1.本发明涉及一种工程菌及其构建方法和应用,涉及基因工程技术领域。
背景技术:
2.外源dna转化技术是基因工程和dna克隆技术发展的基础和核心,是现代分子生物学中最重要的操作手段之一。常见的转化方法包括化学转化和电击转化,对于实验室标准菌株一般具备相对较高的转化效率,但是对于某些环境中筛选的菌株或者工业菌株,外源dna的转化效率一般较低。
3.例如,丁烯基多杀菌素(butenyl-spinosyns)是一种新型大环内酯类天然产物,具有高效生物杀虫活性,是理想的绿色生物杀虫剂。常见的生产方法是利用土壤放线菌须糖多孢菌(saccharopolyspora pogona)的次级代谢产生,但是,野生型须糖多孢菌合成丁烯基多杀菌素的能力较弱、产量较低,需要利用基因工程技术改造须糖多孢菌以获得高产丁烯基多杀菌素的工程化菌株。但是,须糖多孢菌具有很强的限制性修饰系统,当外源基因进入细胞后会被快速降解,导致须糖多孢菌的遗传操作非常困难,限制了高产丁烯基多杀菌素工程菌株的构建。
4.限制性修饰系统通常包括限制酶和修饰酶,限制酶是一种核酸内切酶,能够对外源dna进行切割,修饰酶能够对自身dna进行甲基化修饰。为了克服须糖多孢菌自身的限制性修饰系统,可以通过对外源基因进行甲基化修饰,使得修饰后的外源基因不被须糖多孢菌自身的限制性修饰系统剪切降解,从而提高外源基因的转化效率。因此,如何对外源基因进行甲基化修饰,提高外源基因在须糖多孢菌内的转化效率,受到了本领域技术人员的持续关注。
技术实现要素:
5.本发明提供一种工程菌,其能够过表达如seq id no.1或seq id no.3所示的甲基化酶,能够对外源基因进行甲基化修饰,提高外源基因在须糖多孢菌中的转化效率。
6.本发明还提供上述工程菌的构建方法。
7.本发明还提供一种提高须糖多孢菌外源基因转化效率的方法,通过将外源基因导入上述工程菌内进行甲基化修饰,提高外源基因在须糖多孢菌内的转化效率。
8.本发明第一方面提供一种工程菌,所述工程菌为能够过表达甲基化酶的大肠杆菌,所述甲基化酶用于对外源基因进行甲基化修饰;
9.所述甲基化酶的氨基酸序列如seq id no.1或seq id no.3所示。
10.本发明提供的甲基化酶来源于须糖多孢菌(s.pogona asagf58),通过生物信息学比对分析,发现须糖多孢菌基因组上存在三种甲基化酶,分别命名为甲基化酶04455、28970和29090,甲基化酶04455的氨基酸序列如seq id no.1所示,甲基化酶28970的氨基酸序列如seq id no.3所示,二者均能够对外源基因进行甲基化修饰,并提高外源基因在须糖多孢菌内的转化效率,具体地,甲基化酶28970的转化效率为20.5cfu/ug dna,甲基化酶04455的
转化效率为57.5cfu/ug dna,而甲基化酶29090的氨基酸序列如seq id no.5所示,其对外源基因在须糖多孢菌中的转化效率没有提升,因此,构建能够过表达甲基化酶04455或28970的工程菌,能够对外源基因进行甲基化修饰,提高外源基因在须糖多孢菌内的转化效率,对须糖多孢菌后续的基因工程改造提供重要基础。
11.在一种优选实施例中,基于甲基化酶04455对外源基因在须糖多孢菌中的转化效率较高,构建能够外源表达甲基化酶04455的工程菌,所述甲基化酶04455的氨基酸序列如seq id no.1所示。
12.进一步地,所述大肠杆菌为et12567,大肠杆菌et12567是一种甲基化缺陷型菌株,导入的基因序列不会被菌株的甲基修饰系统降解,是链霉菌基因敲除或者基因倍增通用的菌株,本领域技术人员可根据本领域常规技术手段购买得到。
13.本发明第二方面提供上述任一所述工程菌的构建方法,包括:将编码所述甲基化酶的基因整合到大肠杆菌的基因组上,实现所述甲基化酶的过表达,得到所述工程菌;
14.所述甲基化酶的氨基酸序列如seq id no.1或seq id no.3所示。
15.在一种具体实施方式中,所述方法包括:
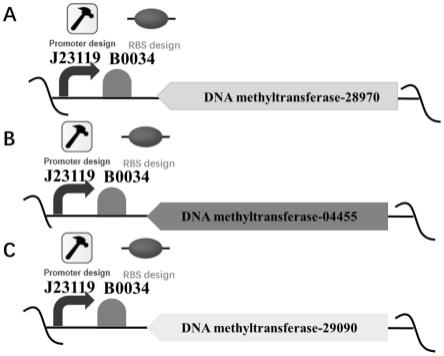
16.步骤1、将编码所述甲基化酶的基因、启动子j23119和核糖体结合位点b0034连接,构建得到甲基化酶的表达盒;
17.具体地,经过密码子优化,编码所述甲基化酶04455的基因的核苷酸序列如seq id no.2所示,编码所述甲基化酶28970的基因的核苷酸序列如seq id no.4所示。
18.编码甲基化酶的基因在大肠杆菌内的表达需要启动子和核糖体结合位点的驱动作用,具体地,本发明所使用的启动子为启动子j23119,核糖体结合位点为b0034,其能够启动下游基因的表达,并具有较高的强度,具体地,启动子j23119和核糖体结合位点b0034可参照文献(systematic analysis of escherichia coli isolates from sheep and cattle suggests adaption to the rumen niche),或者可以通过网站http://parts.igem.org/main_page查询得到。
19.此外,为了阳性转化子的筛选,表达盒的构建还需要加入抗性序列,具体地,本发明使用的抗性序列为kan抗性序列,在转化子筛选过程中,将转化后的大肠杆菌在含有壮观霉素的培养基上培养,而kan抗性序列表达的酶能够降解壮观霉素,使得大肠杆菌正常生长,从而筛选出阳性转化子。
20.通过本领域常规技术手段,按照图1所述的顺序,将启动子j23119、核糖体结合位点b0034、编码甲基化酶的基因序列和kan抗性序列依次连接,构建得到甲基化酶的表达盒。
21.步骤2、培养感受态大肠杆菌,并将所述表达盒导入感受态大肠杆菌中,筛选阳性转化子,得到所述工程菌。
22.本发明通过培养感受态细胞实现外源dna分子的导入,首先,诱导大肠杆菌为感受态细胞,具体地,通过电击转化或者化学转化的方法将ptkred质粒转入大肠杆菌中,在一定条件下培养,挑取单克隆进行液体过夜培养,随后添加iptg进行诱导,制备得到感受态大肠杆菌。
23.其次,将步骤1制备得到的表达盒与感受态大肠杆菌混合,通过电极转化将构建有目的基因的表达盒导入感受态大肠杆菌中,最后对大肠杆菌进行培养,筛选阳性转化子,得到工程菌。
24.本发明第三方面提供上述工程菌在对外源基因进行甲基化修饰并提高所述外源基因在须糖多孢菌中的转化效率中的应用。
25.本发明提供的上述工程菌能够对外源基因进行甲基化修饰,得到甲基化修饰后的外源基因,甲基化修饰后的外源基因即可导入须糖多孢菌中,并提高转化效率。
26.本发明中“外源基因”可以是任何需要转化到须糖多孢菌中的基因,本发明对外源基因不作任何限制。
27.本发明第四方面提供一种提高外源基因在须糖多孢菌中转化效率的方法,包括:
28.将外源基因导入上述任一所述的工程菌中进行甲基化修饰,得到甲基化修饰的外源基因;
29.将所述甲基化修饰的外源基因导入所述须糖多孢菌中。
30.在一种具体实施方式中,该方法包括如下步骤:
31.步骤1、将外源基因导入上述工程菌中进行甲基化修饰,得到甲基化修饰的外源基因;
32.步骤1.1、构建包括所述外源基因的重组载体,将所述重组载体导入所述工程菌中,进行甲基化修饰;
33.将所需转入须糖多孢菌的外源基因插入到质粒上,构建得到包括外源基因的重组载体,通过电极转化法等常规技术手段将重组载体导入工程菌中,通过工程菌表达的甲基化酶对重组载体进行甲基化修饰。
34.步骤1.2、提取所述工程菌的dna,得到甲基化修饰的重组载体;
35.待菌体生长起来后,提取甲基化修饰的重组载体。
36.步骤2、将所述甲基化修饰的外源基因导入所述须糖多孢菌中;
37.采用原生质体转化的方法,将所述甲基化修饰的重组载体导入所述须糖多孢菌中。
38.综上,本发明提供的工程菌能够过表达甲基化酶04455和28970,对外源基因进行甲基化修饰,进而提高外源基因在须糖多孢菌内的转化效率,为须糖多孢菌的基因工程改造提供重要基础。
附图说明
39.图1为表达盒的构建原理图,其中,a为甲基化酶28970表达盒的构建原理图,b为甲基化酶04455表达盒的构建原理图,c为甲基化酶29090表达盒的构建原理图;
40.图2为et12567::04455、et12567::28970和et12567::29090的菌落pcr验证图;
41.图3为pset-159-bpsa转化s.pogona的平板图;
42.图4为甲基化酶04455和28970编码基因的转录水平;
43.图5为sds-page分析甲基化酶04455和28970;
44.图6a为甲基化酶04455的质谱谱图;
45.图6b为甲基化酶28970的质谱谱图。
具体实施方式
46.为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明的实施例,对本
发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
47.实施例1工程菌的构建
48.步骤1、目标片段克隆
49.以ha-f/r为引物,et12567基因组为模板扩增上游同源臂;以hb-f/r为引物,et12567基因组为模板扩增下游同源臂;以k-f/r为引物,质粒pkd3为模板扩增kan抗性序列;04455-f/r为引物,经过密码子优化后的编码甲基化酶04455的基因片段(seq id no.2)为模板进行扩增得到目的序列;利用ha-f/r为引物上述4个片段为模板进行组装,得到表达盒。
50.同理,以ha-f/r为引物,et12567基因组为模板扩增上游同源臂;以hb-f/r为引物,et12567基因组为模板扩增下游同源臂;以k-f/r为引物,质粒pkd3为模板扩增kan抗性序列;以28970-f/r为引物,经过密码子优化后的编码甲基化酶28970的基因片段(seq id no.4)为模板进行扩增得到目的序列;利用ha-f1和hb-r1为引物上述4个片段为模板进行组装,得到表达盒。
51.同理,以ha-f/r为引物,et12567基因组为模板扩增上游同源臂;以hb-f/r为引物,et12567基因组为模板扩增下游同源臂;以k-f/r为引物,质粒pkd3为模板扩增kan抗性序列;以29090-f/r为引物,经过密码子优化后的编码甲基化酶29090的基因片段(seq id no.6)为模板进行扩增得到目的序列;利用ha-f/r为引物上述4个片段为模板进行组装,得到表达盒。
52.上述引物序列见表1。
53.表1实施例1使用的引物名称与序列
54.[0055][0056]
步骤2、通过电击转化的方法将ptkred质粒转入大肠杆菌et12567中,在30℃下在涂布有壮观霉素的lb平板中过夜培养,挑取单克隆进行液体过夜培养。培养结束后,按照1%的接种量转接并添加终浓度为1mm的iptg进行诱导,待od值达到0.8左右开始制备感受态细胞。
[0057]
步骤3、将步骤1构建好的三个表达盒分别缓慢加入到感受态细胞中,混合均匀,进行电击转化后,立即加入lb培养基700μl,将电转完成的菌液转移到新的无菌离心管中,在37℃下恢复培养1h,培养结束后在5000rpm下离心2min,收集菌体沉淀并涂布于含有相应抗生素的lb固体培养基上,37℃过夜培养至生长出单克隆。挑取重组子,分别以引物t-f/t-04455-r(t-28970-r/t-29090-r)进行菌落pcr验证,pcr程序如表2所示,验证结果如图2所示,结果显示dna条带大小符合预期,证明含有目的基因的表达盒已成功整合到大肠杆菌et12567的基因上,构建好的工程菌株分别命名为et12567::04455、et12567::28970和et12567::29090。
[0058]
表2pcr程序
[0059][0060][0061]
实施例2外源基因的转入
[0062]
质粒pset159-bpsa携带有编码合成靛蓝素的基因,转到宿主后在平板上呈现蓝色,则表明质粒转入成功,以此来反应外源基因在须糖多孢菌内的转化效率。
[0063]
步骤1、通过电击转化的方式将质粒pset159-bpsa分别转化至工程菌et12567::04455、et12567::28970和et12567::29090中实现甲基化修饰,待菌落生长完成后,参考质粒小提试剂盒(天根)提取预甲基化质粒pset159-bpsa(m),并对提取的质粒统一定量到100ng/μl,使用质粒pset159-bpsa转化至et12567菌株后提取得质粒作为对照组(control)。
[0064]
步骤2、须糖多孢菌(s.pogona)原生质体转化
[0065]
(1)s.pogona在tsb培养基中培养,添加终浓度为0.2%的甘氨酸,4℃下培养48h,培养结束后将菌液在5000rpm、离心5min,收集菌丝体;
[0066]
(2)菌丝体用10%蔗糖溶液洗两次,并加10ml的p buffer重悬菌丝体;
[0067]
(3)用p buffer配成终浓度为2mg/ml的溶菌酶溶液,30℃保温大约30min,期间每5min轻轻颠倒离心管,促进原生质体的释放;
[0068]
(4)用装有脱脂棉的过滤试管过滤菌液,3000rpm离心5min,收集原生质体;
[0069]
(5)原生质体涂布在r5培养基上以检测原生质体是否污染及其活性;
[0070]
(6)原生质体用1ml的p buffer悬浮,立即分装成小份(每份加入200μl原生质体)置于-80℃保存或进行转化。
[0071]
(7)向200μl制备好的原生质体悬液中分别加入50μl质粒(pset159-bpsa或甲基化修饰后的质粒)和50μl 60%peg 4000,用手指轻轻弹管壁使其混合,将悬液涂于已吹干的r5平板上,于再生培养温度30℃下培养,当再生的原生质体在平板上呈现雾状时(24h),用含适宜浓度的抗生素水溶液涂布于平板上,继续培养至长出菌落。
[0072]
如图3和表3所示,相比对照组,实验组28970和04455中菌落数明显增多,表明甲基化修饰的质粒pset159-bpsa在须糖多孢菌中的转化效率得到大幅度提高,具体地,pset159-bpsa质粒转化到et12567::28970,转化效率为20.5cfu/μg dna,比对照组高5.8倍;pset159-bpsa质粒转化到et12567::04455,转化效率为57.5cfu/μg dna,比对照组高16.4倍;而pset159-bpsa质粒转化到et12567::29090,转化效率低于对照组。
[0073]
表3质粒pset159-bpsa在须糖多孢菌中的转化效率
[0074][0075]
实施例4甲基化酶04455和28970的转录水平验证
[0076]
rna提取方法参照天根rna提取试剂盒说明书,rna反转录为cdna操作方法参照sybr green试剂盒说明书,计算方法采用相对定量算法—δδct,该方法根据检测样本和
校准样本中的标准品(正常)基因的ct调整同样两个样本中的目的基因(goi)的ct,其得出的δδct值可用于测定表达的倍数差异,具体公式如下:
[0077]
倍数差异=2-δδct
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(1)
[0078]
δδc
t
=δc
t样本-δc
t标准品
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(2)
[0079]
δc
t样本
=c
t gois
ꢀ–ct正常s
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(3)
[0080]
δc
t校准品
= c
t goic
ꢀ–ct正常c
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(4)
[0081]
对须糖多孢菌s.pogona asagf58中的甲基化酶04455和28970进行转录水平分析,确认这两个基因均发生转录。培养工程菌et12567::04455和et12567::28970并提取rna,结果如图4所示,甲基化酶04455和28970在工程菌中的转录水平远高于s.pogona asagf58,具体地,甲基化酶28970的转录水平提高了40倍,甲基化酶04455的转录水平提高了180倍。
[0082]
实施例5甲基化酶04455和28970的蛋白水平验证
[0083]
为了验证et12567::04455和et12567::28970中甲基化酶在蛋白水平的表达,为此进行了蛋白质谱鉴定,具体包括如下步骤:
[0084]
(1)取工程菌的发酵液,在4℃,5000rpm下离心10min;
[0085]
(2)去上清,取一定量pbs(ph 7.0)溶液重悬菌体;
[0086]
(3)取80μl上述样品,加入20μl 5
×
sds上样缓冲液,煮沸10min;
[0087]
(4)蛋白检测:取全细胞样品80μl进行电泳,在浓缩胶时,采用8v/cm恒压,当溴酚蓝指示剂进入分离胶时,电压采用12v/cm恒压,待指示剂电泳至分离胶底部0.5cm处,停止电泳后考马斯亮蓝溶液染色后并脱色,结果如图5所示。
[0088]
将胶条送至华大基因基因蛋白质谱检测,其中,胶条04455鉴定结果显示,其分子量为53647.6,丰度为71593.26。胶条28970鉴定结果显示,其分子量为56658.73,丰度为11470.93,表明工程菌et12567::04455中检测到甲基化酶04455,质谱谱图如图6a所示,工程菌et12567::28970中检测到甲基化酶28970,质谱谱图如图6b所示,说明了这两个修饰酶均成功表达,且这两个修饰酶的比对结果为腺嘌呤甲基转移酶与预测结果一致。
[0089]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。