用于鉴定基因组dna中与蛋白质结合的区域的方法、组合物和试剂盒
相关专利申请的交叉引用
1.本技术要求于2020年4月2日提交的美国临时申请63/004,361的优先权,该申请公开的内容据此以引用方式全文并入本文。序列表
2.序列表以文本文件“alti-730wo seq list_st25.txt”随本技术提供,创建于2020年4月2日,大小为10kb。该文本文件的内容据此以引用方式全文并入本文。
背景技术:
3.染色质的初级结构包括核小体阵列,该阵列被含有转录因子和其他非组蛋白的短调控区域截断。这种结构是基因组功能的基础,但在单根染色质纤维(基因调控的基本单位)层面尚不明确。例如,尽管核小体是限制转录因子接近dna的主要屏障,但沿着单根染色质纤维排列的核小体在体内的定位和占位尚未阐明。因此,目前尚不清楚以下方面:核小体如何沿着相同的延伸染色质模板精确排列;单根染色质纤维上可及的调控dna与核小体之间的相互作用;给定dna编码的调控区域在细胞群内不同染色质纤维上被驱动的程度;以及在相同染色质模板上,附近的调控区域被协调驱动的程度。要解决这些问题,需要对单根染色质纤维进行测序,这是目前单细胞或批量分析法所无法实现的。
4.需要以单核苷酸分辨率将染色质的初级结构记录到其基础dna模板上的方法,从而能够同时鉴定基因组中多千碱基片段的遗传特征和表观遗传特征。本公开解决了这些需求和其他需求。
技术实现要素:
5.提供了用于鉴定基因组dna中与蛋白质结合的区域的方法、组合物、试剂盒和系统。在某些方面,该方法包括使基因组dna与腺嘌呤甲基转移酶(a-mtase)接触,其中该a-mtase使得该基因组dna中不与蛋白质结合的区域中的腺嘌呤残基甲基化;以及对所接触的基因组dna进行单分子长读测序,以检测该基因组dna中缺少甲基化腺嘌呤残基的位置,进而鉴定该基因组dna中与该蛋白质结合的区域。在某些方面,所结合的区域是核小体位置。因此,该方法包括测定基因组dna中核小体位置的方法。此外,提供了可用于例如实施本公开的方法的组合物、系统和试剂盒。
6.还提供了通过在使细胞与腺嘌呤甲基转移酶(a-mtase)接触后可视化甲基化腺嘌呤的位置来使染色质区域可视化的方法,该染色质区域不与蛋白质结合,并且在空间上可作为该细胞内该a-mtase的底物。
附图说明
7.当结合附图阅读下文的具体实施方式时,有助于最好地理解本发明。包括以下附图:
8.图1a-图1f示出了非特异性m6a-mtase选择性标记染色质可及性位点。
9.图2a-图2h示出了fiber-seq揭示单个染色质纤维结构的碱基对分辨率图谱。
10.图3a-图3b示出了相邻调控元件在同一染色质纤维上的协调驱动。
11.图4a-图4d示出了调控dna驱动对核小体定位的影响。
12.图5a-图5e示出了果蝇与人类之间染色质结构的守恒。
13.图6a-图6e示出了使用细胞穿透肽(cpp)标记的m6a-mtase鉴定体内染色质结构。
14.图7示出了使用fiber-seq鉴定功能基因调控dna改变。
15.图8示出了使用特异性结合m6a的三种不同抗体检测到的n6-甲基腺苷(m6a)以点状模式随着hia5剂量的增加而增加
16.图9示出了总核m6a信号(左)以及单点强度(右)的hia5剂量依赖性增加。实施方式
17.提供了用于鉴定基因组dna中与蛋白质结合的区域的方法、组合物、试剂盒和系统。在某些方面,该方法包括使基因组dna与腺嘌呤甲基转移酶(a-mtase)接触,其中该a-mtase使得该基因组dna中不与蛋白质结合的区域中的腺嘌呤残基甲基化;以及对所接触的基因组dna进行单分子长读测序,以检测该基因组dna中缺少甲基化腺嘌呤残基的位置,进而鉴定该基因组dna中与该蛋白质结合的区域。在某些方面,所结合的区域是核小体位置。因此,该方法包括测定基因组dna中核小体位置的方法。此外,提供了可用于例如实施本公开的方法的组合物、系统和试剂盒。在某些方面,使用包括处理器的计算机来执行该方法的至少一些步骤,该处理器包括程序,该程序在由该处理器执行时执行这些步骤。
18.还提供了通过在使细胞与腺嘌呤甲基转移酶(a-mtase)接触后可视化甲基化腺嘌呤的位置来使染色质区域可视化的方法,该染色质区域不与蛋白质结合,并且在空间上可作为该细胞内该a-mtase的底物。
19.在描述本发明的示例性实施方案之前,应当理解的是,本发明不限于所描述的特定实施方案,因为这些实施方案当然可以有所变化。还应当理解的是,本文使用的术语仅用于描述特定实施方案,而不意在加以限制,因为本发明的范围将仅由所附权利要求书限制。
20.在提供数值范围的情况下,应当理解的是,除非上下文另有明确说明,否则还明确公开该范围的上限和下限之间精确至下限单位十分之一的各居中值。在宣称范围内的任何宣称值或居中值与在该宣称范围内的任何其他宣称值或局中值之间的各较小范围包含在本发明范围内。这些较小范围的上限和下限可以独立包含在该宣称范围内或排除在该宣称范围外,并且其中一个或两个限值或没有限值包含在这些较小范围内的各范围也包含在本发明范围内,但以该宣称范围内任何明确排除的限值为限。如果该宣称范围包含这些限值中的一个或两个,则排除该等所包含限值中的一个或两个的范围也包含在本发明范围内。
21.除非另有定义,否则本文使用的所有技术和科学术语与本发明所属领域的普通技术人员通常理解的含义相同。尽管在本发明的实践或测试中可以使用任何与本文所述方法和材料类似或等效的方法和材料,但现在可以对一些潜在和示例性方法和材料进行描述。本文提及的任何和所有出版物以引用方式并入本文,以公开和描述与所引用的出版物相关的方法和/或材料。应当理解的是,如有冲突,本公开取代所并入的出版物的任何公开内容。
22.应当指出的是,如本文和所附权利要求书中所使用的,除非上下文另有明确说明,否则单数形式“一个”、“一种”和“该”包括复数指称。因此,例如,提及“一个细胞”时,包括多
个此类包细胞,提及“该细胞”时,包括一个或多个细胞,以此类推。
23.还应当指出的是,权利要求可能撰写为排除任何可选的要素。因此,该声明意在结合权利要求要素的表述而用作使用诸如“单独”、“仅”等排他性术语或使用“否定”限制的先行依据。
24.本文所讨论的出版物仅为了说明它们在本技术提交日期之前公开而提供。本文中的任何内容都不应被解释为承认本发明无权因在先发明而早于此类出版物。此外,所示出版日期可能与实际出版日期不同,可能需要对日期单独进行确认。就此类出版物可能列出与本公开的明示或暗示定义相冲突的术语定义而言,以本公开的定义为准。
25.如本领域技术人员在阅读本公开后将显而易见的,本文描述和说明的各单独实施方案都具有分立组件和特征,这些组件和特征可以简单地与其他几个实施方案中的任何一个特征分离或组合而不脱离本发明的范围或精神。任何列举的方法都可以按照所列举事件的顺序或逻辑上可行的任何其他顺序实施。定义
26.术语“hia5”是指与来自流感嗜血杆菌(h.influenzae)的hia5多肽的氨基酸序列(seq id no:1)至少80%相同(例如,至少85%、至少90%、至少95%、至少96%、至少97%、至少98%、至少99%或100%相同)的多肽。
27.manqntfkqaplpfigqkrmflkqfeqilnenisdngegwtildtfggsgllshtakrlkpkarviyndfdgyaerlahiddinqlraelysvvgnatsknkrmtkdckaeciriiqnfkgykdlnclaswllfsgqqvatlddlfqhnfwhcirqsdypkadgyldgveivkesfhtllpkfsndpkalfvldppylctkqesykqatyfdlidflrlvnitrppyvffsstksefirfvnymledkvdnwqafenakritvnaklnyqvayednlvykf(seq id no:1)。
28.术语“hin1523”是指与来自流感嗜血杆菌的hin1523基因编码的多肽(seq id no:2)至少80%相同(例如,至少85%、至少90%、至少95%、至少96%、至少97%、至少98%、至少99%或100%相同)的多肽。
29.mseyleyqnaiegktmankktfkqaplpfigqkrmflkhveivlnkhidgegegwtivdvfggsgllshtakqlkpkatviyndfdgyaerlnhiddinrlrqiifnclhgiipkngrlskeikeeiinkindfkgykdlnclaswllfsgqqvgsvealfakdfwncvrqsdyptaegyldgievisesfhklipryqnqdkvlllldppylctrqesykqatyfdlidflrlinltkppyiffsstksefirylnymqesktdnwrafenykrivvkasaskdgiyednmiykf(seq id no:2)。
30.如本文中所使用的,术语“m.btr192iv”是指与来自海藻糖比伯斯坦杆菌usda-ars-usmarc-192的wqg_17550基因编码的多肽(seq id no:3)至少80%相同(例如,至少85%、至少90%、至少95%、至少96%、至少97%、至少98%、至少99%或100%相同)的多肽。
31.mkktltalavaslasatqtkqqaskqaskqaskqaskecemakvfkqaplpfigqkrmflkhfeqvlahipddgngwtivdvfggsgllshtakrlkpkarviyndydnyserlqhiddinrlrriiadlmadtpkykrl dnakklqiieaieafqgykdlhilcswlafsgqqvssfdelykqnfwhcirqsdyltadgyldgveivresfhqlvprftgqpntllvldppylcthqesykqeryfdlvdflrlihltkppyvffsstksefvrfidamvedkwdnwqafddaqrivvqtsasyngkyednmvykf(seq id no:3)
32.如本文中所使用的,术语“ecog1”是指与来自大肠杆菌(e.coli)的phk08_22基因编码的多肽(seq id no:4)至少80%相同(例如,至少85%、至少90%、至少95%、至少96%、至少97%、至少98%、至少99%或100%相同)的多肽。
33.msrfilgdcvrvmatfp dnavdfiltdppylvgfrdrsgrtiagdvnddwlqpasnemyrvlkkdalmvsfygwnridrfmaawkragfsvvghlvftknytskaayvgyrhecayilakgrpalpqkplpdvlgwkysgnrhhptekpvtslqpliesfthpnaivldpfagsgstcvaalqsgrryigielleqyhragqqrlaavqramqqgaandnwfepeaa(seq id no:4)
34.hia5、ecogii、btr192iv和ecogi具有腺嘌呤甲基转移酶活性。这些甲基转移酶可以通过密码子优化来增加大肠杆菌(e.coli)细胞的表达。
35.本文所公开的诸如n6-腺嘌呤甲基转移酶(m6a-mtase)等a-mtase包括经修饰的hia5、ecogii、btr192iv和ecogi,例如具有不同于本文所公开序列的氨基酸序列的变体以及包含插入、取代、缺失和融合蛋白的突变体。融合蛋白包含与细胞穿透肽、标签等融合的a-mtase。该细胞穿透肽可以是具有净正电荷的肽,以使融合蛋白质膜可渗透。该细胞穿透肽可以是hiv-1tat易位结构域、8-精氨酸(8r)、穿透蛋白,以及它们的变体等。融合蛋白包含与核定位序列(nls)融合的a-mtase,以将该a-mtase靶向细胞核。a-mtase可以与nls和细胞穿透肽融合。
36.术语“抗体”和“免疫球蛋白”包括任何同型抗体或免疫球蛋白、保留与抗原特异性结合的抗体片段,包括但不限于fab、fv、scfv、fd、fab'、fv和f(ab')2、嵌合抗体、人源化抗体、单克隆抗体、单链抗体,包括仅包含重链的抗体(例如,vhh骆驼科抗体)、双特异性抗体以及包含抗体和非抗体蛋白的抗原结合部分的融合蛋白。根据其重链恒定结构域的氨基酸序列,免疫球蛋白可以被划分为不同类别。主要有五类免疫球蛋白:iga、igd、ige、igg和igm,其中几类可进一步划分为亚类(同型),例如igg1、igg2、igg3、igg4、iga和iga2。术语“抗体”和“免疫球蛋白”具体包括但不限于igg1、igg2、ig3和igg4抗体。这些抗体可被检测标记,例如通过使用放射性同位素、产生可检测产物的酶、荧光蛋白等。这些抗体可以进一步与其他部分缀合,诸如特异性结合对的组成部分,例如生物素(生物素-亲和素特异性结合对的组成部分)等。
[0037]“抗体片段”包含完整抗体的一部分,例如该完整抗体的抗原结合区或可变区。抗体片段示例包括fab、fab'、f(ab')2和fv片段;双抗体;线性抗体(zapata et al.,protein eng.8(10):1057-1062(1995));单链抗体分子,包括仅包含重链的抗体(例如,vhh骆驼科抗体);以及由抗体片段形成的多特异性抗体。抗体的木瓜蛋白酶消化产生两个相同的抗原结合片段,称为“fab”片段,每个片段具有单一抗原结合位点和一个残留的“fc”片段,此命名反映了其快速结晶的能力。胃蛋白酶处理产生f(ab')2片段,该片段具有两个抗原结合位点,并且仍然能够与抗原交联。
[0038]“单链fv”、“sfv”或“scfv”抗体片段包含抗体的vh和vl结构域,其中这些结构域存在于单一多肽链中。在一些实施方案中,fv多肽还包含vh和vl结构域之间的多肽连接子,这使得sfv能够形成用于抗原结合的所需结构。关于sfv的综述,请参见pluckthun in the pharmacology of monoclonal antibodies,vol.113,rosenburg and moore eds.,springer-verlag,new york,pp.269-315(1994)。
[0039]
如本文中所使用的,术语“治疗”、“处理”等是指获得期望的药理和/或生理效果。就完全或部分预防疾病或其症状而言,该效果是预防性的,和/或就部分或完全治愈疾病和/或由该疾病引发的副作用而言,该效果是治疗性的。如本文中所使用的,术语“治疗”涵盖对包括人类在内的哺乳动物所患疾病的任何治疗,包括:(a)在可能易患但尚未确诊患有
该疾病的受试者中防止该疾病发生;(b)抑制该疾病,即阻止其发展;以及(c)缓解该疾病,即使该疾病消退。
[0040]
本文中可互换使用的术语“个体”、“受试者”、“宿主”和“患者”是指哺乳动物,包括但不限于鼠科动物(大鼠、小鼠)、非人灵长类动物、人类、犬科动物、猫科动物、有蹄类动物(例如,马、牛、绵羊、猪、山羊)等。
[0041]“生物样本”包含从个体获得的各种样本类型,可用于诊断或监测分析。该定义包含生物来源的血液和其他液体样本、固体组织样本,诸如活检样本或组织培养物或由此衍生的细胞及其后代。该定义还包括采集后以任何方式处理的样本,诸如通过试剂处理、溶解或富集某些组分(诸如多核苷酸)。术语“生物样本”包含临床样本,还包括培养物中的细胞、细胞上清液、细胞溶解液、血清、血浆、生物体液和组织样本。方法
[0042]
本公开提供了鉴定基因组dna中与蛋白质结合的区域的方法。该蛋白质可以是任何限制腺嘌呤甲基转移酶(a-mtase)接近存在于由该蛋白质结合的基因组序列中的腺嘌呤碱基的蛋白质。该蛋白质可以是核小体、转录因子、转录抑制因子等中的一种或多种。下文将对该方法的各个步骤和方面进行更详细地描述。
[0043]
本公开的方法包括使基因组dna与腺嘌呤甲基转移酶(a-mtase)接触,其中该a-mtase使得该基因组dna中不与蛋白质结合的区域中的腺嘌呤残基甲基化;以及对所接触的基因组dna进行单分子长读测序,以检测该基因组dna中缺少甲基化腺嘌呤残基的位置,进而鉴定该基因组dna中与该蛋白质结合的区域。
[0044]
在某些方面,该a-mtase为n6-腺嘌呤甲基转移酶(m6a-mtase)。在某些方面,该m6a-mtase为hia5。在某些方面,该m6a-mtase为ecogii。在某些方面,该m6a-mtase为btr192iv。在某些方面,该m6a-mtase为ecogi。
[0045]
该接触可能涉及使分离的基因组dna与该a-mtase接触。在某些方面,该接触可能涉及将编码该a-mtase的核酸引入细胞或将该a-mtase引入细胞。
[0046]
在某些方面,该基因组dna来自单个细胞、多个细胞(例如,培养的细胞)、组织、器官或生物体(例如,细菌、酵母菌等)。在某些方面,该基因组dna来自动物的细胞、组织、器官等。在一些实施方案中,该动物是指哺乳动物(例如,人属哺乳动物、啮齿动物(例如,小鼠或大鼠)、犬、猫、马、牛或任何其他相关哺乳动物)。在某些方面,该基因组dna来自人类的细胞、组织、器官等。在其他方面,该基因组dna来自哺乳动物以外的其他来源,诸如细菌、酵母菌、昆虫(例如,果蝇)、两栖动物(例如,青蛙(例如,爪蟾))、病毒、植物或任何其他非哺乳类来源。在某些方面,高基因组dna来自癌细胞。
[0047]
在一些实施方案中,自基因组dna是无细胞的。这种无细胞基因组dna可能存在于任何合适的来源中或从其中获得。在某些方面,该无细胞基因组dna存在于体液样本中或从其中获得,该体液样本取自全血、血浆、血清、羊水、唾液、尿液、胸腔积液、支气管灌洗液、支气管抽吸液、母乳、初乳、眼泪、精液、腹膜液、胸腔液和粪便。在一些实施方案中,该基因组dna是无细胞的胎儿dna。在某些方面,该基因组dna是循环肿瘤dna。在一些实施方案中,该基因组dna包含感染原dna。在一些实施方案中,该基因组dna包含来自移植物的dna。如本文中所使用的,术语“无细胞基因组dna”可以指不含细胞或基本上不含细胞的基因组dna组合物。该基因组dna并不一定意味着细胞的所有遗传物质都存在,确切而言,基因组dna可以包
括细胞基因组物质的一部分。例如,该基因组dna包含分离的染色质片段,该染色质片段可以是任何从细胞中分离出来的与核蛋白相关的基因组dna片段。示例性染色质片段可以是寡核苷酸小体、单核小体、着丝粒、端粒或由转录因子或染色质重构因子结合的基因组dna。
[0048]
在某些方面,细胞可以是外周血单核细胞(pbmc)、白细胞,或者可以是从骨髓、胸腺、组织活检、肿瘤、淋巴瘤、淋巴结、肠道淋巴组织、粘膜相关淋巴组织、脾、其他淋巴组织、肝、肺、胃、肠、结肠、肾、胰腺、乳腺、骨、前列腺、子宫颈、睾丸、卵巢、扁桃体或其他器官中分离出的细胞,和/或从它们衍生的细胞。在一些实施方案中,待评估的核酸(例如,基因组dna、染色体dna)来自血细胞,例如来自全血样本或全血中细胞亚群的血细胞。全血中的细胞亚群包括血小板、红细胞(红血球)、血小板和白细胞(即外周血白细胞,其由中性粒细胞、淋巴细胞、嗜酸性粒细胞、嗜碱性粒细胞和单核细胞组成)。白细胞可进一步分为两组,即粒细胞(也称为多形核白细胞,包括中性粒细胞、嗜酸性粒细胞和嗜碱性粒细胞)和单核白细胞(包括单核细胞和淋巴细胞)。淋巴细胞可进一步分为t细胞、b细胞和nk细胞。外周血细胞存在于循环血池中,而不是隔离在淋巴系统、脾脏、肝脏或骨髓中。
[0049]
在某些方面,标的方法可能涉及分析治疗前获得的基因组dna和治疗后获得的基因组dna。例如,在治疗后1天、1周、10天、15天、1个月、3个月、6个月或更长时间后,以比较不与蛋白质结合并因此受腺嘌呤甲基化影响的dna区域。可以使用腺嘌呤甲基化模式比较来评估基因组转录谱的改变。
[0050]
在某些方面,标的方法可用于生成一种类型细胞的参考染色质结构和调控区域。例如,多种类型的人类细胞。可以将患病受试者的细胞中的染色质结构和调控区域与该细胞类型的参考染色质结构和调控区域进行比较,以测定任何差异。这些差异可能揭示先前未知的染色质结构和调控区域变化,这些变化可用于诊断、预判或治疗该受试者。
[0051]
在一些实施方案中,用于该方法的细胞群可以由任何数量的细胞组成,例如,约500-106个或更多细胞、约500-100,000个细胞、约500-50,000个细胞、约500-10,000个细胞、约50-1,000个细胞、约1-500个细胞、约1-100个细胞、约1-50个细胞或单个细胞。在一些实施方案中,细胞样本中包含的细胞少于约1000、2000、3000、4000、5000、6000、7000、8000、9000、10,000、15,000、20,000、25,000、30,000、40,000、50,000、60,000、70,000、80,000、90,000、100,000、120,000、140,000、160,000、180,000、200,000、250,000、300,000、350,000、400,000、450,000、500,000、600,000、700,000、800,000、900,000或1,000,000个。在一些实施方案中,细胞样本中包含的细胞多于约1000、2000、3000、4000、5000、6000、7000、8000、9000、10,000、15,000、20,000、25,000、30,000、40,000、50,000、60,000、70,000、80,000、90,000、100,000、120,000、140,000、160,000、180,000、200,000、250,000、300,000、350,000、400,000、450,000、500,000、600,000、700,000、800,000、900,000或1,000,000个。
[0052]
在某些方面,该基因组dna在暴露于甲基转移酶期间存在于其原生环境中。例如,在暴露于甲基转移酶期间,该基因组dna可存在于细胞(例如,完整细胞或透化细胞)中。在一些实施方案中,可使用跨完整或透化细胞膜的细胞渗透性甲基转移酶。在一些实施方案中,可使用标准技术将甲基转移酶引入细胞。在某些方面,该基因组dna在暴露于甲基转移酶期间存在于细胞溶解液中。
[0053]
在某些方面,该基因组dna是从生物体(例如,动物、人)的细胞、组织、器官等分离出的核酸样本的一部分。用于分离、纯化和/或浓缩来自相关来源的核酸分子的方法、试剂
和试剂盒是本领域已知的并且可商购。例如,从相关来源中分离dna的试剂盒包括由qiagen,inc.(马里兰州日耳曼敦)生产的和核酸分离/纯化试剂盒;由life technologies,inc.(加利福尼亚州卡尔斯巴德)生产的dna核酸分离/纯化试剂盒;以及由clontech laboratories,inc.(加利福尼亚州芒廷维尤)生产的和核酸分离/纯化试剂盒。在某些方面,从固定生物样本(例如,甲醛固定石蜡包埋(ffpe)组织)中分离核酸。从ffpe组织中分离基因组dna可以使用商购试剂盒,诸如由qiagen,inc.(马里兰州日耳曼敦)生产的ffpe组织dna/rna试剂盒、由life technologies,inc.(加利福尼亚州卡尔斯巴德)生产的ffpe组织总核酸分离试剂盒以及由clontech laboratories,inc.(加利福尼亚州芒廷维尤)生产的的ffpe组织试剂盒。
[0054]
在某些方面,在使该基因组dna与甲基转移酶接触之后且在测序之前,可以对该基因组dna进行处理以备测序。例如,该方法可包括对该基因组dna的末端进行处理以产生平齐末端。平齐化是指“填充”单链悬垂的过程,通过将该悬垂作为聚合模板,在互补链上添加核苷酸,或利用核酸外切酶活性“反咬”该悬垂完成。dna聚合酶,诸如dna聚合酶i和t4 dna聚合酶的klenow片段,可用于填充(5
′→3′
)和反咬(3
′→5′
)。可以用诸如绿豆核酸酶等核酸酶去除5
′
悬垂。
[0055]
在某些方面,可以在使用甲基转移酶处理后剪切或酶消化该基因组dna。
[0056]
单分子实时测序系统可用于通过分析衍生自此类系统的序列和/或动力学数据来检测甲基化腺嘌呤。特别地,甲基化腺嘌呤可以以各种方式改变核酸聚合酶的酶活性,例如,通过增加结合的核碱基掺入的时间和/或增加掺入事件之间的间隔时间。在某些实施方案中,使用单分子核酸测序技术检测聚合酶活性。在某些实施方案中,使用核酸测序技术检测聚合酶活性,该技术实时检测核苷酸掺入新生链的情况。在优选实施方案中,单分子核酸测序技术能够实时检测核苷酸掺入事件。此类测序技术是本领域已知的,包括例如测序和纳米孔测序技术。有关纳米孔测序的更多信息,请参见例如美国专利9,175,348;美国专利5,795,782;kasianowicz,et al.(1996)proc natl acad sci usa 93(24):13770-3;ashkenas,et al.(2005)angew chem int ed engl 44(9):1401-4;howorka,et al.(2001)nat biotechnology 19(7):636-9;以及astier,et al.(2006)j am chem soc 128(5):1705-10,所有这些文献出于所有目的以引用方式全文并入本文。关于核酸测序,术语“模板”是指进行模板定向合成新生链的核酸分子。如本文其他部分所述,模板可以包含例如dna或类似物、模拟物、衍生物,或它们的组合。此外,模板可以是单链、双链,或者可以包含单链和双链区域。双链模板中的修饰可以在与新合成的新生链互补的链中进行,也可以在与新合成的链相同的链(即被聚合酶取代的链)中进行。
[0057]
本文所述的优选直接甲基化测序通常可以使用单分子实时测序系统进行,即,随着时间推移连续照射和观察单个反应复合物(诸如为dna测序开发的那些反应复合物)的系统(请参见例如p.m.lundquist,et al.,optics letters 2008,33,1026,该文献出于所有目的以引用方式全文并入本文)。前述测序仪通常同时检测来自数千个零模波导(zmw)阵列的荧光信号,从而实现高度并行操作。每个zmw与其他zmw相隔几微米的距离,该距离代表一个独立的测序室。
[0058]
实时检测单个分子或分子复合物,例如在分析反应过程中,通常涉及直接或间接处理分析反应,使得待检测的每个分子或分子复合合物可单独解析。通过这种方式,可以单独监测每个分析反应,即使多个这样的反应被固定在单个底物上亦如此。可以通过多种机制实现分析反应的单独可解析配置,并且通常涉及在反应位点固定反应中的至少一种组分。提供这种单独可解析配置的各种方法是本领域已知的,例如,请参见balasubramanian等人的欧洲专利1105529;以及公开的国际专利申请wo 2007/041394,所有这些文献的全部内容出于所有目的以引用方式全文并入本文。底物上的反应位点通常是在该底物上执行和监测单个分析反应的位置,优选地实时进行。反应位点可以位于该底物的平面上,或位于该底物的表面上的孔中,例如,孔板、纳米孔或其他孔。在优选实施方案中,这种孔是指“纳米孔”,即纳米级孔或孔板,将相关分析材料的结构限制在纳米级直径(例如,约1-300nm)范围内。在一些实施方案中,这种孔包含光学限制特性,诸如零模波导,其也是纳米级孔,在本文其他部分有进一步描述。通常,这种孔的观察体积(即反应检测发生的体积)在阿升(10-18l)至仄升(10-21l)级,该体积适合于检测和分析单分子和单分子复合物。
[0059]
分析反应组分的固定可以各种方式进行设计。例如,酶(例如,聚合酶、逆转录酶、激酶等)可以附着在反应位点(例如,在光学限制结构或其他纳米级孔内)的底物上。在其他实施方案中,分析反应中的底物(例如,核酸模板,例如dna及其衍生物和模拟物,或激酶的靶分子)可以附着在反应位点的底物上。例如于2009年9月18日提交的美国专利申请12/562,690(现为美国专利8,481,264)中提供了固定模板的某些实施方案,该专利申请出于所有目的以引用方式全文并入本文。本领域技术人员将理解,将核酸和蛋白质固定到光学限制结构中的方法有很多,无论是共价的还是非共价的,都通过连接部分进行固定,或将它们系链在固定部分上。这些方法在固相合成和微阵列领域是众所周知的(beier et al.,nucleic acids res.27:1970-1-977(1999))。用于将核酸或聚合酶连接到固相载体的非限制性示例性结合部分包括链霉亲和素或亲和素/生物素键、氨基甲酸酯键、酯键、酰胺、硫醚、(n)-官能化硫脲、官能化马来酰亚胺、氨基、二硫化物、酰胺、腙键等。特异性结合一种或多种反应组分的抗体也可充当结合部分。此外,甲硅烷基部分可以使用本领域已知的方法直接连接到与底物(诸如玻璃)结合的核酸上。
[0060]
可以采用其他有用的处理步骤,利用纳米孔检测该基因组dna中甲基化腺嘌呤的位置。例如,该方法可以包括将一个或多个纳米孔测序接头或其亚区添加到该基因组dna的一个或多个末端。“纳米孔测序接头”是指一个或多个核酸结构域,其包括由相关纳米孔测序平台(诸如由oxford nanopore technologies提供的纳米孔测序平台,例如miniontm、gridionx5tm、promethiontm或smidgiontm纳米孔测序系统)应用的核酸序列(或其补充物)的至少一部分。相关纳米孔测序接头可以通过化学或酶连接或任何其他可用于将一个或多个核酸分子连接到双链核酸分子的一个或多个末端的方法进行添加。进行连接反应的合适试剂(例如,连接酶)和试剂盒是已知且可获取的,例如,可从new england biolabs(马萨诸塞州伊普斯威奇)获取的instant sticky-end ligase master mix。可采用的连接酶包括t4 dna连接酶(例如,低浓度或高浓度)、t4 dna连接酶、t7 dna连接、大肠杆菌dna连接酶、等。进行连接反应的适当条件将取决于所用连接酶的类型。
[0061]
在某些方面,可以使用单分子循环共有测序(ccs)生成精确的长读序列。在某些方面,ccs可能涉及使dna在拓扑上呈圆形,以及对该dna进行多次测序,以创建共有序列。在某
些方面,循环dna测序可能多达20次(如5-20次、5-15次、10-20次或10-15次)。
[0062]
在测序之前,可对该基因组dna进行处理以生成长片段,例如最长达100kb、最长达50kb、最长达40kb、最长达30kb、最长达20kb、最长达10kb、最长达5kb或最长达1kb。例如,测序片段的长度范围可以为1-100kb、1-50kb、1-40kb、1-30kb或1-20kb。
[0063]
在某些方面,在双链核酸分子链的连续延伸段中检测到甲基化腺嘌呤的位置,该连续延伸段长500碱基或更多、1千碱基(kb)或更多、2kb或更多、3kb或更多、4kb或更多、5kb或更多、6kb或更多、7kb或更多、8kb或更多、9kb或更多、10kb或多、15kb或更多、20kb或更多、25kb或更多、30kb或更多、35kb或更多、40kb或更多、45kb或更多、50kb或更多、55kb或更多、60kb或更多、65kb或更多、70kb或更多、75kb或更多、80kb或更多、85kb或更多、90kb或更多、95kb或更多,或者100kb或更多。
[0064]
可以采用计算方法(例如,以软件的形式)来检测单链和/或双链核酸分子中甲基化腺嘌呤的位置,根据检测到的甲基化腺嘌呤的位置确定该核酸分子中的蛋白结合区域,然后例如使用单分子实时测序和可选地ccs以及它们的任何组合对该核酸分子进行测序。
[0065]
如上所述,用于确定基因组核酸分子中结合区域的方法包含用于确定基因组dna中核小体位置的方法。此类方法利用来自所用甲基化酶(例如,n6-腺嘌呤dna甲基转移酶)的核小体相关基因组dna的保护性/不可及性,使得该核小体相关基因组dna中不发生甲基化。该方法包括检测基因组dna中甲基化腺嘌呤的位置,其标记该基因组dna中连接基因组dna的位置。该基因组dna中的核小体位置是根据甲基化腺嘌呤的缺失来确定的。该方法还可以揭示与基因组dna结合的某些转录因子的存在或缺失情况。
[0066]
本文所公开的方法可以在一个或多个正常细胞中进行,以生成所测序的基因组dna区域的染色质可及性图谱,其中该图谱指示不与蛋白质结合而因此使该a-mtase可及的染色质区域以及与蛋白质结合而因此使该a-mtase不可及的染色质区域。
[0067]
本文所公开的方法可以在一个或多个测试细胞上进行,以生成该测试细胞的基因组dna的染色质可及性图谱。该测试细胞可以来自受试者,诸如哺乳动物,例如人类患者。在某些情况下,该受试者可能患有疾病或可能疑似患有疾病。该疾病可能是癌症。
[0068]
本文所公开的方法可进一步包括将该测试细胞的染色质可及性图谱与正常细胞的染色质可及性图谱进行比较,其中该测试细胞与该正常细胞为相同细胞类型;以及比较该测试细胞和该正常细胞的基因组dna序列,其中染色质可及性图谱存在差异即表明该测试细胞中的染色质结构发生了变化,其中染色质可及性图谱没有差异但基因组dna序列存在差异表明序列差异与染色质结构的变化无关,并且其中染色质可及性图谱和基因组dna序列同时存在差异表明序列差异与染色质结构的变化有关。
[0069]
该方法进一步包括生成数据库,该数据库包含关于该染色质可及性图谱、潜在基因组dna序列以及与病症或疾病的相关性(如有)的信息。在某些方面,该正常细胞和该测试细胞可能是上皮细胞、白细胞、胶质细胞、成骨细胞或软骨细胞。在某些方面,该正常细胞和该测试细胞包含多个细胞。在某些方面,该多个细胞包含至少10个细胞、至少30个细胞、至少100个细胞、至少300个细胞或至少10,000个细胞。
[0070]
在某些方面,该染色质可及性图谱覆盖至少10%的染色质,例如,至少30%、至少50%或至少80%的染色质。在某些方面,该染色质可及性图谱覆盖该细胞的至少10%的基因组。在某些方面,该染色质可及性图谱覆盖该细胞的至少20%、至少30%、至少50%或至
少80%的基因组。在某些方面,与该基因组dna结合的蛋白质包括核小体、转录调控因子诸如转录抑制因子和转录激活因子,或这两者。
[0071]
本文还提供了使染色质区域可视化的方法,该染色质区域不与蛋白质结合,并且在空间上可作为细胞内腺嘌呤甲基转移酶(a-mtase)的底物。例如,在空间上可作为该a-mtase的底物的染色质区域可以是基因组dna中不与组蛋白和/或转录调控因子(例如,激活因子或抑制因子)结合的区域。该方法可以包括使细胞与a-mtase接触;以及检测该细胞中是否存在甲基化腺嘌呤。
[0072]
使用本文所公开的方法使细胞中甲基化腺嘌呤可视化,随后通过对完整细胞染色质中的甲基化腺嘌呤(m6a)进行选择性荧光标记,可以生成单细胞层面调控基因组的可视化图谱。该方法可用于以高通量方式可视化细胞。例如,可以通过所公开的方法对至少10个、100个、1,000个、10,000个、100,000个、100万个、300万个、1,000万个、3,000万个、1亿个或更多细胞进行分析。
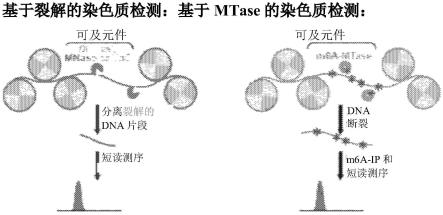
[0073]
该m6a成像法可进一步包括检测细胞内的dna和蛋白质靶标。例如,该方法可包括对细胞内其他dna和蛋白质靶标的ma进行多重检测。
[0074]
该方法可包括生成细胞调控状态的定量图像表示。该方法可进一步包括分析不同细胞和/或不同时间点同类型细胞的图像。
[0075]
该方法可包括根据含有或疑似含有病变细胞的组织样本生成存在于细胞中的甲基化腺嘌呤模式的定量图像,以及将该模式与正常细胞的代表模式进行比较。
[0076]
该方法可包括生成存在于接受诸如治疗药物等刺激的细胞中的甲基化腺嘌呤模式的定量图像,以及将该模式与在接受此类刺激之前的细胞模式进行比较。
[0077]
该细胞可以是任何相关细胞,诸如哺乳动物细胞、人细胞、t细胞、b细胞、病变细胞(例如,癌细胞)。该细胞可以是如本文所述的细胞。
[0078]
在某些方面,多个同类型细胞可以与a-mtase接触。例如,该多个细胞可以是上皮细胞、白细胞、胶质细胞、成骨细胞或软骨细胞。该细胞可来自单个个体,并且在一些实施方案中可来自单个组织,诸如胰腺、血液、皮肤、肠道等。
[0079]
该可视化方法可包括在检测之前执行点击化学以标记甲基化腺嘌呤。该点击化学可以向甲基化腺嘌呤添加荧光标记。该可视化方法可包括添加一个标记的甲基作为a-mtase的底物。该标记的甲基可以进行荧光标记。或者,可以标记a-mtase,例如,使用甲基转移酶的荧光团缀合版。
[0080]
在一些实施方案中,检测细胞中是否存在甲基化腺嘌呤包括使该细胞与特异性结合甲基化腺呤的抗体接触。该抗体可被检测标记。该检测标记可以是荧光团。该方法可进一步包括对细胞中的基因组dna进行染色。
[0081]
在一些实施方案中,该方法可进一步包括使细胞与荧光标记部分接触以靶向其他特定基因组区域或相关细胞蛋白质。
[0082]
在一些实施方案中,该方法可进一步包括使细胞与特异性结合rna聚合酶ii(pol ii)的抗体接触,例如pol ii ser5phos或pol ii ser2phos。
[0083]
在一些实施方案中,该方法可进一步包括测量甲基化腺嘌呤特异性信号的平均核强度和/或核点强度。在一些实施方案中,该甲基化腺嘌呤特异性信号可以是来自直接或间接与ma结合的荧光标记抗体的荧光信号。
[0084]
在一些实施方案中,a-mtase为m6a-mtase,例如hia5、ecogii、btr192iv或ecogi。在一些实施方案中,检测细胞中是否存在甲基化腺嘌呤包括检测m6a。
[0085]
在某些方面,基因组dna中ma的可视化可用于生成一种细胞类型的参考ma模式。例如,多种类型的人类细胞。可以将患病受试者的细胞中的ma模式与该细胞类型的参考ma模式进行比较,以测定任何差异。这些差异可能揭示先前未知的染色质结构和调控区域变化,这些变化可用于诊断、预判或治疗该受试者。该参考ma模式可包括附加信息,诸如存在或缺失某些转录调控因子、rna聚合酶等。
[0086]
在一些实施方案中,用于该方法的细胞群可以由任何数量的细胞组成,例如,约500-106个或更多细胞、约500-100,000个细胞、约500-50,000个细胞、约500-10,000个细胞、约50-1,000个细胞、约1-500个细胞、约1-100个细胞、约1-50个细胞或单个细胞。在一些实施方案中,细胞样本中包含的细胞少于约1000、2000、3000、4000、5000、6000、7000、8000、9000、10,000、15,000、20,000、25,000、30,000、40,000、50,000、60,000、70,000、80,000、90,000、100,000、120,000、140,000、160,000、180,000、200,000、250,000、300,000、350,000、400,000、450,000、500,000、600,000、700,000、800,000、900,000或1,000,000个。在一些实施方案中,细胞样本中包含的细胞多于约1000、2000、3000、4000、5000、6000、7000、8000、9000、10,000、15,000、20,000、25,000、30,000、40,000、50,000、60,000、70,000、80,000、90,000、100,000、120,000、140,000、160,000、180,000、200,000、250,000、300,000、350,000、400,000、450,000、500,000、600,000、700,000、800,000、900,000或1,000,000个。
[0087]
检测细胞中是否存在甲基化腺嘌呤(ma,例如m6a)可能涉及使与ma结合的抗体可视化。当使用直接或间接与抗体结合的荧光标记时,该可视化可以为荧光成像。可以利用超分辨率显微镜法使与ma结合的抗体可视化。该超分辨率显微镜法可以是一种确定性超分辨率显微镜法,其利用荧光团对激发的非线性响应来提高分辨率。示例性确定性超分辨方法可包括受激发射损耗(sted)、基态清空(gsd)、可逆饱和光学荧光跃迁(resolft)和/或饱和结构照明显微镜(ssim)。超分辨率显微镜法还可包括一种随机超分辨率显微镜法,该方法利用荧光团的复杂时间行为来提高分辨率。示例性随机超分辨率方法可包括超分辨率光学波动成像(sofi)、诸如光谱精确测定显微镜(spdm)等全单分子定位显微镜(smlm)、spdmphymod、光敏定位显微镜(palm)、荧光光敏定位显微镜(fpalm)、随机光学重建显微镜(storm),以及dstorm。
[0088]
该检测可包括生成甲基化腺嘌呤在细胞的基因组中的空间位置图谱。该检测可包括生成甲基化腺嘌呤在细胞的基因组中的时空位置图谱。
[0089]
该方法可包括使多个同类型细胞与a-mtase接触,以及生成甲基化腺嘌呤在这些细胞的基因组中的空间位置图谱。
[0090]
该方法可包括使至少两个不同时间点上的多个同类型细胞与a-mtase接触,以及生成甲基化腺嘌呤在这些细胞的基因组中的时空位置图谱。这两个不同时间点可包括第一时间点和第二时间点,其中该第一时间点和该第二时间点由对这些细胞施用疗法的时间点间隔。这些细胞可从受试者获得,并且对该受试者施用该疗法。
[0091]
通过所公开的方法可视化的细胞可以是活细胞或固定和透化细胞。系统
[0092]
本公开还提供了可用于例如实施标的方法的系统,包括执行本公开“方法”部分中
描述的任何一个或多个步骤。
[0093]
用于测定基因组dna中结合区域的系统包含用于测定基因组dna中核小体位置的系统。此类系统的指令使得系统对经腺嘌呤甲基化酶处理的基因组dna进行测序,并记录该基因组dna中甲基化腺嘌呤的位置。此类系统的指令可进一步使得系统根据该基因组dna中甲基化腺嘌呤的确定位置来评估基因组中某些区域的转录可及性。此类系统的指令可进一步使得系统评估基因启动子附近的不同核小体占位或定相。
[0094]
该系统可被调整(例如,包括指令)以对基因组dna的连续延伸段进行测序,该连续延伸段长500碱基或更多、1千碱基(kb)或更多、2kb或更多、3kb或更多、4kb或更多、5kb或更多、6kb或更多、7kb或更多、8kb或更多、9kb或更多、10kb或更多、15kb或更多、20kb或更多、25kb或更多、30kb或更多、35kb或更多、40kb或更多、45kb或更多、50kb或更多、55kb或更多、60kb或更多、65kb或更多、70kb或更多、75kb或更多、80kb或更多、85kb或更多、90kb或更多、95kb或更多,或者100kb或更多,并记录这些甲基化腺嘌呤的位置。
[0095]
在一些实施方案中,该系统包括测序装置,诸如可商购的测序仪,例如pacbio测序仪。
[0096]
本公开包括计算机可读介质,其中包含非暂态计算机可读介质,其上存储有用于本文所述方法或其部分的指令,并且可以是本公开的系统的一部分。本公开的各个方面包括其上存储有指令的计算机可读介质,该指令在被执行时使得系统执行如本文所述的方法的一个或多个步骤。
[0097]
在一些实施方案中,根据本文所述的方法和系统的指令可以“编程”形式被编码到计算机可读介质中,其中如本文中所使用的,术语“计算机可读介质”是指参与向计算机提供指令和/或数据以供执行和/或处理的任何存储或传输介质。存储介质示例包括软盘、硬盘、光盘、磁光盘、cd-rom、cd-r、磁带、非易失性存储卡、rom、dvd-rom、蓝光光盘、固态硬盘和网络附接存储器(nas),无论此类装置在计算机内部还是外部。包含信息的文件可以“存储”在计算机可读介质上,其中“存储”意味着记录信息,使得日后可通过计算机进行访问和检索。
[0098]
本公开的任何方法步骤或由本公开的系统执行的方法步骤可以通过编程来执行,其中编程可以通过任意数量的计算机编程语言中的一种或多种来编写。此类语言包括例如java(sun microsystems,inc.,加利福尼亚州圣塔克拉拉)、visual basic(microsoft corp.,华盛顿州雷德蒙德)和c (at&t corp.,新泽西州贝德明斯特),以及其他许多语言。试剂盒
[0099]
本公开还提供了试剂盒。该试剂盒包括一种或多种可用于实施本公开的方法的试剂。在某些方面,该试剂盒包括任何可用于实施本公开的方法的试剂、装置、指令(例如,位于一个或多个非暂态计算机可读介质上)等,包括在本公开上文“方法”和“系统”部分中描述的任何试剂、装置、指令等。
[0100]
在一些实施方案中,提供的试剂盒包括使基因组dna中的腺嘌呤甲基化的腺嘌呤甲基化酶(例如,n6-腺嘌呤dna甲基转移酶),从而标记该基因组dna中未结合区域(例如,未与蛋白质结合)的位置,还包括使用该甲基化酶的方法指令,其通过单分子测序检测该基因组dna中甲基化腺嘌呤的位置,从而测定该基因组dna中的结合区域。
[0101]
该试剂盒的组件可以分别放在单独容器中,或者多个组件可以放在单个容器中。
合适容器包括单管(例如,小瓶)、单孔或多孔板(例如,96孔板、384孔板等)等。
[0102]
该试剂盒包括例如使用腺嘌呤甲基转移酶的方法指令,其通过测序仪检测甲基化腺嘌呤的位置来测定基因组dna中的结合区域。在一些实施方案中,该试剂盒包括使用腺嘌呤甲基转移酶的方法指令,其根据甲基化腺嘌呤的位置测定基因组dna中的核小体位置。
[0103]
该指令可以记录在合适的记录媒质上。例如,该指令可以打印在诸如纸或塑料等基材上。因此,该指令可以作为包装说明书放入试剂盒中,或标示在试剂盒容器或其组件(即包装或分包装)上等。在其他实施方案中,该指令作为电子存储数据文件呈现在合适的计算机可读存储介质上,例如便携式闪存驱动器、dvd、cd-rom、软盘等。在另一些实施方案中,实际指令不在试剂盒中,而是提供用于从远程源(例如,通过互联网)获得该指令的方式。该实施方案的一个示例是一种试剂盒,其包括可以查看指令和/或从中下载指令的网址。与该指令一样,用于获得该指令的方式被记录在合适的基材上。实用性
[0104]
本文公开的方法、试剂盒和系统可用于生成数据库,该数据库包含关于染色质结构、基因组dna序列以及染色质结构与存在或缺失特定病症或疾病的相关性的信息。该信息可用于疾病诊断和/或预后。
[0105]
在某些方面,该数据库可包含与治疗前相比,治疗后染色质结构变化的相关信息。在某些方面,这些信息可用于监测治疗的效果,并在需要时对该治疗作出调整。该治疗可以是免疫治疗,例如抗体治疗或小分子治疗。该治疗可以是针对癌症的。
[0106]
尽管有所附权利要求书,本公开也由下列编号的条款定义:1.一种用于鉴定基因组dna中与蛋白质结合的区域的方法,所述方法包括:使所述基因组dna与腺嘌呤甲基转移酶(a-mtase)接触,其中所述a-mtase使得所述基因组dna中不与所述蛋白质结合的区域中的腺嘌呤残基甲基化;对所接触的基因组dna进行单分子长读测序,以检测所述基因组dna中缺少甲基化腺嘌呤残基的位置,进而鉴定所述基因组dna中与所述蛋白质结合的所述区域。2.根据条款1所述的方法,其中所述a-mtase为n6-腺嘌呤甲基转移酶(m6a-mtase)。3.根据条款2所述的方法,其中所述m6a-mtase为hia5。4.根据条款2所述的方法,其中所述m6a-mtase为ecogii。5.根据条款2所述的方法,其中所述m6a-mtase为btr192iv。6.根据条款2所述的方法,其中所述m6a-mtase为ecogi。7.根据条款1-6中任一项所述的方法,其中所述接触包括使分离的基因组dna与所述a-mtase接触。8.根据条款1-6中任一项所述的方法,其中所述接触包括与含有所述基因组dna的细胞接触。9.根据条款8所述的方法,其中所述接触包括将对所述a-mtase编码的核酸引入所述细胞。10.根据条款9所述的方法,其中所述a-mtase与细胞穿透肽融合,所述细胞穿透肽使所述a-mtase的质膜可渗透。11.根据条款1-10中任一项所述的方法,其中所述测序在所述基因组dna的至少1
千碱基(kb)长延伸段上进行。12.根据条款1-10中任一项所述的方法,其中所述测序在所述基因组dna的至少3kb长延伸段上进行。13.根据条款1-12中任一项所述的方法,其中所述测序包括通过纳米孔使所述基因组dna易位。14.根据条款1-13中任一项所述的方法,其中所述测序包括将一个或多个纳米孔测序接头连接到所述基因组dna的一个或多个末端。15.根据条款1-14中任一项所述的方法,其中所述测序包括检测指示甲基化腺嘌呤的信号。16.根据条款15所述的方法,其中所述信号为电信号。17.根据条款1-16中任一项所述的方法,其中所述测序包括多轮再测序。18.根据条款17所述的方法,其中所述多轮再测序包括最多20轮测序。19.根据条款1-18中任一项所述的方法,其中所述测序包括循环共有测序。20.根据条款1-19中任一项所述的方法,其中所述测序包括单分子实时(smrt)循环共有测序(ccs)。21.根据条款1-20中任一项所述的方法,其中所述基因组dna来自哺乳动物细胞。22.根据条款1-21中任一项所述的方法,其中所述基因组dna来自癌细胞。23.根据条款1-22中任一项所述的方法,其中所述细胞为正常细胞,所述方法进一步包括生成所测序的基因组dna区域的染色质可及性图谱,其中所述图谱指示不与所述蛋白质结合而因此使所述a-mtase可及的染色质区域以及与所述蛋白质结合而因此使所述a-mtase不可及的染色质区域。24.根据条款1-23中任一项所述的方法,所述方法进一步包括生成测试细胞的基因组dna的染色质可及性图谱。25.根据条款24所述的方法,其中所述测试细胞为来自受试者的细胞。26.根据条款25所述的方法,其中所述受试者患有或疑似患有疾病。27.根据条款26所述的方法,其中所述疾病为癌症。28.根据条款24-27中任一项所述的方法,所述方法包括将所述测试细胞的所述染色质可及性图谱与所述正常细胞的所述染色质可及性图谱进行比较,其中所述测试细胞与所述正常细胞为相同细胞类型;以及比较所述测试细胞和所述正常细胞的基因组dna序列,其中所述染色质可及性图谱存在差异即表明所述测试细胞中的染色质结构发生了变化,其中所述染色质可及性图谱没有差异但所述基因组dna序列存在差异表明序列差异与染色质结构的变化无关,并且其中所述染色质可及性图谱和所述基因组dna序列同时存在差异表明序列差异与染色质结构的变化有关。29.根据条款28所述的方法,所述方法进一步包括生成数据库,所述数据库包含关于所述染色质可及性图谱、潜在基因组dna序列以及与病症或疾病的相关性(如有)的信息。30.根据条款24-29中任一项所述的方法,其中所述正常细胞和所述测试细胞为上皮细胞、白血细胞、胶质细胞、成骨细胞或软骨细胞。31.根据条款24-30中任一项所述的方法,其中所述正常细胞和所述测试细胞包含多个细胞。
32.根据条款31所述的方法,其中所述多个细胞包含至少10个细胞、至少30个细胞、至少100个细胞、至少300个细胞或至少10,000个细胞。33.根据条款24-32中任一项所述的方法,其中所述染色质可及性图谱覆盖至少10%的染色质。34.根据条款24-33中任一项所述的方法,其中所述染色质可及性图谱覆盖至少30%、至少50%或至少80%的染色质。35.根据条款24-34中任一项所述的方法,其中所述染色质可及性图谱覆盖所述细胞的至少10%的基因组。36.根据条款24-35中任一项所述的方法,其中所述染色质可及性图谱覆盖所述细胞的至少20%、至少30%、至少50%或至少80%的基因组。37.根据条款1-36中任一项所述的方法,其中所述蛋白质包含核小体。38.根据条款1-36中任一项所述的方法,其中所述蛋白质包含转录调控因子。39.根据条款38所述的方法,其中所述转录调控因子是一种转录抑制因子。40.根据条款38所述的方法,其中所述转录调控因子是一种转录激活因子。41.一种试剂盒,所述试剂盒包括:腺嘌呤甲基转移酶(a-mtase);测序接头;和使基因组dna与所述a-mtase接触的指令,其中所述a-mtase使得所述基因组dna中不与蛋白质结合的区域中的腺嘌呤残基甲基化,从而将所述测序接头连接到所述基因组dna,并且对所接触的基因组dna进行单分子长读测序,以检测所述基因组dna中缺少甲基化腺嘌呤残基的位置,进而鉴定所述基因组dna中与所述蛋白质结合的区域。42.根据条款41所述的试剂盒,其中所述a-mtase为n6-腺嘌呤甲基转移酶(m6a-mtase)。43.根据条款42所述的试剂盒,其中所述m6a-mtase为hia5。44.根据条款42所述的试剂盒,其中所述m6a-mtase为ecogii。45.根据条款42所述的试剂盒,其中所述m6a-mtase为btr192iv。46.根据条款42所述的试剂盒,其中所述m6a-mtase为ecogi。47.根据条款41-46中任一项所述的试剂盒,其中所述接触包括使分离的基因组dna与所述a-mtase接触。48.根据条款41-46中任一项所述的试剂盒,其中所述接触包括与含有所述基因组dna的细胞接触。49.根据条款41-46中任一项所述的试剂盒,其中所述a-mtase包含融合到其n端或c端的细胞穿透肽,并且其中所述a-mtase的质膜可渗透。50.根据条款41-46中任一项所述的试剂盒,其中所述接触包括将对所述a-mtase编码的核酸引入所述细胞。51.根据条款41-50中任一项所述的试剂盒,其中所述测序在所述基因组dna的至少1千碱基(kb)长延伸段上进行。52.根据条款41-50中任一项所述的试剂盒,其中所述测序在所述基因组dna的至少3kb长延伸段上进行。
53.根据条款41-52中任一项所述的试剂盒,其中所述基因组dna来自癌细胞。54.一种使染色质区域可视化的方法,所述染色质区域不与蛋白质结合,并且在空间上可作为细胞内腺嘌呤甲基转移酶(a-mtase)的底物,所述方法包括:55.使所述细胞与所述a-mtase接触;以及56.检测所述细胞中是否存在甲基化腺嘌呤。57.根据条款54所述的方法,其中所述检测所述细胞中是否存在甲基化腺嘌呤包括使所述细胞与特异性结合甲基化腺呤的抗体接触。58.根据条款54所述的方法,其中所述抗体可被检测标记。59.根据条款56所述的方法,其中所述可检测标记包括荧光团。60.根据条款54-57中任一项所述的方法,其中所述方法进一步包括对所述细胞中的基因组dna进行染色。61.根据条款54-58中任一项所述的方法,其中所述方法进一步包括使所述细胞与特异性结合rna聚合酶ii(pol ii)的抗体接触。62.根据条款59所述的方法,其中所述抗体与pol ii ser5phos或pol ii ser2phos特异性结合。63.根据条款54-60中任一项所述的方法,包括测量所述甲基化腺嘌呤的特异性信号的平均核强度和/或核点强度。64.根据条款54-61中任一项所述的方法,其中所述a-mtase为n6-腺嘌呤甲基转移酶(m6a-mtase)。65.根据条款62所述的方法,其中所述m6a-mtase为hia5、ecogii、btr192iv或ecogi。66.根据条款62或63所述的方法,其中所述检测所述细胞中是否存在甲基化腺嘌呤包括检测m6a。
实施例
[0107]
从上文公开的内容可以理解,本公开具有广泛应用。因此,列出以下实施例,以向本领域普通技术人员提供有关如何制作和使用本发明的完整公开和描述,而非旨在限制发明人的发明范围,也非旨在表示以下实验是所执行的全部或唯一实验。本领域技术人员将能够轻松识别各种非临界参数,可以更改或修改这些非临界参数以产生基本相似的结果。因此,列出以下实施例,以向本领域普通技术人员提供有关如何制作和使用本发明的完整公开和描述,而非旨在限制发明人的发明范围,也非旨在表示以下实验是所执行的全部或唯一实验。已努力确保所用数字(例如,数量、尺寸等)的准确性,但应考虑一些实验误差和偏差。材料和方法
[0108]
分离和克隆m6a-mtase。phia5et和phinet由monika radlinska友情提供(m.drozdz,et al.,nucleic acids res.40,2119
–
2130(2012))。通过idt将密码子优化版btr192iv(genbank:cp003745)和ecogi(genbank:afst01000004)合成为gblocks,并利用ndei和xhoi限制位点克隆到上述pet载体上,分别生成pbtr192ivet和pecogiet载体。在5-αf’iq感受态大肠杆菌细胞(neb c2992h)中进行克隆。
[0109]
生成mtase蛋白。将phia5et、phinet、pbtr192ivet和pecogiet载体转化到t7 express lysy感受态大肠杆菌细胞(neb c3010i)中进行重组蛋白表达。将过夜培养物添加到补充有100μg ml-1氨苄青霉素的两个1l lb培养基中,并在37℃下振荡培育,直至od600达到0.8-1.0。以1mm的最终浓度加入异丙基-β-d-1-硫代半乳糖苷(iptg),并在20℃下振荡培育4小时。在4℃温度下以5000xg的速度使细胞成团10分钟(所有后续步骤均在4℃温度下进行),并在35ml裂解缓冲液(50mm hepes,ph 7.5;300mm nacl;10%的甘油;0.5%的triton x-100;10mmβ-巯基乙醇)中进行重悬,该缓冲液中补充有2x完全无edta的蛋白酶抑制剂混合液(roche 11873580001)。通过超声探针(qsonica q125)在冰上以50%振幅对细胞裂解10分钟,开/关30秒(历时20分钟),然后在40,000xg的速度下离心1小时。通过用30ml平衡缓冲液(50mm hepes,ph 7.5;300mm氯化钠;20mm咪唑)冲洗5ml浆液制备ni-nta琼脂糖(qiagen 30210),并在500xg的速度下离心3分钟,将此步骤重复一次。将澄清的细胞裂解液和琼脂糖混合,并在4℃下旋转1小时,然后倒入色谱柱内。用20ml缓冲液1(50mm hepes,ph 7.5;300mm氯化钠;50mm咪唑)和15ml缓冲液2(50mm hpes,ph 7.5;300mm氯化钠;70mm咪唑)冲洗色谱柱,然后加入15ml洗脱缓冲液(50mm hepes,ph 7.5;300mm氯化钠;250mm咪唑)。将洗脱液加入10k amicon ultra-15超滤管中,在3,220xg的速度下以15分钟增量离心处理后,用15ml蛋白质重悬缓冲液(50mm tris,ph 7.5;50mm氯化钾;1mm dtt;10mm edta;2x完全无edta的蛋白酶抑制剂混合液)交换eb缓冲液。几次15分钟旋转后,体积降至500μl以下,将液体转移至1.5ml eppendorf lobind管中。用过滤灭菌的牛血清白蛋白(bsa)溶液和30%甘油将蛋白质补充至200μg/ml,储存在-20℃温度下。
[0110]
体外mtase活性评估。通过用含有4个gatc序列的羟甲基胆色烷合成酶(hmbs)基因启动子中的759碱基对区域的引物对k562基因组dna进行pcr,制备底物dna。根据制造商说明,用monarch pcr&dna清理试剂盒(neb t1030s)纯化pcr片段。使用1μg底物dna和在补充有0.8mm s-腺苷-甲硫氨酸(neb b9003s)的缓冲液a(15mm tris,ph 8.0;15mm氯化钠;60mm氯化钾;1mm edta,ph 8.0;0.5mm egta,ph 8.0;0.5mm亚精胺)中交替进行两倍和五倍酶稀释(10、5、1、0.5、0.1、0.05、0.01、0.005、0.001、0.0005和0.0001μl mtase),制备11个60μl mtase反应物。制备无mtase的阴性对照物。通过轻敲pcr排管和快速旋转使反应物混合,然后在37℃下孵育1小时。用monarch pcr&dna清理试剂盒终止每个反应,并在20μl eb缓冲液中洗脱纯化dna。通过将15μl的每个纯化dna样本与1μl dpni(neb r0176s)和4μl 10x cutsmart缓冲液(neb)在40μl反应物中进行混合,制备12种限制性酶消化物。通过轻敲小心混合反应物,并在37℃下孵育1.5小时。将1μl的每种反应物与2μl 6x紫色凝胶上样染料(neb b7024s)和11μl h2o混合,并在含有1x gelgreen核酸染剂(biotium 41005)的1.2%琼脂糖凝胶上在130v下运行约1.5小时。在ge typhoon fla 9500激光扫描仪上对凝胶进行成像。mtase活性通过最高mtase稀释度测定,该稀释度使1μg dna底物甲基化,导致dpni消化后dna分子不完整。
[0111]
mtase-seq。在室温下将果蝇s2细胞培育在补充有10% hi fbs(gibco 16140-063)和1%青链霉素(gibco 15140-122)的1x schneider’s drosophila培养基(gibco21720-024)中,在75cm2烧瓶中进行,以达到约90%的融合率和95%以上的存活率。用pbs冲洗细胞,然后在pbs中重悬,并在countess全自动细胞计数仪上进行计数。以250xg的速度使每个样本中的300万个果蝇s2细胞成团5分钟。将细胞团块重悬在相当于每个样本
60μl的缓冲液a中,并等分到pcr排管中。向排管中加入60μl冷2x裂解缓冲液(缓冲液a中加入0.1% igepal ca-630),轻敲排管以进行混合,然后在冰上静置10分钟。在4℃下以350xg的速度使样本成团5分钟,并移除上清液。如前所述分离出k562和hela细胞核(r.e.thurman,et al.,nature.489,75-82(2012))。用大口径移液管吸头将核团块分别轻轻重悬在57.5μl缓冲液a中,并转移至37℃热循环仪中。加入1μl稀释于缓冲液a中的mtase和1.5μl sam(最终0.8mm),然后通过用大口径吸头和多通道移液管上下移液10次,仔细混合。将反应物孵育20分钟,然后用3μl 20% sds(最终1%)进行终止,并转移到新的1.5ml微量离心管中。通过添加130μl缓冲液a和额外7μl 20% sds来增加样本体积。将所有样本与2μl rnase a(invitrogen am2271)混合,并在37℃下孵育1小时,然后与2μl蛋白酶k(neb p8107s)混合,在55℃下再孵育一小时。加入200μl(1:1)的苯酚、氯仿和异戊醇(比例为25:24:1)(用10mm tris饱和,ph 8.0,1mm edta)以纯化dna,然后剧烈颠倒混匀并在室温下孵育10分钟。在17,900xg的速度下将提取物离心10分钟,然后将上层水相转移到新的微量离心管中。向所有样本中加入200μl氯仿和异戊醇(比例为24:1)并重复提取程序,在第二次提取时去除残留苯酚。将水相转移到新的微量离心管中,通过添加0.1体积的3m乙酸钠、1μl glycoblue沉淀剂(invitrogen am9515)和2.5体积的100%冰冷乙醇对dna进行沉淀。将所有样本颠倒混匀数次,然后快速旋转并在-20℃下储存过夜。通过在4℃下以20,000xg的速度离心10分钟使dna成团,然后用1ml冰冷的70%乙醇重复离心以进行冲洗。将管倒置在管架上并风干15分钟,然后在54μl的10mm tris(ph 7.5)中进行重悬。
[0112]
将所有样本转移至microtube-50afa纤维螺旋帽超声管(covaris 520166),并在covaris m220聚焦超声波破碎仪(峰值功率:75.0;占空比:15.0%;循环/脉冲:200;持续时间:720秒;水浴温度:20℃)上单独进行超声波破碎,以获得约100个碱基对的片段长度。然后在含有1x gelgreen核酸染剂的1.2%琼脂糖凝胶上运行样本,并用qiaquick凝胶提取试剂盒(28704)纯化切下的条带。然后合并2个重复样本为文库构建提供充足的输入,并通过qubit ds dna hs检测试剂盒(invitrogen,q32851)进行定量。所有文库均使用用于illumina的nebnext ultra ii dna文库制备试剂盒(neb e7645s)和用于illumina索引引物集1和2的nebnext multiplex oligos(neb e7335s&e7500s)进行制备。将1μg凝胶提取的dna在50μl缓冲液eb(10mm tris-cl,ph 8.5)中与7μl末端修复反应缓冲液和3μl末端修复酶混合液在pcr排管中进行混合,以对dna进行末端修复。将样本混合,然后置于热循环仪上,运行末端修复程序(在20℃下放置30分钟,在65℃下放置30分钟,然后保持在4℃)。为终止修复样本,添加2.5μl用于illumina的接头,再添加30μl ligation master mix连接反应液,然后添加1μl likation enhancer连接反应增强剂。在20℃下孵育样本15分钟,随后添加3μl user酶,接着在37℃下孵育样本15分钟。将116μl(1.2x)agencourt ampure xp磁珠(beckman coulter a63880)添加到每个文库中,然后在室温下孵育10分钟并磁分离10分钟。丢弃上清液,用200μl新鲜的80%乙醇在磁铁上洗涤磁珠两次,然后风干4分钟。从磁铁上取下样本,向每个样本中加入50μl的10mm tris(ph 7.5),然后在室温下孵育10分钟。磁分离10分钟后,将上清液转移到新的lobind管中。
[0113]
将20μl的100μm封闭寡核苷酸(agatcggaagcgtc(seq id no:5))添加到每个文库中,并在95℃下孵育样本10分钟以使dna变性,然后转移到冰/水混合冰沙中快速冷却10分钟。向每个样本中添加325μl的0.1x te缓冲液、100μl的5x ip缓冲液(50mm tris,ph 7.5;
750mm氯化钠;0.5% igepal ca-630)和5μl(5μg)抗-n6-甲基腺苷抗体(millipore sigma abe572)。在4℃下在旋转器中孵育样本12小时。在磁分离后除去上清液并在1x ip缓冲液(10mm tris,ph 7.5;150mm氯化钠;0.1% igepal ca-630)中冲洗4次,然后在25μl 1x ip缓冲剂中重悬,为每个样本制备25μl的蛋白a免疫磁珠(invitrogen 10001d)。将25μl制备的蛋白a磁珠添加到每个样本中,然后在4℃下旋转培养4小时。用750μl 1x ip缓冲液冲洗样本六次,每次冲洗过程中在4℃下旋转并在洗涤缓冲液中孵育5分钟。向磁珠中加入48μl蛋白酶k消化缓冲液(20mm hepes,ph 7.5;1mm edta;0.5% sds)和2μl蛋白酶k,并在50℃下以1200rpm转速振荡孵育1小时,进而对dna进行洗脱。然后使用磁铁分离样本,并将上清液转移到新管中。按照上述相同程序,将90μl(1.8x)ampure xp磁珠添加到每个样本中,但在17μl的10mm tris(ph 7.5)中进行洗脱,然后用qubit ss dna检测试剂盒(invitrogen,q10212)进行定量。通过向每个样本中添加5μl通用pcr引物、5μl索引引物(均在illum ina索引引物集1和2的nebnext multiplex oligos中提供)和25μl nebnext ultra ii q5 master mix来扩增文库,然后进行pcr扩增(程序:在98℃下扩增30秒;在98℃下循环5-7次,持续10秒,然后在65℃下扩增75秒;最后在65℃下孵育5分钟)。按照上述相同程序,将60μl(1.2x)ampure xp磁珠添加到每个样本中,但在33μl的10mm tris(ph 7.5)中进行洗脱。使用qubit ds dna hs检测试剂盒对文库进行定量,并在agilent生物分析仪高灵敏度dna芯片上检查粒度分布。使用illumina hiseq 4000对文库进行测序,使用配对端76bp读取长度,读取深度为1,000-3,000万次。
[0114]
dna sei-seq。按上述方法培养果蝇s2细胞,并按上述方法分离细胞核。在补充有ca2
的缓冲液a中,在37℃下用极限浓度的dna sei(sigma)培育细胞核3分钟。用终止缓冲液(50mm tris-hcl、100mm氯化钠、0.1% sds、100mm edta、1mm亚精胺、0.5精胺,ph 8.0)终止消化反应,并用蛋白酶k和rnase a处理样本。使用ampure xp磁珠恢复小的“双命中”片段(《750bp),并使用illumina文库试剂盒如前所述制备样本(s.john,et al.,current protocols in molecular biology(john wiley&sons,inc.,hoboken,nj,usa,2013;vol.chapter 27,pp.21.27.1-21.27.20))。先前发表的k562和hela dna sei-seq数据集用于分析(r.e.thurman,et al.,2012(见上文))。
[0115]
mtase-seq和dna sei-seq分析。如前所述,读数被映射到dm6基因组(j.vierstra,et al.,science(80).346,1007-1012(2014))。使用bedops生成信号轨迹(s.neph,et al.,bioinformatics.28,1919
–
20(2012)),并将信号归一化为100万次读数。使用热点算法(s.john,et al.nat.genet.43,264
–
268(2011))以fdr 5%截止值识别染色质可及性区域(热点)。对于使用s2细胞dna sei数据量化的每个热点,我们对该热点中包含的归一化读取的总数进行了量化,以识别该元件的信号强度。使用每个mtase-seq库重复执行上述程序。当对文库进行相互比较时,我们使用s2 dna sei-seq热点调用作为基因组调控元件列表,并如上所述在不同文库中对这些区域内的信号强度进行量化。启动子近端元件被定义为ncbi refseq curated基因列表中注释的转录起始位点 /-500bp内的元件。
[0116]
m6a斑点杂交。如上所述分离用不同浓度mtase处理的果蝇s2细胞核的dna,并通过nanodrop对样本进行定量。在96孔板中用20x ssc缓冲液稀释这些dna样本,然后在95℃下变性10分钟。在20x sss缓冲液中润湿硝化纤维素膜,然后将其固定在hybri-dot多管吸印仪(life technologies)中。将膜固定在多管吸印仪中后,使多管吸印仪处于真空状态,向
所有孔板中加入150μl的20x ssc缓冲液,然后加入变性的dna样本。在除去所有气泡后,终止真空状态,将膜面朝上放置在干燥的whatman滤纸上,并在gs gene linker uv chamber紫外交联仪(bio-rad)中使用c-l设置与125mjoule交联。然后用20ml的1x tbs-t(10mm tris,ph 7.5;0.25mm edta;150mm氯化钠;0.1%tween-20)冲洗膜,并在室温下用15ml 1x tbs-t 5%脱脂奶粉封闭1小时。将兔多克隆抗-n6-甲基腺苷抗体(millipore sigma abe572)以1:1000稀释在10ml 1x tbs-t 5%脱脂奶粉中,并于4℃下在缓震器上与印迹孵育过夜。在20ml 1x tbs-t中冲洗印迹3次,共15分钟。将抗兔igg、hrp连接的二级抗体(cell signaling technology 7074)以1:1000稀释在10ml 1x tbs-t 5%脱脂奶粉中,并在室温下与印迹孵育1小时。重复冲洗3次,用pierce ecl plus western印迹底物(thermo scientific 32132)显影印迹,并用胶片成像。
[0117]
fiber-seq。如上所述分离250万果蝇s2细胞或30万人k562细胞的细胞核,除使用20个单位(1μl)的hia5外,其余mtase反应均按上述步骤进行。还产生了未经处理的果蝇s2复制品用于比较。如上所述纯化dna,然后转移至microtube-50 afa纤维螺旋帽超声管(covaris 520166),并在covaris m220聚焦超声波破碎仪(峰值功率:75.0;占空比:5.0%;循环/脉冲:200;持续时间:8秒;水浴温度:20℃)上单独进行超声波破碎,以获得约1.5kb的片段长度。使用测序试剂盒3.0(pacific biosciences,美国加利福尼亚州门洛帕克),利用剪切后的样本生成pacific biosystems测序文库。使用bluepippin(sage science,美国马萨诸塞州贝弗利)去除小文库片段,并将每个文库加载到单个smrt细胞上。
[0118]
fiber-seq m6a甲基化识别。使用pbalign将读数映射到dm6基因组,并使用bamseive提取文库中每个zmw的子读数,以生成per-zmw bam文件。然后,使用-identify m6a通过ipdsummary处理这些per-zmw bam文件。对于果蝇s2分析,使用p值截止值0.001进行甲基化识别,并丢弃含有14个或更少子读数的读数。为了在人类样本中获得更长的读数,在k562分析中使用p值截止值0.02(即phred得分》16)进行甲基化识别,并丢弃含有10个以下子读数的读数。使用ncbi refseq curated基因列表注释每次读取中的启动子位置,并使用先前发布的rrna-seq数据(43)记录每个基因的表达。使用上述热点调用对dna sei超敏元件(dhs)进行标定,并使用repeatmasker定义重复。除非另有说明,否则从分析中去除线粒体读数和与重复元件重叠大于25%的读数,k562数据例外,在此情况下去除与重复元件重叠大于50%的读数。
[0119]
甲基化酶可及dna序列(mad)鉴定。将以上识别的每次读取的m6a甲基化事件集合,以识别mtase敏感区。具体地,对于每个m6a,将距离50bp内的所有m6a事件连接在一起,以调用更大的甲基化酶可及dna序列(mad)。mtase保护位点(mps)被定义为每次读取过程中没有包含在mad中的区域。识别与dhs重叠的所有mad,然后捕获最宽的mad,从而识别与dhs重叠的mad。该最宽mad用于识别dhs在单独读取时是封闭状态还是开放/可及状态。在比较重叠读数时,对每个重叠读数重复上述过程,并计算每个dhs的每个读数的mad大小中位数差异。
[0120]
量化核小体定相/定位。每个核小体的位置被定义为宽度在65-200个碱基之间的每个mps的中心。所有与该位置重叠的读数都被识别出来,并且每个核小体在这些重叠读数上的位置也被类似定义。为了计算核小体定相,确定一个读数上的核小体位置与每个重叠读数上最近的核小体位置之间的距离。如果存在多个重叠读数,则使用中位距离。首先识别与dhs重叠的tss,然后识别与这些dhs重叠的最大mad,进而相对于tss定位核小体。然后根
据基因的阅读框确定mps相对于该mad的位置。从该分析中删除与2个或更多tss重叠的读数。用偏移0-39bp的核小体的数量除以偏移40-79bp的核小体的数量,然后对所得数进行log2转换,即计算得出其中每个位置的核小体的相对定相。实施例1:非特异性m6a-mt
ase
选择性标记染色质可及性位点
[0121]
染色质的初级结构包括核小体阵列,该阵列被含有转录因子和其他非组蛋白的短调控区域截断。这种结构是基因组功能的基础,但在单根染色质纤维(基因调控的基本单位)层面尚不明确。例如,尽管核小体是限制转录因子接近dna的主要屏障,但沿着单根染色质纤维排列的核小体在体内的定位和占位尚未阐明。因此,目前尚不清楚以下方面:核小体如何沿着相同的延伸染色质模板精确排列;单根染色质纤维上可及的调控dna与核小体之间的相互作用;给定dna编码的调控区域在细胞群内不同染色质纤维上被驱动的程度;以及在相同染色质模板上,附近的调控区域被协调驱动的程度。要解决这些问题,需要对单根染色质纤维进行测序,这是目前单细胞或批量分析法所无法实现的。
[0122]
开发了一种以单核苷酸分辨率将染色质的初级结构记录到其基础dna模板上的方法,从而能够同时鉴定基因组中多千碱基片段的遗传特征和表观遗传特征。目前绘制染色质和调控结构的方法取样了大量染色质纤维,并依赖于使用诸如dna se i(d.s.gross,w.t.garrard,annu.rev.biochem.57,159
–
97(1988);r.e.thurman,et al.,2012(见上文))、微球菌核酸酶(m.noll,r.d.kornberg,j.mol.biol.109,393
–
404(1977);d.e.schones,et al.,cell.132,887
–
898(2008))、限制性内切酶(e.lieberman-aiden,et al.,science(80).326,289
–
293(2009))、转座酶(j.d.buenrostro,et al.,nat.methods.10,1213
–
1218(2013))或机械剪切(t.kouzarides,cell.128,693-705(2007))等核酸酶溶解染色质。cpg和gpc甲基转移酶能够在不消化dna的情况下在二核苷酸环境中标记可及胞嘧啶(t.k.kelly,et al.,genome res.22,2497
–
2506(2012);a.r.krebs,et al.,mol.cell.67,411-422.e4(2017))。然而,由于动物基因组中cpg和gpc二核苷酸的散发出现、突变和选择的线性聚类以及内源性胞嘧啶甲基化机制的混杂影响,平均分辨率较低(a.p.bird,nature.321,209-13(1986))。
[0123]
与胞嘧啶不同,dna中的腺嘌呤碱基在真核生物中几乎完全没有内源性甲基化(q.xie,et al.,cell.175,1228-1243.e20(2018)),并且在动物基因组中以接近每2个dna碱基对中1个的平均频率发生,而未出现胞嘧啶-鸟嘌呤二核苷酸聚集和扩展沙漠特征。因此,寻求具有高效、高稳定性且分子量类似于诸如dna sei(约30kd)等非特异性核酸酶的非特异性(即,非序列上下文相关)n
6-腺嘌呤dna甲基转移酶(m6a-mtase),其能够在核苷酸分辨率下接近蛋白质-dna界面(图1a)。分离出五种不同的非特异性dna m6a-mtase(m.drozdz,et al.,nucleic acids res.40,2119
–
2130(2012);b.p.anton,et al.,plos one.11,e0161499(2016);i.a.murray,et al.,nucleic acids res.46,840
–
848(2018);g.fang,et al.,nat.biotechnol.30,1232
–
1239(2012)),并证明了随着每种酶量的增加,非染色质化和染色质化dna模板的处理(图1b)导致腺嘌呤甲基化的单调增加,与单命中动力学相容。
[0124]
为了确定m6a-mtase对核染色质内可及dna模板的选择性,对用dna se i处理果蝇s2细胞核后dna se i裂解的分布(标记可及dna模板的既定标准(d.s.gross,w.t.garrard,1988(见上文);r.e.thurman,et al.,2012(见上文))和s2核暴露于五种增加浓度的腺嘌呤
甲基转移酶后m6a-dna的分布进行了比较(图1c)。从提取的基因组dna(mtase-seq)中对含有m6a的短(中位数110bp)dna片段进行免疫沉淀和大规模平行测序,显示了m6a-dna的基因组分布,这反映了以dna se-seq定量的dna se i裂解密度(图1d)。m6a-mtase对可及dna显示出高选择性,并对dna se i超敏位点(dhs)进行定量表征(图1d)。此外,m6a-mtase表明,随着酶量增加,对dhs的选择性降低(图1e),类似于dna se i(或微球菌核酸酶(mnase))对染色质底物的酶促作用,这是由于对更多但更短的核小体间连接区的消化增加(h.weintraub,m.groudine,science(80).193,848
–
856(1976);k.s.bloom,j.n.anderson,cell.15,141
–
150(1978);j.mieczkowski,et al.,nat.commun.7,11485(2016))。mtase-seq对dna可及性的定量在启动子近端和远端调控元件处都具有高度可复制性(图1f),其中酶hia5的效率最高。这些结果表明,非特异性m6a-mtase提供了染色质内dna可及性的定量探针,并且表明m6a以接近核苷酸的分辨率分布,有效地将染色质结构复制到dna上。
[0125]
图1a-图1f示出了非特异性m6a-mtase选择性标记染色质可及性位点。图1a示出了基于裂解和m6a-mtase标记染色质可及性位点的方法的示意图。图1b示出了在用不同量的m6a-mtase hia5处理后,对来自果蝇s2细胞核的m6a修饰dna进行斑点杂交定量。图1c示出了mtase-seq的实验示意图。图1d-图1e示出了在用5种不同的m6a-mtase处理s2细胞核(图1d)后或增加m6a-mtase hia5的量(图1e)后,比较dna sei-seq信号与mtase-seq信号之间关系的基因组基因座。显示未处理核和输入控制上的m6a-ip-seq进行比较。图1f示出了用hia5处理的s2细胞dhs的mtase-seq信号与用ecogii(顶部)处理的细胞中的mtase-seq信号或dna sei-seq信号(底部)的比较。实施例2:f
iber-seq
揭示单个染色质纤维结构的碱基对分辨率图谱
[0126]
以核苷酸分辨率对m6a沿多千碱基染色质模板的线性模式进行测序,以重建染色质纤维的初级结构;该过程被称为“fiber seq”(图2a)。为实施fiber-seq,使用单分子dna测序仪在测序过程中基于该碱基的dna聚合酶动力学来区分甲基化和非甲基化腺嘌呤残基(g.fang,et al.,nat.biotechnol.30,1232
–
1239(2012))。通过对每根染色质纤维进行15次以上的再测序(称为循环共有测序(ccs)(k.j.travers,et al.,nucleic acids res.38(2010),doi:10.1093/nar/gkq543)),获得了高度精确的核苷酸分辨率,从而能够以与未修饰核苷酸相当的精确度对修饰核苷酸进行碱基识别。
[0127]
为了创建染色质模板,用m6a-mtase对s2细胞核进行处理(使用与mtase-seq类似的条件),然后对提取自经处理或未处理细胞核的高分子量dna进行无pcr文库构建。在pacific biosciences单分子dna测序仪上对所得文库进行ccs,该测序仪提供非常高的碱基识别精确度。尽管未处理细胞核显示出最低m6a信号,但来自m6a-mtase处理细胞的98%以上的单分子读数显示出某种程度的腺嘌呤甲基化(图2b),51%的m6a在四个核苷酸内显示出另一个m6a标记。这些结果以及上述结果表明,fiber-seq能够将染色质模板转化为dna可及性的线性读数。
[0128]
为了重建染色质的初级结构,对m6a核苷酸沿基因组的分布进行了分析。观察到m6a明显聚集成跨越数十至数百个碱基对的短邻接区域,由未修饰的核苷酸延伸段分隔(图2c)。对两类甲基化酶可及dna序列(mad)进行了鉴定:(1)与dna sei超敏位点一致的平均长度为174bp的序列元件(图2c,d);以及(2)平均长度为51bp且间距规则的更多短序列元件,与预期核小体间连接区大小和分布相似(r.v.chereji,et al.,nucleic acids res.44,
1036
–
1051(2016))(图2c,d)。在第一类中,与dhs重叠的所有纤维的m6a标记碱基的聚集数量与从体细胞核获得的dna se i裂解的聚集密度成正比(图2e)。
[0129]
可以通过强标记连接区之间m6a的明显缺失来简单定义核小体占据的dna,这表明m6a mtase通常无法接近核小体包裹的dna(图2c,f),可能是因为这些酶通过碱基翻转修饰腺嘌呤(j.r.horton,et al.,j.mol.biol.358,559-570(2006))。fiber-seq数据精确地记录了最多达数千碱基的核小体位置,高质量的纤维序列平均产生7个以上界限分明的核小体(图2g),这使我们能够评估关键的核小体占位特征。
[0130]
接下来,使用fiber-seq数据探索染色质结构以及核小体占位与调控dna可及性之间的相互作用方面的一些基本问题。观察到核小体重复序列(nr)平均长度为179bp,与先前报道的一致(r.v.chereji,et al.,2016(见上文))。然而,单根纤维上的nr长度变化显著,75%在157-202bp之间(图2g)。一般认为,紧邻启动子和增强子两侧的区域具有排列紧凑、间隔规则的核小体(b.lai,et al.,nature.562,281
–
285(2018);k.struhl,e.segal,nat.struct.mol.biol.20,267-273(2013))。然而,尽管fiber-seq数据显示启动子和远端dhs两侧的核小体阵列内的nr长度更短,但这些区域的结构异质性明显大于离dhs更远的核小体阵列(图2h)。因此,核小体压缩似乎不会产生或加强结构一致性;确切而言,在单个模板层面,调控区域周围的染色质组装似乎呈高度动态性。
[0131]
图2a-图2h示出了fiber-seq揭示单个染色质纤维结构的碱基对分辨率图谱。图2a示出了fiber-seq示意图。图2b示出了从分别未经处理和经hia5处理的s2细胞核中分离出的dna经pacbio ccs得出的含m6a甲基化碱基的染色质纤维的百分比。图2c示出了比较dna sei-seq、mtase-seq和fiber-seq之间关系的基因组位点。单个pacbio读数/染色质纤维用灰色线标记,m6a甲基化碱基用紫色虚线标记。插入按碱基着色的dhs,其中m6a-敏感碱基为灰色(例如所有a/t残基),m6a-甲基化碱基为紫色。图2d示出了显示dna sei-seq信号与每个dhs的fiber-seq m6a信号之间关系的豆荚图。对所有dhs均进行了皮尔逊相关性分析。*p值《0.001(秩和检验)。图2e示出了在dhs外(灰色)、tss远端dhs内(蓝色)和启动子dhs内(绿色)识别的所有mad的mad宽度柱状图。各箱须图显示在下方。*p值《0.001(秩和检验)。图2f示出了未与dhs重叠的5-100bp长mad周围的单个m6a标记的热图(顶部)。上方柱状图显示了对核小体结合碱基免受甲基化的强力保护(底部)。图2g示出了所有nr长度的柱状图(左)以及在单根染色质纤维上识别的nr数量(右)。图2h示出了对于不含dhs(灰色)、含有tss远端dhs(蓝色)或含有启动子dhs(绿色)的纤维,每根纤维的平均nr长度(左)和每根纤维的nr长度的平均bp差异(右)的柱状图。箱须图显示在下方。*p值《0.001(秩和检验)。实施例3:相邻调控元件在同一染色质纤维上的协调驱动
[0132]
转录因子和辅助蛋白如何激活基因组dna中编码的调控信息这一问题对于理解细胞状态和命运决定(a.b.stergachis,et al.,cell.154,888
–
903(2013))以及定义调控dna内的遗传变异影响表型特征和疾病风险的机制至关重要(m.t.maurano,r.et al.,science(80).337,1190-5(2012))。为此,需要回答下述由来已久的问题:是调控dna在任何给定模板上(代替标准核小体)以全部或全无方式驱动,还是致动元件以交替结构的形式存在,同时中间dna可及性由间歇变量核小体占位事件介导?如果是前者,那么使某一给定调控区域内的遗传变异具有渗透性的主要机制可能涉及改变其同源区域被驱动的频率,而不涉及创建替代的调控结构。目前从细胞群(r.e.thurman,et al.,2012(见上文))中收集数据的方
法无法解决任何调控元件是否在单个染色质模板上以全部或全无方式驱动的问题,单细胞采样法也无法解决该问题,因为这些方法产生的数据极为稀少,并且无法在等位基因特异性基础上连续查询染色质的任何区域(j.d.buenrostro,et al.,nature.523,486-490(2015))。
[0133]
在所有14,432个具有多个重叠fiber seq读数的s2细胞dhs中(每个dhs中平均有5个高质量读数),只有64%的重叠染色质纤维显示出一致的mad,且处于开放状态,其余显示出核小体分离,且处于封闭状态(图3a)。更宽的dhs表明采用可及/开放状态的可能性更高(图3b),这与沿着dna片段的额外tf的协同结合可以更有效地在dna占位方面与核小体竞争的概念一致(c.c.adams,j.l.workman,mol.cell.biol.15,1405
–
21(1995);j.a.miller,j.widom,mol.cell.biol.23,1623
–
32(2003);l.a.mirny,proc.natl.acad.sci.u.s.a.107,22534
–
22539(2010))。对长度超过500bp的延伸元件的分析表明,尽管与这些dhs重叠的85%的染色质纤维采用可及状态,但其中65%的可及纤维被一个或多个共同占位的核小体截断,不同纤维上记录了高度的位置可变性。根据fiber seq数据得出结论,调控dna的可及性主要通过全部或全无过程来驱动,在单根染色质纤维层面上,大多数调控dna以两种状态之一(可及或不可及)存在,大元件由共同占位的核小体额外进行调控。
[0134]
解决了驱动一个调控元件上的dna可及性是否会影响同一染色质纤维上相邻元件的行为这一问题。调控dna沿基因组高度聚集,并且许多控制区域似乎是作为多个独立元件的组合物进行组织,诸如在整个基因座(例如,β-珠蛋白基因座控制区)或基因特异性控制簇(例如,bcl11a增强子区域)上起作用的基因座控制区/“超级增强子”(w.a.whyte,et al.,cell.153,307
–
319(2013);p.diaz,et al.,immunity.1,207
–
17(1994);l.madisen,m.groudine,genes dev.8,2212
–
26(1994);f.grosveld,et al.,cell.51,975-85(1987))。如果驱动一个调控元件促进驱动同一染色质纤维上的邻近元件,那么它将提供基因调控元件簇的机制基础,并表明基因调控结构和进化的当前模型并未考虑顺式集成功能水平。它还将通过对邻近元件的连锁效应扩大调控性遗传变异的潜在影响。
[0135]
6%的fiber-seq读数与多个dhs重叠,从而能够量化同一染色质纤维上调控dna可及性的共同驱动(图3c)。观察到远端元件对、启动子对以及它们的组合的共同驱动,这表明相邻调控dna元件可以沿着同一染色质纤维驱动(图3d)。其中,相邻远端调控元件在同一纤维上被共同驱动的背景下显著富集(图3d),这表明驱动一个远端元件处的调控dna可及性促进了相邻元件处的可及性。因此,这些结果为观察动物基因组中远端调控元件的聚集提供了物理化学基础,并表明影响调控dna可及性和功能的遗传变异体可能在顺式中产生局部连锁效应,进而以当前基因组查询技术无法捕捉到的方式扩大其潜在影响力。
[0136]
图3a-图3d示出了相邻调控元件在同一染色质纤维上的协调驱动。图3a示出了比较dhs处dna sei-seq、mtase-seq和fiber-seq之间关系的基因组基因座,其揭示该dhs处具有开放染色质和封闭染色质的重叠染色质纤维。图3b示出了对于根据其宽度(左)或与tss的接近程度(右)划分的dhs,与可及纤维和封闭纤维重叠的dhs的比例。*p值《0.01(z检验)。图3c示出了比较相邻dhs处dna sei-seq、mtase-seq和fiber-seq之间关系的基因组基因座。图3d示出了对于包含两个dhs的染色质纤维,在两个不同元件类别的dhs处含有可及mad的纤维的百分比与预期百分比的比较。*p值《0.01(z检验)。
实施例4:调控dna=驱动对核小体定位的影响
[0137]
核小体定位是基因调控的关键,由多种因素共同决定,这些因素包括dna序列;形成边界的序列特异性dna结合蛋白的竞争性占位;核小体重塑复合物的作用;以及与rna聚合酶的相互作用(k.struhl,e.segal,nat.struct.mol.biol.20,267-273(2013))。这些因素的相对贡献度目前在全球范围内尚不清楚,也无法在特定基因组位置进行研究。现有基于大量细胞数据的分析(s.baldi,et al.,mol.cell.72,661-672.e4(2018);g.c.yuan,et al.,science(80).309,626
–
630(2005);c.jiang,b.f.pugh,nat.rev.genet.10,161
–
172(2009))表明,可及启动子周围的核小体通常定位良好,而远端调控元件周围的核小体定位则不太明确。然而,这种定位是否是由于因子占位(因此可及)调控dna施加的边界条件所致尚不清楚。
[0138]
我们推断,可以通过比较其中调控元件处于可及状态的重叠纤维上与其中调控元件处于交替核小体占位(即封闭)状态的重叠纤维上的调控元件周围的核小体位置来直接测试核小体定位的边界模型。虽然dhs周围的核小体总体上定位良好(图4a),但对单纤维数据的分析表明,这些定位良好的核小体主要来源于调控元件在远端元件(图4b)或dna se i超敏启动子上游处于可及状态的纤维(图4c),这表明核小体在这些位置的定位在很大程度上取决于调控dna的驱动情况,而不是dna序列本身。相比之下,dna sei超敏启动子下游的核小体则定位良好,这与启动子处于可及还是封闭状态无关(图4c)。因此,在大多数情况下,核小体定位似乎是由调控dna对单个染色质模板的驱动所施加的边界条件而导致的。
[0139]
图4a-图4d示出了调控dna驱动对核小体定位的影响。图4a示出了演示重叠读数中核小体定相/定位计算的示意图,以及位于不同类别读数上的核小体的单个核小体偏移的柱状图和箱须图。*p值《0.001(秩和检验)。图4b-图4c示出了邻近tss远端dhs(图4b)和表达基因的启动子dhs(图4c)的核小体的同相与异相核小体富集,根据染色质纤维是否包含与dhs重叠的可及mad(红色)与封闭mad(灰色)进行划分。*p值《0.01;ns=p值》0.05(z检验)。图4d示出了演示调控元件周围核小体布置的边界模型的示意图。实施例5:果蝇与人类之间染色质结构的守恒
[0140]
我们接下来试图确定这些染色质特征在果蝇与人类之间是否守恒。在验证m6a-mtase可以选择性标记人类细胞类型中的细胞类型特异性可及dna(图5a)后,我们用m6a-mtase处理人类k562细胞的细胞核,然后进行pacific biosystems ccs单分子dna测序,从而创建染色质模板。这使得可及调控元件和核小体间连接区(图5b-图5c)均发生稳健甲基化,其中与dhs重叠的m6a标记碱基的聚集数量反映了从体细胞核获得的dna se i裂解的聚集密度(平均每个dhs有0.3个高质量读数),高质量纤维平均产生8个以上界限分明的核小体(图5d)。与我们在果蝇中的发现一致(图2h),k562细胞中启动子和远端dhs两侧的核小体阵列nr长度更短,但与距dhs更远的核小体阵列相比,表现出明显更大的结构异质性(图5e)。我们同样发现,k562细胞中的调控dna可及性主要通过全部或全无过程驱动(图5f),并且启动子和远端dhs周围的核小体均定位良好,这表明这些可能是调控dna驱动以及周围核小体定位和压缩的普遍特征。
[0141]
总之,研究表明,有可能以核苷酸分辨率将染色质和调控结构复制到dna模板上,并将其与长读单分子dna分析相结合,以描绘单根染色质纤维的初级结构(fiber-seq)。调控dna的驱动是具有高度细胞选择性的,所以应该可以将来自复杂细胞混合物的单分子数
据分解为来自组成细胞亚群的调控模板状态。随着读取长度和吞吐量的进一步增加,在不久的将来,应该有可能转录大基因座的初级调控结构,并通过将fiber-seq与遗传变异体的精确碱基识别相结合来组装整个染色质单倍型。通过同时绘制单个调控等位基因的遗传(初级序列)和表观遗传(染色质结构)状态,对单根染色质纤维进行测序,从而为直接分析罕见和常见调控dna变异的功能影响提供统一的工具。
[0142]
图5a-图5e示出了果蝇与人类之间染色质结构的守恒。图5a-图5b示出了对hela细胞和k562细胞中人类β-珠蛋白基因座控制区的dna sei-seq信号和mtase-seq信号的比较(图5a),以及与k562细胞中fiber-seq数据的比较(图5b)。图5c示出了在dhs外(灰色)、tss远端dhs内(蓝色)和启动子dhs内(绿色)识别的所有mad的mad宽度柱状图。各箱须图显示在下方。*p值《0.001(秩和检验)。图5d示出了对于不含dhs(灰色)、含有tss远端dhs(蓝色)或含有启动子dhs(绿色)的纤维,每根纤维的平均nr长度(左)和每根纤维的nr长度的平均bp差异(右)的柱状图。箱须图显示在下方。*p值《0.001(秩和检验)。图5e示出了对于根据其与tss的接近程度划分的dhs,与可及纤维和封闭纤维重叠的dhs的比例。*p值《0.01(z检验)。实施例6:使用细胞穿透肽(cpp)标记的m6a-mt
ase
鉴定体内染色质结构
[0143]
生成了修饰的m6a-mtase,其包含与细胞穿透肽(cpp)和核定位序列(nls)缀合的m6a-mtase hia5。具体地,cpp标签使得m6a-mtase能够渗透活细胞质膜,而nls标签使得mtase能够随后游离入细胞核。在用该试剂处理活细胞后,对分离的dna进行单分子染色质纤维测序和直接碱基修饰测定(即体内fiber-seq),使我们能够在细胞存活时鉴定其染色质结构和动态(图6a)。
[0144]
该方法可以使用多个cpp标记。cpp标签tat、8-精氨酸(8r)和渗透素均显示出类似的效率(图6b)。该方法成功应用于原代血细胞(图6b)。体内fiber-seq分布图反映了从分离的细胞核获得的结果,不仅具有无需核分离步骤的额外优势,还可以量化体内发生的染色质动力学(图6c-图6e)。实施例7:使用f
iber-seq
鉴定功能基因调控dna改变
[0145]
通过将每个分子的m6a标记染色质结构与在单分子测序期间获得的潜在每个分子的高质量dna测序信息相结合,可以使用fiber-seq简单阐明功能性调控dna的改变。如图7所示,在原代人cd4 细胞中进行了fiber-seq,并根据这些dna变体对纤维上重叠染色质结构的影响鉴定功能性调控dna变体。实施例8:原位甲基化腺嘌呤(m6a)位点的可视化
[0146]
开发了原位显示甲基化腺嘌呤(m6a)位点(即在完整哺乳动物细胞中)的成像分析。
[0147]
用1x pbs冲洗处理k562细胞,并将细胞团块重悬于缓冲液a中。然后用0.1%igepal在冰上使重悬的细胞渗透5分钟。使细胞样本成团并重悬于缓冲液a中,用0u、1u或40u的hia5腺嘌呤甲基转移酶处理细胞,随后立即以每毫升100万个细胞的密度接种在pll涂层的玻璃表面上,在37℃下孵育15分钟,然后在室温下用过量的4%多聚甲醛溶液固定10分钟。用2x pbs洗涤固定细胞,用0.25% triton渗透10分钟,然后在37℃下用rnasea处理30分钟,用2% bsa封闭1小时,然后根据标准免疫荧光法用m6a抗体(abe572、abe572-i、sab5600251)标记。在m6a标记后,用dapi对细胞进行复染,并用prolong gold防褪色剂进行固色。然后,使用60x1.4na油浸物镜通过落射荧光显微镜对细胞进行3d成像。对细胞图像进
行去卷积以移除离焦模糊部分,然后进行处理以描绘单个细胞核和m6a标记的核区域。
[0148]
用抗m6a抗体标记以显示明确的点状染色,其随着hia5腺嘌呤甲基转移酶剂量的增加而增加。
[0149]
图8示出了用dapi和m6a染色的k562细胞核的代表性图像,其显示了所有三种受测m6a抗体的点状m6a模式,其随着hia5剂量的增加而增加。
[0150]
核m6a信号的剂量依赖性增加体现在整个核表达以及单个点中。
[0151]
图9示出了箱形图与小提琴图,其显示了总核m6a信号(左)以及单点强度(右)的hia5剂量依赖性增加。
[0152]
使m6a标记的基因组区域可视化将帮助了解单细胞层面可及基因组的空间组织,从而能够对调控dna的结构-功能相互关系进行深入研究。该可视化还可用于对比分析病变细胞和正常细胞。
[0153]
尽管为了更清晰的理解,已经通过图示和实施例对上述发明进行了详细描述,但对本领域普通技术人员显而易见的是,根据本发明的指导,在不脱离所附权利要求书的精神或范围的情况下,可以对其进行某些更改和修改。还应当理解的是,本文使用的术语仅用于描述特定实施方案,而不意在加以限制,因为本发明的范围将仅由所附权利要求书限制。
[0154]
因此,上述仅说明了本发明的原理。应当理解,本领域技术人员将能够设计出各种布置,尽管本文没有明确描述或示出,但这些布置体现了本发明的原理,并且包括在本发明的精神和范围内。此外,本文所述的所有实施例和条件性说明主要旨在帮助读者理解本发明的原理和发明人对本领域的进一步发展所贡献的概念,并且应理解为不受这些具体列举的实施例和条件的限制。此外,本文中描述本发明的原理、方面和实施方案及其具体实施例的所有陈述旨在包括其结构和功能等效物。此外,此类等效物旨在包括当前已知的等效物和未来开发的等效物,即所开发的执行相同功能的任何元件,无论其结构如何。因此,本发明的范围并非旨在限于本文所示和所描述的示例性实施方案。确切而言,本发明的范围和精神由所附权利要求书体现。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。