他氟前列素的纯化方法
1.本技术是申请日为2021年1月27日、申请号为202110111991.0、发明名称为“他氟前列素的纯化方法”的申请的分案申请。
技术领域
2.本发明涉及他氟前列素的新型纯化方法。
背景技术:
3.他氟前列素用下述式表示:
[0004][0005]
其化学名为(5z)-7-(1r,2r,3r,5s)-2-[(1e)-3,3-二氟-4-苯氧基-1-丁烯基]-3,5-二羟基环戊基]-5-庚烯酸异丙酯,在25℃下具有2440mpa
·
s的粘度,是粘性非常高的二氟前列腺素f
2α
衍生物。他氟前列素具有包含2个双键、不饱和脂肪酸酯部位、及4个不对称中心的不稳定的化学结构,具有其他前列腺素衍生物中存在的c15位的羟基和氢原子被2个氟原子取代而成的结构,因此在前列腺素衍生物之中具有显著的脂溶性高的特异物性,作为前列腺素衍生物,虽然化学稳定性高,但还具有在高温下发生分解的性质。另外,他氟前列素具有强效的眼压下降作用,作为眼药水被用于青光眼、高眼压症的治疗(专利文献1)。专利文献1中记载了包含他氟前列素的二氟前列腺素f
2α
衍生物的制造方法,另外,非专利文献1中也记载了同样的制造方法。
[0006]
专利文献1中记载的制造方法包括wittig反应工序,因此难以避免最终产物中的α链反式异构体的混入。专利文献1记载的制造方法中,作为去除包含α链反式异构体的杂质的方法,报告了利用分级用hplc(high performance liquid chromatography(高效液相色谱法))进行分离纯化的方法(专利文献2)。然而,他氟前列素、及作为其合成前体的下述式(i)所示的羧酸化合物(以下称作“他氟前列素酸”。):
[0007][0008]
均为具有非常高粘性的液状化合物,因此难以纯化,另外,专利文献2记载的他氟前列素的纯化方法中,使用大量的有机溶剂,因此需要很高的成本,并且也难以将残留有机溶剂浓度抑制在医药品的残留溶剂准则(非专利文献2)的浓度限度值以下。分级用hplc的柱通常为高价,通常被反复使用,因此具有累积的杂质、分解物的混入、柱的劣化导致的理论塔板数降低的问题,并且为了降低以这些问题为起因的风险,也经常需要使用大量有机溶剂的清洗、以及与其确认、柱的分离性能的确认等相关的烦杂的有效性验证,因此,作为医药品的制造方法缺乏实用性。
[0009]
另一方面,还报告了通过经由他氟前列素酸的有机胺盐(专利文献3、4)或金属盐(专利文献5)来使α链反式异构体等杂质的混入减少的方法,但随着盐形成工序及从盐的游离化工序的追加,基于与有机胺的缩合、自缩合的二聚化等产生的副产物、脱水物、其他杂质等也有增加的可能性。另外,有机胺、金属大多还存在毒性、致突变性等的顾虑,特别是对于在接近医药品的最终工序的阶段作为纯化法使用而言,在安全性上存在问题。
[0010]
进而,还报告有经过大环内酯环形成、及大环内酯环的开环工序,由此防止α链反式异构体的混入的他氟前列素的制造方法(专利文献6)。然而,所述制造方法的制造工序长且收率低,因此实用性低。
[0011]
现有技术文献
[0012]
专利文献
[0013]
专利文献1:欧州专利申请公开第850926号说明书
[0014]
专利文献2:美国专利申请公开第2014/0051882号说明书
[0015]
专利文献3:国际公开第2013/118058号
[0016]
专利文献4:国际公开第2016/090461号
[0017]
专利文献5:中国专利申请公开第108299192号说明书
[0018]
专利文献6:日本特开2015-36382号公报
[0019]
非专利文献
[0020]
非专利文献1:tetrahedron lett.,2004,45,1527-1529
[0021]
非专利文献2:医药审第307号厚生省药品安全局审查管理课长通知(平成10年3月30日),关于医药品的残留溶剂准则
技术实现要素:
[0022]
发明要解决的问题
[0023]
本发明的目的在于提供一种他氟前列素的纯化方法,其可以将作为粘性非常高的液态化合物的他氟前列素简便且低成本地纯化至能够直接作为医药品的原料药提供的程度的纯度,并且还可以按比例放大(scale up)。
[0024]
用于解决问题的方案
[0025]
本发明人等为了解决上述问题,进行深入研究结果发现:他氟前列素的制造方法中,通过包括利用硅胶柱色谱法对他氟前列素酸的酯化工序所得到的他氟前列素的粗产物进行纯化,并利用hplc分析收集包含他氟前列素的组分的工序的纯化方法(以下有时也称作“本发明的纯化方法”。),可以得到高纯度的他氟前列素。另外,通过进行包括在10~55℃下对利用hplc分析收集的包含他氟前列素的组分进行减压浓缩的工序、将残渣溶解于溶剂并进行过滤的工序、及在10~55℃、最终极限真空度为5torr以下的减压下将滤液的溶剂蒸馏去除的工序的纯化方法(以下,有时也将包括前述全部工序的纯化方法称作“本发明的纯化方法”。),可以将残留有机溶剂浓度抑制在医药品的残留溶剂准则的浓度限度值以下,并得到具有可以直接作为医药品的原料药提供的纯度的他氟前列素,从而完成了本发明。
[0026]
即,本发明如下。
[0027]
[1]一种他氟前列素的纯化方法,其包括:利用硅胶柱色谱法对他氟前列素的粗产物进行纯化,并利用hplc分析收集包含他氟前列素的组分的工序。
[0028]
[2]根据上述[1]所述的他氟前列素的纯化方法,其还包括:在10~55℃下对利用hplc分析收集的包含他氟前列素的组分进行减压浓缩的工序;之后将残渣溶解于溶剂并进行过滤的工序;及在10~55℃、最终极限真空度为5torr以下的减压下,将滤液的溶剂蒸馏去除的工序。
[0029]
[3]根据上述[1]或[2]所述的纯化方法,其中,硅胶柱色谱法中使用的硅胶的粒径(d50)为20~70μm。
[0030]
[4]根据上述[1]~[3]中任一项所述的纯化方法,其中,硅胶柱色谱法中使用的硅胶为球状。
[0031]
[5]根据上述[1]~[4]中任一项所述的纯化方法,其中,硅胶柱色谱法的洗脱液为正己烷与极性溶剂的混合溶剂,或正庚烷与极性溶剂的混合溶剂。
[0032]
[6]根据上述[5]所述的纯化方法,其中,洗脱液为正己烷与极性溶剂的混合溶剂。
[0033]
[7]根据上述[5]或[6]所述的纯化方法,其中,极性溶剂为乙酸乙酯、叔丁基甲基醚、2-丙醇、或乙醇。
[0034]
[8]根据上述[1]~[7]中任一项所述的纯化方法,其中,hplc分析为反相hplc分析。
[0035]
[9]根据上述[1]~[8]中任一项所述的纯化方法,其中,组分为包含98%以上的他氟前列素的组分。
[0036]
[10]根据上述[2]~[9]中任一项所述的纯化方法,其中,使用具有0.5μm以下的孔径的过滤器进行过滤。
[0037]
[11]根据上述[2]~[10]中任一项所述的纯化方法,其中,用于溶解残渣的溶剂为乙酸乙酯、叔丁基甲基醚、2-丙醇或乙醇,或者为乙酸乙酯、叔丁基甲基醚、2-丙醇或乙醇与非极性溶剂的混合溶剂。
[0038]
[12]根据上述[11]所述的纯化方法,其中,用于溶解残渣的溶剂为乙酸乙酯、或乙酸乙酯与非极性溶剂的混合溶剂。
[0039]
[13]根据上述[11]或[12]所述的纯化方法,其中,非极性溶剂为正己烷或正庚烷。
[0040]
[14]根据上述[2]~[13]中任一项所述的纯化方法,其中,最终极限真空度为1torr以下。
[0041]
[15]根据上述[2]~[14]中任一项所述的纯化方法,其中,对滤液的溶剂进行蒸馏去除的工序后的正己烷的残留溶剂浓度为290ppm以下,正庚烷、乙酸乙酯、叔丁基甲基醚、2-丙醇或乙醇的残留溶剂的浓度分别为5000ppm以下。
[0042]
[16]一种他氟前列素的制造方法,其包括将他氟前列素的粗产物供于上述[1]~[15]中任一项所述的纯化方法的工序。
[0043]
[17]一种他氟前列素,其是利用上述[16]所述的制造方法得到的。
[0044]
[18]一种药品,其以上述[17]所述的他氟前列素作为有效成分。
[0045]
[19]一种药品,其以上述[17]所述的他氟前列素作为有效成分,用于眼部疾病的预防或治疗。
[0046]
[20]根据上述[19]所述的药品,其中,眼部疾病为青光眼或高眼压症。
[0047]
发明的效果
[0048]
通过本发明的纯化方法,在他氟前列素的制造的最终工序中,在利用硅胶柱色谱
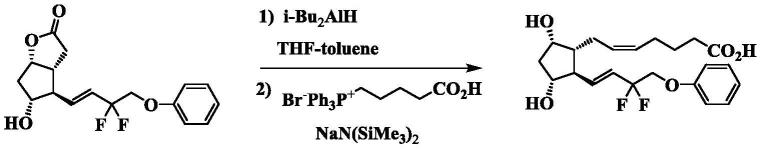
法对他氟前列素的粗产物进行分离纯化时,通过利用hplc分析收集包含他氟前列素的组分,可以将杂质的混入抑制为最小限度。另外,在低温且高真空度的减压条件下,花费时间将溶剂蒸馏去除,由此可以将残留有机溶剂浓度抑制在医药品的残留溶剂准则的浓度限度值以下,并且还可以抑制高温下不稳定的他氟前列素的分解。进而,通过在中途组入过滤器过滤工序,可以去除硅胶的细粉、空气中的浮游颗粒、及细菌,因此在溶剂的蒸馏去除后,可以简便且高效地提供能够直接作为医药品的原料药使用的高纯度的他氟前列素。本发明的纯化方法可以广泛应用于以公知的方法制造的他氟前列素的粗产物,且可以承受按比例放大。
具体实施方式
[0049]
以下对本发明的实施的方式进行详细说明。
[0050]
[用语的定义]
[0051]
本说明书中的用语的含义如下。
[0052]
本说明书中,作为用于“硅胶柱色谱法”的柱,使用常压柱及快速柱均可。
[0053]
本说明书中的“硅胶柱色谱法”为正相系的柱色谱法。
[0054]
本说明书中,“他氟前列素的粗产物”是指在公知的他氟前列素的制造方法中的最终工序的反应的后处理后、纯化前的产物。具体而言,例如,如后述实施例所示,可举出专利文献1中记载的他氟前列素的制造方法中的最终的酯化反应的纯化前的产物等。
[0055]
本说明书中,“杂质”除包括前述他氟前列素的粗产物所包含的残留反应试剂、残留原料化合物、反应的副产物、他氟前列素的分解物等类似物质外,还包括残留有机溶剂、源自填充剂的残留物、细菌等他氟前列素以外的全部物质。
[0056]
本说明书中,“hplc分析”是指利用硅胶柱色谱法对他氟前列素的粗产物进行分离纯化时,使用分析用的高效液相色谱法(high performance liquid chromatography)确认各组分中他氟前列素的有无、含有比率。
[0057]
本说明书中,“过滤”是指过滤器过滤。过滤以去除柱填充剂(硅胶)的细粉、空气中的浮游颗粒、细菌等为目的进行。
[0058]
本说明书中,“极性溶剂”是指介电常数大的溶剂。作为极性溶剂的具体例,可举出例如乙酸乙酯、乙酸丙酯等酯类、二乙醚、叔丁基甲基醚、四氢呋喃等醚类、2-丙醇、乙醇等醇类等。其中,优选乙酸乙酯、叔丁基甲基醚、2-丙醇或乙醇。
[0059]
本说明书中,“非极性溶剂”是指介电常数小的溶剂。作为非极性溶剂的具体例,可举出例如正己烷、正庚烷等链状烃类。其中,优选正己烷。
[0060]
本说明书中,“外温”是指反应容器或浓缩用容器的外侧的温度,通常为外部气温、或者水浴或热水浴的温度。
[0061]
本说明书中,“医药品的残留溶剂准则的浓度限度值”是作为日美eu三方药品许可审查协调国际会议(ich)的课题之一进行研究并为了患者的安全而对医药品中的残留溶剂的容许量进行规定的数值,是指残留溶剂的毒性学上可以容许的限度值。作为医药品的残留溶剂的浓度限度值的具体例,如非专利文献2的记载,例如正己烷为290ppm,正庚烷、乙酸乙酯、叔丁基甲基醚、2-丙醇或乙醇为5000ppm。
[0062]
[本发明的纯化方法]
[0063]
本发明的纯化方法的特征在于,包括利用硅胶柱色谱法对他氟前列素的粗产物进行纯化,并利用hplc分析收集包含他氟前列素的组分的工序(工序1)。另外,为了将残留有机溶剂浓度抑制在医药品的残留溶剂准则的浓度限度值以下,本发明的纯化方法的特征在于,除前述工序1外,还包括在10~55℃下进行减压的浓缩工序(工序2),将残渣溶解于溶剂并进行过滤的工序(工序3),及在10~55℃、最终极限真空度为5torr以下的减压下,将滤液的溶剂蒸馏去除的工序(工序4)。
[0064]
(工序1)
[0065]
本工序为利用硅胶柱色谱法进行纯化,并利用hplc分析收集包含他氟前列素的组分的工序。
[0066]
作为用于硅胶柱色谱法的填充剂,只要是可以用于通常的正相系柱的硅胶就没有特别限定。作为该硅胶的形状,为粉碎状及球状均可,更优选球状。另外,该硅胶的粒径(d50)并没有特别限定,优选为20μm~70μm,更优选为40μm~65μm,特别优选为45μm~60μm。该粒径(d50)为利用激光衍射散射式粒度分布测定,以体积基准制作粒度分布时的该粒度分布的中值粒径。
[0067]
作为用于硅胶柱色谱法的洗脱液,只要是可以将他氟前列素的粗产物中的他氟前列素与杂质分离的溶剂,就没有特别限定,优选正己烷与极性溶剂的混合溶剂、或正庚烷与极性溶剂的混合溶剂,更优选正己烷与极性溶剂的混合溶剂。此处,极性溶剂选自乙酸乙酯、叔丁基甲基醚、2-丙醇及乙醇,其中,优选2-丙醇或乙醇。正己烷与极性溶剂、或正庚烷与极性溶剂的混合比(体积比)可以根据使用的填充剂的种类、形状、和/或粒径来适当设定。作为洗脱液的优选具体例,可举出例如乙酸乙酯、叔丁基甲基醚、2-丙醇或乙醇与非极性溶剂(优选正己烷或正庚烷)的混合溶剂,更优选2-丙醇或乙醇与非极性溶剂(优选正己烷或正庚烷)的混合溶剂,特别优选乙醇与正己烷的混合溶剂。作为洗脱液,使用混合溶剂时的混合比(体积比)并没有特别限定,但从将残留溶剂浓度控制在基准值以下的观点来看,在乙醇与正己烷的混合溶剂的情况下,优选使用以10:90~1:99混合乙醇:正己烷而成的溶剂,更优选以6:94~2:98混合而成的溶剂,进一步优选以5:95~3:97混合而成的溶剂,特别优选以4:96混合而成的溶剂。
[0068]
利用硅胶柱色谱法纯化确认分离的组分中有无他氟前列素时,使用分析用的hplc。作为该hplc,使用正相hplc及反相hplc均可,但更优选杂质的分离效率、检测灵敏度、及定量性等方面优异的反相hplc。作为该hplc分析中使用的柱、分析条件的具体例,可举出后述实施例中记载的条件,但并不限定于此。
[0069]
通常,进行硅胶柱色谱法纯化时,分离的组分中有无目的物的确认使用tlc(薄层色谱)进行(参照实验化学讲座1基本操作i(第4版),平成2年11月5日发行,丸善,5/2/3柱色谱,p.293-296),但发现在他氟前列素的粗产物的纯化中,与惯用的tlc分析相比,在包含他氟前列素的组分及杂质的检测灵敏度的方面,hplc分析显著优异。
[0070]
针对合成的他氟前列素粗产物的多个批次进行硅胶柱色谱法,利用hplc分析对杂质的洗脱图案进行解析,结果确认杂质洗脱图案一直稳定。通过考量所述杂质洗脱图案可知,优选收集各组分的他氟前列素的hplc面积百分比为97%以上的连续组分,特别优选收集98%以上的连续组分。
[0071]
(工序2)
[0072]
本工序为对利用hplc分析确认的包含他氟前列素的组分进行收集,并在10~50℃下进行减压浓缩的工序。
[0073]
对利用hplc分析确认的包含他氟前列素的组分进行收集并减压浓缩时的外温(水浴或热水浴的温度)优选为10℃~55℃,更优选15℃~50℃,特别优选20~45℃。如后述试验例所示,确认了他氟前列素在60℃以上的温度下会随着时间经过而缓缓进行分解,从该方面来看,在上述温度下进行减压浓缩、或将溶剂蒸馏去除也是理想的。
[0074]
(工序3)
[0075]
本工序为将前述工序2得到的残渣溶解在溶剂中并进行过滤的工序。他氟前列素的粘性非常高,因此难以在将溶剂完全蒸馏去除后再进行灭菌过滤。因此,本发明的纯化方法的特征在于,在工序2之后组入过滤工序。
[0076]
作为用于溶解工序2得到的残渣的溶剂,可举出与工序1中的硅胶柱色谱法所使用的洗脱液同样的溶剂,优选充分溶解他氟前列素且沸点比较低的溶剂,也可以为与形成共沸组成的非极性溶剂的混合溶剂。具体而言,优选为乙酸乙酯、叔丁基甲基醚、2-丙醇或乙醇,或者为乙酸乙酯、叔丁基甲基醚、2-丙醇或乙醇与非极性溶剂(优选正己烷或正庚烷)的混合溶剂,更优选乙酸乙酯、或乙酸乙酯与非极性溶剂(优选正己烷或正庚烷)的混合溶剂,特别优选乙酸乙酯与正己烷的混合溶剂。使用混合溶剂时的混合比(体积比)并没有特别限定,从将残留溶剂浓度控制在基准值以下的观点来看,在乙酸乙酯与正己烷的混合溶剂的情况下,特别优选使用以10:1~1:10、优选为4:1~1:4、更优选为2:1~1:2混合乙酸乙酯:正己烷而成的溶剂。
[0077]
作为本工序的过滤中使用的过滤器,只要不因溶剂而溶胀、溶解且可以去除填充剂(硅胶)的细粉、空气中的浮游颗粒等,就没有特别限定,可举出例如玻璃纤维制过滤器、聚丙烯制过滤器、尼龙制过滤器、氟树脂制过滤器等,优选聚偏氟乙烯(pvdf)、聚四氟乙烯(ptfe)等氟树脂制过滤器,其中,特别优选聚四氟乙烯(ptfe)制过滤器。
[0078]
该过滤器的孔径通常为0.5μm以下,优选为0.25μm以下,在还包括灭菌目的的情况下,特别优选为0.22μm以下。
[0079]
(工序4)
[0080]
本工序为将前述工序3得到的滤液的溶剂在10~55℃、最终极限真空度为5torr以下的减压下进行蒸馏去除的工序。
[0081]
他氟前列素的粘性非常高,因此必须尽可能增大溶剂蒸发的表面积并且边避免暴沸边花时间缓缓地将滤液的溶剂蒸馏去除。作为用于达成所述目的的减压浓缩装置,可举出例如旋转蒸发仪、离心蒸发装置、高真空薄膜蒸留装置等。
[0082]
另外,如前所述,从他氟前列素在60℃以上的温度下会随时间经过而缓缓进行分解的方面来看,在减压下将溶剂蒸馏去除时的外温(水浴或热水浴的温度)优选为10℃~55℃,更优选为15℃~50℃,特别优选为20℃~45℃。
[0083]
对于蒸馏去除溶剂时的减压度而言,尽量增大溶剂蒸发的表面积并且边避免暴沸边花费时间来缓缓提高,优选以最终极限真空度成为5torr以下(优选为3torr以下,更优选为1torr以下,特别优选为0.5torr以下)的方式进行控制。另外,在解除减压时,为了防止空气中的浮游颗粒、细菌的侵入,优选使用通过过滤器的空气来恢复至常压。
[0084]
对溶剂进行蒸馏去除的时间优选为10~70小时,更优选为15~60小时,特别优选
为20~60小时。
[0085]
通过使用本发明的纯化方法,可以将残留有机溶剂浓度抑制在医药品的残留溶剂准则(非专利文献2)的浓度限度值以下,如本准则中所记载的“残留溶剂无助于治疗,因此应当将全部残留溶剂降低至能够符合产品标准、gmp或其他品质基准的水平以下。”,可以稳定地制造能够进一步符合严格的各种品质基准的、高品质的他氟前列素。
[0086]
作为用于溶解前述硅胶柱色谱法中使用的洗脱液、残渣的溶剂,理想的是:以优选溶剂的方式进行了例示的乙酸乙酯、叔丁基甲基醚、2-丙醇、乙醇、或正庚烷的残留溶剂浓度更优选可以分别控制为1000ppm以下、特别优选分别控制为100ppm以下而制造,另外,理想的是:正己烷的残留溶剂浓度更优选可以控制为200ppm以下,特别优选控制为20ppm以下而制造。残留溶剂的浓度可以利用气体色谱(gc)等方法测定。
[0087]
本发明还包括将以公知的方法制造的他氟前列素的粗产物供于本发明的纯化方法(包括上述工序1~4的纯化方法)的工序的他氟前列素的制造方法。作为他氟前列素的公知的制造方法,除前述专利文献1及非专利文献1外,还有若干报告例(例如,美国专利申请公开第2014/0046086号说明书;j.org.chem.2016,81,10832-844;molecules,2017,22,217,1-16;org.lett.2020,22,2991-2994等),这些也可通过与本发明的纯化方法组合而包含于本发明。
[0088]
作为本发明中使用的他氟前列素的粗产物的具体例,可举出例如由被羟基等保护的他氟前列素利用脱保护反应得到的他氟前列素的粗产物、利用他氟前列素酸的盐的酯化得到的他氟前列素的粗产物、及利用他氟前列素酸的酯化得到的他氟前列素的粗产物等。其中,可以适宜地使用利用他氟前列素酸的酯化得到的他氟前列素的粗产物。
[0089]
作为本发明的纯化方法的具体特征,可举出以下。
[0090]
(a)利用硅胶柱色谱法对他氟前列素的粗产物进行分离纯化时,利用hplc分析(优选反相hplc分析)收集包含他氟前列素的组分,由此可以将杂质的混入抑制为最小限度。
[0091]
(b)在低温且高真空度的减压条件下花费时间将溶剂蒸馏去除,由此可以抑制在高温下不稳定的他氟前列素的分解,另外,可以将残留有机溶剂浓度抑制在医药品的残留溶剂准则的浓度限度值以下。
[0092]
(c)在中途组入过滤器过滤工序,由此在溶剂的蒸馏去除后能够提供可以直接作为医药品的原料药使用的高纯度的他氟前列素。
[0093]
(d)本发明的纯化方法也可应用于使用公知的他氟前列素的制造方法中的任意方法得到的他氟前列素的粗产物,另外,可以容易地进行按比例放大,因此可以提供简便且高效的纯化方法。
[0094]
本发明的纯化方法如上述(a)的记载,在以提高他氟前列素的纯度为目的而进行时,仅包括上述工序1即可,工序2~4也可以分别根据需要与工序1进行组合。
[0095]
实施例
[0096]
以下举出参考例、实施例及试验例,对本发明进行详细说明,但本发明并不限定于这些。
[0097]
%在收率中表示mol%,在其他中若无特别说明则表示质量%。混合溶剂中示出的比若无特别说明则表示容积比。另外,室温若无特别说明则表示15~30℃的温度。以下的1h-nmr值用核磁共振装置日本电子株式会社制ecp400(400mhz)测定。hplc装置使用
shimadzu lc-10advp或lc-10a。gc装置使用shimadzu gc-2014atf。
[0098]
参考例1:他氟前列素酸的合成
[0099][0100]
在氮气气氛下,在(1s,5r,6r,7r)-6-[(1e)-3,3-二氟-4-苯氧基-1-丁烯基]-7-羟基-2-氧杂双环[3.3.0]辛烷-3-酮(280g)中添加四氢呋喃(1200g)并溶解,在-70℃下滴加二异丁基氢化铝(1m甲苯溶液)(2160ml)。滴加结束后搅拌30分钟,添加1n盐酸,用乙酸乙酯进行提取。合并有机层并水洗后,对滤液进行减压浓缩,由此得到还原体(284g)。在氮气气氛下,在4-羧丁基三苯基溴化鏻(1523g)中添加四氢呋喃(5030g),滴加双(三甲基甲硅烷基)酰胺钠溶液(1m四氢呋喃溶液)(6684ml),搅拌1小时以上。在0℃下滴加溶解于四氢呋喃(970g)的上述的还原体(286g)并搅拌3小时。在反应液中添加水,用乙酸乙酯提取。使水层为酸性后,用乙酸乙酯提取,减压浓缩后,滤除不溶物,用硅胶柱色谱法(己烷/乙酸乙酯=1/1~1/3)进行纯化,由此得到他氟前列素酸(222g)。
[0101]1h nmr(cdcl3)δ1.60(m,1h),1.67(m,2h),1.84(m,1h),2.02-2.16(m,4h),2.25-2.35(m,3h),2.47(m,1h),4.03(m,1h),4.18(m,3h),5.35-5.42(m,2h),5.80(m,1h),6.10(m,1h),6.91(m,2h),7.00(m,1h),7.30(m,2h).
[0102]
参考例2:他氟前列素粗产物的合成
[0103][0104]
在氮气气氛下,于5l的烧瓶中投入参考例1得到的他氟前列素酸(120g),边搅拌边使其溶解于丙酮(600ml)。冷却至5℃,边保持在5℃以下边滴加1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu)(160ml),进而边保持在5℃以下边滴加2-碘丙烷(146ml)后,在30℃下搅拌至反应的转化率为95%以上。在反应混合物中添加乙酸乙酯(1800ml)及5%柠檬酸水溶液(900ml)并分液,用5%柠檬酸水溶液(900ml、1次)、5%碳酸氢钠水溶液(900ml、2次)、及纯化水(900ml、1次)清洗有机层。在减压下,于40℃以下蒸馏去除溶剂,由此得到他氟前列素粗产物(132g,收率100%;hplc纯度:95.5%,α链反式异构体含量:0.73%)。
[0105]
实施例1
[0106]
(工序1)将由硅胶(agc-sitech co.,ltd制、m.s.gel d50-120a,粒径(d50):50μm、球状、50g)和正己烷/乙醇=96/4制备的浆料填充在柱中,将参考例2得到的他氟前列素粗产物(1g)溶解于正己烷/乙酸乙酯=1/1,填充至柱上,用正己烷/乙醇=96/4洗脱。将分级的各组分利用hplc进行分析,收集包含他氟前列素的组分。作为该包含他氟前列素的组分,至少收集他氟前列素的面积百分比为98%以上(减去溶剂峰而算出)的组分。
[0107]
(工序2)在35℃~40℃下对收集的包含他氟前列素的组分进行减压浓缩。
[0108]
(工序3)将残渣溶解于正己烷/乙酸乙酯=3/2,用膜滤器(孔径:0.2μm)过滤,用正
己烷/乙酸乙酯=3/2清洗。
[0109]
(工序4)在35℃~40℃、最终极限真空度为1torr以下的减压条件下,对滤液的溶剂进行一昼夜的蒸馏去除,由此得到他氟前列素(无色~淡黄色粘性液体、收率:82%、hplc纯度:99.5%、α链反式异构体含量:0.25%)。
[0110]
用gc分析得到的他氟前列素的残留溶剂浓度,结果正己烷为0ppm,乙酸乙酯为0ppm,及乙醇为0ppm。
[0111]1h nmr(cdcl3)δ1.22(d,j=6.2hz,3h),1.22(d,j=6.2hz,3h),1.58-1.63(m,1h),1.63-1.69(m,2h),1.84(d,j=14.7hz,1h),2.02-2.08(m,1h),2.10-2.16(m,3h),2.25(t,j=7.3hz,1h),2.26(t,j=7.1hz,1h),2.30-2.35(m,1h),2.46-2.49(m,2h),2.61-2.63(m,1h),4.02-4.03(m,1h),4.18-4.21(m,3h),5.00(heptet,j=6.2hz,1h),5.35-5.42(m,2h),5.80(dt,j=15.8,11.2hz,1h),6.10(dd,j=15.8,8.8hz,1h),6.91(d,j=8.8hz,2h),7.00(t,j=7.3hz,1h),7.30(dd,j=8.8,7.3hz,2h);
[0112]
19
f nmr(cdcl3)δ-102.8(dq,2j
ff
=255.6hz),-103.6(dq,2j
ff
=255.6hz).
[0113]
《hplc(反相)分析条件》
[0114]
柱:ymc-pack ods-am(5μm,6.0
×
150mm)
[0115]
温度:室温
[0116]
流速:1ml/分钟
[0117]
检测波长:220nm
[0118]
洗脱液:(a液)1%三乙胺-磷酸缓冲液(ph6.3),
[0119]
(b液)乙腈
[0120]
梯度条件:a/b=50/50(0~45分钟)、a/b=25/75(45~70分钟)
[0121]
《gc分析条件》
[0122]
柱:g-column g300(1.2mm i.d.,40m)
[0123]
柱温度:50℃
[0124]
检测:氢火焰离子化检测器
[0125]
载气:氦
[0126]
注入器温度:160℃
[0127]
检测器温度:160℃
[0128]
实施例2~6
[0129]
为了研究硅胶柱色谱法中使用的二氧化硅的种类对他氟前列素的纯度及收率造成的影响,与实施例1同样地进行以下实验。
[0130]
使用他氟前列素粗产物(1g;hplc纯度:95.5%)及硅胶(50g),将进行硅胶柱色谱法的结果示于下表1。反相hplc分析以与前述相同条件进行。
[0131]
[表1]
[0132][0133]
比较例1
[0134]
使用他氟前列素粗产物(1g;hplc纯度:95.5%)、实施例2中使用的硅胶(50g)及洗脱液,进行硅胶柱色谱法。除利用tlc而不是hplc对分级的各组分进行分析以外,与实施例1同样,用目视收集仅包含他氟前列素的组分,结果得到的他氟前列素的hplc纯度为97.3%,未达到作为医药品的纯化品所要求的品质水准(限度值:98%)。
[0135]
使用的硅胶的粒径(d50)为65μm以下时,虽然形状不同,但实施例1~6中均得到具有超过98%的纯度的他氟前列素。其中,发现使用球状的硅胶时,可以以高收率得到纯度特别高的他氟前列素。
[0136]
实施例7(按比例放大的研究)
[0137]
(工序1)
[0138]
与实施例1的工序1同样地,将由硅胶(agc-sitech co.,ltd制、m.s.gel d50-120a,粒径(d50):50μm、球状、6.0kg)和正己烷/乙醇=96/4制备的浆料填充在柱中,将参考例2得到的他氟前列素粗产物(120g)溶解于正己烷/乙酸乙酯=1/1,填充至柱上,用正己烷/乙醇=96/4洗脱。将分级的各组分利用hplc进行分析,收集包含他氟前列素的组分。作为该包含他氟前列素的组分,至少收集他氟前列素的面积百分比为98%以上(减去溶剂峰而算出)的组分。
[0139]
(工序2~4)
[0140]
在29℃~35℃下对工序1收集的包含他氟前列素的组分进行减压浓缩(工序2)。
[0141]
将残渣溶解于正己烷/乙酸乙酯=3/2,用膜滤器(孔径:0.2μm)过滤,用正己烷/乙酸乙酯=3/2清洗(工序3)。
[0142]
在32℃~36℃、最终极限真空度为0.30torr的减压条件下,对滤液的溶剂进行26小时蒸馏去除(工序4),由此得到他氟前列素(无色~淡黄色粘性液体、收率:86%、hplc纯度:99.7%、α链反式异构体含量:0.24%、微生物含量:10cfu/0.1g以下)。
[0143]
用gc分析得到的他氟前列素的残留溶剂浓度,结果正己烷为0ppm,乙酸乙酯为0ppm,及乙醇为0ppm。
[0144]
实施例8
[0145]
将与实施例7的工序1同样地进行纯化并收集的包含他氟前列素的组分在与实施
例7的工序2同样的条件下进行减压浓缩。
[0146]
将得到的残渣溶解于乙酸乙酯,用膜滤器(孔径:0.2μm)过滤,用乙酸乙酯清洗(工序3)。
[0147]
在23℃~37℃、最终极限真空度为0.26torr的减压条件下,对滤液的溶剂进行27小时蒸馏去除(工序4),由此得到他氟前列素(无色~淡黄色粘性液体、收率:85%、hplc纯度:99.7%、α链反式异构体含量:0.27%、微生物含量:10cfu/0.1g以下)。
[0148]
用gc分析得到的他氟前列素的残留溶剂浓度,结果正己烷为0ppm,乙酸乙酯为0ppm,及乙醇为0ppm。
[0149]
实施例9
[0150]
将与实施例7的工序1同样地进行纯化并收集的包含他氟前列素的组分在与实施例7的工序2同样的条件下进行减压浓缩,将残渣溶解于正己烷/乙酸乙酯=3/2,用膜滤器(孔径:0.2μm)过滤,用正己烷/乙酸乙酯=3/2清洗(工序3)。
[0151]
在20℃~36℃、最终极限真空度为2.6torr的减压条件下,对滤液的溶剂进行3小时蒸馏去除(工序4),由此得到他氟前列素(无色~淡黄色粘性液体、收率:75%、hplc纯度:99.4%、α链反式异构体含量:0.30%、微生物含量:10cfu/0.1g以下)。
[0152]
用gc分析得到的他氟前列素的残留溶剂浓度,结果正己烷为36ppm,乙酸乙酯为4803ppm,及乙醇为66ppm。
[0153]
实施例10
[0154]
将与实施例7的工序1同样地进行纯化并收集的包含他氟前列素的组分在与实施例7的工序2同样的条件下进行减压浓缩,将残渣溶解于正己烷/乙酸乙酯=3/2,用膜滤器(孔径:0.2μm)过滤,用正己烷/乙酸乙酯=3/2清洗(工序3)。
[0155]
在34℃~37℃、最终极限真空度为2.1torr的减压条件下,对滤液的溶剂进行5小时蒸馏去除(工序4),由此得到他氟前列素(无色~淡黄色粘性液体、收率:78%、hplc纯度:99.5%、α链反式异构体含量:0.32%、微生物含量:10cfu/0.1g以下)。
[0156]
用gc分析得到的他氟前列素的残留溶剂浓度,结果正己烷为2ppm,乙酸乙酯为785ppm,及乙醇为0ppm。
[0157]
实施例11
[0158]
将与实施例7的工序1同样地进行纯化并收集的包含他氟前列素的组分在与实施例7的工序2同样的条件下进行减压浓缩,将残渣溶解于正己烷/乙酸乙酯=3/2,用膜滤器(孔径:0.2μm)过滤,用正己烷/乙酸乙酯=3/2清洗(工序3)。
[0159]
在32℃~36℃、最终极限真空度为0.92torr的减压条件下,对滤液的溶剂进行8小时蒸馏去除(工序4),由此得到他氟前列素(无色~淡黄色粘性液体、收率:82%、hplc纯度:99.6%、α链反式异构体含量:0.26%、微生物含量:10cfu/0.1g以下)。
[0160]
用gc分析得到的他氟前列素的残留溶剂浓度,结果正己烷为0ppm,乙酸乙酯为86ppm,及乙醇为0ppm。
[0161]
实施例12
[0162]
将与实施例7的工序1同样地进行纯化并收集的包含他氟前列素的组分在与实施例7的工序2同样的条件下进行减压浓缩,将残渣溶解于正己烷/乙酸乙酯=3/2,用膜滤器(孔径:0.2μm)过滤,用正己烷/乙酸乙酯=3/2清洗(工序3)。
[0163]
在35℃~39℃、最终极限真空度为0.09torr的减压条件下,对滤液的溶剂进行50小时蒸馏去除(工序4),由此得到他氟前列素(无色~淡黄色粘性液体、收率:80%、hplc纯度:99.5%、α链反式异构体含量:0.26%、微生物含量:10cfu/0.1g以下)。
[0164]
用gc分析得到的他氟前列素的残留溶剂浓度,结果正己烷为0ppm,乙酸乙酯为0ppm及乙醇为0ppm。
[0165]
实施例13
[0166]
将与实施例7的工序1同样地进行纯化并收集的包含他氟前列素的组分在与实施例7的工序2同样的条件下进行减压浓缩,将残渣溶解于正己烷/乙酸乙酯=3/2,用膜滤器(孔径:0.2μm)过滤,用正己烷/乙酸乙酯=3/2清洗(工序3)。
[0167]
在36℃~45℃、最终极限真空度为0.24torr的减压条件下,对滤液的溶剂进行60小时蒸馏去除(工序4),由此得到他氟前列素(无色~淡黄色粘性液体、收率:79%、hplc纯度:99.5%、α链反式异构体含量:0.26%、微生物含量:10cfu/0.1g以下)。
[0168]
用gc分析得到的他氟前列素的残留溶剂浓度,结果正己烷为0ppm,乙酸乙酯为0ppm,及乙醇为0ppm。
[0169]
将上述实施例7~13中的溶剂蒸馏去除的条件、他氟前列素的收率、纯度及α链反式异构体含量、及残留溶剂浓度的结果示于下表2。
[0170]
[表2]
[0171][0172]
根据表2可知,实施例7~13均以良好的纯度和高收率得到他氟前列素,并且可以将残留有机溶剂浓度抑制在医药品的残留溶剂准则的浓度限度值以下,本发明的纯化方法是可以承受按比例放大的通用性高的纯化方法。另一方面确认了:以最终极限真空度为8torr进行减压浓缩的比较例中,残留有机溶剂浓度超过医药品的残留溶剂准则的浓度限度值。
[0173]
试验例
[0174]
对他氟前列素的热稳定性的研究
[0175]
在玻璃容器中分别量取约120mg实施例1得到的他氟前列素,在40℃的恒温槽中保管,利用反相hplc分析进行定量,研究他氟前列素含量的基于时间经过的变化。同样地,在玻璃容器中分别量取约20mg他氟前列素,在60℃或80℃的恒温槽中保管,研究他氟前列素
含量的基于时间经过的变化。
[0176]
《hplc(反相)分析条件》
[0177]
柱:ymc-pack proc18 as-303(5μm,4.6
×
250mm)
[0178]
温度:50℃
[0179]
流速:1ml/分钟
[0180]
检测波长:220nm
[0181]
洗脱液:(a液)10mmol/l磷酸(钠)缓冲液(ph6.9),
[0182]
(b液)乙腈
[0183]
梯度条件:a/b=50/50(0~45分钟)、a/b=25/75(45~70分钟)
[0184]
将研究各温度下的他氟前列素含量的经时变化的结果示于下表3~5。
[0185]
[表3]
[0186]
40℃下的他氟前列素的稳定性
[0187]
时间(月)036他氟前列素含量(%)101.399.899.4
[0188]
[表4]
[0189]
60℃下的他氟前列素的稳定性
[0190]
时间(天)03714他氟前列素含量(%)99.098.998.595.4
[0191]
[表5]
[0192]
80℃下的他氟前列素的稳定性
[0193]
时间(天)0137他氟前列素含量(%)99.599.194.486.7
[0194]
根据表3~5的结果可确认:他氟前列素在60℃以上的温度下,即使是数天~2周左右的保存期间,也会随着时间经过而缓缓分解,特别是在80℃下显著分解,但发现在40℃下即使经过6个月,也稳定地存在。
[0195]
根据以上的结果可知:本发明的纯化方法中,在55℃以下(特别优选为45℃以下)的温度下进行减压浓缩、溶剂的蒸馏去除,由此可以抑制源自他氟前列素的分解的杂质(类似物质)的混入。
[0196]
产业上的可利用性
[0197]
通过本发明的纯化方法,在他氟前列素的制造中最终工序中,利用硅胶柱色谱法对他氟前列素的粗产物进行分离纯化时,利用hplc分析收集包含他氟前列素的组分,由此可以将杂质的混入抑制为最小限度。另外,在低温且高真空度的减压条件下,花费时间将溶剂蒸馏去除,由此可以将残留有机溶剂浓度抑制在医药品的残留溶剂准则的浓度限度值以下,并且可以抑制在高温下不稳定的他氟前列素的分解。进而,在中途组入过滤器过滤工序,由此可以去除硅胶的细粉、空气中的浮游颗粒、及细菌,因此具有在溶剂的蒸馏去除后能够简便且高效地提供可以直接作为医药品的原料药使用的高纯度的他氟前列素的优点。另外,本发明的纯化方法可以广泛应用于以公知的方法制造的他氟前列素的粗产物,且是也可以承受按比例放大的通用性高的方法。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。