唾液酸转移酶基因缺失的mdck细胞株及构建方法和应用
技术领域
1.本发明涉及细胞技术领域,具体涉及一种部分唾液酸转移酶基因缺失的mdck细胞株及其构建方法和应用。
背景技术:
2.目前,mdck细胞广泛用于多种呼吸道肠道病毒的分离、培养和纯化。mdck细胞是正常犬肾来源的传代细胞,是由madin和darby从考克斯班尼犬的肾脏组织中分离培养获得。mdck细胞在培养平板上培养时可形成紧密连接的蛋白,能形成致密的单细胞层,同时具有接触抑制的特性。
3.但是,天然mdck细胞对呼吸道肠道病毒的敏感性相对较低,有必要进行改造研究。
技术实现要素:
4.基于此,有必要提供一种部分唾液酸转移酶基因缺失的mdck细胞株及其构建方法和应用,能够显著提升对人流感病毒等呼吸道肠道病毒的敏感性。
5.本发明采用如下技术方案:
6.本发明首先涉及株唾液酸转移酶基因敲除mdck-st3galko细胞株。具体提供一种唾液酸转移酶基因st3gal1缺失的mdck细胞株,保藏编号为c2022178。
7.唾液酸转移酶基因st3gal1缺失的mdck细胞株的构建方法,采用基于crisprcas9基因编辑系统来敲除mdck细胞中的唾液酸转移酶基因st3gal 1,所针对靶点序列为:gcccgtaccaggactctcgg。
8.在其中一些实施例中,crisprcas9基因编辑系统所采用的grna载体质粒为带有grna骨架、cas9表达序列、nls核定位序列以及抗氨苄基因的px330,同源臂载体质粒选择带有多个临近的常用酶切位点、loxp片段以及嘌呤霉素抗性基因的py75。
9.在其中一些实施例中,针对性敲除基因st3gal 1的grna序列如seq id no.1和seq id no.2所示。5’同源臂序列如seq id no.5所示;3’同源臂序列如seq id no.6所示。
10.在其中一些实施例中,构建过程所采用的嘌呤霉素筛选条件为:嘌呤霉素溶液的浓度6μg/ml;和/或电转工艺条件为:单脉冲,脉冲电压110v,脉冲宽度25ms。
11.本发明还提供一种唾液酸转移酶基因st3gal 1和st3gal 2均缺失的mdck细胞株,保藏编号为c2022131。
12.本发明还提供唾液酸转移酶基因st3gal 1和st3gal 2均缺失的mdck细胞株的构建方法,包括如下步骤:获取唾液酸转移酶基因st3gal 1缺失的mdck细胞株;采用cre-loxp系统将puro序列剪切;进一步采用crispr cas9基因编辑系统敲除mdck细胞中的唾液酸转移酶基因st3gal 2。
13.在其中一些实施例中,针对性敲除基因st3gal 2的grna序列如seq id no.7和seq id no.8所示;5’同源臂序列如seq id no.11所示;3’同源臂序列如seq id no.12所示。
14.本发明还可以提供唾液酸转移酶基因st3gal 1缺失的mdck细胞株或唾液酸转移
酶基因st3gal 1和st3gal 2均缺失的mdck细胞株在培养流感病毒中的应用。
15.与现有技术相比,本发明的有益技术效果:
16.本发明采用crispr-cas9基因编辑方法成功得到了贴壁培养的mdck-st3gal1ko细胞株以及st3gal1、st3gal2基因联合敲除的mdck-st3galko细胞株,并且经验证mdck-st3gal1ko和mdck-st3galko细胞株对人流感病毒的敏感性显著性增强。
附图说明
17.图1为作为grna载体质粒px330的图谱。
18.图2为同源臂载体质粒py75的图谱。
19.图3为实施例1中的grna质粒pcr产物测序图。
20.图4为实施例1中的同源臂载体质粒构建过程中的pcr核酸电泳验证图;其中,泳道1、2、3、4分别代表grna25’同源臂、grna23’同源臂、grna15’同源臂、grna23’同源臂。
21.图5为实施例1中的5'同源臂酶切连接示意图。
22.图6为实施例1中的py75 5'同源臂质粒的酶切验证图;其中,a图为菌液pcr结果,其中释放1500bp条带为阳性,而释放500bp左右条带则为阴性;b图为为质粒酶切验证结果,其中释放1000bp的同源臂条带为阳性。
23.图7、8及9均为实施例1中的py75 5'同源臂质粒的5'同源臂测序比对测序图。
24.图10为实施例1中的3'同源臂酶切连接示意图。
25.图11为实施例1中的py75 5' 3'同源臂质粒的酶切验证图;其中,a图为菌液pcr结果,其中释放1300bp条带为阳性,而250bp左右条带则为阴性,b图为酶切验证结果,其中释放1000bp的同源臂条带为阳性。
26.图12、13及14均为实施例1中的py75 5' 3'同源臂质粒的3'同源臂测序比对图。
27.图15为实施例1中嘌呤霉素细胞杀灭实验中5-7天细胞活率统计图。
28.图16为实施例1中不同嘌呤霉素浓度下第六天细胞生长状态图。
29.图17为实施例1中不同电转染条件下细胞的生长状况图。
30.图18为实施例1中极限稀释法筛选流程图。
31.图19为实施例1中基因组全长pcr验证图。
32.图20为实施例1中基因组定位pcr验证图。
33.图21、22及23均为实施例1中定位pcr产物测序比对图。
34.图24为实施例1中mdck-st3gal1ko细胞生长曲线图。
35.图25为实施例1中mdck细胞和mdck-st3gal1ko细胞的q-pcr结果图。
36.图26为实施例2中grna质粒pcr产物测序图。
37.图27为实施例2中5'同源臂连接菌液pcr核酸电泳结果图。
38.图28、29为实施例2中py75 5'同源臂序列测序比对图。
39.图30为实施例2中3'同源臂连接菌液pcr核酸电泳结果图。
40.图31、32以及33为实施例2中py75 5' 3'同源臂序列测序比对图。
41.图34为实施例2中基因组定位pcr验证图;其中,1、c4样,引物1(5
’‑
puro);2、c4样,引物2(5
’‑
puro);3、e8样,引物1(5
’‑
puro);4、e8样,引物2(5
’‑
puro)5、f5样,引物1(5
’‑
puro)6、f5样,引物2(5
’‑
puro)。
42.图35及36为实施例2中细胞基因组pcr产物测序比对图。
43.图37为实施例2中mdck-st3gal2ko细胞生长曲线图。
44.图38为实施例3中细胞基因组全长pcr验证结果图。
45.图39为实施例3中细胞基因组定位pcr验证结果图;其中,编号1-4,5-8分别为a3、a10、c7、g2四个细胞样的5’和3’的定位pcr结果,其中5’定位pcr阳性条带为1288bp,3’定位pcr阳性条带为1340bp。
46.图40为实施例3中q-pcr结果图。
47.图41为实施例3中mdck-st3galko的生长曲线图。
48.图42为实施例4中三种基因缺失的mdck细胞的血凝实验统计图。
49.图43为实施例5中mdck、mdck-st3gal1ko、mdck-st3galko三种细胞的胞唾液酸凝集素-生物素结合流式结果;其中,上下两图分别为α-2,3唾液酸 mal
‑ⅱ
pe标记亲和素(红色)平均荧光强度流式结果和α-2,6-唾液酸 snp-fitc(绿色荧光)平均荧光强度流式结果,从左至右分别为mdck细胞、mdck-st3gal1ko细胞p92、p97、p102、p107以及mdck-st3galko细胞p101。
50.图44为实施例5中mdck、mdck-st3gal1ko、mdck-st3galko三种细胞不同代次的唾液酸表达的平均荧光强度结果统计图;其中,a表示α-2,3-唾液酸 mal
‑ⅱ
pe标记亲和素(红色)平均荧光强度,图b表示α-2,6-唾液酸 snp-fitc(绿色荧光)平均荧光强度的对比,两图种横轴分别代表1(mdck p72),2(mdck-st3gal1ko p92),3(mdck-st3gal1ko p97),4(mdck-st3gal1ko p102),5(mdck-st3gal1ko p107),3(mdck-st3galko p101)*p《0.05,**p《0.01,***p《0.001,****p《0.0001,图中无显著性差异ns存在于mdck-st3gal1ko细胞的不同代次间。
51.图45为实施例中基因组测序差异图。
52.图46为实施例中转录组差异基因火山图。
53.图47为实施例中转录组go功能富集分析。
54.图48为实施例中转录组kegg功能富集分析。
55.图49为实施例中转录组在st3gal1和st3gal2上表达差异对比。
具体实施方式
56.下面结合具体实施例对本发明作进一步的详细说明,以使本领域的技术人员更加清楚地理解本发明。
57.以下各实施例,仅用于说明本发明,但不止用来限制本发明的范围。基于本发明中的具体实施例,本领域普通技术人员在没有做出创造性劳动的情况下,所获得的其他所有实施例,都属于本发明的保护范围。
58.在本发明实施例中,若无特殊说明,所有原料组分均为本领域技术人员熟知的市售产品;在本发明实施例中,若未具体指明,所用的技术手段均为本领域技术人员所熟知的常规手段。
59.值得首先说明的是,研究表明:细胞表面唾液酸分为α-2,3-唾液酸和α-2,6-唾液酸。调控α-2,3-唾液酸转移酶合成的基因是st3gal基因家族,包括st3gal 1、st3gal2、st3gal3、st3gal 4、st3gal 5、st3gal6和类st3gal2。调控α-2,6-唾液酸转移酶合成的基因
是st6gal家族,包括st6gal 1和st6gal2。
60.关键试验材料及试验方法:
61.细胞:mdck细胞,atcc引进,mdck ccl-34,p57。
62.病毒:三者均为nibsc引进。病毒h1n1:来自于h1n1(ivr215)工作种子库,保存于武汉生物制品研究所病毒性疫苗研究二室,批号:202103w01h1。病毒bv:来自于bv(bvr-11)工作种子库,保存于武汉生物制品研究所病毒性疫苗研究二室,批号:202103w01bv。病毒by:来自于by(bvr-1b)工作种子库,保存于武汉生物制品研究所病毒性疫苗研究二室,批号:202103w01by。
63.vp-sfm培养基购于gibco公司,货号:12559019;胎牛血清购于gibco公司,货号:16140071;tpck-trypsin购自sigma公司。
64.完全培养基的配制:在无菌条件下,向vp-sfm培养基中加入5%的fbs/nbs,4℃下保存备用。若培养基配制后存放时间超过三个月,则需添加1%谷氨酰胺添加剂。
65.单克隆细胞培养基的配制:无菌条件下,将vp-sfm无血清培养基和24h后的细胞培养液进行1:1混合,在1500rpm、5min条件下离心去除混合液中的细胞碎片后,在0.22μm滤膜滤过去除杂质成分,4℃条件下备用,若需要进行抗性筛选,则在使用之前向其中加入一定浓度的筛选抗生素(加入筛选抗生素后即配即用,不宜长时间保存)。
66.细胞维持液的配制:无菌条件下,向vp-sfm培养基中加入2μg/ml的tpck胰酶(加入tpck胰酶后即配即用,不宜长时间保存)。
67.细胞冻存液的配制:根据细胞冻存时所需的全部冻存液体积,无菌条件下按照以下配比配制细胞冻存液:70%vp-sfm 20%fbs 10%dmso,即配即用。
68.磷酸缓冲液(pbs)正常ph范围是7.2~7.4。
69.mdck细胞生长曲线绘制:将培养瓶中的细胞按照上述步骤消化成细胞悬液后,再按照上述步骤进行细胞计数,根据计数结果将细胞悬液稀释至密度为1
×
105/ml,将稀释好的细胞悬液加入t25中(每瓶加入5ml,起始细胞总量即为5
×
105/瓶),至少需要30个t25培养瓶,置于37℃孵箱培养。每隔12h对细胞进行计数,每次计数时取两个t25按照上述方法进行计数,若两瓶计数结果相差不到10%则记录数据计算平均值,若两瓶细胞计数结果相差大于10%,则需再取一个t25进行计数根据三个细胞瓶的计数结果进行分析。连续计数7天(168h),根据每次细胞计数结果,以小时(h)为横坐标,活细胞密度(
×
105/ml)为纵坐标,绘制细胞生长曲线。
70.grna载体质粒:选择带有grna骨架、cas9表达序列、nls核定位序列以及抗氨苄基因的crispr-cas9系统专用哺乳动物细胞表达载体px330作为grna载体质粒,其图谱如图1所示。
71.同源臂载体质粒:选择带有多个临近的常用酶切位点、loxp片段以及嘌呤霉素抗性基因的py75作为同源臂载体质粒,其图谱见图2。
72.质粒、dna、rna提取试剂盒均购自takara公司。酶切酶连实验的工具酶均购自neb公司。
73.grna载体质粒的构建:根据设计好的grna序列合成如下形式的两个单链:
[0074]5′‑
caccgnnnnnnnnnnnnnnnnnnnn-3
′
(oligos 1)
[0075]3′‑
cnnnnnnnnnnnnnnnnnnnncaaa-5
′
(oligos 2)
[0076]
得到以上的oligos后,按照以下体系进行进行磷酸化和退火:
[0077]
grna磷酸化和退火体系
[0078]
体积组分1μloligo1(100μm)1μloligo 2(100μm)1μl10
×
t4 ligation buffer6.5μlddh2o0.5μlt4 pnk(磷酸激酶)10μltotal
[0079]
按照上述体系进行pcr反应,具体程序为:37℃,30min;95℃,5min;每分钟降低5℃进行反应,最终25℃;25℃保温,反应液250倍稀释后备用
[0080]
px330(grna载体质粒)的酶切和连接反应体系如下表:
[0081]
px330酶切连接一步法体系
[0082]
体积组分xμlpx330(100ng)2μl1∶250稀释后的反应液2μl10
×ꢀ
tango buffer1μldtt(10mm)1μlatp(10mm)1μlbbsl0.5μlt7 dna连接酶yμlddh2o20μltotal
[0083]
按上述体系进行pcr反应,具体程序为:37℃,30min;23℃,30min;4度保存。
[0084]
提取mdck细胞基因组后,分别pcr得到5’和3’同源臂,对5’或3’同源臂以及含有同样酶切位点的py75质粒进行酶切,使用neb酶切体系37℃孵育过夜(由于酶切位点相距较近,因此酶切过夜保证充分酶切);酶切产物用pcr纯化试剂盒纯化;按t4 dna连接酶相应体系进行酶连反应(设立空白对照),于16℃酶连孵育过夜;取少量酶连产物转化约100μl top10感受态细胞,静置冰上30min后,转入42℃水浴1min,冰浴5min,加入800μllb液体培养基37℃摇菌1h后取150μl涂lb固体平板。
[0085]
鉴定成功的质粒进行去内毒素大提(使用去内毒素大提试剂盒),保证无内毒素避免转染对细胞的伤害,并且大提质粒浓度高,有利于提高转染效率。
[0086]
细胞电转:由于实验室之前尚未进行过mdck细胞,相关的电转实验,因此对细胞的电转条件,尤其是电转仪的参数需进行探索。为避免电转杯导致的污染,首先将清洗后的电转杯置于无水乙醇中浸泡6-8h。电转前,将电转杯放在紫外下照射杀菌1h,无菌条件下风干1h,使电转杯中残留的乙醇挥发,4℃无菌保存备用。在6孔细胞培养板中加入vp-sfm完全培养基2ml/孔,放入37℃孵箱中预热备用,为了避免质粒带来的污染,在每孔中加入1:1000双抗。按照细胞计数的实验步骤记录细胞浓度,根据电转需要的细胞总量,计算所取的细胞悬液体积按照1500rpm,5min离心,弃上清,收取细胞沉淀,加入定量无血清培养基。用无血清
培养基按照每个电转体系中细胞总量为1
×
106/ml,体系体积为0.2ml重悬细胞后,向其中加入grna载体质粒以及同源臂载体质粒各8μg/体系,将细胞悬液轻轻吹打混匀备用。设置电转电压强度,脉冲宽度以及脉冲次数,取0.2ml细胞悬液轻轻加入电转杯两个电极片中间的凹槽中,吸取前应充分混匀细胞悬液,不应存在细胞沉淀,盖上电转杯,打开电源,开始电转。电转脉冲程序完成后,每次在凹槽中吸取180μl电转后的细胞悬液平均加入六孔板中的三个孔,每孔60μl,轻轻吹打将培养板中的细胞混匀,置于37℃孵箱培养。
[0087]
单克隆细胞筛选及基因组pcr鉴定,包括如下步骤:依据嘌呤霉素细胞杀灭实验优选的参数、优化后的电转参数条件进行嘌呤霉素细胞初筛结果以及极限释法单克隆细胞再筛,再进行单克隆细胞的鉴定。
[0088]
其中,嘌呤霉素细胞初筛的步骤为:电转完成后密切观察细胞生长状况,由于电转时细胞表面电穿孔导致部分细胞逐渐死亡,第二天六孔板镜下观察可发现部分细胞凋亡漂浮,贴壁细胞长至60%汇合度时需要换液。六孔板中每2-3天换一次液,镜下观察可发现绝大部分细胞会逐渐停止生长脱落。有嘌呤霉素抗性的细胞大约可在第5-7天镜下观察到单个细胞或2-3个细胞形成小细胞团,继续筛选培养镜下可见细胞克隆数量逐渐增加。
[0089]
极限释法单克隆细胞再筛的步骤为:当细胞培养7-10天后,观察六孔板中出现单克隆细胞长成细胞团,计数单个细胞孔中等单克隆细胞团的数目,弃去培养基,将其充分消化。将消化后的细胞悬液进行细胞计数,计算细胞总量后进行极限稀释至96孔板。极限稀释后每天密切观察单克隆细胞生长状况,4-5天后镜下观察,标记仅含有单个单克隆细胞团的孔。一般单克隆细胞正常情况下在培养10-14天可长满96孔板的细胞孔,当细胞长满后,传代至24孔板进行扩大培养,依次扩增至两个6孔板,一份继续培养保种,另一份提细胞基因组进行pcr鉴定。
[0090]
单克隆细胞的鉴定的步骤为:将需鉴定的六孔板进行细胞消化后提取mdck细胞基因组,分别设计两对引物来对单克隆细胞进行鉴定:首先分别在靶基因的5’同源臂和3’同源臂位置设计上下游引物(或是在5’同源臂的上游以及3’同源臂的下游设计引物),且其引物的结合位置应和grna结合位点的距离大于100bp以上,此法设计的全长pcr可同时鉴别阴性、单等位基因敲除和双等位基因敲除的不同种类单克隆;其次,分别在5’同源臂序列和嘌呤霉素抗性基因序列或是抗性基因序列和3’同源臂序列上分别设计两组上下游引物,这样设计的定位pcr用来进一步确认克隆的位置,以排除细胞中残留质粒以及脱靶位点的干扰,由于全长pcr片段太长,pcr难以得到结果,因此以定位pcr为主。pcr反应体系应采用高保真酶体系,可用于后续的pcr产物测序以及pcr产物电泳分析,对应的阳性克隆(单敲除或双敲除)进行传代扩增及冻存,传代后取细胞样品提取dna重复上述pcr检测。对最终确定的阳性克隆进行后续表达以及功能的鉴定。
[0091]
cre-loxp体系的构建以及loxp标签的剪切:由于同源臂载体质粒(px75)质粒中两段同源臂序列之间含有两边带有loxp的嘌呤霉素抗性基因序列,因此若想在基因敲除的基础上重复利用嘌呤霉素的抗性进行细胞筛选,首先要将敲入的loxp-puro-loxp这一段序列去掉使细胞失去原有的嘌呤霉素抗性。其中cre重组酶表达序列位于cre-loxp系统专用工具质粒py48质粒上。
[0092]
具体试验步骤为:将完成嘌呤霉素筛选的已编辑细胞电转入py48质粒,具体电转方式同上,py48质粒采用去内毒素大提,电转体系中质粒浓度保持8μg/ml。电转后第二天换
液后正常培养,不加任何抗生素筛选,待细胞正常生长3-5天,由于细胞已经适应环境,3-5天细胞汇合度可达60%以上。对细胞进行极限稀释至96孔板,标识出单细胞的孔,进行单克隆细胞培养。1-2周后待96孔板中单克隆细胞长至汇合度》80%,将每孔分板至两个孔中,一孔正常培养,另外一孔加入一定浓度的嘌呤霉素进行筛选。观察加入嘌呤霉素进行筛选的细胞孔,一一对应观察,一周后,在筛选孔中细胞完全死亡,但原始孔中细胞正常生长的细胞样即为阳性细胞,扩大培养保存后,提取基因组进行定位测序验证。
[0093]
实施例1构建st3gal1基因缺失的st3gal1ko细胞株
[0094]
本实施例提供一种构建st3gal1基因缺失的st3gal1ko细胞株,其包括如下步骤:
[0095]
s1,设计并评估grna序列:
[0096]
从ncbi下载st3gal1的基因序列(基因id:482050),舍弃st3gal1基因中是3的倍数的exon1,针对st3gal1基因中的exon2序列设计grna:
[0097]
依据网页评估特异性s、文献引用率a、切割效率e的评分,进行grna初步筛选,得到五个关于exon2的grna序列,其后进行脱靶位点off-target评估,具体评估见下表:
[0098]
st3gal1ko细胞的grna脱靶性评估
[0099]
grna编号潜在脱靶位点有义脱靶位点pam序列相符脱靶风险位点e评分2-120697521772.9562-21908241470.7912-31888343966.0962-41774625560.1782-51616434156.072
[0100]
根据上表设计2对grna序列:
[0101]
grna1:5'-caccgcccgtaccaggactctcgg-3'(seq id no.1)
[0102]
3'-cgggcatggtcctgagagcccaaa-5'(seq id no.2)。
[0103]
grna2:5'-caccgaagatgaagaggacgagga-3'(seq id no.3)
[0104]
3'-cttctacttctcctgctcctcaaa-5'(seq id no.4)。
[0105]
将两对grna进行ncbi-blast定位验证:
[0106]
grna1定位chromosome13,canfam3.1,29863779-29863798,blast匹配率100%。
[0107]
grna2定位chromosome13,canfam3.1,29871706-29871725,blast匹配率100%。
[0108]
表明两对grna序列均有望100%定位于靶基因的目标位置。
[0109]
s2,针对构建的grna载体质粒进行测序:
[0110]
按照上述步骤方法构建grna载体质粒。
[0111]
设计grna相关测序引物为:
[0112]
grna forward primer:ggcctatttcccatgattcc;
[0113]
grnareverse primer:cttgatgtactgccaagtgg。
[0114]
由于grna构建质粒中插入的grna序列过短,核酸电泳检测难以鉴别,因此直接进行质粒pcr产物测序,测序结果如图3和下表:
[0115]
样本编号序列匹配率px330-1100%(符合)px330-2100%(符合)
px330-3测序失败px330-410%(不符)px330-5100%(符合)px330-6100%(符合)px330-715%(不符)px330-8100%(符合)
[0116]
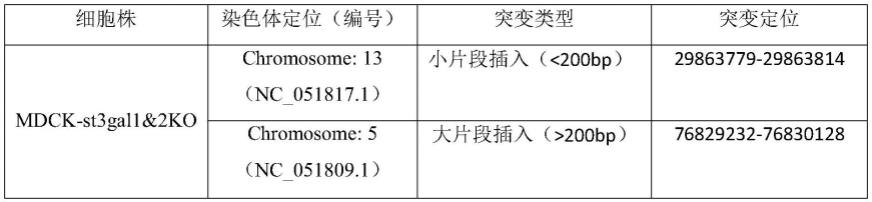
由上表可以看出,8个样本中有5个完全匹配,表明grna载体质粒构建成功。选取px330-1质粒大提备用。
[0117]
s3,针对构建的同源臂载体质粒进行测序:
[0118]
按照上述步骤方法构建同源臂载体质粒。
[0119]
在grna两侧30bp左右的1kbp序列设计为同源臂序列,预留引物的空间将同源臂扩展至1.2kbp,这两端的序列即为同源臂序列,具体序列如下:
[0120]5’
同源臂序列:
[0121]5’
cctggtgggcctctccctcggagacctgattccttccatctcggggccgggggtgctgcctgcctctggatctccccatagaccccatcacgaatctcctggacggtggtcaaagcaggccctaggctatcctttcttttctcagccactcagccctgggccagggggctgggagaggctagtgagactcaccccacagccaacctccccacccgtgaccccccaccccggcccctacaaccccatggactgggctttctgaaaggtcaaggttccaagggtctgagatgggggcagtattgtgggagagactttgaggtgggtttctaaaagcctgggttcaaatcctggtttgggatttgggacctgtggttcagccagcccgggccccagagcatcctcctcgtccacagttcagatgtggtgtggtatagatgttgcggcacggcgacggggtcagaggtgataactgcttaggaaatgtctccaggttttaccgtgagtgaggaggtgggaagtctggagctaggtcttcaaggatgaaaacagtttgccaggcagacatcggaggccttgtgcatccagcaaagggctggttttttggtcttgctggagcccagtggtcccctgggaggagtggcgggggcaggaggatggagggagcagaagcagggaggtgggctcagaggtcacctgacccttctcctatcacccgatgtcattgcctccggctgatggaggagctagcacatagtaggtgctcaataagcctttgcagaatgagtgagggagctctctcccgtctcctggagtgggccctgggcttctggaccagcggggaggtggctggctgagcctcctggggcctccggcgccttgcagaagggaaggtactggagaagggggcccagggagggggctcaggggccccggccagaggcagcacagcccggctgacctttctccttccaccctccgcccgcagaggctccagcgggagaagcaacc 3’(seq id no.5)
[0122]3’
同源臂序列:
[0123]5’
caggtgagcgcctggagcccctcggagccgcgggaggcgcctgggctcagggccaggagggcaagacccagtcctagcaggcctagactccccttcggaccttgggggctccctgtgggcctcagtttccccatctgtgcattataggaaggaggtagaccagcagtgcctctctggaccccgatgggagaccataaacgggaggaagccagcaggtcgggagctttgcacagagaggcctggagggagggcaagaggcagttgcagacccttgtctgatctttaaaattcagccccattttgcaacctaggctgtaataaaactatgtgcttttaatgtaaatgcatttccctgtcaccaaggagaatagtttccctgggaagagatgtattctgggtttcttataatctccacgctgttcactcagtagtgaggccgctgctccccaggcaccttctagaatagtctcctgtctctgcgatctctgcatttatgtccttagactgctctcacccggcttctcggagcctccgtgtgcccggcgtgcggcaggaacatccgtgggggagggaaagcatcgggtcccaggtcagacagacctgagttcaaggctgcacacttgccagctccgtgtcctgggactaattcttggcgttcagatttctcccatcatagaataggaccagagaatacctcccgggcgcagtcatcattcggatgtaatgaggcgcgcctcttggccaggacggggccttgcgcacagtaggtcagcaggagtggccacggtcaggctcgctccagagcgcgtaagagatggaaagcagc
aagagcatttgagctgtgtcctgatcgtctccatcgaagccgcccccttgcgacgtgatgcgatgcgtgtcccgctgtcccttccctcccaggatgaacaaggcccccacggcgggcttcgagatggatgtcgggagcaagaccacccaccacctggtgtaccccgagagcttcagggagctggcggagaatgtcagcatggtcctggtgcccttcaagaccaccgacctggagtgggtggtcagtgccaccaccacaggcaccatctctcagtgagtccccggggtggagagcccccacgccctcactggtcctccgtgcccccaccatgtggtgcccgcaccaggatgcctcccccgctggacttccttctg 3’(seq id no.6)
[0124]
其中,5'同源臂可用酶切位点有:ecori,clai,ecor
ⅴ
;3'同源臂可用酶切位点为:sall,acci,hinci,bglii,bamhi,hindiii。由于3'同源臂含acci,为了避开同尾酶以及前后相同的酶切位点以保持酶切特异性。实验设计结果如下:5'酶切位点:ecorⅰ、ecor
ⅴ
;3'酶切位点:bamhⅰ、hindⅲ,连接顺序为5'同源臂-3'同源臂。
[0125]
以上设计的同源臂两端加入适当酶切位点以及相应的保护碱基,使用酶切位点 保护碱基的引物进行pcr核酸电泳验证,结果如下图4:在1000bp左右都出现了阳性同源臂条带
[0126]
同源臂pcr核酸电泳显示,在mdck细胞上进行同源臂pcr均可释放1kbp左右的条带。与设计相符,5’和3’同源臂均通过pcr获得。
[0127]
按照图5所示的5'同源臂酶切连接示意图进行酶切反应,构建含5'同源臂的py75质粒。由此通过转化摇菌、质粒小提得到py75 5'同源臂的质粒,菌液pcr和酶切验证结果如图6所示。a图为菌液pcr结果,其中释放1500bp条带为阳性,而释放500bp左右条带则为阴性。b图为为质粒酶切验证结果,其中释放1000bp的同源臂条带为阳性。得到1、4样本的菌液pcr以及酶切验证均为阳性,进行下一步的测序验证。测序验证结果见针对5'同源臂测序比对的图7至9。
[0128]
进一步按照图10所示的3'同源臂酶切连接示意图进行酶切反应,构建含3'同源臂的py75质粒,命名为py75 5' 3'同源臂。菌液pcr和酶切验如图11所示(a图为菌液pcr结果,其中释放1300bp条带为阳性,而250bp左右条带则为阴性,b图为酶切验证结果,其中释放1000bp的同源臂条带为阳性),结果可见仅有3号样本质粒的菌液pcr验证与酶切验证均为阳性。
[0129]
针对3'同源臂测序比对的图12至14,错配碱基出现概率低,其余序列基本一致,不同样本测序出现相同的错配碱基可认为是细胞中碱基突变,不影响实验准确性。
[0130]
s4,转染及筛选获得st3gal1基因敲除细胞(mdck-st3gal1ko细胞):
[0131]
按照每隔2.5μg/ml的浓度梯度设置0~22.5μg/ml范围内的10个浓度梯度的嘌呤霉素溶液,探究mdck细胞存活情况。结果表明:对照组以及2.5、5μg/ml实验组中的mdck细胞部分存活,其余细胞全部死亡。
[0132]
更改按照每隔0.5μg/ml的浓度梯度设置3~8μg/ml范围内的嘌呤霉素溶液,实验结果如图15(嘌呤霉素细胞杀灭实验5-7天细胞活率)和图16(不同嘌呤霉素浓度下第六天细胞生长状态)所示:最终确认mdck细胞杀灭的最低浓度为6μg/ml(到第6天即可完全杀灭细胞)。
[0133]
设计三组不同的电转条件,分别是:100v,25ms;100v,30ms;110v,25ms,三者均为单脉冲。不同电转移条件下细胞的生长状况见图17。
[0134]
在设计的三组电转方案中,研究结果表明:第一组(100v,25ms)和第二组(100v,
30ms)在两周内仍有一定量的细胞存活,且夹杂着大量的细胞碎片聚集成团,不易分辨,消化后镜下观察有大量正常状态的活细胞,因此前两种电转条件不利于细胞单克隆筛选。第三组(110v,25ms)在一周内细胞基本能够被全部杀灭,有利于单细胞克隆的挑取。因此最终确定最优电转参数为脉冲电压110v,脉冲宽度25ms,单脉冲。
[0135]
按照图18的极限稀释法筛选流程对macka细胞单克隆进行筛选,得到了784个单细胞孔,培养两周后排除未能生长成明显的单克隆细胞团后还剩余13个样孔,对其进行细胞基因组pcr验证。
[0136]
dna水平的验证(细胞基因组pcr)分别用两种引物设计方案进行细胞基因组pcr验证:
[0137]
(a)在5'同源臂和3'同源臂两侧进行全长的pcr,结果如图19(基因组全长pcr验证)所示。
[0138]
pcr产物核酸电泳显示:仅9c5样有阴性(2600bp)及阳性(4000bp)条带,属杂合的突变。
[0139]
(b)在5'同源臂和puro上设计引物进行细胞基因组定位pcr。
[0140]
采用定位pcr将两个引物分别设计在嘌呤霉素抗性序列以及5'同源臂上游或者嘌呤霉素抗性序列以及3'同源臂下游,可通过核酸电泳条带显示鉴定其基因编辑的确切位置,若出现脱靶情况则不会出现条带。结果图20(基因组定位pcr验证)所示。基因组定位pcr验证结果显示,mdck-9c5细胞二次极限稀释后,得到60个样孔扩大培养,后进行定位pcr验证,其中45个样出现阳性条带(1400bp左右)。定位pcr产物测序见图21~23。
[0141]
s5,绘制st3gal1基因敲除细胞的生长曲线,结果如图24(mdck细胞与mdck-st3gal1ko细胞生长曲线对比)所示。细胞生长曲线显示mdck细胞的倍增时间为26h,达到平台期时间为60h,平台期活细胞平均密度为5.3
×
105/ml,mdck-st3gal1ko细胞倍增时间为36h,达到平台期的时间为72h,平台期活细胞平均密度为5.2
×
105/ml。
[0142]
s6,针对st3gal1基因设计q-pcr实验引物及探针,q-pcr实验结果如图25(mdck细胞和mdck-st3gal1ko细胞的q-pcr)所示。其中,wt和ko组分别代表mdck细胞和mdck-st3gal1ko细胞。
[0143]
结果显示:q-pcr结果显示相较于mdck细胞,mdck-st3gal1ko细胞的st3gal1的转录水平显著降低(p《0.01)。
[0144]
实施例2构建st3gal2基因缺失的st3gal2ko细胞株
[0145]
本实施例提供一种构建st3gal2基因缺失的st3gal2ko细胞株,其包括如下步骤:
[0146]
s1,设计并评估grna序列:
[0147]
从ncbi下载st3gal2的基因序列(基因id:489714),舍弃st3gal2中是3的倍数的exon1(339bp)以及exon3(180bp),由于exon4位置偏后,因此对st3gal2基因中的exon2、exon1的序列在网页上设计grna序列:
[0148]
依据网页评估特异性s、文献引用率a、切割效率e的评分,进行grna初步筛选,在exon2、exon1上各得到五个grna序列,其后进行脱靶位点off-target评估,具体评估如下:
[0149]
st3gal2-ko的grna脱靶性评估
[0150]
grna编号潜在脱靶位点脱靶风险点e评分2-139275.858
2-230266.2112-312266.1492-427564.6212-541164.2861-119472.2411-233558.6671-3601357.431-448456.3391-514272.334
[0151]
根据上表设计2对grna序列:
[0152]
grna2-1:5
’‑
caccgtgttgtgtgacttgaactg-3’(seq id no.7)
[0153]3’‑
cacaacacactgaacttgaccaaa-5’(seq id no.8)。
[0154]
grna1-5:5
’‑
caccgtggctgtcaaaccagtcag-3’(seq id no.9)
[0155]3’‑
caccgacagtttggtcagtccaaa-5’(seq id no.10)。
[0156]
将两对grna进行ncbi-blast定位验证:
[0157]
grna2-1定位chromosome5,canfam3.1,76829417to 76829436,blast匹配率100%
[0158]
grna1-5定位chromosome5,canfam3.1,76829709 to 76829728,blast匹配率100%。两个grna序列均能100%定位于靶基因的目标位置。
[0159]
s2,针对构建的grna载体质粒进行测序:
[0160]
grna载体质粒构建后设计grna相关测序引物为:
[0161]
grnaforwardprimer:ggcctatttcccatgattcc;
[0162]
grnareverse primer:cttgatgtactgccaagtgg。
[0163]
由于grna构建质粒中仅仅插入20bp的grna序列,因此核算电泳检测难以鉴别,因此直接进行质粒pcr产物测序,测序结果如图26。
[0164]
s3,针对构建的同源臂载体质粒进行测序:
[0165]
在grna两侧距离30bp左右的1kbp序列设计为同源臂序列,同时也要考虑预留引物的空间将同源臂扩展至1.2kbp,这两端的序列即为同源臂序列,具体序列如下:
[0166]5’
同源臂序列:
[0167]5’
aaaatggggatgatgattacagccctgtgtgagtctcctggggctgccaccacaaagttctacaactgggtgacttaaacaacagaagcgtatttttttgtaattctagaatcaagatcagggtgccatcaaaattggtgtctcgtgagacttctcttcctggcctgcagactggcaccattctgctgcatcctcatgggcccttcttgtctgggtgcactgcgagggctcttcggtgtctcttccttttcttataaggatgattagggccgtactcttatgacctcatcaaacctcagttacctccccataggcccgatctccaaaatacagtcacattgaggtgctgaacaaaagctaggtctctattagtgcttggctggggcttgtcccacattatactgattgggcatttccctgcagcagtgattcctcaattccatctatctttatgtccttggcacttatcccttggcacagaccaggtgctcagaaaacacatttttggaatcctacagtgaaccaggaggcagttaatggcagccaaccagatttggggagccctgagcattgctaacctgccaagacttaaggcatgtcactgtctccaggctccaccttgcaagacaagggttttgcaaatggcaagatttcattctttttatggctgaatagtattccattgtatttctacaaccacatctttctccattcatttattgatggaaaatttggactgcttccataatttggctattgtcaataatgctacactaggtaacctatagaattcttgaattactacattgtgcacctgaaactaatg
taacactgtatattaactacactgaattttttttttttttaaagaaaaaggccaaagattttgccagcaaaatctcatcagattgcattttcgtacagccttttgtggctacaccagggtaactgggcaatcttcaggaccaaaatagactcggattcttttttacaaagatgctgttgtaaacttcaaagtggcagttgtaattctccgaacccctcccagaaatactgtctcccaccaataaagcaccacaatgaccct 3’。(seq id no.11)
[0168]3’
同源臂序列:
[0169]5’
gtgagaggatgaggtcagagatgtcactaagaccctgggactgaccagtgtatgcagggacaggtgggagcccccagcctctgtcccctttctctccagtctcccacctgcgttcccatgggccacacccaccgcaagccccccggggtgggagcccattgatgcaggccctagggtcagcccgggacacggagcaaggcgggaaagacagagggaaagcccagatgcccagctccccgcctgaacacccccgcccccccaggaaggctcgggtgcacacgttcagggctcctcgacagcagggccaggagaggtggcgccgtgggaggtgcccttctgggggccctgactatgcatcccagcagcgtggcccaggcaggccatgacctcccttagcctgacttggaaacctcaccatgtcataccctgccaggggcttgctgtggcccccagggtgtggcccagcccgccctccagctccattttgcatctttctctctggggttctgctgggcttggccttctctctttcagttccttcggactcacccactcttcccccacagaccttttttgcaccagcagctggcccggaggctcatcttttctgccccgtgacccatctgtcgtgcagacctcaggccattaccacttcctggggacacttgccagctgcagcccatgcccacttccctgtggcactgcaggtggccgttatcagtcagggttcttgagggaaacagaccctacagaataggcgtacacatattcaggaattggcttaagtgttgtgggactggcaacttcaaaatccatagggctggatggcaggctgtaaactcaggcaggattagatttctctgttacggtgctgagatgaaattccctctcctggaagcctctgtttccttcgcacctagggccttcaactgattagatgaggcccacccacattttggaggataatctcctttacttcatggcaaccacatctataaaaatgccttcacggccacacctagactagcatttgaccaagcatccgggtgccatggcctagctgagtcgactcatggatgtgatccatctcagcctttcttaattct 3’。(seq id no.12)
[0170]
py75酶切位点选择。5'酶切位点:ecorⅰ、ecor
ⅴ
;3'酶切位点:salⅰ、hindⅲ,连接顺序为5'同源臂-3'同源臂。
[0171]
根据同源臂序列以及酶切位点的选取,加上相应酶切位点的保护碱基后,得到5’和3’同源臂的引物设计方案如下表:
[0172]
引物名称序列酶切位点5up primercggaattcatagcagcctgcctgcaacgecorⅰ5down primergggatatccgctgcagaccagcatagccecorv3up primeracgcgtcgactgggagtcactcttcaagccsali3down primercccaagcttaagctccaggctgttacagghindⅲ[0173]
进行酶切酶连反应后,可构建含有5'同源臂的py75质粒通过转化摇菌、质粒小提得到py75 5'同源臂的质粒,菌液pcr结果如图27(5'同源臂连接菌液pcr核酸电泳结果)。py75 5'同源臂序列测序比对结果见图28~29,序列比对一致。
[0174]
进一步继续进行酶切连接反应,构建含有3'同源臂的py75 5’ 3’质粒,菌液pcr,结果如图30(3'同源臂连接菌液pcr核酸电泳结果)。py75 5' 3'同源臂序列测序比对结果见图31~33。
[0175]
s4,转染及筛选获得st3gal2基因敲除细胞(mdck-st3gal2ko细胞):
[0176]
按照实施例1的步骤方法进行转染和筛选。最终,将六孔板中全部的30个样孔极限稀释至96孔板中,培养两周后排除未能生长成明显的单克隆细胞团后还剩余3株,对其进行细胞基因组pcr验证。
[0177]
dna水平的验证(细胞基因组pcr)分别用两种引物设计方案进行细胞基因组pcr验证:采用定位pcr将两个引物分别设计在嘌呤霉素抗性序列以及5’同源臂上游或者嘌呤霉素抗性序列以及3’同源臂下游,可通过核酸电泳条带显示鉴定其基因编辑的确切位置,若只出现脱靶情况则不会出现条带。结果如图34(基因组定位pcr验证)所示。上述定位pcr验证中设计两对引物,对c4、e8、f5三株细胞进行了5’定位pcr验证。
[0178]
结果可以看出,在引物2的体系中,杂带少,阳性条带明显(2600bp),c4、e8、f5三株细胞都出现阳性条带,将三者在引物2体系得到的pcr产物纯化后送测序,测序结果见图35和36,测序显示puro序列成功插入。
[0179]
s5,绘制st3gal2基因敲除细胞的生长曲线,结果如图37(mdck细胞与mdck-st3gal2ko细胞生长曲线对比)所示。细胞生长曲线显示mdck细胞的倍增时间为26h,达到平台期时间为60h,平台期活细胞平均密度为5.3
×
105/ml,mdck-st3gal2ko细胞倍增时间为34h,达到平台期的时间为70h,平台期活细胞平均密度为4.7
×
105/ml。
[0180]
实施例3构建st3gal1和st3gal2联合敲除的mdck细胞株(mdck-st3galko或者mdck-st3gal 1&2ko)
[0181]
本实施例是在mdck-st3gal1 ko基础上进一步敲除st3gal2基因,并需要去除对嘌呤霉素的抗性。采用cre-loxp的体系,用py48来切除loxp-puro-loxp这一段序列,然后再进行st3gal2的基因编辑。
[0182]
将去内毒素大提的py48质粒电转入已完成嘌呤霉素筛选及验证的mdck-st3gal1ko细胞株,电转后第二天待换液后正常培养,培养基中不加抗生素,待细胞正常生长3-5天汇合度达60%左右时进行极限稀释至96孔板,1周后96孔板中单克隆细胞汇合度》80%,将细胞分板,另一板加入嘌呤霉素进行筛选,注意两个板中样孔一一对应。继续培养一周后,在筛选孔中细胞完全死亡,但原始孔中细胞正常生长的细胞样即为阳性细胞,共得到两个阳性细胞样孔(b1、h11),扩大培养保存后,提取基因组进行全长pcr(设计全长pcr的两对引物分别来自3’和5’同源臂,两个细胞样分别采用两组不同的引物)以及测序验证,全长pcr验证结果如图38,表明两个细胞样基因中插入的loxp-puro的序列被剪切,只剩下同源臂序列,因此条带均为1000-1200bp之间,将此pcr产物纯化后测序。
[0183]
将上述的细胞进行扩大培养并冻存后,再进行st3gal2的基因敲除,具体步骤与实施例2中的实验步骤相同,所用引物、质粒以及同源臂都相同,筛选后得到4个单细胞克隆样品,取样提取基因组进行定位pcr验证及测序,结果如图39:四个细胞样的5’和3’定位pcr均出现阳性条带,取pcr产物纯化后测序。
[0184]
针对st3gal2基因设计q-pcr实验引物及探针,进行q-pcr实验,其中wt和ko组分别代表mdck-st3gal1ko细胞和mdck-st3gal1&2ko细胞,实验结果如图40。其中,选取的阴性对照是经过cre-loxp剪切loxp-puro-loxp片段后的mdck-st3gal1ko细胞株,检测st3gal2敲除前后细胞st3gal2的转录水平,结果表明st3gal2转录水平显著降低(p《0.01)。
[0185]
绘制mdck-st3galko的生长曲线结果如图41所示。细胞生长曲线显示mdck细胞的倍增时间为26h,达到平台期时间为60h,平台期活细胞平均密度为5.3
×
105/ml,mdck-st3gal1&2ko细胞倍增时间为34h,达到平台期的时间为72h,平台期活细胞平均密度为4.8
×
105/ml。
[0186]
实施例4人流感病毒敏感性验证
[0187]
分别对mdck-st3gal1ko、mdck-st3gal2ko、mdck-st3galko三种细胞按照moi=0.01接种h1n1、by、bv三种流感病毒,72h测其血凝滴度进行对比,从而验证各细胞对人流感病毒的敏感性。
[0188]
结果如图42所示,血凝实验结果表明:在mdck-st3gal2ko细胞中培养h1、bv、by三种毒株获得的病毒滴度与mdck细胞无显著差异。mdck-st3gal1ko细胞和mdck-st3gal1&2ko细胞在h1n1、bv、by三种病毒中的滴度相较于mdck细胞有显著升高:相较于mdck细胞,在mdck-st3gal1ko细胞中收获的h1n1、bv、by病毒滴度分别提高了4倍、4倍、2倍,在mdck-st3gal1&2ko细胞中收获的h1n1、bv、by病毒滴度分别提高了8倍、8倍、4倍。
[0189]
实施例5唾液酸表达情况验证
[0190]
由于唾液酸表达在细胞表面,而不同的唾液酸可以与不同的凝集素特异性结合,因此可通过凝集素-生物素与唾液酸特异性结合后用亲和素进行荧光标记,从而通过流式分析平均荧光强度来定量唾液酸丰度。其中,生物素化怀槐凝集素ii(mal
‑ⅱ
凝集素)可特异性识别细胞表面的α-2,3唾液酸,而接骨木凝集素(sna凝集素)特异性识别α-2,6唾液酸。
[0191]
1)mal
‑ⅱ
凝集素特异性识别α-2,3-唾液酸检测:
①
mdck细胞消化后进行细胞计数后将其配制成1
×
106/ml的细胞悬液(此时用pbs 2%nbs作为孵育液),分装于1.5ml的ep管中孵育20min。
②
向每个ep管中加入10~15μl的生物素化mal ii,室温孵育30min。
③
离心弃去上清,用pbs进行细胞重悬并吹打混匀,重复离心重悬三次后向细胞悬液中加入荧光标记的streptavidin protein(pe标记),避光室温孵育30min。
④
离心弃去上清,用pbs进行细胞重悬,重复三次后向细胞悬液中加入1mlpbs,充分吹打混匀后,观察ep管中不存在细胞团块后用流式检测荧光信号,统计不同组别的荧光强度。
[0192]
2)sna凝集素特异性识别α-2,6-唾液酸检测:
①
将mdck细胞消化后进行细胞计数后将其配制成1
×
106/ml的细胞悬液(此时用pbs 2%nbs作为孵育液),分装于1.5ml的ep管中孵育20min。
②
向每个ep管中加入10~15μl的fitc标记的sna,避光室温孵育30min。
③
离心去上清,用pbs进行细胞重悬,重复三次后向细胞悬液中加入1mlpbs,充分吹打混匀后,观察ep管中不存在细胞团块后用流式检测荧光信号,统计不同组别的荧光强度,计算其平均荧光强度。
[0193]
分别研究mdck、mdck-st3gal1ko、mdck-st3gal1&2ko三种细胞的唾液酸表达情况:通过凝集素-生物素与唾液酸特异性结合后,生物素与带有荧光标记的亲和素进行反应,采用流式分析细胞表面的平均荧光强度以定量唾液酸丰度。每组做五个重复对照,通过设门对细胞碎片以及细胞团进行了两步筛选,得到的细胞群再进行相应荧光标记的阳性细胞进行圈门,分析阳性细胞的平均荧光强度。
[0194]
三种细胞唾液酸凝集素-生物素结合后的平均荧光强度流式结果如图图43所示。三种细胞不同代次唾液酸的表达的平均荧光强度测试结果如图44所示。
[0195]
mdck细胞、mdck-st3gal1ko细胞以及mdck-st3gal1&2ko细胞三者互相存在显著性差异,而mdck-st3gal1ko细胞各代次之间差异不显著。其中mdck细胞p72对照组α-2,6-唾液酸 snp-fitc(绿色荧光)平均荧光强度(16325单位荧光);mdck-st3gal1ko p92、p97、p102、p107实验组α-2,6-唾液酸 snp-fitc平均荧光强度分别为25433、24810、26917、24828单位荧光,均值为25497单位荧光,是对照组的1.56倍;mdck-st3gal1&2kop101实验组α-2,6-唾液酸 snp-fitc平均荧光强度为29100单位荧光,是对照组的1.78倍。
[0196]
mdck细胞、mdck-st3gal1ko细胞以及mdck-st3gal1&2ko细胞三者互相存在显著性差异,而mdck-st3gal1ko细胞各代次之间差异不显著,其中mdck细胞p72对照组α-2,3-唾液酸 mal
‑ⅱ
pe标记亲和素(红色)平均荧光强度为721561单位荧光;mdck-st3gal1ko p92、p97、p102、p107实验组α-2,3-唾液酸 mal
‑ⅱ
pe标记亲和素平均荧光强度分别为541008、501920、539661、584379单位荧光,均值为541742单位荧光,是对照组的0.75倍;mdck-st3gal1&2kop101实验组α-2,3-唾液酸 mal
‑ⅱ
pe标记亲和素平均荧光强度为481677单位荧光,是对照组的0.67倍。
[0197]
实施例6组学测序及分析
[0198]
提取mdck-st3galko(p102)细胞基因组,进行二代基因组测序并与mdck细胞参考基因组进行比对。
[0199]
二代测序实验流程为:细胞基因组dna的提取及纯化—dna片段化—末端修复并选择文库大小—3’末端加a—连接接头—文库扩增质检—上机测序。
[0200]
基因组测序结果分析流程为:测序原始数据—数据质控筛选高质量数据—比对参考基因组—进行变异检测,其中变异检测包括:单核苷酸多态性(snp)、核苷酸插入或缺失(indel)、结构变异(sv)以及基因拷贝数变异(cnv)。最终得到基因组中基因敲除相关突变情况如图45。
[0201]
mdck-st3galko细胞基因组二代测序分析结果
[0202][0203]
注:二代测序结果中出现小片段插入以及大片段插入,在本实验中小片段插入为插入puro序列后进行了cre-loxp剪切留下的残端loxp(30-35bp);大片段插入为整个puro序列的插入(1kbp-1.3kbp)。
[0204]
经测序比对,13号染色体上的小片段插入序列与loxp序列匹配(st3gal1),5号染色体上的大片段插入序列与puro序列匹配(st3gal2),证明在两个位点上的插入位点符合预期。
[0205]
转录组学分析
[0206]
为了探究st3gal1、st3gal2联合敲除后影响mdck对流感病毒敏感性的作用机制。进一步对mdck-st3galko进行了转录组测序。两种细胞各送检了3个样本(分别命名为mdck-normal1、2、3和mdck-st3gal1、2、3)。
[0207]
转录组测序流程为:细胞rna提取质检、cdna合成、文库制备、文库扩增质检以及上机测序。转录组数据分析流程为:数据质控、比对参考基因组、进行变异检测(snp、indel)、基因定量及表达差异分析(kegg富集分析、go富集分析)。具体分析结果如图46-49。
[0208]
转录组测序结果显示,与mdck细胞相比,在mdck-st3galko细胞中st3gal1和st3gal2表达水平显著降低,p1《0.01,p2《0.05。
[0209]
本发明针对基因编辑获取的新mdck细胞株进行了一系列验证性实验,可以具体看
出:
[0210]
1、定位pcr及测序结果显示mdck-st3gal1ko细胞为杂合突变,mdck-st3gal2ko细胞为纯合突变。q-pcr结果显示相较于mdck细胞,mdck-st3gal1ko细胞的st3gal1的转录水平显著降低;而相较于mdck-st3gal1ko细胞,mdck-st3gal1&2ko细胞的st3gal2转录水平显著降低。
[0211]
2、基因组测序结果显示mdck-st3gal1&2ko细胞中,st3gal1所在的13号染色体29863779-29863814位置上的小片段插入序列与loxp序列匹配,st3gal2所在5号染色体76829232-76830128位置上的大片段插入序列与puro序列匹配。转录组测序结果显示相较于mdck细胞,mdck-st3gal1&2ko细胞中st3gal1和st3gal2的表达水平均显著降低。
[0212]
3、细胞生长曲线显示mdck细胞的倍增时间为26h,达到平台期时间为60h,平台期活细胞平均密度为5.3
×
105/ml;mdck-st3gal1ko细胞倍增时间为36h,达到平台期的时间为72h,平台期活细胞平均密度为5.2
×
105/ml;mdck-st3gal2ko细胞倍增时间为34h,达到平台期的时间为70h,平台期活细胞平均密度为4.7
×
105/ml;mdck-st3gal1&2ko细胞倍增时间为34h,达到平台期的时间为72h,平台期活细胞平均密度为4.8
×
105/ml。
[0213]
4、细胞表面唾液酸凝集素结合平均荧光强度的差异体现了唾液酸含量的差异,其中mdck细胞、mdck-st3gal1ko细胞以及mdck-st3gal1&2ko细胞三者存在显著差异。而mdck-st3gal1ko细胞各代次之间差异不显著。mdck-st3gal1ko细胞中α-2,6-唾液酸的表达量是mdck细胞的1.56倍;mdck-st3gal1&2ko细胞中α-2,6-唾液酸的表达量是mdck细胞的1.78倍;mdck-st3gal1ko细胞中α-2,3-唾液酸的表达量mdck细胞的0.75倍;mdck-st3gal1&2ko细胞中α-2,3-唾液酸的表达量是mdck细胞的0.67倍。
[0214]
5、血凝实验结果表明mdck-st3gal1ko细胞和mdck-st3gal1&2ko细胞在h1n1、bv、by三种病毒中的滴度相较于mdck细胞有显著升高。相较于mdck细胞,在mdck-st3gal1ko细胞中收获的h1n1、bv、by病毒滴度分别提高了4倍、4倍、2倍,在mdck-st3gal1&2ko细胞中收获的h1n1、bv、by病毒滴度分别提高了8倍、8倍、4倍,而在mdck-st3gal2ko细胞中培养h1、bv、by三种毒株获得的病毒滴度与mdck细胞无显著差异。
[0215]
综上,基因编辑得到的mdck-st3galko细胞株具有对人流感病毒高度敏感的特性,具有用于大流行流感疫苗生产的潜力。
[0216]
在此有必要指出的是,以上实施例仅限于对本发明的技术方案做进一步的阐述和说明,并不是对本发明的技术方案的进一步的限制,本发明的方法仅为较佳的实施方案,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。