本申请要求于2019年3月22日提交的美国临时专利申请62/822,730的优先权,出于所有目的,通过引用将其全部内容并入本文。
技术领域:
本公开涉及用于评估对象的临床状况的机器学习分类器的训练和实施。
背景技术:
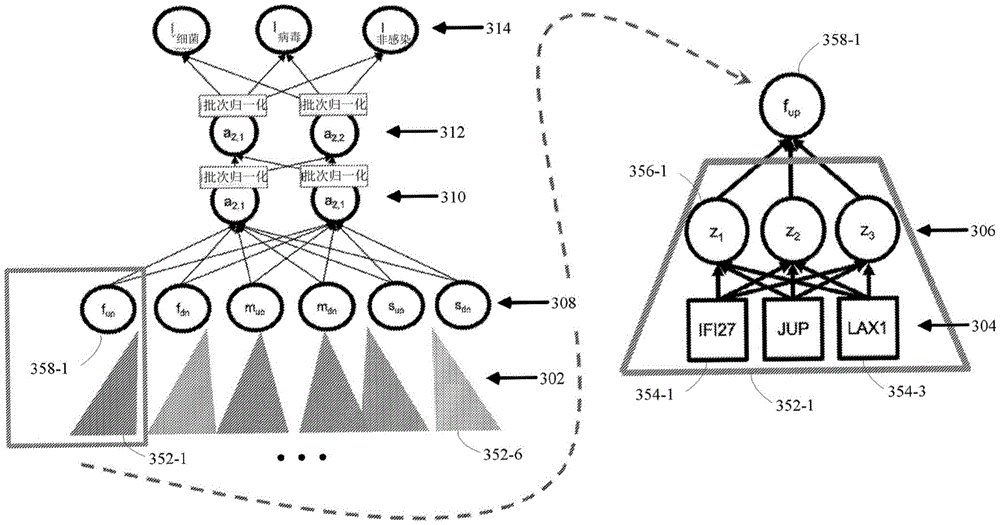
依赖转录组学和/或其他基于‘组学’的数据(例如基因组学、蛋白质组学、代谢组学、脂质组学、糖组学等)的生物建模方法可用于为医疗状况提供有意义且可操作的诊断和预后。例如,一些商业基因组诊断测试用于指导癌症治疗决策。OncotypeIQ测试套件(GenomicHealth)是此类基于基因组的检测的示例,其可提供指导各种癌症治疗的诊断信息。例如,这些测试之一,用于乳腺癌的ONCOTYPE(GenomicHealth)查询患者肿瘤中的21个基因组等位基因,以提供指导早期浸润性乳腺癌治疗的诊断信息,例如,通过提供可能获益于化疗和复发可能性的预后。参见例如Paiketal.,2004,NEnglJMed.351,pp.2817-2825和Paiketal.,2016,JClinOncol.24(23),pp.3726-3734。高通量‘组学’技术,例如基因表达微阵列,通常用于发现较小的靶向生物标志物组套(panel)。但是,此类数据集的变量总是比样本多,因此容易出现不可重复的过拟合结果。参见例如Shietal.,2008,BMCBioinformatics,9(9),p.S10和Ioannidisetal.,2001,NatGenet.29(3),pp.306–09。此外,为了增加统计功效,生物标志物的发现通常使用单一类型的测定法,例如单一类型的微阵列,在临床同质群组中进行。尽管这种同质设计确实产生了更大的统计功效,但结果不太可能在使用不同实验室技术的不同临床群组中保持真实。因此,任何源自高通量研究的新分类器都需要多次独立验证。幸运的是,技术进步导致了许多不同类型的高通量生物数据分析的发展。这反过来又导致对许多不同医学疾病的生物学效应进行大型临床研究。大量基于组学的数据集可以在线找到,例如,在国家生物技术信息中心(NCBI)主持的基因表达综合数据库(GeneExpressionOmnibus)(GEO)和欧洲生物信息学研究所(EMBL-EBI)主持的ArrayExpressArchiveofFunctionalGenomic中。这些数据集和其他数据集(其中许多是公开可用的)是训练机器学习分类器以区分各种疾病状态和预期治疗结果的良好来源,特别是因为它们使用不同的临床群组和不同的实验室技术。理论上,可以使用这些不同的数据集训练更好的分类器,因为可以识别和忽略个体患者群组和检测技术的检测特异性和批次特异性效应,同时强调由潜在生物学引起的表型效应。然而,针对异质数据集的分类器训练,例如从多项研究和/或使用多个分析平台收集的,是有问题的,因为特征值,例如表达水平,在不同的研究和分析平台之间是不可比较的。也就是说,包含来自不同技术和生物学背景的多个数据集会导致包含的数据集之间存在很大的异质性。如果不去除,这种异质性会混淆跨数据集的分类器的构建。使用异质数据集训练分类器的传统方法只是在单个群组中优化参数化分类器,然后将其应用到外部。然而,不同的技术背景阻碍了在外部数据集中的直接应用,因此分类器经常在本地重新训练,导致对性能的严重偏差估计。参见Tsaliketal,2016;和SciTranslMed8,322ra311。在另一种方法中,非参数化分类器在未共归一化的多个数据集上进行优化,因为无法在汇集设置中也优化这些分类器。参见Sweeneyetal,2015,SciTranslMed7(287),pp.287ra71;和Sweeneyetal.,2016,SciTranslMed8(346),pp.346ra91。最后,在最近发表的工作中,来自SageBionetworks的一个小组试图学习跨多个未正确共归一化的汇集数据集的参数化模型。然而,据报道,这些模型在验证中表现不佳。参见Sweeneyetal.,2018,NatureCommunications9,694。技术实现要素:鉴于上述背景,本领域需要用于开发和实现更稳健和可推广的机器学习分类器的改进方法和系统。有利地,本发明提供了解决这些问题和在医学诊断领域中的其他问题的技术方案(例如,计算系统、方法和非瞬时计算机可读存储介质)。例如,在一些实施方案中,本公开提供了使用输入分子(例如基因组、转录组、蛋白质组、代谢组)和/或具有相关临床表型的临床数据的异质库来生成机器学习分类器的方法和系统,例如用于诊断、预后或临床预测,它们比传统分类器更稳健和可推广。重要的是,如本文所述,已经开发了非传统的共归一化技术,其减少了数据集差异的效应并将数据带入单一汇集格式。通过整合和克服临床异质性来产生可推广的、准确的分类器,适当地共归一化的异质数据集释放了机器学习的潜力。因此,本文描述的方法和系统允许在使用多个数据集的新型分类器的开发方面取得突破。下文呈现了本发明的概述,以便提供对本发明的一些方面的基本理解。此概述不是本发明的广泛概要。此概述并不旨在标识本发明的关键/关键要素或描绘本发明的范围。此概述的唯一目的是以简化的形式呈现本发明的概念中的一些作为对之后所呈现的更加详细的说明的序言。在一些实施方案中,本公开提供了用于实施基于输入分子(例如基因组、转录组、蛋白质组、代谢组)和具有相关临床表型的临床数据的异质库来训练神经网络分类器的那些方法的方法和系统。在一些实施方案中,该方法包括先验地识别在感兴趣的临床状况中具有统计学显著差异特征值(例如,基因表达值)的生物标志物,并且确定每个生物标志物的特征值在该临床状况中的符号或方向,例如正或负。在一些实施方案中,收集通常检查相同临床状况的多个数据集,例如医学状况,例如急性感染的存在。然后使用特定于研究的程序对来自这些数据集的每一个的原始数据进行归一化,例如使用稳健的多阵列平均(RMA)算法归一化基因表达微阵列数据或Bowtie和Tophat算法归一化RNA测序(RNA-Seq)数据。然后将来自这些数据集的每一个的归一化数据映射到一个公共变量并与其他数据集共归一化。最后,共归一化和映射的数据集然后用于构建和训练神经网络分类器,其中对应于已识别的生物标志物的输入单元(其统计上显著的差异特征值对临床状况状态具有共享的效应符号,例如正或负)每个都使用统一符号系数分组为‘模块’,以保留模块基因效应的方向。例如,在一方面,本公开提供了用于执行使用先验特征分组来评估物种的测试对象的临床状况的此类方法的方法和系统,其中先验特征分组包括多个模块。多个模块中的每个模块包括独立的多个特征,其对应的特征值各自和与临床状况相关联的独立表型的不存在、存在或阶段相关联。该方法包括以电子形式获得第一训练数据集,其中对于物种的第一多个训练对象中的每个相应的训练对象,第一训练数据集包括:(i)通过第一技术背景,使用相应的训练对象的生物样本,对于独立的多个特征,以第一形式获得的第一多个特征值,第一形式是多个模块中的至少第一模块的转录组学、蛋白质组学或代谢组学中的一个,以及(ii)在相应的训练对象中,对应于第一模块的第一独立表型的不存在、存在或阶段的指示。该方法然后包括以电子形式获得第二训练数据集,其中对于物种的第二多个训练对象中的每个相应的训练对象,第二训练数据集包括:(i)通过第一技术背景以外的第二技术背景,使用相应的训练对象的生物样本,对于独立的多个特征,以与至少第一模块的第一形式相同的第二形式获得的第二多个特征值,以及(ii)在相应的训练对象中,第一独立表型的不存在、存在或阶段的指示。然后,该方法包括对至少第一和第二训练数据集中存在的特征的特征值跨至少第一和第二训练数据集进行共归一化,以去除数据集间批次效应,从而针对第一多个训练对象的每个相应训练对象和针对第二多个训练对象的每个相应训练对象计算相应训练对象的至少第一模块的共归一化特征值。该方法然后包括针对复合训练集训练主分类器以评估测试对象的临床状况,对于第一多个训练对象中的每个相应训练对象和对于第二多个训练对象中的每个相应训练对象,所述复合训练集包括:(i)第一模块的共归一化特征值的汇总,以及(ii)相应训练对象中第一独立表型的不存在、存在或阶段的指示。在另一方面,本公开提供了用于执行评估物种的测试对象的临床状况的此类方法的方法和系统。该方法包括以电子形式获得第一训练数据集,其中对于物种的第一多个训练对象中的每个相应的训练对象,第一训练数据集包括:(i)使用相应的训练对象的生物样本,对于多个特征获得的第一多个特征值,以及(ii)在相应的训练对象中第一独立表型的不存在、存在或阶段的指示。第一个独立表型代表患病状况,并且第一训练数据集的第一子集由没有患病状况的对象组成。该方法然后包括以电子形式获得第二训练数据集,其中对于物种的第二多个训练对象中的每个相应的训练对象,第二训练数据集包括:(i)使用相应的训练对象的生物样本,对于所述多个特征获得的第二多个特征值,以及(ii)在相应的训练对象中第一独立表型的不存在、存在或阶段的指示。第二训练数据集的第一子集由没有患病状况的对象组成。该方法然后包括对至少第一和第二训练数据集的多个特征的子集的特征值进行共归一化以去除数据集间批次效应,其中特征子集存在于至少第一和第二训练数据集中。共归一化包括仅使用相应的第一和第二训练数据集的第一子集来估计第一和第二训练数据集之间的数据集间批次效应。数据集间批次效应包括加性分量和乘性分量,并且共归一化为跨各自的第一和第二训练数据集的第一子集的特征值求解普通最小二乘模型并使用经验贝叶斯估计器缩小表示加性分量和乘性分量的所得参数,从而使用所得参数计算:对于第一多个训练对象中的每个相应训练对象和第二多个训练对象中的每个相应训练对象,多个特征的子集的共归一化特征值。该方法然后包括针对复合训练集训练主分类器以评估测试对象的临床状况,对于第一多个训练对象中的每个相应训练对象和对于第二多个训练对象中的每个相应训练对象,所述复合训练集包括:(i)多个特征的子集的共归一化特征值,以及(ii)相应训练对象中第一独立表型的不存在、存在或阶段的指示。所附权利要求范围内的系统、方法和装置的各个实施方案各自均具有若干个方面,其中并非仅靠任何单一方面来负责本文所述的期望的属性。在不限制所附权利要求的范围的情况下,本文描述了一些突出的特征。在考虑了这一讨论之后,特别是在阅读了题为“具体实施方式”的部分之后,人们将理解如何使用各个实施方案的特征。通过引用结合本说明书中所提到的所有出版物、专利和专利申请均通过引用其全文结合在此,其程度就如同明确且单独地指明了每一个单独的出版物、专利或专利申请通过引用结合。附图说明在附图的图中,通过实例而非限制的方式示出了本文公开的实施方式。贯穿附图的若干视图,相似的附图标记指代对应的部分。图1A、1B、1C和1D共同示出了根据本公开的一些实施方案的计算装置的示例框图。图2A、2B、2C、2D、2E、2F、2G、2H和2I图示了根据本公开的一些实施方案的对对象进行分类的方法的示例流程图,其中可选步骤由虚线框指示。图3图示了一个网络拓扑,其中底部的多个模块每个都贡献了先验已知基因的几何平均值,所有都在感兴趣的临床状况下平均在相同的方向上移动。根据本公开的一些实施方案,网络顶部的输出是感兴趣的临床状况(细菌感染-Ibac,病毒感染Ivira,无感染-Inon)。图4图示了一个网络拓扑,其中每个模块都使用了minispoke网络(图的右侧部分更详细地显示了其中一个)。单个生物标志物由局部网络汇总(而不是按其几何平均值汇总),然后传入主分类网络。图5A和5B图示了根据本公开的实施方案的迭代COCONUT比对,其中“参考”是微阵列数据,“目标”是NanoString数据。图表显示了NanoString基因表达和微阵列基因表达的健康样本的分布,针对的是29个基因的组中的两个基因(5A–HK3、5B–IFI27)。微阵列分布显示在基于共归一化的比对过程中的三个不同迭代中。虚线表示中间迭代时的分布,实线表示程序终止时的分布。图6A和6B图示了在本公开的实例中使用的训练数据集中29个基因的组的选定基因(6A-发烧标志物)(6B-严重性标志物)的细菌、病毒和非感染训练集样本的共归一化表达值的分布。图7A和7B分别图示了根据本公开的实施方案的训练数据集中29个基因的共归一化表达值的二维(7A)和三维(7B)t-SNE投影,其中每个对象被标记为细菌、病毒或未感染。图8A和8B分别图示了根据本公开的实施方案的训练数据集中29个基因的共归一化表达值的二维(8A)和三维(8B)主成分分析图,其中每个对象被标记为细菌、病毒或未感染。图9图示了根据本公开内容的实施方案的跨训练数据集的29个基因的共归一化表达值的二维主成分分析图,其中每个对象由源研究标记。图10A和10B分别图示了根据本公开的实施方案的使用6个几何平均分数代替29个基因的直接表达值的验证性能偏差分析,其中图10A上小图是逻辑回归,图10A下小图是XGBoost,图10B上小图是带有RBF核的支持向量机,图10B下小图是多层感知器。x轴是每个模型类型的前10个模型的外折叠和内折叠平均成对ROC下面积(APA)曲线之间的差异,按交叉验证APA排序。每个点对应一个模型。y轴对应于外折叠APA。垂直虚线表示内环和外环中的APA之间没有区别。图11A和11B分别图示了根据本公开的实施方案的使用29个基因的直接表达值的验证性能偏差分析,其中图11A上小图是逻辑回归,图11A下小图是XGBoost,图11B上小图是带有RBF核的支持向量机,图11B下小图是多层感知器。x轴是每个模型类型的前10个模型的外折叠和内折叠平均成对ROC下面积(APA)曲线之间的差异,按交叉验证APA排序。每个点对应一个模型。y轴对应于外折叠APA。垂直虚线表示内环和外环中的APA之间没有区别。图12图示了根据本公开的一些实施方案的用于COCONUT算法的迭代应用的伪代码。图13图示了根据本公开的一些实施方案的用于训练分类器以评估对象的临床状况的方法的示例流程图。图14图示了根据本公开的一些实施方案的评估对象的临床状况的方法的示例流程图。具体实施方式现将详细参照实施方案,在附图中展示其实例。在以下详细描述中,阐述了许多具体细节以便彻底理解本公开。然而,对本领域普通技术人员而言将显而易见的是,本公开可以在没有这些具体细节的情况下实施。在其它例子中,并未详细描述熟知的方法、程序、组件、电路以及网络,以免不必要地模糊实施方案的各个方面。这里描述的实现方式提供了用于生成和使用机器学习分类器来诊断、提供预后或提供对医学状况的临床预测的各种技术方案。特别地,本文提供的方法和系统促进使用分子(例如基因组、转录组、蛋白质组、代谢组)和/或具有相关临床表型的临床数据的异质储库来训练具有改进性能的机器学习分类器。在一些实施方案中,如本文所述,所公开的方法和系统通过估计异类训练数据集之间的数据集间批次效应来实现具有改进性能的机器学习分类器。在一些实施方案中,本文描述的系统和方法利用开发来将多个离散数据集带入单个汇集数据框架中的共归一化方法。这些方法提高了在整体汇集精度、汇集框架内单个数据集精度的一些平均函数或两者上的分类器性能。本领域技术人员将认识到,这种能力需要改进异质数据集的共归一化,这不是传统的基于组学的数据科学管道的特征。在一些实施方案中,本文描述的分类器训练方法中的初始步骤是先验识别要训练所针对的生物标志物。可以使用文献搜索或在‘发现’数据集中识别感兴趣的生物标志物,其中使用统计测试来选择与感兴趣的临床状况相关的生物标志物。在一些实施方案中,然后根据感兴趣的临床决策中它们的变化方向的符号将感兴趣的生物标志物分组。在一些实施方案中,用于训练这些分类器的变量子集从存在于异质数据集中的已知分子变量(例如,基因组、转录组、蛋白质组、代谢物组的数据)选择。在一些实施方案中,使用诸如微阵列显著性分析(SAM)、或数据集之间的荟萃分析、或与类别的相关性或其他方法的工具对差异表达使用统计阈值法来选择这些变量。在一些实施方案中,可用数据通过基于分子图谱模式设计新特征来扩展。可以使用无监督分析(例如去噪自动编码器)或监督方法(例如使用现有本体或路径数据库(例如KEGG)进行路径分析)来发现这些新特征。在一些实施方案中,用于训练分类器的数据集是从公共或私人来源获得的。在公共领域,可以使用NBCIGEO或ArrayExpress(如果使用转录组数据)等储库。数据集必须至少存在一个感兴趣的类别,并且如果使用需要健康对照的共归一化函数,则它们必须具有健康对照。在一些实施方案中,只有单一的生物类型的数据被收集(例如,仅转录数据,但不是蛋白质组数据),但可以来自广泛不同的技术背景(例如RNAseq和DNA微阵列二者)。在一些实施方案中,输入数据被分层以确保在每个输入数据集中存在大约相等比例的每个类别。这一步避免了在跨汇集数据集学习单个分类器时异质数据源的混淆。分层可以进行一次、多次或根本不进行。在一些实施方案中,当获得来自原始技术格式的原始数据时,执行标准化的数据集内归一化程序,以最小化不同归一化方法对最终分类器的影响。来自相同类型技术平台的数据优选地以相同的方式进行归一化,通常使用一般程序,如背景校正、log2转换和分位数归一化。特定于平台的归一化程序也很常见(例如用于阳性匹配对照的Affymetrix平台的gcRMA)。结果是每个数据集的单个文件或其他数据结构。在一些实施方案中,然后在两个步骤中执行共归一化,可选的平台间公共变量映射,然后是必要的共归一化。平台间公共变量映射在涉及数据集的平台不遵循相同的命名约定和/或测量与多种变体相同的目标的那些情况下是需要的(例如,许多RNA微阵列具有用于单个基因的简并探针)。选择一个共同的参考(例如映射至RefSeq基因),并重新标记变量(在单个情况或汇总(在多变量情况下;例如通过采取集中趋势的测度,例如中值、平均值等,或同一基因简并探针的固定效应荟萃分析)。共归一化是必要的,因为在确定了数据集之间具有通用名称的变量后,通常情况下这些变量在数据集之间具有显著不同的分布。从而这些值被转换为匹配数据集之间的相同的分布(例如,平均值和方差)。可以使用多种方法进行共归一化,例如COCONUT(Sweeneyetal.,2016,SciTranslMed8(346),pp.346ra91;和Abouelhodaetal.,2008,BMCBioinformatics9,p.476),位数归一化,ComBat,汇集RMA,汇集gcRMA,或不变异的基因(例如,持家基因)归一化,等等。在一些实施方案中,使用本文描述的改进方法共归一化的数据经受机器学习,以针对感兴趣的临床状况的类别,例如疾病诊断或预后分类,训练主分类器。在非限制性实例中,这可以利用线性回归、惩罚线性回归、支持向量机、基于树的方法(例如随机森林或决策树)、集成方法(例如adaboost、XGboost)或其他弱或强分类器的集成、神经网络方法(例如多层感知器)或其他方法或其变体。在一些实施方案中,主分类器可以直接从所选变量、工程化特征或两者中学习。在一些实施方案中,主分类器是分类器的集成。在一些实施方案中,这些方法和系统通过借助生成函数从汇集数据生成新样本而得到进一步扩充。在一些实施方案中,这包括向每个样本添加随机噪声。在一些实施方案中,这包括更复杂的生成模型,例如玻尔兹曼机、深度置信网络、生成不利网络、对抗性自动编码器、其他方法或其变体。在一些实施方案中,用于分类器开发的方法和系统包括交叉验证、模型选择、模型评估和校准。初始交叉验证估计固定分类器的性能。模型选择使用超参数搜索和交叉验证来识别最准确的分类器。模型评估用于评估所选模型在独立数据中的性能,并且可以使用数据集留一法(LODO)交叉验证、嵌套交叉验证或bootstrap校正的性能评估等来执行。校准根据临床实践中观察到的表型分布调整分类器分数,目的是将分数转换为直观的、人可解释的值。它可以使用诸如Hosmer-Lemeshow检验和校准斜率等方法来执行。在一些实施方案中,诸如多层感知器的神经网络分类器用于对共归一化数据中的感兴趣的结果(例如感染的存在)进行监督分类。已知在感兴趣的临床条件下平均一起移动的变量被分组到‘模块’中,并且解释这些分组模块的神经网络架构在上面学习。在一些实施方案中,‘模块’以两种方式之一构建。在第一种方式中,模块内的生物标志物是通过取它们的中心倾向,如几何平均值的度量,并且将其馈送到主分类分组是(例如,如在图3中示出)。在另一实施方案中,构建‘辐条’网络,其中在模块中输入是生物标志物,并且它们经由馈送到主分类器的分量分类器解释(例如,如在图4中示出)。定义在本公开中使用的术语仅用于描述特定实施方案,并不旨在限制本发明。如在本发明的说明书和所附权利要求中所使用的,单数形式“一”、“一个”和“所述”也旨在包括复数形式,除非上下文另外清楚地指示。还应当理解,这里使用的术语“和/或”是指并涵盖相关联的所列项目中的一个或多个的任何和所有可能的组合。应进一步理解的是,当在本说明书中使用时,术语“包括(comprises和/或comprising)”指定所陈述的特征、整数、步骤、操作、要素和/或组件的存在,但不排除存在或添加一个或多个其它特征、整数、步骤、操作、要素、组件和/或其群组。如本文中所使用,取决于上下文,术语“如果”可以被解释成意指“当……时”或“在……时”或者“响应于确定”或“响应于检测”。类似地,取决于上下文,短语“如果确定”或“如果检测到[所陈述的条件或事件]”可以被解释为意指“在确定……时”或“响应于确定”或“在检测到[所陈述的条件或事件]时”或“响应于检测到[所陈述的条件或事件]”。还将理解,虽然术语“第一”、“第二”等可在本文中用于描述各种元件,但这些元件不应受这些术语限制。这些术语仅用于将一个元件与另一元件区分开来。例如,在不脱离本公开的范围的情况下,第一主体可以被称为第二主体,并且类似地,第二主体可以被称为第一主体。虽然第一对象和第二对象都是对象,但他们不是同一对象。进一步地,术语“对象”、“用户”和“患者”在本文中可互换使用。如本文所公开,术语“核酸”和“核酸分子”可互换使用。该术语是指任何组成形式的核酸,例如脱氧核糖核酸(DNA)、核糖核酸(RNA)和/或DNA或RNA类似物(例如,含有碱基类似物,糖类似物和/或非天然主链等),所有这些都可以是单链或双链形式。除非另有限制,核酸可以包含已知的天然核苷酸类似物,其中一些可以以与天然存在的核苷酸类似的方式起作用。核酸可以是用于进行本文的过程的任何形式(例如,线性、环状、超螺旋、单链、双链等)。在一些实施方案中,核酸可以来自单个染色体或其片段(例如,核酸样本可以来自从二倍体生物获得的样本的一个染色体)。在某些实施方案中,核酸包括核小体、核小体的片段或部分或核小体样结构。核酸有时包含蛋白质(例如,组蛋白、DNA结合蛋白等)。通过本文所述方法分析的核酸有时基本上是分离的并且基本上不与蛋白质或其他分子相关联。核酸还包括从单链合成、复制或扩增的DNA的衍生物、变体和类似物(“有义”或“反义”、“正”链或“负”链、“正向”阅读框或“反向”阅读框)和双链多核苷酸。脱氧核糖核苷酸包括脱氧腺苷、脱氧胞苷、脱氧鸟苷和脱氧胸苷。可以使用从受试者获得的核酸作为模板来制备核酸。如本文所公开,术语“受试者”是指任何有生命的或无生命的生物,包括但不限于人(例如,男人、女人、胎儿、怀孕女性、儿童等)、非人类动物、植物、细菌、真菌或原生生物。任何人类或非人类动物可用作对象,包括但不限于哺乳动物、爬行动物、鸟类、两栖动物、鱼类、有蹄动物、反刍动物、牛科动物(例如家牛(cattle))、马科动物(例如马)、山羊类和绵羊类(例如绵羊、山羊)、猪类(例如猪)、骆驼科动物(例如骆驼、美洲驼、羊驼)、猴类、猿类(例如大猩猩、黑猩猩)、熊科动物(例如熊)、家禽类、狗类、猫类、鼠类、鱼类、海豚类、鲸类和鲨鱼类。在一些实施方案中,对象是任何阶段的雄性或雌性(例如,男性、女性或儿童)。如本文所用,术语“对照”、“对照样本”、“参考”、“参考样本”、“正常”和“正常样本”描述来自不具有特定状况或以其他方式是健康的对象的样本。在一个例子中,本文公开的方法可以对患有肿瘤的对象进行,其中参考样本是取自对象的健康组织的样本。参考样本可以从对象或从数据库中获得。参考可以是例如参考基因组,其用于绘制从对对象样本测序获得的序列读段。如本文所用,本文所用的术语“测序”、“序列确定”等一般指可用于确定诸如核酸或蛋白质的生物大分子的顺序的任何和所有生化方法。例如,测序数据可以包括核酸分子中的全部或部分核苷酸碱基,例如mRNA转录本或基因组基因座。示例性系统实施方案既然已经提供了本公开的一些方面的概述和本公开中使用的一些定义,现在结合图1描述示例性系统的细节。图1是展示根据一些实施方案的系统100的框图。在一些实施方案中,装置100包含一个或多个处理单元CPU102(也称为处理器)、一个或多个网络接口104、用户接口106、非永久性存储器111、永久性存储器112和一个或多个用于互连这些组件的通信总线114。一个或多个通信总线114可选地包含互连并控制系统组件之间的通信的电路系统(有时称为芯片组)。非永久性存储器111通常包含高速随机存取存储器,如DRAM、SRAM、DDRRAM、ROM、EEPROM、闪速存储器,而永久性存储器112通常包括CD-ROM、数字多功能盘(DVD)或其它光学存储装置、磁带盒、磁带、磁盘存储装置或其它磁性存储装置、磁盘存储装置、光盘存储装置、闪速存储器装置或其它非易失性固态存储装置。永久性存储器112可选地包含一个或多个布置的远离CPU102的存储装置。永久性存储器112和非永久性存储器112内的一个或多个非易失性存储装置包括非暂时性计算机可读存储介质。在一些实施方案中,非永久性存储器111或者(替代性地)所述非暂时性计算机可读存储介质(有时与永久性存储器112结合)存储以下程序、模块和数据结构或者其子集:●操作系统116,所述操作系统包含用于处理各种基本系统服务和用于执行硬件相关任务的程序;●网络通信模块(或指令)118,所述网络通信模块(或指令)用于将可视化系统100与其它装置连接,或与通信网络连接;●变量选择模块120,用于识别感兴趣的表型的信息特征;●原始数据归一化模块122,用于归一化每个原始训练数据集132内的原始特征数据136;●数据共归一化模块124,其用于共归一化跨异质训练数据集例如内部归一化数据构造138的特征数据,例如归一化特征数据142;●分类器训练模块126,用于基于跨异质数据集的共归一化特征数据148来训练机器学习分类器;●训练数据集存储器130,其用于存储一个或多个数据结构,例如用于训练对象的一个或多个样本的原始数据构造132、内部归一化数据构造138和/或共归一化数据构造144,每个这样的数据构造对于多个训练对象中的每个相应的训练对象包括多个特征值,例如原始特征值136、内部归一化特征值142和/或共归一化特征值148;●数据模块集存储器150,其用于存储用于训练分类器的一个或多个模块152,每个这样的相应模块150包括(i)独立的多个差异调节特征154的标识,(ii)相应的汇总算法或分量分类器156,和(iii)与研究的临床状况相关的独立表型157(例如,临床状况本身或是决定性或与临床状况相关的表型);和●测试数据集存储器160,其用于存储用于测试对象164的一个或多个样本的一个或多个数据构造162,每个这样的数据构造包括多个特征值166。在一些实施方案中,一个或多个上述元件存储在一个或多个先前提及的存储装置中,并且对应于用于执行上述功能的指令组。上述模块、数据或程序(例如指令集)不需实施为单独分开的软件程序、过程、数据集或模块,并且因此这些模块和数据的各个子集可以在各种实施方案中被组合或以其它方式重新布置。在一些实施方案中,非永久性存储器111可选地存储上述模块和数据结构的子集。此外,在一些实施方案中,所述存储器存储以上未描述的另外的模块和数据结构。在一些实施方案中,上述元件中的一个或多个存储在可视化系统100的计算机系统之外的计算机系统中,所述计算机系统可由可视化系统100寻址,使得可视化系统100可在需要时检索所有或部分这样的数据。尽管图1描绘了“系统100”,但是该图更多地旨在作为可能存在于计算机系统中的各种特征的功能描述,而不是作为本文所述的实施方案的结构示意图。在实践中,并且如本领域普通技术人员所认识的,可以将单独示出的项目组合,并且可以将一些项目分离。此外,尽管图1描绘了非永久性存储器111中的某些数据和模块,但是这些数据和模块中的一些或全部可以存在于永久性存储器112中。示例性方法实施方案虽然已经参照图1公开了根据本公开的系统,但是现在参照图2详细描述根据本公开的方法。参考图2A的方框202-214,在一些实施方案中,在计算机系统(例如图1的系统100)处提供使用特征的先验分组来评估物种的测试对象的临床状况的方法,该系统具有一个或多个处理器102和存储一个或多个程序的存储器111/112,例如变量选择模块120,它们由一个或多个处理器执行。特征的先验分组包括多个模块152。多个模块152中的每个相应模块152包括独立的多个特征154,其对应的特征值每个和与临床状况相关联的独立表型157的不存在、存在或阶段相关联。例如,表1提供了六个败血症相关模块(基因组)的非限制性示例定义和组成,每个模块与败血症相关的独立表型157的不存在、存在或阶段相关。表1的模块152-1和152-2分别针对在严格的病毒感染中具有升高(模块152-1)和降低(模块152-2)表达的基因。表1的模块152-3和152-4分别针对败血症相比于无菌炎症的患者中表达升高(模块152-3)和降低(模块152-4)的基因。模块152-5和152-6分别针对在入院30天内死亡的患者中具有升高(模块152-5)和降低(模块152-6)表达的基因。表1:败血症相关模块的定义和组成参考方框204,在一些实施方案中,对象是人或哺乳动物。在一些实施方案中,对象是任何有生命的或无生命的生物,包括但不限于人(例如,男人、女人、胎儿、怀孕女性、儿童等)、非人类动物、植物、细菌、真菌或原生生物。在一些实施方案中,对象是哺乳动物、爬行动物、鸟类、两栖动物、鱼类、有蹄动物、反刍动物、牛科动物(例如家牛(cattle))、马科动物(例如马)、山羊类和绵羊类(例如绵羊、山羊)、猪类(例如猪)、骆驼科动物(例如骆驼、美洲驼、羊驼)、猴类、猿类(例如大猩猩、黑猩猩)、熊科动物(例如熊)、家禽类、狗类、猫类、鼠类、鱼类、海豚类、鲸类和鲨鱼类。在一些实施方案中,对象是任何阶段的雄性或雌性(例如,男性、女性或儿童)。参见方框206,在一些实施方案中,临床状况是二分的临床状况(例如,具有败血症相对于不具有败血症,具有癌症相对于不具有癌症,等等)。参考方框208,在一些实施方案中,临床状况是多类临床状况。例如,参考方框210,在一些实施方案中,临床状况由三类临床状况组成:(i)严格的细菌感染,(ii)严格的病毒感染,和(iii)非感染性炎症。参看方框212,在一些实施方案中,多个模块152包括至少三个模块或至少六个模块。上面的表1提供了其中多个模块152由六个模块组成的实例。在一些实施方案中,多个模块152包括三到一百个模块。在一些实施方案中,多个模块152由两个模块组成。此外,参考方框214,在一些实施方案中,多个模块中的每个模块152的每个独立的多个特征154包括至少三个特征或至少五个特征。上面的表1提供了其中多个模块152由六个模块组成的实例。在一些实施方案中,多个模块152包括三到一百个模块。在一些实施方案中,多个模块152由两个模块组成。此外,不要求每个模块包括相同数量的特征。上面表1的实例证明了这一点。因此,例如,在一些实施方案中,一个模块152可以具有两个特征154,而另一个模块可以具有超过五十个特征。在一些实施方案中,每个模块152具有两个到五十个特征154。在一些实施方案中,每个模块152具有三到一百个特征。在一些实施方案中,每个模块152具有四到两百个特征。在一些实施方案中,每个模块152中的特征154是唯一的。即,任何给定特征仅出现在模块152之一中。在其他实施方案中,不要求每个模块152中的特征是唯一的,即,在这样的实施方案中,给定特征154可以在多于一个模块中。参见图2B的方框216,获得第一训练数据集(例如,图1A的原始数据构造132-1)。对于物种的第一多个训练对象中的每个相应的训练对象134,第一训练数据集包括:(i)通过第一技术背景,使用相应的训练对象的生物样本,对于独立的多个特征,以第一形式获得的第一多个特征值136,第一形式是多个模块中的至少第一模块152的转录组学、蛋白质组学或代谢组学中的一个,以及(ii)在相应的训练对象中,对应于第一模块的第一独立表型157的不存在、存在或阶段的指示。在实践中,因为这是训练数据集,该数据集将提供每个对象的临床状况的指示。然而,在一些实施方案中,第一独立表型和临床状况是相同的。在它们不同的实施方案中,训练集提供第一独立表型和临床状况二者。例如,在第一模块是上表1的模块152-1的情况下,第一数据集将为第一数据集中的每个训练对象提供:(i)基因IFI27、JUP和LAX1的测量表达值,其使用相应训练对象的生物样本通过第一技术背景获得,(ii)对象是否发烧的指示,以及(iii)对象是否患有败血症。在一些实施方案中,每个模块158与和临床状况相关的独立表型的不存在、存在或阶段唯一地关联,但是对于每个训练对象,第一训练数据集仅提供临床状况本身而不是每个相应模块的独立表型157的不存在、存在或阶段的指示。例如,在表1的情况下,在一些实施方案中,第一训练数据集包括临床状况(败血症)的不存在、存在或阶段的指示,但不指示每个训练对象是否具有表型发热。也就是说,在一些实施方案中,本公开依赖于先前的工作,该工作已经确定了关于给定表型(例如发烧)哪些特征被上调或下调,并且因此不需要指示训练数据集中的每个训练对象是否具有模块的表型。在不提供对应于模块的表型的情况下,提供关于训练对象中临床状况的不存在、存在或阶段的指示。在一些实施方案中,第一训练数据集仅为每个训练对象提供临床状况的不存在或存在。即,在这样的实施方案中不提供临床状况的阶段。参考图2B的方框218,在一些实施方案中,第一模块中的每个相应特征对应于与第一独立表型相关联的生物标志物,与在物种的对象群组中没有表现出独立的表型的对象相比,其在表现出第一独立表型的对象中在统计上显著更丰富。所述物种的对象群组不必是第一个数据集的对象。该物种的对象群组是满足选择标准并且包括具有临床状况的对象和不具有临床状况的对象的任何对象组。在败血症的情况下群组的非限制性实例的选择标准是:1)是医师裁定为感染的存在和类型(例如严格细菌感染,严格病毒感染,或非感染性炎症),2)在所述多个模块中具有特征的特征值,3)超过18岁,4)被看见在医院环境中(例如急诊科,重症监护),5)社区或医院获得性感染,和6)在最初怀疑感染和/或败血症的24小时内采集了血液样本。在一些这样的实施方案中,通过将标准t检验、Welcht检验、Wilcoxon检验或置换检验应用于生物标志物的丰度来评估关于生物标志物是否“统计上显著更丰富”的确定,所述生物标志物的丰度如在表现出第一独立表型的群组(第1组)中的对象中和在未表现出第一独立表型以达到p值的群组(第2组)中的对象中测量的。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,生物标志物在统计学上显著更丰富。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,生物标志物在统计学上显著更丰富,这使用错误发现率程序例如Benjamini-Hochberg或Benjamini–Yekutieli针对多个测试进行调整。参见例如Benjamini和Hochberg,JournaloftheRoyalStatisticalSociety,SeriesB57,pp.289-300;和Benjamini和Yekutieli,2005,JournalofAmericanStatisticalAssociation100(469),pp.71-80,在此通过引用将每一篇并入本文。在一些实施方案中,通过多个数据集(群组或训练数据集)的固定效应或随机效应荟萃分析,认为生物标志物在统计学上显著更丰富。例如,参见Sianphoeetal.,2019,BMCBioinformatics20:18,在此通过引用并入。在一些实施方案中,每个模块152与和临床状况相关的独立表型157的不存在、存在或阶段唯一地关联,但是对于第一训练集中的每个训练对象,第一训练数据集仅提供临床状况本身的不存在、存在或阶段以及多个模块中的一些而非全部模块的独立表型的不存在、存在或阶段的指示。例如,在表1的情况下,在一些实施方案中,第一训练数据集包括临床状况/表型“败血症”的不存在、存在或阶段的指示,表型“严重性”的不存在、存在或阶段的指示,但不指示每个训练对象是否发烧。参考图2B的方框222,在一些实施方案中,第一模块中的每个相应特征对应于与第一独立表型157相关联的生物标志物,与在物种的对象群组中没有表现出独立的表型的对象相比,其在表现出第一独立表型的对象中在统计上显著更不丰富。在一些实施方案中,通过将标准t检验、Welcht检验、Wilcoxon检验或置换检验应用于生物标志物的丰度来评估关于生物标志物是否“统计上显著更不丰富”的确定,所述生物标志物的丰度如在表现出第一独立表型的群组(第1组)中的对象中和在未表现出第一独立表型以达到p值的群组(第2组)中的对象中测量的。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,生物标志物在统计学上显著更不丰富。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,生物标志物在统计学上显著更不丰富,这使用错误发现率程序例如Benjamini-Hochberg或Benjamini–Yekutieli针对多个测试进行调整。参见例如Benjamini和Hochberg,JournaloftheRoyalStatisticalSociety,SeriesB57,pp.289-300;和Benjamini和Yekutieli,2005,JournalofAmericanStatisticalAssociation100(469),pp.71-80,在此通过引用将每一篇并入本文。在一些实施方案中,通过多个数据集(群组或训练数据集)的固定效应或随机效应荟萃分析,认为生物标志物在统计学上显著更不丰富。例如,参见Sianphoeetal.,2019,BMCBioinformatics20:18,在此通过引用并入。参考图2B的方框224,在一些实施方案中,第一模块中的每个相应特征与第一独立表型157相关联,因为与在物种的对象群组中没有表现出独立的表型的对象相比,其特征值在表现出第一独立表型的对象中在统计上显著更大。在一些实施方案中,通过将标准t检验、Welcht检验、Wilcoxon检验或置换检验应用于特征的丰度来评估关于该特征是否“统计上显著更丰富”的确定,所述特征的丰度如在表现出第一独立表型的群组(第1组)中的对象中和在未表现出第一独立表型以达到p值的群组(第2组)中的对象中测量的。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,特征值在统计学上显著更大。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,特征在统计学上显著更大(更丰富),这使用错误发现率程序例如Benjamini-Hochberg或Benjamini–Yekutieli针对多个测试进行调整。参见例如Benjamini和Hochberg,JournaloftheRoyalStatisticalSociety,SeriesB57,pp.289-300;和Benjamini和Yekutieli,2005,JournalofAmericanStatisticalAssociation100(469),pp.71-80,在此通过引用将每一篇并入本文。在一些实施方案中,通过多个数据集(群组或训练数据集)的固定效应或随机效应荟萃分析,认为该特征在统计学上显著更大。例如,参见Sianphoeetal.,2019,BMCBioinformatics20:18,在此通过引用并入。参考图2B的方框226,在一些实施方案中,第一模块中的每个相应特征与第一独立表型157相关联,因为与在物种的对象群组中没有表现出独立的表型的对象相比,其特征值在表现出第一独立表型的对象中在统计上显著更少。在一些实施方案中,通过将标准t检验、Welcht检验、Wilcoxon检验或置换检验应用于特征的丰度来评估关于该特征是否“统计上显著更少”的确定,所述特征的丰度如在表现出第一独立表型的群组(第1组)中的对象中和在未表现出第一独立表型以达到p值的群组(第2组)中的对象中测量的。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,特征在统计学上显著更少。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,特征在统计学上显著更少,这使用错误发现率程序例如Benjamini-Hochberg或Benjamini–Yekutieli针对多个测试进行调整。参见例如Benjamini和Hochberg,JournaloftheRoyalStatisticalSociety,SeriesB57,pp.289-300;和Benjamini和Yekutieli,2005,JournalofAmericanStatisticalAssociation100(469),pp.71-80,在此通过引用将每一篇并入本文。在一些实施方案中,通过多个数据集(群组或训练数据集)的固定效应或随机效应荟萃分析,认为该特征在统计学上显著更少。例如,参见Sianphoeetal.,2019,BMCBioinformatics20:18,在此通过引用并入。参考图2C的方框228,在一些实施方案中,多个模块中的模块152中的第一特征的特征值通过参考对象的生物样本中的相应成分的物理测量来确定。参考方框230,成分的实例包括但不限于组分(例如,核酸、蛋白质、或代谢产物)。参考图2C的方框232,在一些实施方案中,多个模块中的模块152中的第一特征的特征值是通过物理测量参考对象的生物样本中每一个相应的成分(例如,核酸、蛋白质、或其代谢物)获得的一组成分中的每个相应成分的特征值的线性或非线性组合。关于方框216,注意到,对于独立的多个特征,第一训练集是使用相应训练对象的生物样本以转录组学、蛋白质组学或代谢组学中的一种的第一形式获得的。参考方框234,在一些实施方案中,第一形式是转录组学的。参考方框236,在一些实施方案中,第一形式是蛋白质组学的。关于方框216,注意到,对于第一多个训练对象中的每个相应训练对象,第一训练集包括通过第一技术背景获取的第一多个特征值。参看方框238,在一些实施方案中,第一技术背景是DNA微阵列、MMChip、蛋白质微阵列、肽微阵列、组织微阵列、细胞微阵列、化合物微阵列、抗体微阵列、聚糖阵列,或反相蛋白质裂解物微阵列。在一些实施方案中,从每个对象收集的生物样本是对象的血液、全血、血浆、血清、尿液、脑脊液、粪便、唾液、汗液、眼泪、胸水、心包液或腹膜液。在一些实施方案中,生物样本是对象的特定组织。在一些实施方案中,生物样本是对象的特定组织或器官(例如,乳房、肺、前列腺、直肠、子宫、胰腺、食道、卵巢、膀胱等)的活检。在一些实施方案中,特征是对应于从测序序列读段获得的物种基因的核酸的核酸丰度值,而该序列读数又来自生物样本中的核酸并且代表生物样本中此类核酸以及它们所代表的基因的丰度。任何形式的测序均可用于从生物样本获得的核酸获得序列读段,包括但不限于高通量测序系统,例如Roche454平台,AppliedBiosystemsSOLID平台,HelicosTrueSingleMoleculeDNA测序技术,AffymetrixInc.的杂交测序平台,PacificBiosciences的单分子实时(SMRT)技术,454LifeSciences、Illumina/Solexa和HelicosBiosciences的合成测序平台,以及AppliedBiosystems的连接测序平台。LifeTechnologies的IONTORRENT技术和纳米孔测序也可用于从生物样本中获得的无细胞核酸获得序列读段140。在一些实施方案中,合成测序和基于可逆终止子的测序(例如,Illumina的基因组分析仪;基因组分析仪II;HISEQ2000;HISEQ2500(Illumina,SanDiegoCalif.))用于从生物样本中获得的核酸获得序列读段。在一些这样的实施方案中,平行地对数以百万计的无细胞核酸(例如,DNA)片段测序。在这种类型的测序技术的一个例子中,使用流动池,其含有具有八个单独的泳道的光学透明载玻片,在其表面上结合了寡核苷酸锚(例如,接头引物)。流动池通常是固体支撑物,其被配置为保留和/或允许试剂溶液在结合的分析物上有序通过。在某些情况下,流动池是平面形状、光学透明的,通常为毫米或亚毫米级,并且通常具有其中发生分析物/试剂相互作用的通道或泳道。在一些实施方案中,无细胞核酸样本可包括促进检测的信号或标签。在一些这样的实施方案中,从生物样本获得的核酸获得序列读段包括通过多种技术获得信号或标签的量化信息,例如流式细胞术、定量聚合酶链反应(qPCR)、凝胶电泳、基因芯片分析、微阵列、质谱、细胞荧光分析、荧光显微镜、共聚焦激光扫描显微镜、激光扫描细胞计数、亲和层析、手动分批模式分离、电场悬浮、测序及其组合。参看方框240,在一些实施方案中,模块的第一独立表型和临床状况相同。这针对表1的模块152-3和152-4进行了说明,其中临床状况是败血症,模块152-3的第一个独立表型是“败血症消退”,模块152-4的第一个独立表型是败血症消退。因此,对于模块152-3和152-4,训练集中所需的全部(除了特征值丰度)是每个训练对象被标记为是否患有败血症。参考方框242,在一些实施方案中,获得第二训练数据集。对于该物种的第二多个训练对象中的每个相应的训练对象,第二训练数据集包括:(i)通过第一技术背景以外的第二技术背景,使用相应的训练对象的生物样本,对于独立的多个特征,以与至少第一模块的第一形式相同的第二形式获得的第二多个特征值,以及(ii)在相应的训练对象中,第一独立表型的不存在、存在或阶段的指示。参见方框244,在一些实施方案中,第一技术背景(通过其获得第一训练集)是RNAseq,而第二技术背景(通过其获得第二训练集)是DNA微阵列。在一些实施方案中,第一技术背景是选自cDNA微阵列、寡核苷酸微阵列、BAC微阵列和单核苷酸多态性(SNP)微阵列的第一形式的微阵列实验并且第二技术背景是选自cDNA微阵列、寡核苷酸微阵列、BAC微阵列和SNP微阵列的不同于第一形式的微阵列实验的第二形式的微阵列实验。在一些实施方案中,第一技术背景是使用第一制造商的测序技术的核酸测序,第二技术背景是使用第二制造商的测序技术的核酸测序(例如,Illumina珠芯片相对于Affymetrix或Agilent微阵列)。在一些实施方案中,第一技术背景是使用第一测序仪进行第一测序深度的核酸测序,第二技术背景是使用第二测序仪进行第二测序深度的核酸测序,其中第一测序深度不是第二测序深度和第一测序仪器与第二测序仪器的品牌和型号相同,但第一和第二仪器是不同的仪器。在一些实施方案中,第一技术背景是第一类型的核酸测序(例如,基于微阵列的测序)和第二技术背景是第一类型的核酸测序以外的第二类型的核酸测序(例如,下一代测序)。在一些实施方案中,第一技术背景是配对末端核酸测序,而第二技术背景是单读段核酸测序。以上是不同技术背景的非限制性实例。一般来说,当特征丰度数据是在不同的技术条件下,如不同的机器、不同的方法,或不同的技术条件下,如不同的试剂,或不同的技术参数(如在核酸测序的情况下,不同的覆盖范围等)下捕获时,两个技术背景是不同的。参看方框248,在一些实施方案中,第一训练数据集和第二训练数据集的每个相应生物样本属于对应训练对象的指定组织或指定器官。例如,在一些实施方案中,每个生物样本是血液样本。在另一个例子中,每个生物样本是乳房活检、肺活检、前列腺活检、直肠活检、子宫活检、胰腺活检、食道活检、卵巢活检或膀胱活检。参考图2D的方框252,在一些实施方案中,基于第一训练数据集中相应特征的特征值的每个相应分布对第一训练数据集执行第一归一化算法。进一步地,基于第二训练数据集中相应特征的特征值的每个相应分布,对第二训练数据集进行第二归一化算法。参考图2D的方框254,在一些实施方案中,第一归一化算法或第二归一化算法是稳健的多阵列平均算法、GeneChipRMA算法或用于背景校正的正态指数卷积算法,然后是分位数归一化算法。在一些实施方案中,在公开的方法中不执行这种归一化。作为非限制性实例,在这样的实施方案中,不执行方框252的归一化,因为数据集已经归一化。作为另一个非限制性实例,在一些实施方案中,不执行方框252的归一化,因为这样的归一化被确定为没有必要。参见方框256,至少第一和第二训练数据集中存在的特征的特征值跨至少第一和第二训练数据集进行共归一化,以去除数据集间批次效应,从而针对第一多个训练对象的每个相应训练对象和针对第二多个训练对象的每个相应训练对象计算相应训练对象的至少第一模块的共归一化特征值。在一些这样的实施方案中,这样的归一化为相应的训练对象提供多个模块中的每一个的共归一化的特征值。参见方框258,在一些实施方案中,(第一模块的)第一独立表型代表患病状况。此外,第一训练数据集的第一子集由没有患病状况的对象组成,而第二训练数据集的第一子集由没有患病状况的对象组成。此外,存在于至少第一和第二训练数据集中的特征值的共归一化包括仅使用相应的第一和第二训练数据集的第一子集来估计第一和第二训练数据集之间的数据集间批次效应。参见方框260,在一些此类实施方案中,数据集间批次效应包括加性分量和乘性分量,并且共归一化为跨各自的第一和第二训练数据集的第一子集的特征值求解普通最小二乘模型并使用经验贝叶斯估计器缩小表示加性分量和乘性分量的所得参数。例如,参见Sweeneyetal.,2016,SciTranslMed8(346),pp.346ra91,在此引入作为参考。参考方框264,在一些实施方案中,跨至少第一和第二训练数据集的在至少第一和第二训练数据集中存在的特征值的共归一化包括估计第一和第二训练数据集之间的数据集间批次效应。参见方框266,在一些实施方案中,数据集间批次效应包括加性分量和乘性分量,并且共归一化为跨各自的第一和第二训练数据集的第一子集的特征值求解普通最小二乘模型并使用经验贝叶斯估计器缩小表示加性分量和乘性分量的所得参数。例如,参见Sweeneyetal.,2016,SciTranslMed8(346),pp.346ra91,在此引入作为参考。参考图2E的方框266,在一些实施方案中,跨至少第一和第二训练数据集的在至少第一和第二训练数据集中存在的特征值的共归一化包括利用非变量特征、分位数归一化或等级归一化。参见Qiuetal.,2013,BMCBioinformatics14,p.124;和Hendriketal.,2007,PLoSOne2(9),p.e898,其中的每一个通过引用并入本文。参考图2F的方框258,在一些实施方案中,第一和第二数据集中的每个特征是核酸。第一技术背景是选自cDNA微阵列、寡核苷酸微阵列、BAC微阵列和单核苷酸多态性(SNP)微阵列的第一形式的微阵列实验。第二技术背景是选自cDNA微阵列、寡核苷酸微阵列、BAC微阵列和SNP微阵列的不同于第一形式的微阵列实验的第二形式的微阵列实验。参见,例如,Bumgarner,2013,Currentprotocolsinmolecularbiology,第22章,所述文献据此通过引用并入。在一些这样的实施方案中,共归一化是稳健多阵列平均(RMA)、GeneChip稳健多阵列平均(GC-RMA)、MAS5、探针对数强度误差(Plier)、dChip或芯片校准。参见,例如Irizarry,2003,Biostatistics4(2),pp.249-264;Welshetal.2013,BMCBioinformatics14,p.153;以及Therneau和Ballman,2008,CancerInform6,pp.423-431;和Oberg,2006,Bioinformatics22,pp.2381-2387,每篇文献均通过引用并入本文。参考图2F,该方法继续针对复合训练集训练主分类器,以评估测试对象的临床状况。对于第一多个训练对象中的每个相应训练对象和第二多个训练对象中的每个相应训练对象,复合训练集包括:(i)第一模块的共归一化特征值的汇总,以及(ii)相应训练对象中第一独立表型的不存在、存在或阶段的指示。参见方框270,在一些这样的实施方案中,对于第一和第二多个训练对象中的每个相应的训练对象,第一模块的共归一化特征值的汇总是从相应训练对象获得的生物样本中第一模块的共归一化特征值的集中趋势的量度(例如,算术平均值、几何平均值、加权平均值、中间范围、中枢纽(midhinge)、三均值、Winsorized平均值、中值或众数)。例如,在一些这样的实施方案中,在从相应训练对象获得的生物样本中,对于第一和第二多个训练对象中的每个相应的训练对象,第一模块的共归一化特征值的汇总是多个模块中的每个相应模块的共归一化特征值的集中趋势的量度(例如,算术平均值、几何平均值、加权平均值、中间范围、中枢纽、三均值、Winsorized平均值、中值或众数)。这在图3中进行了说明,其中每个模块fup、fdn、mup、mdn、sup和sdn分别为给定的训练对象提供了它们相应的共归一化特征值的集中趋势的度量。参见方框274,在替代实施方案中,对于第一多个训练对象中的每个相应训练对象和对于第二多个训练对象中的每个相应训练对象,第一模块的共归一化特征值的汇总是在输入从相应训练对象获得的生物样本中第一模块的共归一化特征值时,与第一模块相关联的分量分类器的输出。这在图4中进行了说明,其中每个模块都使用了迷你的网络‘辐条’。单个特征由局部网络汇总(而不是由其几何平均值汇总),然后传入主分类网络(主分类器)。参考方框276,在一些实施方案中,分量分类器是神经网络算法、支持向量机算法、决策树算法、无监督聚类算法、监督聚类算法、逻辑回归算法、混合模型或隐马尔可夫模型。如本文所用,主分类器指具有固定(锁定)参数(权重)和阈值的模型,其准备应用于以前未见过的样本(例如,测试对象)。在这种情况下,模型是指机器学习算法,例如逻辑回归、神经网络、决策树等(类似于统计学中的模型)。因此,参考图2G的方框278,在一些实施方案中,主分类器是神经网络。也就是说,在这样的实施方案中,主分类器是具有固定(锁定)参数(权重)和阈值的神经网络。在一些这样的实施方案中,参考方框280,第一独立表型和临床状况相同。参见方框282,在主分类器是神经网络的一些实施方案中,对于物种的第一多个训练对象中的每个相应训练对象,第一训练数据集进一步包括:(iii)多个特征值,其是使用多个模块中的第二模块的相应训练对象的生物样本通过第一技术背景获得的,以及(iv)相应训练对象中第二独立表型的不存在、存在或阶段的指示。对于物种的第二多个训练对象中的每个相应的训练对象,第二训练数据集还包括:(iii)多个特征值,其是使用第二模块的相应训练对象的生物样本通过第二技术背景获得的,以及(iv)相应训练对象中第二独立表型的不存在、存在或阶段的指示。换言之,如图3和图4所示,可以有多于一个模块。在方框282的情况中,有两个模块。根据方框284,在一些这样的实施方案中,第一独立表型和第二独立表型与临床状况相同(例如,败血症)。第一模块中的每个相应特征通过具有在物种群组中与不表现出独立表型的对象相比在表现出第一独立表型的对象中统计上显著更大的特征值而与第一独立表型相关联。这在图3中作为模块mup进行了说明。在一些实施方案中,通过将标准t检验、Welcht检验、Wilcoxon检验或置换检验应用于特征的丰度来评估关于该特征是否“统计上显著更大”的确定,所述特征的丰度如在表现出第一独立表型的群组(第1组)中的对象中和在未表现出第一独立表型以达到p值的群组(第2组)中的对象中测量的。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,特征在统计学上显著更少(更不丰富)。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,特征在统计学上显著更少,这使用错误发现率程序例如Benjamini-Hochberg或Benjamini–Yekutieli针对多个测试进行调整。参见例如Benjamini和Hochberg,JournaloftheRoyalStatisticalSociety,SeriesB57,pp.289-300;和Benjamini和Yekutieli,2005,JournalofAmericanStatisticalAssociation100(469),pp.71-80,在此通过引用将每一篇并入本文。在一些实施方案中,通过多个数据集(群组或训练数据集)的固定效应或随机效应荟萃分析,确定该特征在统计学上显著更少。例如,参见Sianphoeetal.,2019,BMCBioinformatics20:18,在此通过引用并入。第二模块中的每个相应特征通过具有在物种群组中与不表现出第一独立表型的对象相比在表现出第一独立表型的对象中统计上显著更少的特征值而与第一独立表型相关联。这在图3中作为模块mdn进行了说明。在一些实施方案中,通过将标准t检验、Welcht检验、Wilcoxon检验或置换检验应用于特征的丰度来评估关于该特征是否“统计上显著更少”的确定,所述特征的丰度如在表现出第一独立表型的群组(第1组)中的对象中和在未表现出第一独立表型以达到p值的群组(第2组)中的对象中测量的。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,特征在统计学上显著更少(更不丰富)。在一些此类实施方案中,当此类测试中的p值为0.05或更小、0.005或更小或0.001或更小时,特征在统计学上显著更少,这使用错误发现率程序例如Benjamini-Hochberg或Benjamini–Yekutieli针对多个测试进行调整。参见例如Benjamini和Hochberg,JournaloftheRoyalStatisticalSociety,SeriesB57,pp.289-300;和Benjamini和Yekutieli,2005,JournalofAmericanStatisticalAssociation100(469),pp.71-80,在此通过引用将每一篇并入本文。在一些实施方案中,通过多个数据集(群组或训练数据集)的固定效应或随机效应荟萃分析,确定该特征在统计学上显著更少。例如,参见Sianphoeetal.,2019,BMCBioinformatics20:18,在此通过引用并入。参见方框286,在框282的实施方案的一些实施方案中,第一独立表型和第二独立表型是不同的(例如,如在图3中示出,模块fup相对于模块sup)。参考方框288,在一些实施方案中,神经网络是前馈人工神经网络。关于前馈人工神经网络的公开,参见例如Svoziletal.,1997,ChemometricsandIntelligentLaboratorySystems39(1),pp.43-62,其在此通过引用并入。参考图2H的块290,在一些实施方案中,主分类器包括线性回归算法或惩罚线性回归算法。关于线性回归算法和惩罚线性回归算法的公开,例如参见Hastieetal.,2001,TheElementsofStatisticalLearning,纽约Springer-Verlag。在一些实施方案中,主分类器是神经网络。参见例如Hassoun,1995,FundamentalsofArtificialNeuralNetworks,麻省理工学院,其在此以引用的方式并入本文中。在一些实施方案中,主分类器是支持向量机算法。SVM在以下文献中描述:Cristianini和Shawe-Taylor,2000,“AnIntroductiontoSupportVectorMachines”,剑桥市剑桥大学出版社;Boseretal.,1992,“Atrainingalgorithmforoptimalmarginclassifiers”,第5届年度ACM计算学习理论研讨会论文集,宾夕法尼亚州匹兹堡市ACM出版社,第142-152页;Vapnik,1998,StatisticalLearningTheory,纽约Wiley出版社;Mount,2001,Bioinformatics:sequenceandgenomeanalysis,纽约州冷泉港市冷泉港实验室出版社;Duda,PatternClassification,第二版,2001,JohnWiley&Sons,Inc.,第259、262-265页;和Hastie,2001,TheElementsofStatisticalLearning,纽约Springer;以及Fureyetal.,2000,Bioinformatics16,906-914,在此通过引用将每篇文献全文并入。在一些实施方案中,主分类器是基于树的算法(例如,决策树)。参考图2H的方框292,在一些实施方案中,主分类器是选自随机森林算法和决策树算法的基于树的算法。决策树一般由Duda,2001,PatternClassification,JohnWiley&Sons,Inc.,NewYork,pp.395-396描述,在此通过引用并入。参见图2H的方框294,在一些实施方案中,主分类器由经受集成优化算法的分类器的集成(例如,adaboost、XGboost或LightGBM)组成。参见Alafate和Freund,2019,“FasterBoostingwithSmallerMemory”,arXiv:1901.09047v1,在此通过引用并入。参考图2H的方框295,在一些实施方案中,主分类器由神经网络的集成组成。参见Zhouetal.,2002,ArtificialIntelligence137,pp.239-263,在此通过引用并入。参考图2H的方框296,在一些实施方案中,临床状况是多类临床状况并且主分类器输出多类临床状况中每类的概率。例如,参考图3,在一些实施方案中,临床状况是细菌感染(Ibac)、病毒感染(Ivira)或非病毒、非细菌型感染(Inon)的三类状况,并且分类器提供对象具有Ibac的概率、对象具有Ivira的概率以及对象具有Inon的概率(其中概率总和为百分之百)。参见方框297,在一些实施方案中,获得多个附加训练数据(例如,3个或更多,4个或更多,5个或更多,6个或更多,10个或更多,或30个或更多)。对于物种的独立的相应多个训练对象中的每个相应的训练对象,多个附加数据集中的每个相应的附加数据集包括:(i)通过独立的相应技术背景,使用相应的训练对象的生物样本,对于多个模块中的相应模块的第一形式的独立的多个特征获得的多个特征值,以及(ii)对应于相应模块的相应训练对象中相应表型的不存在、存在或阶段的指示。在这样的实施方案中,方框256的共归一化还包括跨越包括第一训练数据集、第二训练数据集和多个附加训练数据集的训练组中的至少两个或更多个相应的训练数据集,共归一化在该训练组中的相应两个或更多个训练数据集中存在的特征的特征值,以去除数据集间批次效应,从而为多个训练数据集中的每个相应的两个或更多个训练数据集中的每个相应的训练对象计算在多个模块中的每个模块的共归一化的特征值。进一步地,对于训练组中每个训练数据集中的每个相应训练对象,复合训练集还包括:(i)对所述多个模块中相应训练对象中的一个模块的共归一化特征值的汇总和(ii)相应训练对象中相应独立表型的不存在、存在或阶段的指示。参考方框298,在一些实施方案中,获得包括多个特征值的测试数据集。对于至少第一模块中的特征,以第一形式(转录组学、蛋白质组学或代谢组学),在测试对象的生物样本中测量多个特征值。测试数据集被输入到主分类器中,从而评估测试对象的临床状况。即,主分类器响应于输入主分类器提供对测试对象的临床状况的确定。在一些实施方案中,临床状况是多类的,如图3所示,并且由主分类器提供的测试对象的临床状况的确定是在多类临床状况中测试对象具有每个组成类的概率。在一些实施方案中,本公开涉及用于训练用于评估测试对象的临床状况的分类器的方法1300,下面参考图13进行详述。在一些实施方案中,方法1300在如本文所述的系统处执行,例如,如上文关于图1所述的系统100。在一些实施方案中,方法1300在具有如关于系统100所描述的模块和/或数据库的子集的系统处执行。方法1300包括获得(1302)第一群组训练对象的特征值和临床状态。在一些实施方案中,特征值是从来自第一群组中的训练对象的生物样本中收集的,例如,如上文关于方法200所描述的。生物样本的非限制性实例包括固体组织样本和液体样本(例如,全血或血浆样本)。关于对方法1300有用的样本的更多细节在上面参考方法200进行了描述,并且为了简洁在此不再重复。在一些实施方案中,本文描述的方法包括测量各种特征值的步骤。在其他实施方案中,本文描述的方法例如以电子方式获得先前测量的特征值,例如存储在一个或多个临床数据库中的特征值。测量技术的两个例子包括核酸测序(例如,qPCR或RNAseq)和微阵列测量(例如,使用DNA微阵列、MMChip、蛋白质微阵列、肽微阵列、组织微阵列、细胞微阵列、化合物微阵列、抗体微阵列、聚糖阵列或反相蛋白质裂解物微阵列)。然而,技术人员将知道用于测量来自生物样本的特征的其他测量技术。关于对方法1300有用的特征测量技术(例如,技术背景)的更多细节在上面参考方法200进行了描述,并且为了简洁在此不再重复。在一些实施方案中,使用相同的测量技术收集第一群组中每个训练对象的特征值。例如,在一些实施方案中,每个特征是相同类型的,例如蛋白质、核酸、碳水化合物或其他代谢物的丰度,并且用于测量每个值的特征值的技术在整个第一群组中是一致的。例如,在一些实施方案中,特征是mRNA转录物的丰度并且测量技术是RNAseq或核酸微阵列。在其他实施方案中,例如,在一些实施方案中,当特征值未跨不同训练对象群组共归一化时,使用不同技术来测量跨第一训练对象群组的特征值。然而,在特征值没有跨不同群组共归一化的一些实施方案中,例如,在使用单个训练对象群组来训练分类器的情况下,使用相同的技术来测量跨第一群组的特征值。在一些实施方案中,方法1300包括获得(1304)附加训练对象群组的特征值和临床状态。在一些实施方案中,为至少2个附加群组收集特征值。在一些实施方案中,为至少3、4、5、6、7、8、9、10或更多个附加群组收集特征值。在一些实施方案中,使用相同技术测量为每个群组获得的特征值。也就是说,针对第一群组获得的所有特征值都是使用第一种技术测量的,针对第二群组获得的所有特征值都是使用不同于第一种技术的第二种技术测量的,针对第三群组获得的所有特征值都是使用与第一种技术和第二种技术不同的第三种技术测量的,等等。关于使用对方法1300有用的不同特征测量技术(例如,技术背景)的更多细节在上文中参考方法200进行了描述,此处不再赘述。在一些实施方案中,例如,在其中获得多个训练对象群组的特征值的一些实施方案中,方法1300包括在第一群组和任何附加群组之间共归一化(1306)特征值。在一些实施方案中,至少在第一和第二训练数据集(例如,对于第一和第二训练对象群组)中存在的特征的特征值跨至少第一和第二训练数据集共归一化以去除数据集间批次效应,从而为第一多个训练对象中的每个相应训练对象和第二多个训练对象中的每个相应训练对象计算相应训练对象的多个模块的共归一化特征值。在一些实施方案中,至少跨越第一和第二训练数据集的在至少第一和第二训练数据集(例如,以及任何附加训练数据集)中存在的共归一化特征值包括估计第一和第二训练数据集之间的数据集间批次效应。在一些实施方案中,数据集间批次效应包括加性分量和乘性分量,并且共归一化为跨各自的第一和第二训练数据集的第一子集的特征值求解普通最小二乘模型并使用经验贝叶斯估计器缩小表示加性分量和乘性分量的所得参数。在一些实施方案中,跨至少第一和第二训练数据集的在至少第一和第二训练数据集中存在的特征值的共归一化包括利用非变量特征或分位数归一化。在一些实施方案中,多个模块中相应模块的第一表型代表患病状况,第一训练数据集的第一子集由没有患病状况的对象组成,第二训练数据集的第一子集(例如,以及任何附加训练数据集)由没有患病状况的对象组成。在一些实施方案中,存在于至少第一和第二训练数据集中的特征值的共归一化则包括仅使用相应的第一和第二训练数据集的第一子集来估计第一和第二训练数据集之间的数据集间批次效应。在一些实施方案中,数据集间批次效应包括加性分量和乘性分量,并且共归一化为跨各自的第一和第二训练数据集的第一子集的特征值求解普通最小二乘模型并使用经验贝叶斯估计器缩小表示加性分量和乘性分量的所得参数。上面参考方法200描述了关于用于跨对应于各个训练群组的各个数据集的对方法1300有用的共归一化技术的更多细节,并且为了简洁在此不再重复。在一些实施方案中,方法1300包括汇总(1308)与多个模块的临床病症的表型相关的特征值。即,在一些实施方案中,将各自与一类或多类临床状况的特定表型相关联的亚多个所获得的特征值(例如,亚多个mRNA转录物丰度值)分组至模块中,并将这些分组的特征值汇总,以形成每个训练对象的相应模块的特征值的相应汇总。例如,图3和图4说明了一个示例分类器,其经过训练以区分与细菌感染、病毒感染以及既非细菌感染也非病毒感染有关的三类临床状况。具体地,图3图示了作为前馈神经网络的主分类器300的实例。输入层308被配置为接收多个模块352的特征值354的汇总358。例如,如图4的右侧所示,模块352-1包括特征值354-1、354-2和354-3,其对应于基因IFI27、JUP和LAX1的mRNA丰度值,它们每一种都以类似的方式与一类或多类临床状况的表型相关。在这种情况下,IFI27、JUP和LAX1都是在对象感染病毒时上调的基因。如图4所示,特征值通过在输入层304将它们输入到馈线神经网络中来进行汇总,其中该神经网络包括隐藏层306和输出汇总358-1,该汇总用作主分类器300的输入值。其他模块302-2至302-6中的每一个还包括为对象获得的亚多个特征,例如,其不同于每个其他模块中的亚多个特征,每个模块类似地与和一类或多类临床状况相关的不同表型相关联。例如,当对象感染病毒时,模块302-2中的基因被下调。类似地,模块302-3和302-4中的基因在败血症患者中分别上调和下调,与无菌炎症相反。同样,在因败血症入院30天内死亡的患者中,模块302-5和302-6中的基因都分别上调和下调。在一些实施方案中,方法1300使用至少3个模块,每个模块包括与由主分类器评估的一类或多类临床状况的表型类似地相关联的特征。在一些实施方案中,方法1300使用至少6个模块,每个模块包括与由主分类器评估的一类或多类临床状况的表型类似地相关联的特征。在其他实施方案中,方法1300使用至少2、3、4、5、6、7、8、9、10、15、20或更多个模块,每个模块包括与由主分类器评估的一类或多类临床状况的表型类似地相关联的特征。关于模块的更多细节,特别是关于对方法1300有用的与特定表型相关联的特征的分组,在上文参考方法200进行了描述,并且为了简洁在此不再重复。尽管图4中所示的汇总方法使用馈线循环网络,但也考虑了用于汇总相应模块的特征的其他方法。汇总模块特征的示例方法包括神经网络算法、支持向量机算法、决策树算法、无监督聚类算法、监督聚类算法、逻辑回归算法、混合模型或隐马尔可夫模型。在一些实施方案中,汇总是相应模块的特征值的集中趋势的度量。集中趋势的度量的非限制性实例包括相应模块的特征值的算术平均值、几何平均值、加权平均值、中间范围、中枢纽、三均值、Winsorized平均值、中值和众数。关于对方法1300有用的用于汇总模块的特征值的方法的更多细节在上面参考方法200进行了描述,为了简洁在此不再重复。方法1300然后包括针对(i)来自一个或多个训练对象群组的特征值的衍生值和(ii)一个或多个训练群组中的对象的临床状态来训练(1310)主分类器。在一些实施方案中,主分类器是神经网络算法、支持向量机算法、决策树算法、无监督聚类算法、监督聚类算法、逻辑回归算法、混合模型或隐马尔可夫模型。在一些实施方案中,主分类器是神经网络算法、线性回归算法、惩罚线性回归算法、支持向量机算法或基于树的算法。在一些实施方案中,主分类器由经受集成优化算法的分类器集成组成。在一些实施方案中,集成优化算法包括adaboost、XGboost或LightGBM。训练分类器的方法是本领域公知的。关于对方法1300有用的分类器类型和用于训练那些分类器的方法的更多细节在上文参考方法200进行了描述,并且为了简洁在此不再重复。在一些实施方案中,特征值衍生值是共归一化的特征值(1312)。即,在一些实施方案中,方法1300包括跨两个或多个训练数据集共归一化特征值的步骤,例如,所述数据集由使用如上文关于方法200和1300所述的不同测量技术获取的特征值形成,但是不是将细分至不同模块的特征值的组汇总的步骤。在一些实施方案中,特征值衍生值是特征值的汇总(1314)。即,在一些实施方案中,方法1300不包括跨两个或多个训练数据集共归一化特征值的步骤,例如,在使用单个测量技术来获取所有特征值的情况,但确实包括将细分至不同模块的特征值的组汇总的步骤,例如,如上文关于方法200和1300所述。在一些实施方案中,特征值衍生值是共归一化特征值的汇总(1316)。即,在一些实施方案中,方法1300包括跨两个或多个训练数据集共归一化特征值的步骤,例如,所述数据集由使用不同测量技术获取的特征值形成,如上文关于方法200和1300所述,以及将细分至不同模块的共归一化特征值的组汇总的步骤,例如,如上文关于方法200和1300所述。在一些实施方案中,特征值衍生值是特征值的共归一化汇总(1318)。即,在一些实施方案中,方法1300包括对细分至不同模块的特征值的组汇总的第一步骤,例如,如上文关于方法200和1300所描述的,以及使用如上文关于方法200和1300所述的共归一化技术对来自模块的汇总跨两个或多个训练数据集进行共归一化的第二步骤,例如,所述数据集由使用不同测量技术获取的特征值形成。应当理解,描述图13中的操作的特定顺序仅仅是示例,并不旨在表明所描述的顺序是可以执行操作的唯一顺序。本领域的普通技术人员将认识到对本文描述的操作重新排序的各种方式。例如,在一些实施方案中,每个模块的特征值的汇总(1308)在跨群组的共归一化(1306)之前执行,其中使用不同的测量技术收集特征数据。另外,应当注意,关于本文描述的其他方法(例如,上面关于图2描述的方法200和下面关于图14描述的方法1400)的本文描述的其他过程的细节也以类似的方式适用于上面关于图13描述的方法1300。例如,上文参考方法1300描述的特征值、模块、临床状况、临床表型、测量技术等可选地具有参考本文描述的其他方法(例如,方法200或1400)在本文描述的特征值、模块、临床状况、临床表型、测量技术等的一个或多个特征。类似地,上文参考方法1300描述的在各个步骤(例如,数据收集、共归一化、汇总、分类器训练等)使用的方法任选地具有参考本文描述的其他方法(例如,方法200或1400)在本文描述的数据收集、共归一化、汇总、分类器训练等的一个或多个特征。为简洁起见,这里不再重复这些细节。在一些实施方案中,本公开涉及用于评估测试对象的临床状况的方法1400,下面参考图14详述。在一些实施方案中,方法1400在如本文所述的系统处执行,例如,如上文关于图1所述的系统100。在一些实施方案中,方法1400在具有如关于系统100所描述的模块和/或数据库的子集的系统处执行。方法1400包括获得(1402)测试对象的特征值。在一些实施方案中,特征值是从来自测试对象的生物样本中收集的,例如,如上文关于方法200和1300所描述的。生物样本的非限制性实例包括固体组织样本和液体样本(例如,全血或血浆样本)。关于对方法1400有用的样本的更多细节在上文参考方法200和1300进行了描述,并且为了简洁在此不再重复。在一些实施方案中,本文描述的方法包括测量各种特征值的步骤。在其他实施方案中,本文描述的方法例如以电子方式获得先前测量的特征值,例如存储在一个或多个临床数据库中的特征值。测量技术的两个例子包括核酸测序(例如,qPCR或RNAseq)和微阵列测量(例如,使用DNA微阵列、MMChip、蛋白质微阵列、肽微阵列、组织微阵列、细胞微阵列、化合物微阵列、抗体微阵列、聚糖阵列或反相蛋白质裂解物微阵列)。然而,技术人员将知道用于测量来自生物样本的特征的其他测量技术。关于对方法1400有用的特征测量技术(例如,技术背景)的更多细节在上面参考方法200和1300进行了描述,并且为了简洁在此不再重复。在一些实施方案中,例如,分类器被训练以评估从各种不同的测量方法(例如,技术背景)获得的特征值的一些实施方案中,方法1400包括针对预定模式共归一化(1404)特征值。在一些实施方案中,预定模式源自跨两个或更多个训练数据集的特征数据共归一化,例如,使用不同的测量方法。上面参考方法200和1300详细描述了用于跨不同训练数据集进行共归一化的各种方法,为简洁起见,此处不再赘述。在一些实施方案中,为测试对象获得的特征值不进行归一化,该归一化说明用于获取这些值的测量技术。在一些实施方案中,方法1400包括将对象的特征值或归一化特征值分组(1406)至多个模块,其中相应模块中的每个特征值以类似方式与和正在评估的临床状况的一个或多个类别相关联的表型相关联。即,在一些实施方案中,将各自与一类或多类临床状况的特定表型相关联的亚多个所获得的特征值(例如,亚多个mRNA转录物丰度值)分组至模块中。在一些实施方案中,方法1400使用至少3个模块,每个模块包括与由主分类器评估的一类或多类临床状况的表型类似地相关联的特征。在一些实施方案中,方法1400使用至少6个模块,每个模块包括与由主分类器评估的一类或多类临床状况的表型类似地相关联的特征。在其他实施方案中,方法1400使用至少2、3、4、5、6、7、8、9、10、15、20或更多个模块,每个模块包括与由主分类器评估的一类或多类临床状况的表型类似地相关联的特征。关于模块的更多细节,特别是关于对方法1400有用的与特定表型相关联的特征的分组,在上文参考方法200和1300进行了描述,并且为了简洁在此不再重复。在一些实施方案中,特征值没有被分组到模块中,而是直接输入到主分类器中。在一些实施方案中,方法1400包括汇总(1408)每个相应模块中的特征值,以形成测试对象的相应模块的特征值的对应汇总。例如,如上文针对图3和4中所示的模块352-1所述。尽管图4中所示的汇总方法使用馈线循环网络,但也考虑了用于汇总相应模块的特征的其他方法。汇总模块特征的示例方法包括神经网络算法、支持向量机算法、决策树算法、无监督聚类算法、监督聚类算法、逻辑回归算法、混合模型或隐马尔可夫模型。在一些实施方案中,汇总是相应模块的特征值的集中趋势的度量。集中趋势的度量的非限制性实例包括相应模块的特征值的算术平均值、几何平均值、加权平均值、中间范围、中枢纽、三均值、Winsorized平均值、中值和众数。关于对方法1400有用的用于汇总模块的特征值的方法的更多细节在上面参考方法200和1300进行了描述,为了简洁在此不再重复。方法1400然后包括将特征值的衍生值输入(1410)到被训练以区分不同类别的临床状况的分类器中。在一些实施方案中,分类器被训练以区分两类临床状况。在一些实施方案中,分类器被训练以区分临床状况的至少3个不同类别。在其他实施方案中,分类器被训练以区分临床状况的至少4、5、6、7、8、9、10、15、20或更多个不同类别。参考方法200和1300如上所述训练主分类器。简而言之,针对(i)来自一个或多个训练对象群组的特征值的衍生值和(ii)一个或多个训练群组中的训练对象的临床状态来训练主分类器。在一些实施方案中,主分类器是神经网络算法、支持向量机算法、决策树算法、无监督聚类算法、监督聚类算法、逻辑回归算法、混合模型或隐马尔可夫模型。在一些实施方案中,主分类器是神经网络算法、线性回归算法、惩罚线性回归算法、支持向量机算法或基于树的算法。在一些实施方案中,主分类器由经受集成优化算法的分类器集成组成。在一些实施方案中,集成优化算法包括adaboost、XGboost或LightGBM。训练分类器的方法是本领域公知的。关于对方法1400有用的分类器类型和用于训练那些分类器的方法的更多细节在上文参考方法200和1300进行了描述,并且为了简洁在此不再重复。在一些实施方案中,特征值衍生值是依赖于测量平台的归一化特征值(1412)。即,在一些实施方案中,方法1400包括基于用于获取特征测量的方法对特征值进行归一化的步骤,这与在训练群组中使用的其他测量方法相反,如上文关于方法200和1300所描述的,但是不是将细分至不同模块的特征值的组汇总的步骤。在一些实施方案中,特征值衍生值是特征值的汇总(1414)。即,在一些实施方案中,方法1400不包括基于用于获取特征测量的方法对特征值进行归一化的步骤,这与在训练群组中使用的其他测量方法相反,但是的确包括将细分至不同模块的特征值的组汇总的步骤,如上文关于方法200和1300所描述的。在一些实施方案中,特征值衍生值是归一化特征值的汇总(1416)。即,在一些实施方案中,方法1400包括基于用于获取特征测量的方法对特征值进行归一化的步骤,这与在训练群组中使用的其他测量方法相反,如上文关于方法200和1300所描述的,以及将细分至不同模块的归一化特征值的组汇总的步骤,例如,如上文关于方法200和1300所描述的。在一些实施方案中,特征值衍生值是特征值的共归一化汇总(1418)。即,在一些实施方案中,方法1400包括将细分至不同模块的特征值的组汇总的第一步骤,例如,如上文关于方法200和1300所描述的,以及基于用于获取特征测量的方法对特征值进行归一化的第二步骤,这与在训练群组中使用的其他测量方法相反,如上文关于方法200和1300所描述的。在一些实施方案中,方法1400还包括基于分类器的输出治疗测试对象的步骤。在一些实施方案中,分类器提供对象具有被评估的临床状况的多个类别之一的概率。当从分类器输出的概率肯定地识别出一种类别的临床状况,或肯定地排除特定类别的临床状况时,治疗决策可以基于所述输出。例如,在分类器的输出指示对象具有第一类别的临床状况的情况下,通过向对象施用针对第一类别的临床状况定制的第一疗法来治疗对象。相反,在分类器的输出指示对象具有第二类别的临床状况的情况下,通过向对象施用针对第二类别的临床状况定制的第二疗法来治疗对象。例如,参考图4中所示的分类器,该分类器经过训练以评估对象是否患有细菌感染、病毒感染或患有与细菌或病毒感染无关的炎症。在将测试数据输入分类器后,当分类器指示对象患有细菌感染时,向对象施用抗菌剂,例如抗生素。然而,当分类器指示对象患有病毒感染时,对象不会被施用抗生素但可以被施用抗病毒剂。类似地,当分类器指示对象患有与细菌或病毒感染无关的炎症时,对象不会被施用抗生素或抗病毒剂,但可能被施用抗炎剂。应当理解,描述图14中的操作的特定顺序仅仅是示例,并不旨在表明所描述的顺序是可以执行操作的唯一顺序。本领域的普通技术人员将认识到对本文描述的操作重新排序的各种方式。例如,在一些实施方案中,每个模块的特征值的汇总(1408)在跨群组的归一化(1404)之前执行,其中使用不同的测量技术收集特征数据。另外,应当注意,关于本文描述的其他方法(例如,上面关于图2描述的方法200和上面关于图13描述的方法1300)的本文描述的其他过程的细节也以类似的方式适用于上面关于图14描述的方法1400。例如,上文参考方法1400描述的特征值、模块、临床状况、临床表型、测量技术等可选地具有参考本文描述的其他方法(例如,方法200或1300)在本文描述的特征值、模块、临床状况、临床表型、测量技术等的一个或多个特征。类似地,上文参考方法1400描述的在各个步骤(例如,数据收集、共归一化、汇总、分类器训练等)使用的方法任选地具有参考本文描述的其他方法(例如,方法200或1300)在本文描述的数据收集、共归一化、汇总、分类器训练等的一个或多个特征。为简洁起见,这里不再重复这些细节。实施例1临床感染基因表达研究的系统检索和纳入标准从NCBIGEO(www.ncbi.nlm.nih.gov/geo/)和EMBL-EBIArrayExpress(www.ebi.ac.uk/arrayexpress)数据库获得用于临床感染研究的符合定义的纳入标准的IMX训练数据集。具体而言,纳入标准包括在研究中的患者1)必须是医师裁定为感染的存在和类型(例如严格细菌感染、严格病毒感染、或非感染炎症),2)具有先前由Sweeney等人鉴定的29种诊断标志物的基因表达测量(Sweeneyetal.,2015,SciTranslMed7(287),pp.287ra71;Sweeneyetal,2016,SciTranslMed8(346),pp.346ra91;和Sweeneyetal.,2018,NatureCommunications9,p.694),3)超过18岁,4)已经被看见在医院环境中(例如急诊科,重症监护),5)具有社区或医院获得性感染,和6)在最初怀疑感染和/或败血症的24小时内采集了血液样本。此外,使用的归一化/批次效应控制方法要求每个包括的研究必须测定至少对照样本(例如,未诊断出考虑的三种状况的任一种的样本)。排除以下研究:患者经历过创伤,或者具有在典型的临床环境不会遇到(例如实验LPS攻击)或与感染混淆(如过敏性休克)的状况。实施例2表达数据的归一化和COCONUT共归一化然后在每项研究中进行归一化,取决于平台,采用两种方法之一。对于Affymetrix阵列,使用RobustMulti-arrayAverage(RMA)(Irizarryetal.,2003,Biostatistics,4(2):249-64)或gcRMA(Wuetal.,2004,JournaloftheAmericanStatisticalAssociation,99:909–17)归一化表达数据。使用指数卷积方法对来自其他平台的表达数据进行归一化,以进行背景校正,然后进行分位数归一化。在对原始表达数据进行归一化之后,使用COCONUT算法(Sweeneyetal.,2016,SciTranslMed8(346),pp.346ra91;和Abouelhodaetal.,2008,BMCBioinformatics9,p.476)共归一化这些测量值,并确保它们在研究中具有可比性。COCONUT建立在ComBat(Johnsonetal.,2007,Biostatistics,8,pp.118-127)经验贝叶斯批量校正方法的基础上,计算来自健康患者的每个基因的预期表达值并针对基因表达中研究特定性位置修改(平均值)和尺度(标准偏差)进行调整。对于该分析,使用ComBat的参数先验,其中基因表达分布被假定为高斯分布,以及研究特定性位置和方差修改参数的经验先验分布分别是高斯和逆伽马分布。实施例3通过机器学习开发败血症分类器为了开发败血症分类器,采用了机器学习方法。该方法包括指定候选模型,使用训练数据和指定的性能统计评估不同分类器的性能,然后选择性能最佳的模型来评估独立数据。在这种情况下,模型是指机器学习算法,例如逻辑回归、神经网络、决策树等(类似于统计学中使用的模型)。类似地,在这种情况下,主分类器指具有固定(锁定)参数(权重)和阈值的模型,其准备应用于以前未见过的样本。分类器使用两种类型的参数:由核心学习算法(例如XGBoost)学习的权重,以及作为核心学习器输入的附加的用户提供的参数。这些附加参数称为超参数。分类器开发需要学习(固定)权重和超参数。权重由核心学习算法学习;以学习超参数。对于本研究,采用随机搜索方法(Bergstraetal.,2012,JournalofMachineLearningResearch13,pp.281-305)。比较了四种不同类型预测模型的性能:1)带有lasso(L1)惩罚的逻辑回归,2)带有径向基函数内核(RBF)的支持向量机(SVM)分类器,3)极端梯度提升树(XGBoost),和4)多层感知器(MLP)。评估每种类型的预测模型在将患者样本分类为以下之一时的准确性:a)严格的细菌感染,b)严格的病毒感染,或c)非感染性炎症。为了在这个三类分类任务上评估每个预测模型,开发了一种称为平均成对ROC曲线下面积(APA)的指标。APA被定义为三个一类对所有(OVA)ROC曲线下面积的平均值;即,细菌对其他AUC、病毒对其他AUC以及非感染对其他AUC的平均值。多种用于评估特定分类器的性能的方法(例如,用一组固定的权重和超参数的模型)可以在机器学习中使用。在这里,采用了交叉验证(CV),这是一种用于败血症研究等小样本场景的成熟方法。使用了两种CV变型,如下所述。实施例4模型交叉验证方法最初考虑了两种不同类型的CV方案:传统的5折交叉验证和留一研究(LOSO)交叉验证。对于5折CV的试验,使用标准方法将所有IMX样本随机划分为样本大小大致相似的五个不重叠子集。对于LOSOCV的试验,每项研究都被视为CV分区。以这种方式,在LOSOCV的每一步(“折叠”),将候选模型在所有研究但除了一个中训练,然后将训练的模型用于为剩余的研究生成预测。使用LOSOCV的基本原理如下。简而言之,k-折CV的假设是交叉验证训练和验证样本来自相同的分布。然而,由于败血症研究的异常异质性,这一假设甚至不能大致满足。LOSO旨在支持根据经验对这种异质性最稳健的模型;换句话说,最有可能很好地推广到以前未见过的研究的模型。这是败血症分类器临床应用的关键要求。LOSO方法与先前的工作有关,该先前的工作提出在交叉验证之前对训练数据进行聚类,作为核算异质性的一种手段(Tabe-Bordbar,2018,etal.,SciRep8(1),pp.6620)。在这种情况下,不需要聚类,因为从训练数据的划分到研究,聚类自然而然地遵循。在k-折CV和LOSO中,预测都集中在所有折叠的左侧折叠中,以评估模型性能。或者,可以通过估计每个折叠感兴趣的统计数据,然后平均每个折叠的结果,计算CV统计数据。在本研究中,LOSO需要汇集,因为大多数研究没有来自所有三个类别的样本,因此大多数感兴趣的统计数据无法在单个LOSO折叠上计算。在这种情况下,为了与k折CV进行公平比较,统一应用汇集方法。为了确定用于诊断分类器的选择和前瞻性验证的适当交叉验证方案和特征集,使用了分层交叉验证(HCV)。HCV在技术上等同于嵌套CV(NCV)。但是,这里将其称为HCV,因为它用于与NCV不同的目的。具体来说,在NCV中,目标是估计已选择模型的性能。相比之下,这里使用HCV评估和比较模型选择过程的组成部分(步骤)。HCV将IMX数据集分为三个折叠;每个折叠构建为使得来自给定研究的所有样本仅出现在一个折叠中。这三个HCV折叠是手动构建的,以具有相似的细菌、病毒和未感染样本的组成。为了在此框架中评估5折叠和LOSOCV,对来自两个HCV折叠(内折叠)的样本执行每种CV方法。然后根据它们在内折叠上的CV性能(根据APA)对模型进行排名,并在剩余的第三个HCV折叠(外折叠)上评估来自每种CV方法的前100个模型。该程序进行三次,每次将外折叠设置为一个HCV折叠,将内折叠设置为剩余的两个HCV折叠。实施例5预测模型评估和超参数搜索发现有前景的候选预测模型涉及识别每个模型的超参数值,这些值会导致稳健的泛化性能。这里评估的四个预测模型可以大致分为具有小(低维)或大(高维)超参数数量的模型。更具体地说,具有低维超参数空间的预测模型是带有lasso惩罚SVM的逻辑回归,而具有高维超参数空间的预测模型是XGBoost和MLP。对于具有低维超参数空间的预测模型,抽样了5000个模型实例(模型对应超参数的不同值)进行交叉验证评估。对于具有高维超参数空间预测模型(例如xgboost和MLP),随机抽样了100000个模型实例。在逻辑回归的情况下,只考虑一个超参数:lasso惩罚系数。对于SVM,对C惩罚项的值和核系数γ进行抽样。对于XGBoost,抽样了以下超参数:1)伪随机数生成器种子,2)学习率,3)在分类器树中引入分裂所需的最小损失减少,4)最大树深度,5)最小子节点权重,6)每个子节点所需的最小实例权重总和,7)最大增量步长,8)权重正则化的L2惩罚系数,9)树方法(精确或近似),以及10)轮数。对于MLP,批大小固定为128,优化算法为ADAM。然后抽样下面的超参数:1)隐藏层数,2)每个隐藏层的节点数,3)每个隐藏层的激活函数类型(例如ReLU和变体,直链,sigmoid,双曲正切),4)学习率,5)训练迭代次数,6)权重正则化的类型(L1、L2、无),和7)输入层和隐藏层的丢弃(dropout)的存在(是否启用)和数量(概率)。每个隐藏层的节点数在所有隐藏层中都是相同的。ADAM的β1、β2和ε参数分别固定为0.9、0.999和1e-08。在XGBoost和MLP的情况下,一些超参数是从网格中均匀采样的,而另一些则是按照上面Bergstra&Bengio的方法从连续范围中采样的。实施例6神经网络超参数的微调在神经网络分析中,观察到的结果的显著变化与用于初始化网络权重的种子值有关。为了解释这种可变性,考虑了多种方法,包括各种集成模型。根据经验证据,采用了将种子作为附加超参数包含在搜索中的方法。“核心”超参数是随机搜索的,而种子是使用固定的预定义的1000个值列表进行详尽搜索的。添加随机种子显著增加了超参数搜索空间。为了减少计算量,使用大网格超参数(种子除外)作为起始点。对于来自网格的每个随机样本,搜索了超过250个种子值。完成初始搜索后,选择了最有希望的超参数的较小网格。然后通过在有希望的超参数配置附近搜索来细化超参数值。对于每个随机采样的微调点,搜索额外的更大的一组种子值(例如,750个)。选择具有最大APA的配置作为最终的、锁定的超参数值组。该组包括随机数生成器种子。实施例7诊断标记和几何平均特征组在这些分析中考虑了两组输入特征。第一组由29个基因标记组成,这些基因标记先前被鉴定为对感染的存在、类型和严重程度具有高度辨别力(Sweeneyetal.,2015,SciTranslMed7(287),pp.287ra71;Sweeneyetal,2016,SciTranslMed8(346),pp.346ra91;和Sweeneyetal.,2018,NatureCommunications9,p.694)。第二组输入特征基于模块(相关基因的子集)。这29个基因被分在6个模块中,这样每个模块由在给定感染或严重程度条件下共享表达模式(趋势)的基因组成。例如,发烧模块中的基因在发烧患者中过度表达(上调)。模块的组成如表1所示。表1.败血症相关模块(基因的组)的定义和组成。发烧/烧退:在严格的病毒感染中表达升高/降低的基因。败血症上升/下降:败血症患者相对于无菌炎症患者的表达升高/降低的基因。严重性升高/降低:在入院30天内死亡的患者中表达升高/降低的基因。这些分析中使用的基于模块的特征是根据每个模块中基因的表达值计算的几何平均值,从而导致每个患者样本的六个几何平均分数。这种方法可以被视为“特征工程”的一种形式,这是已知有时可以显著提高机器学习分类器的性能的方法。实施例8通过COCONUT的迭代应用比对IMX和ICU数据集以验证临床数据集外部验证在IMX上训练的预测模型需要在不同技术平台上相当的第一制作表达水平(例如,微阵列用于IMX和NanoString用于验证临床数据)用于生成两个数据集。在对原始表达数据进行归一化之后,我们使用COCONUT算法(Sweeneyetal.,2016,SciTranslMed8(346),pp.346ra91)共归一化这些测量值,并确保它们在研究中具有可比性。COCONUT建立在ComBat(Johnsonetal.,2007,Biostatistics,8,pp.118-127)经验贝叶斯批量校正方法的基础上,计算来自健康患者的每个基因的预期表达值并针对基因表达中研究特定性位置修改(平均值)和尺度(标准偏差)进行调整。对于该分析,我们使用ComBat的参数先验,其中基因表达分布被假定为高斯分布,以及研究特定性位置和方差修改参数的经验先验分布分别是高斯和逆伽马分布。有利地,COCONUT算法被迭代地应用,对IMX数据集的健康样本应用共归一化,同时在每个步骤保持验证临床数据集的健康样本不被修改。在此设置中,NanoString健康样本代表目标数据集,因为它在整个过程中保持不变,而IMX健康样本代表与目标数据集相似的查询数据集。当IMX和NanotString中29种诊断标志物的平均表达向量之间的平均绝对偏差(MAD)在连续迭代中变化不超过0.001时,该过程终止。该过程的更详细伪代码显示在图12中。根据图1和图12,本公开提供了一种用于数据集共归一化的计算机系统100,该计算机系统包括至少一个处理器102和存储由至少一个处理器执行的至少一个程序(例如,数据共归一化模块124)的存储器111/112)。该至少一个程序还包括用于(A)以电子形式获得第一训练数据集的指令。对于物种的第一多个训练对象中的每个相应的训练对象,第一训练数据集包括:(i)使用相应的训练对象的生物样本,对于多个特征获得的第一多个特征值,以及(ii)在相应的训练对象中临床状况的不存在、存在或阶段的指示,并且其中所述第一训练数据集的第一子集由不呈现临床状况的对象组成(例如,图12的Q数据集)。该至少一个程序还包括用于(B)以电子形式获得第二训练数据集的指令。对于物种的第二多个训练对象中的每个相应的训练对象,第二训练数据集包括:(i)使用相应的训练对象的生物样本,对于多个特征获得的第二多个特征值,以及(ii)在相应的训练对象中临床状况的不存在、存在或阶段的指示,并且其中所述第二训练数据集的第一子集由不呈现临床状况的对象组成(例如,图12的T数据集)。所述至少一个程序还包括用于(C)估计(i)跨越第一多个对象的多个特征的子集的平均表达的向量和(ii)跨第二多个对象的多个特征的子集的平均表达的向量之间的初始平均绝对偏差的指令(例如,图12,步骤2)。例如,如图12的步骤2所示,在一些实施方案中,(C)估计(i)跨越第一多个对象的多个特征的子集的平均表达的向量和(ii)跨第二多个对象的多个特征的子集的平均表达的向量之间的初始平均绝对偏差包括将初始平均绝对偏差设置为零。所述至少一个程序还包括用于(D)跨至少第一和第二训练数据集共归一化多个特征的子集的特征值以去除数据集间批次效应的指令,其中至少在第一和第二训练数据集中存在特征子集,所述共归一化包括仅使用相应的第一和第二训练数据集的第一子集来估计第一和第二训练数据集之间的数据集间批次效应,以及数据集间批次效应包括加性分量和乘性分量,并且所述共归一化为跨相应的第一和第二训练数据集的第一子集的特征值求解普通最小二乘模型,并使用经验贝叶斯估计器缩小表示所述加性分量和所述乘性分量的所得参数,从而使用所得参数计算:对于在第一多个训练对象中每个相应的训练对象,多个特征中的每个特征值的共归一化特征值(例如图12步骤3a以及如在Sweeneyetal.,2016,SciTranslMed8(346),pp.346ra91中公开的)。所述至少一个程序还包括用于(F)估计(i)跨第一训练数据集的多个特征的共归一化特征值的平均表达的向量和(ii)跨第二训练数据集的多个特征的子集的平均表达的向量之间的共归一化后平均绝对偏差的指令(例如,图12步骤3b、3c、3d和3e)。所述至少一个程序还包括用于(G)重复共归一化(E)和估计(F)直至共归一化平均绝对偏差收敛的指令(例如,图12步骤3f和3g以及步骤3的while条件τ>0001)。实施例9用于与NanoString表达数据进行一般比对的商业健康样本在临床环境中部署上述迭代COCONUT程序是不可行的,因为它需要在部署地点获取健康样本并重新比对所有健康样本(以前获取的和新获取的)。为了在健康患者中建立NanoString表达的通用模型,确定了一组40个市售健康对照样本,其中鉴定了十个PAXGENETM全血RNA样本,每个样本均获自美国大陆的四个不同地点。提供这些样本的捐献者自我报告为健康,HIV和丙型肝炎检测结果均呈阴性。在性别方面,12份健康样本来自女性捐献者,其余28份样本来自男性捐献者。实施例10验证临床研究样本描述和NanoString表达谱本研究招募了因疑似败血症入院的患者。为了生成ICU样本的NanoString表达,在QIAcube(Qiagen)上使用RNeasyPlusMicroKit(Qiagen,部件#74034)分离RNA,然后提取每个样本的PAXgeneRNA,使用自定义脚本用于QIAcube进行RNA分离。每个表达谱反应由每个样本150ngRNA组成。根据制造商的说明,用于检测我们的生物标志物组的表达的定制探针代码集和样本RNA在65℃下杂交16小时。然后使用nCounterSPRINT标准方案生成NanoString表达,从而产生原始RCC表达文件。没有对这些原始表达值进行归一化。处理后,共有104个数据样本可供分析。如上所述,在公共领域确定了18项符合纳入标准并用于分类器训练的研究。这些研究包括1069个不同的患者样本。研究的组成和主要特征显示在表2中。表2.训练研究的特征。ED=急诊科;ICU=重症监护室。ED/ICU是在ED收集的样本数量(百分比)(其余来自ICU)。平台=基因表达平台。括号中的数字表示百分比。1平台:A=Agilent,I=Illumina归一化根据上述程序,使用COCONUT、PROMPT数据和在NanoString仪器上处理的40个商业对照样本对研究归一化的训练数据进行迭代调整。由此产生的批次调整训练数据进入探索性数据分析和机器学习。为了说明COCONUT共归一化的迭代过程,图5绘制了归一化之前、期间和之后训练集中所选基因的分布图。正如预期的,随着迭代,目标和查询数据集中的分布在视觉上变得更加接近。探索性数据分析然后对算法中使用的29个基因中的每一个的细菌、病毒和未感染样本的共归一化表达值的分布进行可视化,如图6所示。该直方图表明在个体基因水平上类别的适度(细菌相对于病毒)到最小(非感染)分离,并且需要先进的多基因建模以实现败血症分类器的临床效用。接下来,使用t分布随机邻域嵌入(t-SNE)(如图7所示)和主成分分析(PCA)(如图8所示)将三类数据的投影可视化为2维和3维。两项分析都证实了需要开发高维分类器才能达到临床可行性能的初步发现。样本也在二维PCA空间中通过研究绘制,如图9所示。该结果表明,通过COCONUT进行归一化后存在残留的研究效果。这一观察,与该领域现有研究一起表明,分类器必须在不同的、以前看不见的研究进行测试,以避免被研究混淆(例如,为了避免学习批次而不是疾病信号)。鉴于训练集中的一些研究是单一疾病,这一点尤为重要。留一研究与交叉验证疾病异质性和剩余批次效应表明,模型选择的普通交叉验证可能会出现明显的过度拟合。为了检验这一假设,对两种模型选择方法进行了比较分析:5折交叉验证和留一研究交叉验证。该分析使用了3折分层交叉验证(HCV),其中每个外折叠模拟对内循环中选择的最佳分类器的独立验证。这暴露了特定分类器选择方法的潜在过度拟合,而无需单独(且不可用)的验证集。将这些研究结合起来,使每个分区中的类别分布尽可能相似。在HCV中,每个内循环使用标准CV或LOSO执行分类器调整。为了选择最佳模型,我们通过平均成对AUROC统计(APA)对候选者进行排名。选择APA的原因是:(1)在初步分析中,它在所有相关统计数据的训练和测试数据之间表现出最一致的行为,(2)它在诊断败血症方面在临床上高度相关,以及(3)模型选择统计数据的选择并不重要,因为先前的证据表明CV和LOSO的泛化能力之间的差距很大。换句话说,可以使用其他统计数据,但APA是一个直接的选择。使用带有RBF核、深度学习MLP、逻辑回归(LR)和XGBoost分类器的SVM进行比较。使用这些分类器的基本原理是:(1)对于SVM,先前的经验,在现有临床诊断测试中的使用,(2)对于LR,一般医学上的广泛接受,特别是传染病的诊断,(3)对于XGBoost,机器学习社区的广泛接受,以及在重大竞争挑战(如Kaggle)中最佳性能的跟踪记录,和(4)对于深度神经网络,近期在多个应用领域(图像分析、语音识别、自然语言处理、强化学习)的突破性成果。使用29个归一化的表达谱作为输入特征和6个GM分数作为分类器的输入特征进行分析。使用6个GM分数的理由是,在先前的研究和初步分析(内部数据,未显示)中,它显示了非常有希望的结果。结果如图10和11所示。在所有分析中,除了GM逻辑回归运行之一之外,LOSOCVAUC估计值比k折CV估计值更接近测试集值。与红色(k折)点相比,蓝色(LOSO)点与垂直虚线的接近程度证明了这一点。基于这一发现,其余的分析都使用了LOSO。此外,分析表明,与29个基因表达特征相比,使用6个GM分数的测试集性能更优。表3显示了两组特征和不同分类器的测试集APA的比较。本次比较的模型选择标准使用了LOSO,因为之前发现LOSO的偏差要小得多。表3.使用GM分数和基因表达作为输入特征比较测试集性能。该表包含GM分数(GMS)和29个基因表达值(GENEX)的APA值。APA列包含图11中所示的10个模型的平均值,用于三个HCV测试集。最好的模型是使用LOSO交叉验证方法发现的。对于每个GMS/GENEX对,较高的APA用粗体字母表示。如表3所示,GMS分数几乎在所有情况下都产生了更高的性能。基于这一发现,其余的分析使用GM分数作为分类算法的输入特征。这种GM分数的使用是上文结合图1A和1B讨论的模块152/汇总算法156的实例化。分类器开发为了开发分类器,对四种不同的模型进行了超参数搜索。使用LOSO交叉验证方法进行搜索,并使用6个GM分数作为输入特征。对于每个配置,都执行了LOSO学习,并汇集了遗漏数据集中的预测概率。对于每个配置,结果是训练集中所有样本的一组预测概率。然后使用汇集概率计算APA,并使用APA值对超参数配置进行排名。最佳配置是具有最大APA的配置。表4给出了不同算法的汇总的LOSO结果。表4.LOSO训练结果。“APALOSO”列包含相应模型的最佳表现超参数配置的LOSO交叉验证统计数据。模型APALOSO多层感知器0.87支持向量机0.85XGBoost0.77逻辑回归0.76在四个分类器中,MLP给出了最好的LOSO交叉验证APA结果。获胜配置使用了以下超参数:两个隐藏层,每个隐藏层四个节点,250次迭代,线性激活,无丢弃,学习率=1e-5,批大小=128,批归一化,正则化:L1(惩罚=0.1),以及使用权重先验的输入层权重初始化。表5包含使用获胜配置的汇集LOSO概率估计的附加性能统计数据。表5.获胜神经网络分类器的详细LOSO统计数据。该分析表明网络性能对网络权重的伪随机初始化很敏感。为了探索这些初始起点的空间,对具有获胜超参数配置的模型进行了额外的LOSO分析,并使用5000次不同的网络权重随机初始化(使用由所选配置指定的权重先验)。使用与初始运行相同的方法对网络进行训练和评估,例如通过汇集LOSO运行中所有折叠的预测概率并计算汇集概率的APA。获胜种子是对应于具有最高APA的模型的种子。锁定的最终模型应用于验证临床数据。即,通过将锁定分类器应用于验证临床NanoString表达数据来计算验证临床结果。这为每个样本:细菌、病毒和未感染产生了三类概率。使用多个临床相关统计数据,通过将预测与临床判定的诊断进行比较来评估分类器的效用。表6包含结果。表6.应用于独立验证临床样本(n=104)的BVN1分类器的性能统计数据。统计点估计值[95%CI]APA0.83细菌相对其他AUROC0.85病毒相对其他AUROC0.88非感染相对其他AUROC0.77细菌准确度80%病毒准确度50%非感染准确度62%在临床使用中,诊断患者时感兴趣的关键变量预计是细菌和病毒感染的可能性。这些值由神经网络的顶(softmax)层发出。讨论如上所述,开发了一种机器学习分类器,用于诊断疑似具有状况的患者的细菌和病毒败血症,并对独立测试数据进行初步验证。该项目面临几个主要挑战。首先,关于平台转移,分类器是专门使用公共领域数据开发的,在各种微阵列芯片上进行分析。相比之下,测试数据是使用NanoString分析的,这是一个以前在训练中从未遇到过的平台。其次,可用的训练数据集之间存在显著的异质性。第三,训练样本量相对较小,特别是考虑到训练数据的异质性问题。为了应对这些挑战,应用了多个研究方向。首先,研究了选择用于败血症分类的最佳机器学习模型的方法。迄今为止的研究表明,由于败血症数据中非常显著量的技术和生物学异质性,标准随机交叉验证会产生过度的乐观偏差。根据经验发现和对该对象的先前研究,选择了留一研究(LOSO)方法进行分类器开发。接下来,分析了输入特征工程的影响。LOSO一直偏爱由六个几何平均分数组成的定制工程输入,因此将其用作最终锁定分类器的输入。这是一个有点出乎意料的结果,值得进一步研究,包括自动学习和改进特征工程转换的可能性。独立测试数据的概率分布在预期方向上表现出明显的趋势,因为细菌样本的细菌概率往往很高,病毒样本的病毒概率也是如此。此外,未感染的样本倾向于较低的细菌和病毒可能性。这些趋势通过有利的成对AUROC估计和类别条件精度进行量化。然而,分布之间的显著残余重叠也被注意到,这是正在进行的研究的重点。目前平台转移的尝试已经成功。尽管如此,为了提高测试临床性能,我们败血症分类器的未来增强将把NanoString数据添加到训练集中。这项研究证明了使用公共数据成功学习复合败血症分类器的可行性,然后转移到以前看不见的平台上分析的以前看不见的样本。据我们所知,这在之前的败血症文献中没有报道过,在分子诊断学的其他地方也许没有报道过。结束语可以为本文描述为单个实例的组件、操作或结构提供多个实例。最后,各个组件、操作和数据存储之间的边界在某种程度上是任意的,并且在特定说明性配置的上下文中说明了特定操作。设想了其它功能分配,并且可以落入所述(多个)实施方案的范围内。总体上,在示例配置中作为单独分开的组件呈现的结构和功能可以实施为组合结构或组件。类似地,作为单个组件呈现的结构和功能可以实施为单独分开的组件。这些结构和功能及其它变型、修改、添加和改进落入所述(多个)实施方案的范围内。还将理解,虽然术语“第一”、“第二”等可在本文中用于描述各种元件,但这些元件不应受这些术语限制。这些术语仅用于将一个元件与另一元件区分开来。例如,在不脱离本公开的范围的情况下,第一主体可以被称为第二主体,并且类似地,第二主体可以被称为第一主体。虽然第一对象和第二对象都是对象,但他们不是同一对象。在本公开中使用的术语仅用于描述特定实施方案,并不旨在限制本发明。如在本发明的说明书和所附权利要求中所使用的,单数形式“一”、“一个”和“所述”也旨在包括复数形式,除非上下文另外清楚地指示。还应当理解,这里使用的术语“和/或”是指并涵盖相关联的所列项目中的一个或多个的任何和所有可能的组合。应进一步理解的是,当在本说明书中使用时,术语“包括(comprises和/或comprising)”指定所陈述的特征、整数、步骤、操作、要素和/或组件的存在,但不排除存在或添加一个或多个其它特征、整数、步骤、操作、要素、组件和/或其群组。如本文中所使用,取决于上下文,术语“如果”可以被解释成意指“当……时”或“在……时”或者“响应于确定”或“响应于检测”。类似地,取决于上下文,短语“如果确定”或“如果检测到[所陈述的条件或事件]”可以被解释为意指“在确定……时”或“响应于确定”或“在检测到[所陈述的条件或事件]时”或“响应于检测到[所陈述的条件或事件]”。前述描述包含体现说明性实施方案的示例系统、方法、技术、指令序列和计算机器程序产品。出于解释的目的,阐述了许多具体细节,以便提供对本发明主题的各个实施方案的理解。然而对于本领域的技术人员将显而易见的是,本发明的主题可以在没有这些具体细节的情况下实践。总体而言,未详细示出众所周知的说明实例、协议、结构和技术。为了解释的目的,前面的描述已经参照特定的实施方案进行了描述。然而,上述说明性讨论并不旨在穷举或将所述实施方案限制于所公开的精确形式。鉴于以上教导,许多修改和变化是可以的。选择和描述这些实施方案是为了最好地解释这些原理及其实际应用,由此使得本领域其它技术人员能够用适合预期的特定用途的多种修改方案来最好地使用这些实施方案和多种实施方案。当前第1页12
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。