1.本发明属于检测方法领域,具体涉及一种荧光碳量子点的定量检测方法。
背景技术:
2.碳量子点(carbon dots,简称碳点,cds)是一种尺寸小于10nm,自发光的惰性纳米颗粒,因其优异的荧光性能,亦被称为纳米荧光碳点。碳点已有近二十年的发展史,其概念已逐步得到了拓展,目前,碳点成为了一类荧光碳纳米材料的统称。与有机荧光染料和传统半导体量子点相比,碳点的发射波长可调,且其碳成分使其对生物体毒性低,不良反应少。这些优良特性使其在成像方面具有良好的应用潜力。目前,碳点在离子分子检测、生物成像、光催化、光学材料等方面得到广泛研究应用,尤其在生物医学领域展现出强大的发展潜力。同时,由于碳点表面存在易于修饰的官能团,这一特性为癌细胞标记及示踪提供可能,为实现可视化检测提供研究新方向。在碳点细胞成像研究逐渐成熟的同时利用碳点的生物相容性和生物膜穿透性,针对耐药菌治疗和抗药肿瘤治疗的新药研发正逐渐发展并成为新的研究焦点。鉴于碳点在医学诊疗领域和新药研发领域展现潜在的巨大前景,碳点的量效关系要求碳点的质量标准研究更为完善,碳点含量测定方法的建立,特别是针对体内碳点的定量方法的建立势必成为碳点进一步研究的基础。
3.由于碳点为一群粒径在10nm以下的零维聚集物颗粒组成,针对荧光纳米材料的定量研究中大都采用“粗略定量”,如基于主观目视下的视觉亮度进行粗判断,这样的主观判断方式往往有失精准。然而针对未来精准诊断、治疗为目标,以人等生物为服务对象的荧光纳米材料,需要更加准确的质量控制,以确保疗效的重现性和安全的保障,而纳米材料的含量测定方法的建立是碳点质量标准建立重要研究项目之一,是实现药物安全、有效、质量可控基本条件。
4.由于碳点没有纯物质作为对照品,所以关于碳点的定量方法的建立一直是有待开发的课题,目前也未见对碳点进行定量检测的方法报道。如果能够突破碳点无对照品的瓶颈,建立定量碳点的含量测定方法,将具有非常重要的意义。
技术实现要素:
5.本发明的目的在于以纯荧光物质为对照品,通过建立纳米荧光碳点体外和体内含量测定的荧光分光光度法,为碳点质量标准建立和体内定量检测提供含量测定的方法。
6.本发明提供了一种荧光碳量子点的定量检测方法,包括如下步骤:
7.(1)用稀释液配制浓度为c0的待测样品溶液,检测荧光强度,得到待测样品的荧光强度f;
8.(2)用稀释液配制对照品的储备液,储备液用稀释液稀释,得到至少5份梯度浓度的对照品溶液,浓度分别为c1、c2…cn
,检测荧光强度,得到梯度浓度的对照品溶液对应的荧光强度f1、f2…fn
,n为大于等于5的整数;
9.(3)以步骤(2)的对照品溶液浓度c1、c2…cn
和对应的荧光强度f1、f2…fn
绘制标准
曲线,并线性拟合得到对照品荧光强度-浓度的拟合方程;
10.(4)将步骤(1)测得的待测样品荧光强度f代入步骤(3)得到的拟合方程,计算得到浓度c’;
11.(5)按照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%;
12.其中,所述对照品为甲酚紫或罗丹明。
13.进一步地,上述荧光碳量子点为dccds、fa-dccds、acucds、fa-acucds、ucpg-fe或fa-ucpg-fe;
14.所述所述dccds是由柠檬酸、尿素、磷酸和n,n-二甲基甲酰胺为原料制备而成的碳量子点,所述fa-dccds是dccds与叶酸化学键接而成的肿瘤靶向碳量子点;所述acucds是由柠檬酸铵和尿素为原料制备而成的碳量子点,所述fa-acucds是acucds与叶酸化学键接而成的肿瘤靶向碳量子点;所述ucpg-fe是由柠檬酸、尿素、peg400和铁盐为原料制备而成的碳量子点,所述fa-ucpg-fe是ucpg-fe与叶酸化学键接而成的肿瘤靶向碳量子点。
15.进一步地,上述待测样品是含有荧光碳量子点的固体混合物或组织液,或摄取了荧光碳量子点的细胞裂解得到的裂解液;优选地,所述组织液为肝组织液或肾组织液。
16.更进一步地,上述稀释液是超纯水或氯化钠溶液,优选地,所述氯化钠溶液的浓度为0.9%wt。
17.更进一步地,上述检测荧光强度是:采用酶标仪,在激发波长λ下检测发射波长λm处的荧光强度;所述发射波长λm是荧光碳量子点的最大发射波长,所述激发波长λ是荧光碳量子点在获得最大发射波长时所对应的激发波长。
18.更进一步地,上述荧光碳量子点为dccds,所述对照品为甲酚紫;所述检测荧光强度是在570nm激发波长下检测634nm发射波长处的荧光强度,优选地,述所述待测样品是含有荧光碳量子点的固体混合物,稀释液为超纯水,所述c0的范围为1.250~19.99μg/ml,c1、c2…cn
的范围为0.3900~12.50μg/ml;
19.或,所述待测样品是摄取了荧光碳量子点的细胞裂解得到的裂解液,稀释液为氯化钠溶液,所述c0的范围为0.1000~1.602μg/ml,c1、c2…cn
的范围为0.0250~0.8010μg/ml;
20.或,所述待测样品是含有荧光碳量子点的肝组织液,稀释液为氯化钠溶液,所述c0的范围为1.250~19.94μg/ml,c1、c2…cn
的范围为0.3906~6.250μg/ml;
21.或,所述待测样品是含有荧光碳量子点的肾组织液,稀释液为氯化钠溶液,所述c0的范围为0.6231~9.970μg/ml,c1、c2…cn
的范围为0.1900~3.120μg/ml。
22.更进一步地,上述荧光碳量子点为fa-dccds,所述对照品为罗丹明;所述荧光强度是在460nm激发波长下检测560nm发射波长处的荧光强度;优选地,上述待测样品是含有荧光碳量子点的固体混合物,稀释液为超纯水,所述c0的范围为2.005~32.08μg/ml,c1、c2…cn
的范围为0.8500~13.60μg/ml;
23.或,所述待测样品是摄取了荧光碳量子点的细胞裂解得到的裂解液,稀释液为氯化钠溶液,所述c0的范围为0.4988~7.980μg/ml,c1、c2…cn
的范围为0.2262~3.620μg/ml。
24.更进一步地,上述荧光碳量子点为acucds,所述对照品为罗丹明;所述检测荧光强度是在490nm激发波长下检测570nm发射波长处的荧光强度;优选地,上述待测样品是含有荧光碳量子点的固体混合物,所述稀释液为超纯水,所述c0的范围为0.9850~15.74μg/ml,
c1、c2…cn
的范围为0.4300~6.802μg/ml。
25.更进一步地,上述碳量子点为fa-acucds,所述对照品为罗丹明;所述荧光强度是在420nm激发波长下检测534nm发射波长处的荧光强度;优选地,上述待测样品是含有荧光碳量子点的固体混合物,所述稀释液为超纯水,所述c0的范围为7.520~120.3μg/ml,c1、c2…cn
的范围为1.700~27.20μg/ml。
26.更进一步地,上述碳量子点为ucpg-fe,所述对照品为罗丹明;所述检测荧光强度是在480nm激发波长下检测555nm发射波长处的荧光强度;优选地,上述待测样品是含有荧光碳量子点的固体混合物,所述稀释液为超纯水,所述c0的范围为0.7800~12.48μg/ml,c1、c2…cn
的范围为0.2124~3.398μg/ml。
27.更进一步地,上述碳量子点为fa-ucpg-fe,所述对照品为罗丹明;所述荧光强度是在480nm激发波长下检测545nm发射波长处的荧光强度;优选地,上述待测样品是含有荧光碳量子点的固体混合物,所述稀释液为超纯水,所述c0的范围为0.3000~4.801μg/ml,c1、c2…cn
的范围为0.2100~3.402μg/ml。
28.在首次开发研究可用于建立dx1002的荧光定量检测的候选碳点时,不论是制备工艺还是定量方法的建立,都将面临较大的困难,为了满足生物成像和光热治疗研究的应用,如何开发制备出高量子产率的近红外发光碳点是前期工作的难点之一;由于近红外光波能够较好地穿过生物组织,同时能更好地区分于生物体自身的蓝色荧光的干扰因此,近红外发光的碳点备受青睐。发明人通过对不同工艺制备的多种不同荧光特性的碳点进行筛选,最终获得3种适用于体内成像、抗耐药菌或抗耐药肿瘤所需的长波长碳点(λem≥500nm)作为性能优越的候选碳点。
29.此外,如何选择碳点定量用对照品是实验难点之二,介于碳点的制备材料通常包括碳源、氮源、溶剂等物质,同时制得的碳点虽为一种惰性纳米颗粒,但是其自身为多种粒径颗粒的集聚物,很难制得单一粒径碳点作为对照品,这也是制约碳点定量方法建立的瓶颈之一。发明人经过大量探索,才根据制得碳点的荧光特性寻找到合适的纯品荧光参比物作为碳点的替代对照品。
30.申请人以候选碳点为对象,选择了恰当的纯品荧光参比物,首先建立候选碳点体外定量测定方法,在确定碳点具备定量关系的基础上,进一步实现了细胞内候选碳点的定量方法的建立以及体内组织中候选碳点定量方法的建立。本发明突破了碳点无对照品,难以进行定量检测的瓶颈,为碳点质量标准的建立以及生物诊疗方面的深入研究提供定量研究的方法。
31.本发明所述的“梯度浓度”是指:浓度c1、c2、
…cn
之间的关系是固定倍数关系,即c2/c1=c3/c2=c4/c3=
…
=cn/c
n-1,n≥5。
32.显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
33.以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
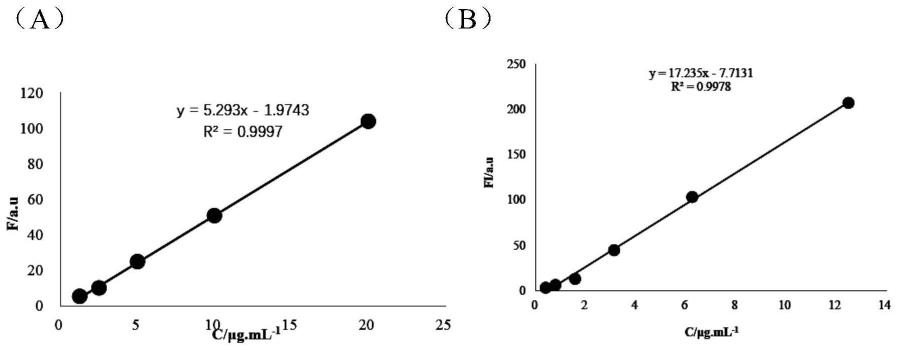
34.图1为(a)dccds荧光强度与浓度线性关系;(b)甲酚紫荧光强度与浓度线性关系图。
35.图2为(a)fa-dccds荧光强度与浓度线性关系图;(b)罗丹明6g荧光强度与浓度线性关系。
36.图3为(a)acucds荧光强度与浓度线性关系;(b)罗丹明6g荧光强度与浓度线性关系。
37.图4为(a)fa-acucds荧光强度与浓度线性关系;(b)罗丹明6g荧光强度与浓度线性关系。
38.图5为(a)ucpg-fe荧光强度与浓度线性关系;(b)罗丹明6g荧光强度与浓度线性关系。
39.图6为(a)fa-ucpg-fe荧光强度与浓度线性关系;(b)罗丹明6g荧光强度与浓度线性关系。
40.图7为(a)细胞环境对dccds的影响;(b)细胞环境对fa-dccds的影响。
41.图8为(a)hela细胞内dccds荧光强度与浓度线性关系;(b)hela细胞内甲酚紫荧光强度与浓度线性关系。
42.图9为(a)hela细胞内fa-dccds荧光强度与浓度线性关系;(b)hela细胞内罗丹明6g荧光强度与浓度线性关系。
43.图10为(a)dccds、肝组织液、dccds-肝组织液荧光发射光谱图;(b)dccds、肾组织液、dccds-肾组织液荧光发射光谱图;(c)dccds、dccds-肝组织液、dccds-肾组织液荧光发射光谱图。
44.图11为(a)肝组织液中dccds荧光强度与浓度线性关系;(b)肝组织液中甲酚紫荧光强度与浓度线性关系。
45.图12为(a)肾组织液中dccds荧光强度与浓度线性关系;(b)肾组织液中甲酚紫荧光强度与浓度线性关系。
具体实施方式
46.除另有说明外,本发明所用原料与设备均为已知产品,通过购买市售产品所得。
47.本发明实施例所用碳点的制备方法如下:
48.1、dccds的制备:
49.分别称取柠檬酸2.0g,尿素1.0g置于烧杯中,再加入掺杂剂磷酸2ml,10ml dmf,搅拌并辅以超声至溶解。将溶液转移至50ml反应釜内胆(聚四氟乙烯)中,并以反应釜金属外壳加固,于180℃反应10h。将碳点原液进行过滤、离心(13000r/min,10min)、透析(500da,24h),并将溶液以油泵进行旋蒸,得到碳点固体。
50.经检测,dccds溶液在日光下呈现深红色,在紫外灯365nm激发下发射蓝色荧光。dccds发射波长相对稳定,随激发波长的增长只有微弱红移,其在激发波长570nm处具有最大发射波长634nm,最大发射波长位于红光区。经细胞成像初试,该碳点具有良好的显像能力。
51.2、fa-dccds的制备:
52.(1)叶酸活性酯的制备:精密称定88.28mg叶酸、115.02mg edc、46.04mg nhs于圆底烧瓶中,加入4ml pbs(ph7.4),充分震荡至溶解完全,在磁力搅拌器上持续搅拌过夜。
53.(2)fa-dccds肿瘤靶向碳量子点的制备:精密称定88.28mg dccds于微量离心管中,加入2ml pbs(7.4)溶解,缓慢滴加至搅拌的2ml叶酸活性酯中,持续搅拌24h后将反应液先后以pbs缓冲液、超纯水进行透析(mwco=500da,24h),冷冻干燥,即得。
54.经检测,fa-dccds溶液在日光下呈现橙红色,在紫外灯下呈现视觉蓝色荧光。fa-dccds具有非激发依赖特性,并在460nm激发波长处具有最大发射波长560nm,最大发射波长位于黄绿光区,且经细胞成像初试,可用作后续细胞成像研究。
55.3、acucds的制备:
56.称取柠檬酸铵1g,尿素1g于研钵中,充分研磨至混匀,后将混合物转入坩埚,于电阻炉150℃条件下加热反应10h,反应结束后,自然放置至室温,加入超纯水40ml并以超声辅助提取,将提取液进行过滤,后将滤液透析(500da,24h)、冻干,得到碳点固体。
57.经检测,acucds溶液在日光下呈现淡黄色,在紫外灯365nm激发下发射黄绿色荧光。acucds具有激发非依赖特性,其在激发波长490nm处具有最大发射波长570nm,最大发射波长位于黄绿光区。经细胞成像初试,该碳点具备显像应用潜力。
58.4、fa-acucds的制备:
59.(1)叶酸活性酯的制备:精密称定88.28mg叶酸、115.02mg edc、46.04mg nhs于圆底烧瓶中,加入4ml pbs(ph=7.4),充分震荡,使其溶解完全,在磁力搅拌器上持续搅拌过夜,以达到活化fa的目的。
60.(2)fa-acucds肿瘤靶向碳量子点的制备:精密称定88.28mg acucds于微量离心管中,加入2mlpbs(ph=7.4)溶解,缓慢滴加至搅拌的2ml叶酸活性酯中,再通过乙二胺调节ph为9,持续搅拌24h后将反应液先后以pbs缓冲液、超纯水进行透析(mwco=500da,24h),冷冻干燥,即得。
61.经检测,fa-acucds溶液在日光下呈现黄色,在紫外灯下呈现视觉绿色荧光。fa-acucds在534nm发射波长处稳定,此时激发波长为420nm。最大发射波长位于绿光区,经细胞成像初试,可用作后续细胞成像研究。
62.5、ucpg-fe的制备:
63.分别称取1.0g柠檬酸、2.0g尿素置于研钵中,先后加入5ml水、15ml peg400进行研磨,待分散均匀后加入0.5g六水合三氯化铁继续研磨至均匀。将样品转移至反应釜内胆(聚四氟乙烯)中,以反应釜金属外壳加固,于240℃反应3h,经丙酮二次萃取、过滤、烘干即得碳点固体。
64.经检测,ucpg-fe溶液在日光下呈现红色,在紫外灯365nm激发下发射白色荧光。其视觉红色可能为铁离子自身颜色。ucpg-fe具有激发光依赖特性,其在激发波长480nm处具有最大发射波长555nm,最大发射波长位于绿光区,该碳点在550~600nm区间吸收较强,可考虑用作细胞成像。经细胞成像初试,该碳点具有良好的显像能力。
65.6、fa-ucpg-fe的制备:
66.(1)叶酸活性酯的制备:精密称定88.28mg叶酸、115.02mgedc、46.04mgnhs于圆底烧瓶中,加入4mlpbs(ph7.4),充分震荡,使其溶解完全,在磁力搅拌器上持续搅拌过夜,以达到活化fa的目的。
67.(2)fa-ucpg-fe肿瘤靶向碳量子点的制备:精密称定88.28mg ucpg-fe于微量离心管中,加入2mlpbs(7.4)溶解,缓慢滴加至搅拌的2ml叶酸活性酯中,再滴加乙二胺调ph为9,持续搅拌24h后将反应液先后以pbs缓冲液、超纯水进行透析(mwco=500da,24h),冷冻干燥,即得。
68.经检测,fa-ucpg-fe溶液在日光下呈现淡黄色,在紫外灯下呈现视觉白色荧光。fa-ucpg-fe具有激发依赖特性,其在激发波长480nm处具有最大发射波长545nm,最大发射波长位于绿光区,经细胞成像初试,可用作后续细胞成像研究。
69.实施例1、碳点及其叶酸偶碳点体外定量检测方法的建立
70.1、储备液配制
71.1.1对照品储备液的配制
72.精密称定0.1002g甲酚紫,加入超纯水溶解,将溶液转移至100ml容量瓶,定容,即得1.002mg/ml甲酚紫对照品溶液。备用。
73.精密称定0.1088g罗丹明6g,加入超纯水溶解,将溶液转移至100ml容量瓶,定容,即得1.088mg/ml罗丹明6g对照品溶液。备用。
74.1.2碳点及其叶酸偶联物储备液的配制
75.精密称定0.1280g dccds,加入超纯水溶解,将溶液转移至至100ml容量瓶,定容,即得1.280mg/ml dccds溶液。备用。
76.精密称定0.1007g acucds,加入超纯水溶解,将溶液转移至至100ml容量瓶,定容,即得1.007mg/ml acucds溶液。备用。
77.精密称定0.9986g ucpg-fe,加入超纯水溶解,将溶液转移至至100ml容量瓶,定容,即得0.9986mg/ml ucpg-fe溶液。备用。
78.精密称定0.1002g fa-dccds,加入超纯水溶解,将溶液转移至至100ml容量瓶,定容,即得0.1002g/ml fa-dccds-fa溶液。备用。
79.精密称定0.1925g fa-acucds,加入超纯水溶解,将溶液转移至至100ml容量瓶,定容,即得1.925mg/ml fa-dccds溶液。备用。
80.精密称定0.9604g fa-ucpg-fe,加入超纯水溶解,将溶液转移至至100ml容量瓶,定容,即得0.9604mg/ml fa-ucpg-fe溶液。备用。
81.2、dccds定量方法的建立
82.2.1测定方法
83.取每一待测溶液100μl加入96孔细胞培养板(以下简称96孔板),使用酶标仪进行荧光测定,测定波长为(激发波长/最大发射波长)ex 570nm/em 634nm。
84.2.2方法学验证
85.2.2.1线性和范围
86.2.2.1.1dccds线性关系的考察
87.取dccds储备液适量,分别配制1.250、2.499、4.998、9.996、19.99μg/ml的dccds溶液。照“2.1测定方法”项下方法进行测定。
88.由图1a可知,dccds在1.250~19.99μg/ml范围内具有良好的线性关系(y=5.293x-1.9743,y为荧光强度(a.u.),x为浓度(μg/ml),下同。r2=0.9997)。
89.2.2.1.2甲酚紫线性关系的考察
90.取甲酚紫对照品储备液适量,分别配制0.3900、0.7800、1.560、3.125、6.250、12.50μg/ml的甲酚紫对照品溶液。照“2.1测定方法”项下方法进行测定。
91.由图1b可知,甲酚紫在0.3900~12.50μg/ml范围内具有良好的线性关系:(y=17.235x-7.7131,r2=0.9978)。
92.可见,dccds与甲酚紫在一定浓度范围内各自具有良好的线性关系表明dccds和甲酚紫的浓度与其发光强度在指定波长下存在定量关系,且在此基础上,dccds荧光强度范围在甲酚紫的线性范围之内,可以确保在甲酚紫浓度范围的定量性能够涵盖dccds的定量范围。
93.2.3定量检测:
94.用超纯水将待测样品dccds的储备液稀释到浓度c0,c0在1.250~19.99μg/ml范围内,按照“2.1测定方法”检测荧光强度,得到待测样品的荧光强度f;
95.将测得的待测样品荧光强度f代入甲酚紫对照品的线性拟合方程:y=17.235x-7.7131,取y=f,计算得到浓度c’=x的值;
96.(5)按照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%。
97.3、fa-dccds定量方法的建立
98.3.1测定方法
99.取每一待测溶液100μl加入96孔细胞培养板(以下简称96孔板),使用酶标仪进行测定,测定波长为ex 460nm/em 560nm。
100.3.2方法学验证
101.3.2.1线性和范围
102.3.2.1.1fa-dccds线性关系的考察
103.取fa-dccds储备液,分别配制2.005、4.010、8.02、16.04、32.08μg/ml的fa-dccds溶液。照“3.1测定方法”项下方法进行测定。
104.由图2a可知,fa-dccds在2.005~32.08μg/ml范围内具有良好的线性关系(y=6.6001x 21.69,r2=0.9972)。
105.3.2.1.2罗丹明6g线性关系的考察
106.取罗丹明6g对照品储备液,分别配制0.8500、1.700、3.400、6.800、13.60μg/ml的罗丹明6g对照品溶液。照“3.1测定方法”项下方法进行测定。
107.由图2b可知,罗丹明6g在0.8500~13.60μg/ml范围内具有良好的线性关系(y=46.02x-44.367,r2=0.9964)。
108.可见,fa-dccds与罗丹明6g在一定浓度范围内各自具有良好的线性关系表明fa-dccds和罗丹明6g的浓度与其发光强度在指定波长下存在定量关系,且在此基础上,fa-dccds荧光强度范围在罗丹明6g的线性范围之内,可以确保在罗丹明6g浓度范围的定量性能够涵盖fa-dccds的定量范围。
109.3.3定量检测:
110.用超纯水将待测样品fa-dccds的储备液稀释到浓度c0,c0在2.005~32.08μg/ml范围内,按照“3.1测定方法”检测荧光强度,得到待测样品的荧光强度f;
111.将测得的待测样品荧光强度f代入罗丹明对照品的线性拟合方程:y=46.02x-44.367,取y=f,计算得到浓度c’=x的值;
112.(5)按照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%。
113.4、acucds定量方法的建立
114.4.1测定方法
115.取每一待测溶液100μl加入96孔细胞培养板(以下简称96孔板),使用酶标仪进行测定,测定激发波长为490nm,发射波长为570nm。
116.4.2方法学验证
117.4.2.1线性和范围
118.4.2.1.1acucds线性关系的考察
119.精密量取acucds储备液适量,分别配制0.9850、1.965、3.935、7.865、15.74μg/ml的acucds溶液。照“4.1测定方法”项下方法进行测定。
120.由图3a可知,acucds在0.9850~15.74μg/ml范围内具有良好的线性关系(y=8.6123x 7.4863,r2=0.994)。
121.4.2.1.2罗丹明6g线性关系的考察
122.精密量取罗丹明6g对照品储备液适量,分别配制0.4300、0.8500、1.700、3.401、6.802μg/ml的甲酚紫对照品溶液。照“4.1测定方法”项下方法进行测定。
123.由图3b可知,罗丹明6g在0.4300~6.802μg/ml范围内具有良好的线性关系(y=68.505x-37.183,r2=0.9966)。
124.综上,acucds与罗丹明6g在一定浓度范围内各自具有良好的线性关系表明acucds和罗丹明6g的浓度与其发光强度在指定波长下存在定量关系,且在此基础上,acucds荧光强度范围在罗丹明6g的线性范围之内,可以确保在罗丹明6g浓度范围的定量性能够涵盖acucds的定量范围。
125.4.3定量检测:
126.用超纯水将待测样品acucds的储备液稀释到浓度c0,c0在0.9850~15.74μg/ml范围内,按照“4.1测定方法”检测荧光强度,得到待测样品的荧光强度f;
127.将测得的待测样品荧光强度f代入罗丹明对照品的线性拟合方程:y=68.505x-37.183,取y=f,计算得到浓度c’=x的值;
128.(5)按照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%。
129.5、fa-acucds定量方法的建立
130.5.1测定方法
131.取每一待测溶液100μl加入96孔细胞培养板(以下简称96孔板),使用酶标仪进行荧光测定,测定激发波长为420nm,发射波长为534nm。
132.5.2方法学验证
133.5.2.1线性和范围
134.5.2.1.1fa-acucds线性关系的考察
135.精密量取fa-acucds储备液适量,分别配制7.520、15.04、30.08、60.16、120.3μg/ml的fa-acucds溶液。照“5.1测定方法”项下方法进行测定。
136.由4a可知,fa-acucds在7.520~120.3μg/ml范围内具有良好的线性关系(y=1.2251x 7.9835,r2=0.9928)。
137.5.2.1.2罗丹明6g线性关系的考察
138.精密量取罗丹明6g对照品储备液,分别配制1.700、3.400、6.800、13.60、27.20μg/ml的罗丹明6g对照品溶液。照“5.1测定方法”项下方法进行测定。
139.由图4b可知,罗丹明6g在1.700~27.20μg/ml范围内具有良好的线性关系(y=8.2362x-9.5705,r2=0.9973)。
140.综上,fa-acucds与罗丹明6g在一定浓度范围内各自具有良好的线性关系表明fa-acucds和罗丹明6g的浓度与其发光强度在指定波长下存在定量关系,且在此基础上,fa-acucds荧光强度范围在罗丹明6g的线性范围之内,可以确保在罗丹明6g浓度范围的定量性能够涵盖fa-acucds的定量范围。
141.5.3定量检测:
142.用超纯水将待测样品fa-acucds的储备液稀释到浓度c0,c0在7.520~120.3μg/ml范围内,按照“5.1测定方法”检测荧光强度,得到待测样品的荧光强度f;
143.将测得的待测样品荧光强度f代入罗丹明对照品的线性拟合方程:y=8.2362x-9.5705,取y=f,计算得到浓度c’=x的值;
144.(5)按照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%。
145.6、ucpg-fe定量方法的建立
146.6.1测定方法
147.取每一待测溶液100μl加入96孔细胞培养板(以下简称96孔板),使用酶标仪进行测定,测定激发波长为480nm,发射波长为555nm。
148.6.2方法学验证
149.6.2.1线性和范围
150.6.2.1.1ucpg-fe线性关系的考察
151.取ucpg-fe储备液适量,分别配制0.7800、1.560、3.120、6.240、12.48μg/ml的ucpg-fe溶液。照“6.1测定方法”项下方法进行测定。
152.由5a可知,ucpg-fe在0.7800~12.48μg/ml范围内具有良好的线性关系(y=0.4149x-0.097,r2=0.9998)。
153.6.2.1.2罗丹明6g线性关系的考察
154.取罗丹明6g对照品储备液,分别配制0.2124、0.4248、0.8495、1.699、3.398μg/ml的甲酚紫对照品溶液。照“6.1测定方法”项下方法进行测定。
155.由图5b可知,在0.2124~3.398μg/ml范围内具有良好的线性关系(y=37.961x-10.664,r2=0.9967)。
156.综上,ucpg-fe与罗丹明6g在一定浓度范围内各自具有良好的线性关系表明ucpg-fe和罗丹明6g的浓度与其发光强度在指定波长下存在定量关系,且在此基础上,ucpg-fe荧光强度范围在罗丹明6g的线性范围之内,可以确保在罗丹明6g浓度范围的定量性能够涵盖ucpg-fe的定量范围。
157.6.3定量检测:
158.用超纯水将待测样品ucpg-fe的储备液稀释到浓度c0,c0在0.7800~12.48μg/ml范围内,按照“6.1测定方法”检测荧光强度,得到待测样品的荧光强度f;
159.将测得的待测样品荧光强度f代入罗丹明对照品的线性拟合方程:y=37.961x-10.664,取y=f,计算得到浓度c’=x的值;
160.(5)按照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%。
161.7、fa-ucpg-fe定量方法的建立
162.7.1测定方法
163.取每一待测溶液100μl加入96孔细胞培养板(以下简称96孔板),使用酶标仪进行测定,测定激发波长为480nm,发射波长为545nm。
164.7.2方法学验证
165.7.2.1线性和范围
166.7.2.1.1fa-ucpg-fe线性关系的考察
167.精密量取fa-ucpg-fe储备液适量,分别配制0.3000、0.6000、1.200、2.400、4.801μg/ml的fa-ucpg-fe溶液。
168.由图6a可知,fa-ucpg-fe在0.3000~4.801μg/ml范围内具有良好的线性关系(y=4.3441x-0.335,r2=0.9989)。
169.7.2.1.2罗丹明6g线性关系的考察
170.精密量取罗丹明6g对照品储备液适量,分别配制0.2100、0.4200、0.850、1.701、3.402μg/ml的甲酚紫对照品溶液。照“7.1测定方法”项下方法进行测定。
171.由图6b可知,罗丹明6g在0.2100~3.402μg/ml范围内具有良好的线性关系(y=43.049x-13.659,r2=0.9924)。
172.综上,fa-ucpg-fe与罗丹明6g在一定浓度范围内各自具有良好的线性关系表明fa-ucpg-fe和罗丹明6g的浓度与其发光强度在指定波长下存在定量关系,且在此基础上,fa-ucpg-fe荧光强度范围在罗丹明6g的线性范围之内,可以确保在罗丹明6g浓度范围的定量性能够涵盖fa-ucpg-fe的定量范围。
173.7.3定量检测:
174.用超纯水将待测样品ucpg-fe的储备液稀释到浓度c0,c0在0.3000~4.801μg/ml范围内,按照“7.1测定方法”检测荧光强度,得到待测样品的荧光强度f;
175.将测得的待测样品荧光强度f代入罗丹明对照品的线性拟合方程:y=43.049x-13.659,取y=f,计算得到浓度c’=x的值;
176.(5)按照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%。
177.实施例2、本发明dccds、fa-dccds细胞内定量检测方法的建立
178.1.储备液的配制
179.1.1对照品储备液的配制
180.精密称定0.1003g甲酚紫,加入氯化钠溶液(0.9%)溶解,将溶液转移至100ml容量瓶,定容,即得1.003mg/ml甲酚紫对照品溶液。备用。
181.精密称定0.1076g罗丹明6g,加入氯化钠溶液(0.9%)溶解,将溶液转移至100ml容量瓶,定容,即得1.076mg/ml罗丹明6g对照品溶液。备用。
182.1.2dccds及fa-dccds储备液的配制
183.精密称定0.1011g dccds,加入氯化钠溶液(0.9%)溶解,将溶液转移至至100ml容量瓶,定容,即得1.011mg/ml dccds溶液。备用。
184.精密称定99.71mg fa-dccds,加入氯化钠溶液(0.9%)溶解,将溶液转移至至100ml容量瓶,定容,即得0.9971mg/ml fa-dccds溶液。备用。
185.2、细胞环境对荧光材料的影响
186.取500μl dccds溶液,加入500ul氯化钠溶液(0.9%),混合均匀,得到待测液-1;取经dmso裂解的细胞液500μl,加入500ul氯化钠溶液(0.9%),混合均匀,得到待测液-2;取经dmso裂解的细胞液500μl,加入500μl dccds溶液,混合均匀,得到待测液-3。将三个待测液分别于激发波长570nm下进行荧光发射光谱扫描,结果见图7a。
187.取500μl fa-dccds溶液,氯化钠溶液(0.9%),混合均匀,得到待测液-4;取经dmso裂解的细胞液500μl,加入500ul氯化钠溶液(0.9%),混合均匀,得到待测液-5;取经dmso裂解的细胞液500μl,加入500μl fa-dccds溶液,混合均匀,得到待测液-6。将三个待测液分别于激发波长460nm下进行荧光发射光谱扫描,结果见图7b。
188.如图7a所示,细胞液在570nm激发下,于dccds的发射光段没有吸收峰,且细胞液对dccds的荧光强度具有增强效应,因此在细胞环境中可以建立dccds的定量方法。
189.如图7b所示,细胞液在460nm激发下,于fa-dccds的发射光段没有吸收峰,且细胞液对fa-dccds的荧光强度具有增强效应,因此在细胞环境中可以建立fa-dccds的定量方法。
190.3.dccds细胞内定量方法的建立
191.3.1测定原理及方法
192.3.1.1测定原理
193.以hela细胞株为培养对象,将hela细胞于96孔板培养48h(细胞含量达到90%),终止培养。将培养基(或细胞外含碳点溶液)吸出,以pbs(ph7.30)清洗3次,将残留培养基(或细胞外多余碳点溶液)洗去。每孔加入50μl dmso进行细胞裂解,获得细胞液。加入50μl氯化钠(0.9%),采用酶标仪进行荧光测定。
194.3.1.2测定方法
195.以hela细胞株为培养对象,将hela细胞株于96孔板培养48h(细胞含量达到90%),终止培养。将培养基吸出,以pbs(ph7.30)清洗3次,每孔加入50μl dmso,另取每一待测溶液50μl加入96孔细胞板,使用酶标仪于570nm激发波长、634nm波长通道进行测定。
196.3.2.方法学验证
197.3.2.1线性和范围
198.3.2.1.1dccds线性关系的考察
199.精密量取dccds储备液适量,以氯化钠溶液(0.9%)分别配制0.1000、0.2000、0.4000、0.8001、1.201、1.602μg/ml的dccds溶液。照“3.1.2测定方法”项下方法进行测定。
200.由图8a可知,dccds在0.1000~1.602μg/ml范围内具有良好的线性关系(y=8.2262x 0.5213,r2=0.9933)。
201.3.2.1.2甲酚紫线性关系的考察
202.精密量取甲酚紫对照品储备液适量,以氯化钠溶液(0.9%)分别配制0.0250、0.0500、0.1000、0.2000、0.4005、0.8010μg/ml的甲酚紫对照品溶液。照“3.1.2测定方法”项下方法进行测定。
203.由图8b可知,甲酚紫在0.0250~0.8010μg/ml范围内具有良好的线性关系(y=23.838x 0.4847,r2=0.9961)。
204.综上,细胞内dccds与甲酚紫在一定浓度范围内各自具有良好的线性关系表明
dccds和甲酚紫的浓度与其发光强度在指定波长下存在定量关系,且在此基础上,dccds荧光强度范围在甲酚紫的线性范围之内,可以确保在甲酚紫浓度范围的定量性能够涵盖dccds的定量范围。
205.3.3.定量检测:
206.以hela细胞株为培养对象,将hela细胞株于96孔板培养36h后,以10mg/ml dccd溶液分别于3h、6h、9h、12h四个实验时间点间断给药及进行细胞摄取。终止培养。将培养基与碳点混合培养液吸出,以pbs(ph7.30)清洗3次,将残留培养基及细胞外多余dccd溶液洗去。每孔加入50μl dmso进行细胞裂解,获得含dccd的细胞液,加入50μl稀释液:氯化钠(0.9%),得到待测溶液,其中,待测样品(细胞裂解液)中dccds的浓度为c0,c0的范围在0.1000~1.602μg/ml范围内。使用酶标仪于570nm激发波长、634nm波长通道进行测定检测荧光强度,得到待测样品的荧光强度f;
207.将测得的待测样品荧光强度f代入甲酚紫对照品的线性拟合方程:y=23.838x 0.4847,取y=f,计算得到浓度c’=x的值;
208.(5)按照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%。
209.4.fa-dccds细胞内定量方法的建立
210.4.1测定原理及方法
211.4.1.1测定原理
212.以hela细胞株为培养对象,将hela细胞于96孔板培养48h(细胞含量达到90%),终止培养。将培养基(或细胞外含碳点溶液)吸出,以pbs(ph7.30)清洗3次,将残留培养基(或细胞外多余碳点溶液)洗去。每孔加入50μl dmso进行细胞裂解,获得细胞液。加入50μl氯化钠(0.9%),采用酶标仪进行荧光测定。
213.4.1.2测定方法
214.以hela细胞株为培养对象,将hela细胞于96孔板培养48h(细胞含量达到90%),终止培养。将培养基吸出,以pbs(ph7.30)清洗3次,每孔加入50μl dmso,另取每一待测溶液50μl加入96孔细胞板,使用酶标仪于460nm激发波长、560nm波长通道进行测定。
215.4.2.方法学验证
216.4.2.1线性和范围
217.4.2.1.1fa-dccds线性关系的考察
218.取fa-dccds储备液适量,以氯化钠溶液(0.9%)分别配制0.4988、0.9975、1.995、3.990、5.990、7.980μg/ml的fa-dccds溶液。照“4.1.2测定方法”项下方法进行测定。
219.由图9a可知,fa-dccds在0.4988~7.980μg/ml范围内具有良好的线性关系(y=2.0915x 0.7486,r2=0.9968)。
220.4.2.1.2罗丹明6g线性关系的考察
221.取罗丹明6g对照品储备液适量,以氯化钠溶液(0.9%)分别配制0.2262、0.4525、0.9050、1.810、2.690、3.620μg/ml的罗丹明6g对照品溶液。照“4.1.2测定方法”项下方法进行测定。
222.由图9b可知,罗丹明6g在0.2262~3.620μg/ml范围内具有良好的线性关系(y=12.996x-2.9095,r2=0.9983)。
223.综上,细胞内fa-dccds与罗丹明6g在一定浓度范围内各自具有良好的线性关系表
明fa-dccds和罗丹明6g的浓度与其发光强度在指定波长下存在定量关系,且在此基础上,fa-dccds荧光强度范围在罗丹明6g的线性范围之内,可以确保在罗丹明6g浓度范围的定量性能够涵盖fa-dccds的定量范围。
224.4.3.定量检测:
225.以hela细胞株为培养对象,将hela细胞株于96孔板培养36h后,以10mg/ml fa-dccd溶液分别于3h、6h、9h、12h四个实验时间点间断给药及进行细胞摄取。终止培养。将培养基与碳点混合培养液吸出,以pbs(ph7.30)清洗3次,将残留培养基及细胞外多余fa-dccd溶液洗去。每孔加入50μl dmso进行细胞裂解,获得含fa-dccd的细胞液,加入50μl稀释液:氯化钠(0.9%),得到待测溶液,其中,待测样品(细胞裂解液)中fa-dccds的浓度为c0,c0的范围在0.4988~7.980μg/ml范围内。使用酶标仪于460nm激发波长、560nm波长通道进行测定检测荧光强度,得到待测样品的荧光强度f;
226.将测得的待测样品荧光强度f代入罗丹明对照品的线性拟合方程:y=12.996x-2.9095,取y=f,计算得到浓度c’=x的值;
227.照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%。
228.实施例3、dccds体内肝肾组织中定量检测方法的建立
229.1.溶液的配制
230.1.1对照品储备液的配制
231.精密称定0.1003g甲酚紫,加入氯化钠溶液(0.9%)溶解,将溶液转移至100ml容量瓶,定容,即得1.003mg/ml甲酚紫对照品溶液。备用。
232.1.2dccds储备液的配制
233.精密称定0.1011g dccds,加入氯化钠溶液(0.9%)溶解,将溶液转移至至100ml容量瓶,定容,即得1.011mg/ml dccds溶液。备用。
234.2.组织液对dccds的影响
235.将肝、肾组织配成10%的生物组织液,以13000r/min转速离心10min,并用0.22nm微孔滤膜过滤,得到肝、肾组织上清液。
236.取500μl dccds溶液,加入500μl肝组织液,混匀,于570nm激发波长下扫描dccds-肝组织液英光发射光谱图。
237.取500μl dccds溶液,加入500μl肾组织液,混匀,于570nm激发波长下扫描dccds-肾组织液英光发射光谱图。
238.并扫描碳点溶液、肝组织液、肾组织液,以考察肝肾组织液对dccds的影响情况。
239.图10a-c表明,肝、肾组织液对dccds存在背景干扰,同时肝组织液与肾组织液对dccds的影响程度不同,因此需要分别对其进行定量分析。
240.3.dccds肝组织液中定量方法的建立
241.3.1测定方法
242.将肝组织以氯化钠(0.9%)配成10%的生物组织液,以13000r/min转速离心10min,并用0.22μm微孔滤膜过滤,得到肝组织上清液。将肝组织上清液与每一碳点溶液混合得到供试品溶液。取每一供试品溶液100μl加入96细胞板,使用酶标仪进行荧光测定,测定激发波长为570nm,发射波长为634nm。
243.3.2方法学验证
244.3.2.1线性和范围
245.3.2.1.1肝组织液中dccds线性关系的考察
246.取dccds储备液适量,以氯化钠溶液(0.9%)分别配制1.250、2.490、4.980、9.970、14.99、19.94μg/ml的dccds溶液。照“3.1测定方法”项下方法进行测定。
247.由图11a可知,肝组织液中dccds在1.250~19.94μg/ml范围内具有良好的线性关系(y=1.0193x 0.4106,r2=0.9992)。
248.3.2.1.2肝组织液中甲酚紫线性关系的考察
249.取甲酚紫对照品储备液,以氯化钠溶液(0.9%)分别配制0.3906、0.7812、1.562、3.125、5.010、6.250μg/ml的甲酚紫对照品溶液。照“3.1测定方法”项下方法进行测定。
250.由图11b可知,肝组织液中甲酚紫在0.3906~6.250μg/ml范围内具有良好的线性关系(y=3.1678x-0.532,r2=0.9980)。
251.综上,肝组织液中dccds和甲酚紫在一定浓度范围内各自具有良好的线性关系表明dccds和甲酚紫的浓度与其发光强度在指定波长下存在定量关系,且在此基础上,dccds荧光强度范围在甲酚紫的线性范围之内,可以确保在甲酚紫浓度范围的定量性能够涵盖dccds的定量范围。
252.3.3含量测定
253.以氯化钠(0.9%)溶液配制10mg/ml dccds溶液,并经0.22μm微孔滤膜过滤,以滤液作为注射液。以正常昆明小鼠为实验对象,取滤液100μl以尾静脉注射方式注入体内,实验设2个平行组。并分别于给药20、30、40、50、60min后将小鼠解剖,分别取其肝、肾。照“3.1测定方法”进行组织处理得到上清液,其中,待测样品(上清液)中dccds的浓度为c0,c0的范围在1.250~19.94μg/ml范围内,上清液中,进行荧光测定,得到荧光强度f。
254.将测得的待测样品荧光强度f代入甲酚紫对照品的线性拟合方程:y=3.1678x-0.532,取y=f,计算得到浓度c’=x的值;
255.(5)按照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%。
256.4.dccds肾组织液中定量方法的建立
257.4.1测定方法
258.将肾组织以氯化钠(0.9%)配成10%的生物组织液,以13000r/min转速离心10min,并用0.22μm微孔滤膜过滤,得到肾组织上清液。将肾组织上清液与每一碳点溶液混合,得到供试品溶液。取每一供试品溶液100μl加入96细胞板,使用酶标仪进行荧光测定,测定激发波长为570nm,发射波长为634nm。
259.4.2方法学验证
260.4.2.1线性和范围
261.4.2.1.1肾组织液中dccds线性关系的考察
262.取dccds储备液适量,以氯化钠溶液(0.9%)分别配制0.6231、1.246、2.492、4.985、7.500、9.970μg/ml的dccds溶液。照“4.1测定方法”项下方法进行测定。
263.由图12a可知,肾组织液中dccds在0.6231~9.970μg/ml范围内具有良好的线性关系(y=1.9374x-0.0693,r2=0.999)。
264.4.2.1.2肾组织液中甲酚紫线性关系的考察
265.取甲酚紫对照品储备液适量,以氯化钠溶液(0.9%)分别配制0.1900、0.3900、
0.7800、1.560、2.480、3.120μg/ml的甲酚紫对照品溶液。照“4.1测定方法”项下方法进行测定。
266.由图12b可知,肝组织液中甲酚紫在0.1900~3.120μg/ml范围内具有良好的线性关系(y=5.1277x-0.031,r2=0.9956)。
267.综上,肾组织液中dccds和甲酚紫在一定浓度范围内各自具有良好的线性关系表明dccds和甲酚紫的浓度与其发光强度在指定波长下存在定量关系,且在此基础上,dccds荧光强度范围在甲酚紫的线性范围之内,可以确保在甲酚紫浓度范围的定量性能够涵盖dccds的定量范围。
268.4.3含量测定
269.以氯化钠(0.9%)溶液配制10mg/ml dccds溶液,并经0.22μm微孔滤膜过滤,以滤液作为注射液。以正常昆明小鼠为实验对象,取滤液100μl以尾静脉注射方式注入体内,实验设2个平行组。并分别于给药20、30、40、50、60min后将小鼠解剖,分别取其肝、肾。照“3.1测定方法”进行组织处理得到上清液,其中,待测样品(上清液)中dccds的浓度为c0,c0的范围在0.6231~9.970μg/ml范围内,上清液中,进行荧光测定,得到荧光强度f。
270.将测得的待测样品荧光强度f代入甲酚紫对照品的线性拟合方程:y=5.1277x-0.031,取y=f,计算得到浓度c’=x的值;
271.(5)按照如下公式计算得到待测样品中的碳量子点含量c:c%=(c’/c0)*100%。
272.以下通过实验例证明本发明方法的有益效果。
273.实验例1、本发明体外定量检测方法的验证
274.1、dccds定量方法的验证:
275.1、仪器精密度
276.为检验仪器的稳定性,取同一dccds供试品溶液,照“实施例1的2.1测定方法”项下方法进行测定,连续测定6次,6次测定荧光强度的rsd为1.46%,结果表明,本实验所使用仪器具有良好的精密度。
277.2、重复性
278.精密量取dccds储备液适量,分别配制含量为低(8.000μg/ml)、中(9.980μg/ml)、高(11.97μg/ml)浓度的dccds溶液,每个浓度样品各3份,照“2.1测定方法”项下方法进行测定。结果低、中、高浓度的dccds平均含量为34.31%,rsd为2.34%。实验结果表明该方法具有良好的重复性。
279.3、中间精密度
280.精密量取dccds储备液(1.280mg/ml)适量,分别配制含量为低(8.000μg/ml)、中(9.980μg/ml)、高(11.97μg/ml)浓度的dccds溶液,每个浓度样品各3份,照“2.1测定方法”项下方法,由两人分别于于不同仪器、不同时间进行测定。结果低、中、高三个实验浓度的中间精密度rsd分别为1.78%,2.03%,2.06%。实验结果表明该方法具有良好的中间精密度。
281.4、样品稳定性
282.取同一dccd供试品溶液,分别设置0、2、4、8、12、24h六个实验时间点,照“2.1测定方法”项下方法,进行含量测定。结果6个时间点样品含量测定的rsd为2.29%。表明在室温下放置24h的dccd样品溶液的稳定性良好。
283.5、回收率
284.对照品溶液的配制:精密量取甲酚紫对照品储备液适量,以超纯水配制为3.40μg/ml的甲酚紫对照品溶液。
285.dccds溶液的配制:精密量取dccds储备液(1.280mg/ml)适量,分别配制含量为低(8.000μg/ml)、中(9.980μg/ml)、高(11.97μg/ml)浓度的dccds溶液,每个浓度样品各3份。
286.精密移取1ml甲酚紫对照品溶液、1ml低、中、高浓度的dccds溶液进行混合,每个浓度各设置3份。照“2.1测定方法”进行测定。结果dccds低、中、高浓度的平均回收率为100.4%,rsd为1.91%。
287.上述结果证实,本发明dccds的体外定量检测方法所检测的dccds样品稳定性好,本发明检测方法的重复性好,中间精密度高且回收率高。
288.2、fa-dccds定量方法的验证:
289.1、仪器精密度
290.为检验仪器的稳定性,取同一fa-dccds供试品溶液,照“实施例1部分3.1测定方法”项下方法测定,连续测定6次,6次测定荧光强度的rsd为1.81%,结果表明,本实验所使用仪器具有良好的精密度。
291.2、重复性
292.精密量取fa-dccds储备液适量,分别配制低(12.78μg/ml)、中(16.04μg/ml)、高(19.30μg/ml)三个浓度的fa-dccds溶液,每个浓度样品各3份,照“3.1测定方法”项下方法进行测定。结果低、中、高浓度的fa-dccds平均含量为22.87%,rsd为2.61%。实验结果表明该方法具有良好的重复性。
293.3、中间精密度
294.精密量取fa-dccds储备液适量,分别配制低(12.78μg/ml)、中(16.04μg/ml)、高(19.30μg/ml)三个浓度的fa-dccds溶液,每个浓度样品各3份,照“3.1测定方法”项下方法,由两位实验人员于不同时间不同仪器进行测定。结果低,中,高浓度的中间精密度rsd分别为1.70%,1.44%,1.82%。实验结果表明该方法具有良好的中间精密度。
295.4、样品稳定性
296.取同一fa-dccd供试品溶液,照“3.1测定方法”项下方法,分别设置0、2、4、8、12、24h等6个实验时间点,进行含量测定。结果的rsd为1.67%。表明在室温下放置24h的fa-dccd样品溶液的含量具有良好的稳定性。
297.5、回收率
298.对照品溶液的配制:取罗丹明6g对照品储备液适量,以超纯水配置为3.59μg/ml的罗丹明6g对照品溶液。
299.fa-dccds溶液的配制:精密量取fa-dccds储备液适量,以超纯水分别配制低(12.78μg/ml)、中(16.04μg/ml)、高(19.30μg/ml)三个浓度的fa-dccds溶液。
300.精密移取1ml罗丹明6g对照品溶液、1ml低中高浓度的fa-dccds溶液混合均匀,每个浓度各设置3份。照“3.2测定方法”项下方法,进行测定。结果低、中、高三个浓度的fa-dccds平均回收率为101.2%,rsd为2.05%,方法回收率良好。
301.本发明fa-dccds的体外定量检测方法所检测的fa-dccds样品稳定性好,本发明检测方法的重复性好,中间精密度高且回收率高。
302.3、acucds定量方法的验证:
303.1、仪器精密度
304.为检验仪器的稳定性,取同一acucds供试品溶液,照“实施例1部分4.1测定方法”项下方法测定,连续测定6次,6次测定荧光强度的rsd为1.88%,结果表明,本实验所使用仪器具有良好的精密度。
305.2、重复性
306.精密量取acucds储备液适量,分别配制分别配制低(6.292μg/ml)、中(7.865μg/ml)、高(9.438μg/ml)三个浓度的acucds溶液,每个浓度样品各3份,照“4.1测定方法”项下方法进行测定。结果低、中、高浓度的acucds含量为21.54%,rsd为1.86%。结果表明该方法重复性良好。
307.3、中间精密度
308.精密量取acucds储备液适量,分别配制分别配制低(6.292μg/ml)、中(7.865μg/ml)、高(9.438μg/ml)三个浓度的acucds溶液,每个浓度样品各3份,照“4.1测定方法”项下方法,由两人分别于不同仪器、不同时间进行测定。结果低,中,高浓度的中间精密度rsd分别为2.76%,2.80%,2.54%。实验结果表明该方法具有良好的中间精密度。
309.4、样品稳定性
310.取同一acucds供试品溶液,照“4.1测定方法”项下方法,分别设置0、2、4、8、12、24h 6个实验时间点,进行含量测定。结果样品稳定性实验的rsd为2.68%。实验结果表明在室温下放置24h的acucd样品溶液具有良好的稳定性。
311.5、回收率
312.对照品溶液的配制:取罗丹明6g对照品储备液适量,以超纯水配置为1.658μg/ml的罗丹明6g对照品溶液。
313.acucd溶液的配制:精密量取acucds储备液适量,分别配制分别配制低(6.292μg/ml)、中(7.865μg/ml)、高(9.438μg/ml)三个浓度的acucds溶液,每个浓度样品各3份,照“4.1测定方法”项下方法进行测定。
314.分别精密移取1ml罗丹明6g对照品溶液、1ml低中高浓度的acucd溶液混合均匀,每个浓度各设置3份,摇匀。照“4.1测定方法”项下方法,进行测定。结果表明,低、中、高三个浓度的acucd平均回收率为99.94%,rsd为2.20%。
315.上述结果证实,本发明acucds的体外定量检测方法所检测的acucds样品稳定性好,本发明检测方法的重复性好,中间精密度高且回收率高。
316.4、fa-acucds定量方法的验证:
317.1、仪器精密度
318.为检验仪器的稳定性,取同一fa-acucds供试品溶液,照“实施例1部分5.1测定方法”项下方法测定,连续测定6次,6次荧光强度的rsd为1.81%,结果表明,本实验所使用仪器具有良好的精密度。
319.2、重复性
320.精密量取fa-acucds储备液适量,分别配制低(48.13μg/ml)、中(60.16μg/ml)、高(72.19μg/ml)浓度的fa-acucds溶液,每个浓度样品各3份,照“5.1测定方法”项下方法进行测定。结果低、中、高三个浓度的fa-acucds含量为18.18%,rsd为2.25%。实验结果表明该方法具有良好的重复性。
321.3、中间精密度
322.精密量取fa-acucds储备液适量,分别配制低(48.13μg/ml)、中(60.16μg/ml)、高(72.19μg/ml)浓度的fa-acucds溶液,每个浓度样品各3份,照“5.1测定方法”项下方法,由两人分别于不同仪器、不同时间进行测定。结果低,中,高浓度的fa-acucds中间精密度rsd分别为2.36%,2.30%,2.06%。实验结果表明该方法具有良好的中间精密度。
323.4、样品稳定性
324.取同一fa-acucds供试品溶液,照“5.1测定方法”项下方法,分别设置0、2、4、8、12、24h 6个实验时间点进行含量测定。结果样品含量测定rsd为2.19%。实验结果表明在室温下放置24h的acucd样品溶液进行含量测定依然能够保证准确性。
325.5、回收率
326.对照品溶液的配制:取罗丹明6g对照品储备液适量,以超纯水配置为11.06μg/ml的罗丹明6g对照品溶液。
327.fa-acucd溶液的配制:精密量取fa-acucd储备液适量,以超纯水分别配制低(48.13μg/ml)、中(60.16μg/ml)、高(72.19μg/ml)浓度的fa-acucds溶液。
328.分别精密移取1ml罗丹明6g对照品溶液、1ml低中高浓度的fa-acucd溶液混合均匀,每个浓度各设置3份。照“5.1测定方法”项下方法,进行测定。结果显示,低、中、高三个浓度的fa-acucd平均回收率为101.7%。平均rsd为2.56%。
329.上述结果证实,本发明fa-acucds的体外定量检测方法所检测的fa-acucds样品稳定性好,本发明检测方法的重复性好,中间精密度高且回收率高。
330.5、ucpg-fe定量方法的验证:
331.1、仪器精密度
332.为检验仪器的稳定性,取同一ucpg-fe供试品溶液,照“实施例1部分6.1测定方法”项下方法测定,连续测定6次,6次测定荧光强度的rsd为1.89%,结果表明,本实验所使用仪器具有良好的精密度。
333.2、重复性
334.精密量取ucpg-fe储备液适量,分别配制低(4.992μg/ml)、中(6.240μg/ml)、高(7.488μg/ml)三个浓度的ucpg-fe溶液,每个浓度样品各设置3份,照“6.1测定方法”项下方法进行测定。结果低、中、高三个浓度的ucpg-fe的含量为26.48%,rsd为2.61%。实验结果表明该方法具有良好的重复性。
335.3、中间精密度
336.精密量取ucpg-fe储备液适量,分别配制低(4.992μg/ml)、中(6.240μg/ml)、高(7.488μg/ml)三个浓度的ucpg-fe溶液,每个浓度样品各设置3份,照“6.1测定方法”项下方法,由两人分别于不同仪器、不同时间进行测定。结果低,中,高三个浓度的ucpg-fe中间精密度rsd分别为2.11%,2.47%,2.11%。实验结果表明该方法具有良好的中间精密度。
337.4、样品稳定性
338.取同一ucpg-fe供试品溶液,分别设置0、2、4、8、12、24h 6个实验时间点,照“6.1测定方法”项下方法,进行含量测定。结果样品稳定性实验rsd为2.02%。实验结果表明在室温下放置24h后的ucpg-fe样品溶液进行含量测定依然能够保证准确性。
339.5、回收率
340.对照品溶液的配制:取罗丹明6g对照品储备液适量,以超纯水配置为1.641μg/ml的罗丹明6g对照品溶液。
341.ucpg-fe溶液的配制:精密量取ucpg-fe储备液适量,以超纯水分别配制低(4.992μg/ml)、中(6.240μg/ml)、高(7.488μg/ml)三个浓度的ucpg-fe溶液。
342.分别精密移取1ml罗丹明6g对照品溶液、1ml低中高浓度的ucpg-fe溶液混合均匀,每个浓度各设置3份。照“6.1测定方法”项下方法,进行测定。实验结果显示,低、中、高三个浓度的ucpg-fe溶液平均回收率为99.61%。rsd为2.33%。
343.上述结果证实,本发明ucpg-fe的体外定量检测方法所检测的ucpg-fe样品稳定性好,本发明检测方法的重复性好,中间精密度高且回收率高。
344.6、fa-ucpg-fe定量方法的验证:
345.1、仪器精密度
346.为检验仪器的稳定性,取同一fa-ucpg-fe供试品溶液,照“实施例1部分7.1测定方法”项下方法测定,连续测定6次,6次测定荧光强度rsd为2.60%,结果表明,本实验所使用仪器具有良好的精密度。
347.2、重复性
348.精密量取fa-ucpg-fe储备液适量,分别配制低(1.920μg/ml)、中(2.400μg/ml)、高浓度(2.880μg/ml)的fa-ucpg-fe溶液,每个浓度样品各设置3份,照“7.1测定方法”项下方法进行测定。结果低、中、高三个浓度的ucpg-fe平均含量为23.47%,rsd为2.04%。实验结果表明该方法重复性良好。
349.3、中间精密度
350.精密量取fa-ucpg-fe储备液适量,分别配制低(1.920μg/ml)、中(2.400μg/ml)、高浓度(2.880μg/ml)的fa-ucpg-fe溶液,每个浓度样品各设置3份,照“7.1测定方法”项下方法,由两人分别于不同仪器、不同时间进行测定。结果低,中,高三个浓度的fa-ucpg-fe溶液中间精密度rsd分别为1.92%,1.42%,1.52%。实验结果表明该方法具有良好的中间精密度。
351.4、样品稳定性
352.取同一fa-ucpg-fe供试品溶液,分别设置0、2、4、8、12、24h 6个实验时间点,照“7.1测定方法”项下方法测定,测定结果样品含量测定rsd为1.69%。实验结果表明在室温下放置24h的fa-ucpg-fe样品溶液进行含量测定能够保证准确性。
353.5、回收率
354.对照品溶液的配制:取罗丹明6g对照品储备液适量,以超纯水配置为0.5612μg/ml的罗丹明6g对照品溶液。
355.fa-acucd溶液的配制:精密量取fa-ucpg-fe储备液适量,以超纯水配制为1.920mg/ml、2.400mg/ml、2.880mg/ml的低中高三个浓度的fa-acucd溶液。
356.分别精密移取1ml罗丹明6g对照品溶液、1ml低中高浓度的fa-ucpg-fe溶液混合均匀,每个浓度各设置3份。照“7.1测定方法”项下方法,进行测定。测定结果实验结果显示,fa-ucpg-fe低、中、高浓度的平均回收率为99.86%。平均rsd为2.12%。
357.上述结果证实,本发明fa-ucpg-fe的体外定量检测方法所检测的fa-ucpg-fe样品稳定性好,本发明检测方法的重复性好,中间精密度高且回收率高。
358.实验例2、本发明dccds和fa-dccds细胞内定量检测方法的验证
359.1、dccds细胞内定量检测方法的验证
360.1、仪器精密度
361.为检验仪器的稳定性,取同一dccds供试品溶液,照“实施例2部分3.1.2测定方法”项下方法测定,连续测定6次,6次检测荧光强度rsd为2.96%,结果表明,本实验所使用仪器具有良好的精密度。
362.2、日内精密度
363.精密量取dccds储备液(以甲酚紫为对照标示含量为34.40%,0.3474mg/ml)适量,分别配制含量低中高三个浓度的溶液,加入已有50μl细胞液的96孔板,得到低(0.2316μg/ml)、中(0.2895μg/ml)、高(0.3474μg/ml)三个浓度的供试品溶液,每个浓度样品各3份,照“实施例2的3.1.2测定方法”项下方法进行测定。结果低、中、高浓度的dccds日内精密度rsd分别为3.61%、4.67%、3.44%。实验结果表明该方法日内精密度良好。
364.3、日间精密度
365.精密量取dccds储备液(以甲酚紫为对照标示含量为34.40%,0.3474mg/ml)适量,分别配制含量低中高三个浓度的溶液,加入已有50μl细胞液的96孔板,得到低(0.2316μg/ml)、中(0.2895μg/ml)、高(0.3474μg/ml)三个浓度的供试品溶液,每个浓度样品各3份,照“实施例2的3.1.2测定方法”项下方法,由两人分别于不同仪器、不同时间进行测定。结果细胞中dccds低,中,高浓度的日间精密度rsd分别为4.15%,4.15%,3.41%。实验结果表明该方法日间精密度良好。
366.4、样品稳定性
367.取同一dccd供试品溶液,分别设置0、2、4、8、12、24h 6个实验时间点,照“实施例2的3.1.2测定方法”项下方法,进行测定,结果样品含量测定的rsd为2.14%。结果表明在室温下放置24h的dccd样品溶液具有良好的稳定性。
368.5、回收率
369.精密量取dccds储备液(以甲酚紫为对照标示含量为34.40%,0.3474mg/ml)适量,分别配制含量低中高三个浓度的溶液各10ml,每个浓度样品各3份。取每一样品50μl加入已有50μl细胞液的96孔板,得到低(0.2316μg/ml)、中(0.2895μg/ml)、高(0.3474μg/ml)三个浓度的供试品溶液。照“实施例2的3.1.2测定方法”项下方法,进行测定。测定结果实验结果显示,低、中、高三个浓度的dccds的平均回收率为102.5%,rsd为3.28%。
370.上述结果证实,本发明dccds的细胞内定量检测方法所检测的dccds样品稳定性好,本发明检测方法的重复性好,日内、日间精密度高且回收率高。
371.2、fa-dccds细胞内定量检测方法的验证
372.1、仪器精密度
373.为了检验仪器的稳定性,取同一fa-dccds供试品溶液,照“4.1.2测定方法”项下测定方法,连续测定6次,6次测定荧光强度rsd为3.84%,结果表明,本实验所用仪器具有良好的精密度。
374.2、日内精密度
375.精密量取fa-dccds储备液(以罗丹明6g为对照标示含量为22.92%,0.2285mg/ml)适量,分别配制含量低中高三个浓度的溶液,加入已有50μl细胞液的96孔板,得到低
(0.7334μg/ml)、中(0.9168μg/ml)、高(1.100μg/ml)三个浓度的供试品溶液,每个浓度样品各3份,每个浓度样品各设置3份,照“4.1.2测定方法”项下方法进行测定。结果低、中、高三个浓度的fa-dccds溶液日内精密度rsd分别为2.04%、2.77%、3.64%。实验结果表明该方法日内精密度良好。
376.3、日间精密度
377.精密量取dccds储备液(以罗丹明6g为对照标示含量为22.92%,0.2285mg/ml)适量,分别配制含量低中高三个浓度的溶液,加入已有50μl细胞液的96孔板,得到低(0.7334μg/ml)、中(0.9168μg/ml)、高(1.100μg/ml)三个浓度的供试品溶液,每个浓度样品各3份,每个浓度样品各设置3份,照“4.1.2测定方法”项下方法,由两人分别于不同仪器、不同时间进行测定。结果低,中,高浓度的日间精密度rsd分别为2.29%,2.46%,1.94%。实验结果表明该方法具有良好的日间精密度。
378.4、样品稳定性
379.取同一fa-dccd供试品溶液,分别设置0、2、4、8、12、24h 6个实验时间点,照“4.1.2测定方法”项下方法,进行测定。结果样品含量的rsd为2.64%。结果表明在室温下放置24h的fa-dccd样品溶液具有良好的稳定性。
380.5、回收率
381.精密量取fa-dccds储备液(以罗丹明6g为对照标示含量为22.92%,0.2285mg/ml)适量,分别配制含量低中高三个浓度的溶液各10ml,每个浓度样品各3份。取每一样品50μl加入已有50μl细胞液的96孔板,得到低(0.7334μg/ml)、中(0.9168μg/ml)、高(1.100μg/ml)三个浓度的供试品溶液。照“4.1.2测定方法”项下方法,进行测定。结果显示低、中、高三个浓度的fa-dccds溶液平均回收率为100.9%,rsd为3.87%,显示出较好的准确度。
382.上述结果证实,本发明fa-dccds的细胞内定量检测方法所检测的fa-dccds样品稳定性好,本发明检测方法的重复性好,日内、日间精密度高且回收率高。
383.上述结果表明,在一定浓度范围内候选的碳点及其fa偶联物均具有良好的线性关系,且细胞液环境不干扰碳点的荧光光谱,体现了碳点抗漂白性的特性,同时能够避免生物样品繁杂的前处理过程,只需要将细胞直接裂解即可实现检测,提示本发明方法具有实现快速检测的潜质。
384.实验例3、本发明dccds组织内定量检测方法的验证
385.1、回收率与精密度
386.肝组织液:精密量取dccds储备液(以甲酚紫为对照标示含量为34.40%,0.3474mg/ml)适量,以氯化钠(0.9%)溶液配制定量下限、低、中、高四个浓度的dccds样品溶液,每个浓度样品各5份。取肝组织液100μl,每一样品100μl,混合均匀,得到定量下限(0.4300μg/ml)、低(1.720μg/ml)、中(3.440μg/ml)、高(5.160μg/ml)四个浓度的供试品溶液,照“3.1测定方法”进行荧光测定。
387.检测结果:肝组织液中dccds回收率在86.61%~108.8%之间,日内精密度rsd在4.83~8.53%之间和日间精密度rsd在0.26~2.80%之间。结果表明,该方法具有较好的精密度。
388.肾组织液:精密量取dccds储备液(以甲酚紫为对照标示含量为34.40%,0.3474mg/ml)适量,以氯化钠(0.9%)溶液配制定量下限、低、中、高四个浓度的dccds样品
溶液,每个浓度样品各5份。取肾组织液100μl,每一样品100μl,混合均匀,得到定量下限(0.2334μg/ml)、低(0.4736μg/ml)、中(1.915μg/ml)高(2.876μg/ml)四个浓度的供试品溶液,照“4.1测定方法”进行荧光测定。
389.检测结果:肾组织液中dccds回收率在89.83~112.9%之间,日内精密度的rsd在3.49~8.76%之间,日间精密度的rsd在0.95~2.64%之间。
390.2、稳定性
391.将dccds储备液与肝组织液、肾组织液分别混合均匀,分别在室温、-20℃冷冻条件下放置一段时间,进行荧光检测,计算平均浓度并判断平均准确度,发现,dccds的组织液样品在室温下可稳定放置8h以上,平均准确度仍接近100%。在-20℃条件下放置30天,平均准确度仍高达100%;并且,在-20℃条件反复冻融3次,检测含量仍然稳定。说明dccds的组织液样品稳定性很好,在实际临床应用时有极高的潜力。
392.上述结果说明,在一定浓度范围内候选碳点在组织液中的线性范围良好,组织液不影响碳点的检测,灵敏度、日内日间精密度均满足检测要求。
393.综上,本发明提供了一种在体外、细胞内、组织内定量检测碳点含量的方法,该方法准确度高、重复性好、精密度高、回收率高。突破了碳点无对照品,难以进行定量检测的瓶颈,为碳点质量标准的建立以及生物诊疗方面的深入研究提供定量研究的方法。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。