本实验实施温度为30℃),在氮气气氛下向反应器中加入18ml的二氯甲烷与极性单体的混合溶液和所需量的nabarf(本实施例中的nabarf加入量为0.04mmol,极性单体为1mol/l),然后通过注射器将含所需非对称苊烯基α
‑
二亚胺钯配合物的二氯甲烷溶液注入聚合体系(本实施例中催化剂加入量为0.02mmol)中。在快速搅拌后,将压力反应器放空,将聚合物在真空下干燥过夜。
[0113]
上述催化反应中,pd1、pd2和pd3分别在40℃条件下进行2次重复,则试验分别得到
pd1、pd2和pd3三份催化剂的6份催化聚合物产物,测得聚合物产物的各项数据如下表2所示。表中pd0项为对照组数据,pd0信息同实施例5。
[0114]
表2 pd1、pd2和pd3催化聚合产物比较
[0115][0116][0117]
插入比是极性丙烯酸酯类单体占所有单体的比值,与功能化聚乙烯关系密切。由表2可以看出,在乙烯
‑
ma共聚反应中,pd1、pd2和pd3的产物,随着单体ma(丙烯酸甲酯)浓度的增加,共聚活性和分子量均降低,而插入比显著提高,同时共聚物的分枝密度随ma浓度的增加而增加。并且,pd1、pd2和pd3还能制备出分子量适中,高插入比(4.0
‑
10.7mol%)的高度支化(105
‑
131)的极性功能化e
‑
ma共聚物,该共聚物的分子量分布范围pdi仅在1.2
‑
1.6之间。
[0118]
而对照组中在较高的ma浓度下,产物插入比和支化度依然较低。其生成低分支密度(31
‑
36/1000c)的聚乙烯,极性基团的插入比仅1mol%左右,所获得的聚乙烯pdi分布在2.7
‑
3.2之间。说明本发明提供的非对称苊烯基α
‑
二亚胺钯配合物作为催化剂具有很好地催化高插入比和高支化度的烯烃共聚反应的能力。
[0119]
以上实施方式仅用以说明本发明的技术方案,而并非对本发明的限制;尽管参照前述实施方式对本发明进行了详细的说明,本领域的普通技术人员应当理解:凡在本发明创造的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明创造的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。